

Abstract
Biomaterials scientists strive to develop polymeric materials with distinct chemical make-up, complex molecular architectures, robust mechanical properties and defined biological functions by drawing inspirations from biological systems. Salient features of biological designs include (1) repetitive presentation of basic motifs; and (2) efficient integration of diverse building blocks. Thus, an appealing approach to biomaterials synthesis is to combine synthetic and natural building blocks in a modular fashion employing novel chemical methods. Over the past decade, orthogonal chemistries have become powerful enabling tools for the modular synthesis of advanced biomaterials. These reactions require building blocks with complementary functionalities, occur under mild conditions in the presence of biological molecules and living cells and proceed with high yield and exceptional selectivity. These chemistries have facilitated the construction of complex polymers and networks in a step-growth fashion, allowing facile modulation of materials properties by simple variations of the building blocks. In this review, we first summarize features of several types of orthogonal chemistries. We then discuss recent progress in the synthesis of step growth linear polymers, dendrimers and networks that find application in drug delivery, 3D cell culture and tissue engineering. Overall, orthogonal reactions and modulular synthesis have not only minimized the steps needed for the desired chemical transformations but also maximized the diversity and functionality of the final products. The modular nature of the design, combined with the potential synergistic effect of the hybrid system, will likely result in novel hydrogel matrices with robust structures and defined functions.
1. Introduction
Nature combined relatively simple building blocks in a modular and repetitive fashion to construct biological materials with complex organizations and diverse functions.1 Many types of cells present multiple copies of glycans in branched structures on the cell surface that contribute to the concerted interactions with the binding partners in cell signaling.2, 3 Many proteins in the natural extracellular matrix (ECM) contain repetitive motifs linked together in a modular and tandem fashion with spatial periodicity, conferring structural and biological roles and maintaining intimate interactions with cell surface receptors.1, 4–7 The ECM of different types of tissues has variable composition and compliance depending on how the modular components are combined and integrated.1 In order to foster desired cellular behaviors for tissue growth and morphogenesis, tissue specific microenvironments must be recreated in vitro. However, a complete replication and reconstitution of the natural proteins and ECMs is technically daunting and economically unrealistic. An attractive alternative is the hybridization of synthetic polymers with molecules of biological origin that are synthetically tractable and can be readily manipulated. Overall, Rational combination of judiciously selected synthetic and biological building blocks has resulted in hybrid systems with enhanced biological functions and improved materials properties. The hybrid materials can be engineered to mimic the natural proteins8 in terms of their molecular architectures, dynamic responsiveness and cell-instructive properties, with the added attributes of tunability and processibility provided by the synthetic polymer constituents.9–11
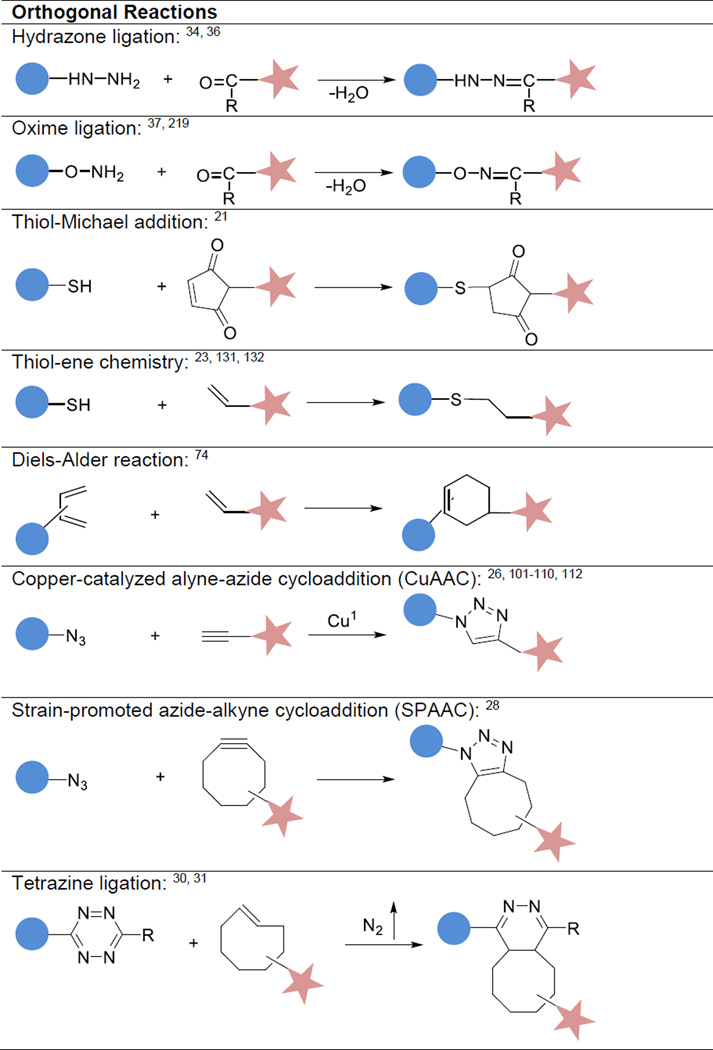
Over the past decade, significant advancement has been made in the development of customized and biomimetic materials using modular building blocks and employing bioorthogonal reactions.12 Covalent integration of modular building blocks with distinct chemical compositions and diverse functionality has resulted in advanced materials with synergistically enhanced properties.13 These developments, originally inspired by biological design principles, have been fuelled by recent advances in polymer chemistry and biomaterials science,14 and most significantly by repurposing of efficient organic chemistries.15 Bioorthogonal chemistry16 refers to chemical transformations that occur between a pair of molecules with mutually reactive functional groups without significant interference from co-existing functionalities in physiological milieu. Additional features of bioorthogonal chemistry include biocompatibility, specificity, high yield and fast reaction kinetics.17, 18 Examples include (Table 1) the hydrazone/oxime chemistry.19, 20 Michael addition,21 radical mediated thiol-ene (or thiol-yne) chemistry,22, 23 Diels–Alder reactions,24, 25 copper-catalyzed alkyne-azide cycloaddition (CuAAC, popularly known as the “click” reaction),26 strain-promoted azide-alkyne cycloaddition (SPAAC)27, 28 and inverse electron demand Diels-Alder reaction (sometimes referred to as the tetrazine ligation).29–31 The reaction rate can be elegantly tuned through subtle changes in structures and certain reactant pairs can undergo mechanistically different reactions depending on the reaction conditions.
Table 1.
Classic orthogonal chemistries used in synthesis of biomaterials.
 |
 |
These chemistries are being exploited by the biomaterials community for efficient and modular fabrication of bioactive and cell-instructive materials. In this feature article, we summarize recent progress in the development of hybrid and biomimetic materials constructed by covalent coupling of discrete modular building blocks. Materials discussed here range from linear polymers, to dendritic structures to crosslinked networks. The building blocks include synthetic polymer, peptide, protein, carbohydrate or polysaccharide. We discuss the materials properties, in the context of their interactions with mammalian cells, as a consequence of the modular coupling of diverse building blocks. We offer some examples of biomedical application of the modular constructs. Topics covered in this review are summarized in Figure 1. We limit our discussions to the de novo construction of functional biomaterials using biorthogonal chemistries, i.e. growing multiblock hybrid copolymers, multifunctional dendritic structures and biologically active hydrogel networks in a step growth fashion. Other usages of bioorthogonal chemistries, i.e. post-polymerization coupling or modification,12, 13, 32, 33 are outside the scope of our discussion. Overall, the modular approach, enabled by powerful chemistries, aids the efficient fabrication of materials with controlled complexity from the bottom up in a plug-and-play fashion. The resultant materials exhibit collective properties that exceed the simple sum of the individual constituents and are readily adaptable to specific biomedical applications.
Figure 1.

Schematic summary of biomaterials synthesized using modular approaches employing orthogonal chemistry.
2. Synthetic Strategies
Popular bioorthogonal chemistries utilized for biomaterials synthesis can be roughly divided into three categories (Table 1): (1) carbonyl-based condensation reactions; (2) addition reactions through enes/ynes and (3) cycloaddition reactions. The reaction of aldehyde or ketone with hydrazine or aminooxy derivatives results in hydrazone or oxime bonds with water being the only by-product.34 This reaction is chemoselective and can be carried out under aqueous conditions without interference from functionalities found in biomolecules and cells, thus widely exploited for bioconjugation purposes. Oxime/hydrazone condensations are relatively slow at low substrate concentrations. The reaction can be accelerated by lowering the pH or by nucleophilic catalysis using aniline through rapid transimination.35 Using carefully chosen carbonyl and hydrazine substrates, hydrazone formation can be rapid at biological pH even in the absence of a catalyst.36 Rate constants for the fastest carbonyl/hydrazine combinations are 2−20 M−1s−1. Alternatively, dialdehydes can react with O-alkylhydroxylamines at rates of 500 M−1s−1 at neutral pH values in the absence of catalysts through an unusually stable cyclic intermediate that ultimately undergoes dehydration to yield an oxime.37 Although hydrolytically more stable than the imine counterparts, hydrazone/oxime linkages are still susceptible to hydrolysis, especially under acidic conditions, reversing back to the starting hydrazide or hydroxyamines. Of note, the rate constant for the acid-catalyzed hydrolysis of the oxime was nearly 103-fold lower than that for simple hydrazones.38
Molecules containing ene functionality can participate in multiple types of orthogonal reactions, including Michael addition with heteroatomic donors and radical-based thiol-ene reaction. Although Michael addition involves the addition of a nucleophile, such as enolates, amines and thiols, to an activated electrophilic olefin,21 the reaction is considered orthogonal only when thiols are used as Michael donors. Acrylate, vinyl sulfone, or maleimide are frequently used as Michael acceptors and under these conditions, thiolate anion is the active species,39 thus the reaction rates increase with pH.17, 34 Although thiol/maleimide reaction is relatively fast, the resultant adduct is labile or exchangeable under physiologically relevant conditions.40
In the presence of thiol-containing molecules, enes can also react with thiyl radicals, frequently generated under light irradiation in the presence of photoinitiators, to form thioether linkage.22, 41, 42 The physicochemical nature of the reaction enables the direct manipulation of materials properties in both space and time through the controlled application of light. Generally speaking, the conversion rate of the thiol– ene reaction is directly related to the electron density on the ene, with electron-rich enes being consumed much more quickly than electron-poor enes. Electron-rich enes, such as (meth)acrylates undergo rapid homopolymerization under thiol-ene conditions. Highly substituted alkenes are less reactive than singly substituted alkenes. For rapid reactions involving multifunctional groups, enes must be located at terminal positions. In addition, thiols based on mercaptopropionate esters and glycolate esters react more quickly than simple alkyl thiols.22, 41, 43 Thiols can also radically add to triple bonds and a single alkyne unit can accommodate the addition of two thiol species in the single “two-step” conjugation reaction.44 Unlike radical polymerization, the thiol-norbornene-type photocoupling reaction is insensitive to O2 inhibition and can tolerate other functional groups. However, thiol can be oxidized by O2 to disulfide, albeit at a slower rate as compared to the thiol-ene reaction. Native cysteine and amine residues can complete with the desired thiols.
Originally developed for use in organic synthesis and chemical biology, the [4+2] cycloaddition between an electron-rich diene and an electron-deficient dienophile, known as the Diels-Alder (DA) reaction,24 is highly selective and proceeds at high yield without any catalyst or byproduct.45 Electron-withdrawing substituents on the alkene and the electron-donating groups on the diene are important for increasing reaction rates. The reaction can be greatly accelerated in water due to increased hydrophobic effects. Retro-DA reaction occurs at an elevated temperature, producing the original diene and dienophile.46, 47
The CuAAC reaction generally proceeds with quantitative yield and complete specificity in the presence of a wide variety of other functional groups.17, 26 The reaction rate is dependent on the concentration of CuI and can be accelerated by heat,48 tailored ligands,49 and microwave irradiation.50 In the presence of 20–500 µM CuI, the second-order rate constants were found in the range of 10–200 M−1s−1.51 Various ligands have been designed to stabilize CuI and to increase its catalytic efficiency.52–54 Work from the Ting group shows that55 copper-chelating azides undergo much faster reactions than nonchelating azides under a variety of biocompatible conditions. This kinetic enhancement increased CuAAC detection of alkyne-modified proteins and allowed for site-specific protein labeling to be performed on the surface of living cells with only 10–40 µM CuI/CuII.55, 56 The toxicity concerns of CuAAC arise from CuI-promoted generation of reactive oxygen species57 and the electrophilic properties of oxidized ascorbate (when used as reducing agent).58
Bertozzi and coworkers developed a Cu-free variant of CuAAC chemistry by promoting ring strain using cyclooctynes.27, 59 The reactivity of the cyclooctyne can be modulated by appending electron withdrawing groups at the propargylic position or by augmentation of strain energy through aryl ring or cyclopropyl ring fusion28, 60 Nitrones, nitrile oxides, diazoalkanes and syndones have been used as alternative 1,3-dipoles. Limitations of SPAAC include slow kinetics (10–100 times slower than CuAAC), hydrophobicity of the cyclooctyne component and instability of fast reacting cyclooctynes under physiological conditions.49, 60
Recently, the Fox group developed the tetrazine ligation, an inverse electron demand D-A reaction that proceeds with unusually fast rates.61 This reaction involves the cycloaddition of s-tetrazine with trans-cyclooctene (TCO) derivatives, with N2 being the only by-product.62 Such reactions are high yielding, do not require a catalyst, do not produce any toxic side products, and are more than three orders of magnitude faster than pre-existing methods for bioorthogonal ligation. Increasing the strain in TCO by cycloproyl fusion led to the discovery of the fastest bioorthogonal reaction reported to date (second-order rate constant up to 3.3 × 106 M–1s–1 in H2O at room temperature). Various TCO derivatives with reactivity towards tetrazine spanning 8 orders of magnitude have been synthesized.31, 63, 64 Other complementary dienophiles (e.g. norbornene,65 cyclopropenes,18, 66–71 cyclooctynes64, 72, 73 and terminal alkenes74) have been utilized for tetrazine ligation, but at significantly lower rates than TCO derivatives. Owing to its ability to create bonds rapidly in high yield and at low concentrations, tetrazine ligation has become an enabling tool for a host of biomedical applications,29–31, 70, 75, 76 as well as biomaterials synthesis and fabrication.30, 77–80
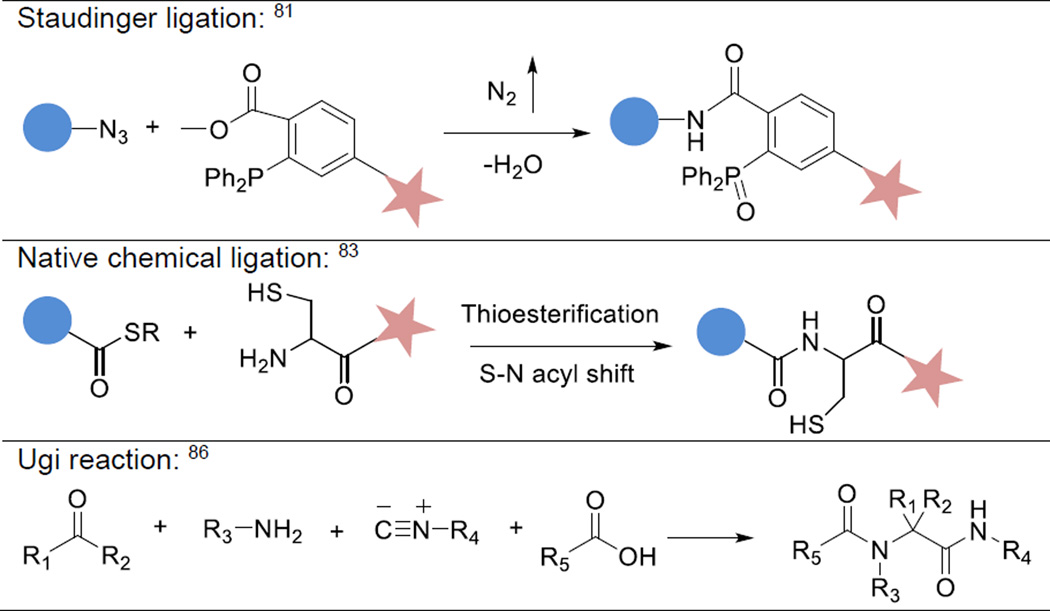
Other chemistries, such as Staudinger ligation81, 82 and native chemical ligation,83 are useful in bioconjugation, but have not been widely employed in biomaterials synthesis. Staudinger ligation refers to the amide bond formation between an azide and a phosphine derivative containing a neighboring electrophilic group. In native chemical ligation, a peptide having a C-terminal thioester reacts with an N-terminal cysteine residue in another peptide to undergo a transthioesterification reaction, resulting in the formation of the neighboring α-amine group on cysteine. A subsequent nucleophilic attack of the electron-rich nitrogen in the ester carbonyl results in an S-N shift, forming a native amide bond. Both reactions proceed at physiological pH under mild conditions without any additional additives.
Reactions described above require two components, Century-old multicomponent reactions, such as the Biginelli reaction and the Ugi reaction,84, 85 have also been explored for biomaterials synthesis where three or more building blocks are rapidly and almost quantitatively combined.86 Although these chemistries are useful in recombinatorial chemistry and polymer synthesis, the abundance of amines and carboxylic acids in the biological environment limits their utility in 3D cell cultures. Enzyme-catalyzed reactions require very specific substrates and proceed with high efficiency and minimal toxicity, thus it is orthogonal and specific.87 For example, transglutaminase (also known as Factor XIII), when activated by thrombin and Ca2+ to factor XIIIa during the blood coagulation cascade, is capable of catalyzing covalent crosslinks between the ε-amine group of lysine side chains and the γ-glutamyl side chain of glutamine residues.
3. Modular Synthesis of Hybrid Linear Copolymers
The orthogonal reactions discussed above, with a favorable equilibrium, fast reaction kinetics, lack of side reactions, high yielding under mild conditions and tolerant of various functional groups, are uniquely suited for building complex polymers with backbone diversity and complexity through step growth polymerization, which is essentially a second-order reaction of the functional groups. In the step growth polymerization, the number-average molecular weight is related to the fractional conversion of functional groups by the Carothers equation. Specifically, molecular species of varying sizes coexist throughout the entire polymerization process and high molecular weight products are not obtained unless the polymerization is driven to close to 100% conversion. Aside from the chemistry, a strict control of the experimental parameters (e.g. reaction stoichiometry, monomer stability, and the purity of reactants and solvents) is critical to achieve moderate and high molecular weight.88 While the reaction by-products must be easily removed to shift the reaction equilibrium to the product, the reaction stoichiometry must be maintained throughout the polymerization.
While abundant literature exists on the orthogonal attachment of biomolecules to synthetic polymers to produce hybrid conjugates in the form of graft copolymers or diblock/triblock copolymers,89, 90 very few groups91–93 investigate the synthesis of step growth hybrid copolymers using orthogonal reactions. This is due, in part, to the challenging conditions required for step growth polymerization. These requirements are even more stringent when the building blocks are oligomeric species of biological origin. So far, the majority of hybrid step growth copolymers reported in the literature contains short peptides. In this regard, this is an unexplored territory with great potential.
3.1. Linear copolymers synthesized via cycloaddition reactions
Although peptide/polymer hybrid copolymers have been synthesized using traditional chemistries, the peptide blocks often do not contain any functional groups or protection/deprotection strategies are required, thereby requiring lengthy synthesis and purification.94–98 Matyjaszewski and coworkers demonstrated the first synthesis of step growth polymer by CuAAC using homo difunctional (α,ω-diazido-terminated) polystyrene (PS) with propargyl ether or heterotelechelic PS (α-azide, ω-alkyne) oligomers prepared by atom transfer radical polymerization (ATRP). The polymerization was carried out at ambient temperature in DMF in the presence of CuBr.99 Subsequently, various poly(triazoles) (PTAs)100 with molecular weights up to 43 kDa have been produced by step growth polymerization of small molecule diyne/bisazide using CuAAC.101–110 CuAAC-mediated interfacial step growth polymerization under oil-in-water miniemulsion conditions has led to the production of stable monodispersed glyconanocapsules having a diameter around 200 nm. The resultant polytriazoles have a degree of polymerization ranging between 2 and 8 and the formation of a nonnegligible amount of low molar mass cyclic species in the course of the interfacial polymerization.111 Our group112 further explored CuAAC for the construction of amphiphilic multiblock copolymers containing alternating blocks of poly(acrylic acid) (PAA) and PS with up to 9 diblock repeats. These multiblock copolymers self-assembled into nanoparticles on 20–40 nm.
Our group has a longstanding interest in the synthesis and characterization of peptide/polymer hybrids.11 In our earlier work, we aimed to recapture the molecular architecture and the mechanical properties of elastin. Elastin achieves its excellent mechanical properties through an alternating molecular architecture containing highly flexible segments rich in non-polar amino acids (such as valine, glycine, and proline), and alanine-rich, lysine-containing segments which form crosslinks between adjacent molecules.4 The combination of highly crosslinked regions with loosely structured hydrophobic segments results in an elastomeric network that is enzymatically stable and mechanically robust.6 Thus, elastin mimetic hybrid polymers (EMHP, Figure 2) have been successfully synthesized via the CuAAC reaction between telechelic, azide-terminated PEG and alkyne-functionalized peptide with a sequence of (AKAAAKA)2 (AK2) that is abundant in the crosslinking region of the natural elastin. PEG was chosen to impart flexibility and solubility to the hybrid copolymer. The resulting multiblock copolymers, [PEG-AK2]n had an estimated molecular weight of 34 kDa. Covalent crosslinking of the hybrid coplymer through the lysine amines in peptide segments using hexamethylene diisocyanate gave rise to hydrogels with mechanical properties comparable to those of the natural elastin.92
Figure 2.

Elastin-mimic hybrid multiblock copolymers synthesized using CuAAC chemistry. (A): Multiblock copolymers contain (AKAAAKA)2 sequence for crosslinking and GGRGDSPG for cell adhesion. (B): The peptide sequence (VPGVG)2 is adopted from elastin that contributes to elasticity.91–93
Because the polymerization is modular and the CuAAC reaction is fully compatible with other functional groups, the composition of the multiblock copolymer can be readily adjusted to include desired peptidic motifs. To foster integrin-mediated cell adhesion, RGD-containing peptide with a sequence of X(AKAAAKA)2XGGRGDSP was used in place of X-AK2-X (X=Fmoc propargyl glycine) for EMHP synthesis. The resultant copolymer (Figure 2), [PEG-AK2(RGD)]n, when covalently crosslinked and tested in hydrated conditions, had a compressive modulus of 150 kPa. Neonatal foreskin fibroblasts plated on the RGD-containing hydrogels attached to the substrate, adopted a fibroblastic morphology and proliferated readily in serum-free culture media. Although cell attachment was also observed on RGD-lacking hybrid hydrogels, possibly through weak and non-specific cell-substrate interactions, cell proliferation over the course of culture (7 days) was not evident. Our results suggest that the integrin-binding motif incorporated in EMHP is essential not only for cell attachment but also for cell proliferation.113
Realizing the essential roles the hydrophobic, VPGVG-rich domains play in controlling the elastin elasticity, we synthesized multiblock block hybrid copolymers consisting of a synthetic polymer with addressable groups alternating with (VPGVG)2 (VG2). Thus, azide-functionalized, telechelic poly(tert-butyl acrylate) (PtBA) was synthesized by ATRP of t-butyl acrylate. Separately, peptide with a sequence X(VPGVG)2X was synthesized by SPPS with high purity. Condensation polymerization of azide-functionalized PtBA with alkyne-terminated (VPGVG)2 resulted in multiblock copolymers with up to five PtBA-VG2 repeats. Treatment of the above polymers with trifluoroacetic acid resulted in an alternating copolymer (Figure 2) of PAA and VG2 that assembled into usually-stable, flower-like micelles under acidic conditions.113
In addition to ATRP, CuAAC has also been combined with reversible addition fragmentation chain transfer polymerization (RAFT) to produce complex modular polymers. Using a bifunctional RAFT chain transfer agent containing alkyne end groups. Luo et al.114 synthesized telechelic poly(N-(2-hydroxypropyl) methacrylamide) (HPMA) with Mn of 10 to 40 kDa and a polydispersity (PDI) <1.1. Enzymatically degradable multiblock HPMA copolymers (Figure 3) were readily prepared by subsequent chain extension reaction with α,ω-diazido-oligopeptide (GFLG) sequence via CuAAC. Upon exposure of high molecular weight fractions of multiblock poly(HPMA) to papain or cathepsin B, the polymer was degraded into segments of molecular weight and narrow polydispersity similar to those of the initial telechelic poly(HPMA).
Figure 3.

Enzymatically degradable multiblock copolymers synthesized by CuAAC between poly(HPMA)-di-alkyne and GFLG-di-azide. Multiblock copolymers can be degraded by Cathepsin B at GFLG peptide blocks.114
CuAAC was recently applied to the synthesis of protein containing hybrid copolymers. Specifically, CuAAC between PEG-dialkyne and diazide functionalized green fluorescent protein (GFP) resulted in oligomeric products (GFP-PEG)n (Figure 4) that formed giant fibrous objects readily. The authors hypothesize that the reversible assembly of giant fibers was driven by the well-established tendency of GFP molecules to aggregate into dimers through the hydrophobic interaction between the localized surface patches of non-polar residues.115
Figure 4.

Protein/polymer hybrid multiblock copolymers synthesized by CuAAC chemistry between green fluorescence protein (GFP)–di-azide and PEO-di-alkyne.115
The Diels-Alder chemistry has been applied to the step growth polymerization between a difunctional diene and a difunctional dithioester. However, under the optimized conditions, only oligomers (Mn 9,600 Da) were obtained. Theoretical and experimental data suggest that 60% of the polymer becomes debonded at a temperature around 220 °C through retro-DA reaction.116 Tetrazine ligation was recently applied to the synthesis of hybrid multiblock copolymers using tetrazine-functionalized PEG with a dangling, integrin-binding peptide and an aliphatic bisTCO as the building blocks. The resultant multiblock hybrid copolymer has a molecular weight up to 263 kDa and provides guidance cues for cell attachment.117
3.2. Linear copolymers synthesized by other orthogonal chemistries
Linear step growth polymers have been synthesized by amine-ene Michael reactions using low molecular weight monomeric building blocks. Reactions of diamines with bisacrylamides, bismaleimides or diacrylates result in poly(amido amine)s, poly(imido amine)s and poly(amino ester)s118, respectively.42 Biodegradable poly(β-thioester)s (PBTs) have been prepared under thiol-ene Michael addition conditions between a variety of diacrylate and dithiol monomers or PS oligomers synthesized from a bifunctional trithiocarbonate RAFT agent (with or without amine catalysis).119 In the presence of UV irradiation, thiol addition across norbornene chemistry yielded multiblock copolymers of at least four or five blocks with different block chemistries.120 Step growth polymers have also been prepared using monoalkyne and dithiol compound. Model reaction shows that UV-initiated thiol-yne polymerization is completed in 2 h. Thiol-yne polymerization of different functional compounds resulted in linear polymers with number averaged molecular weights ranging from 5 to 30 kDa, except for propargylic acid and its methyl ester, where only oligomers formed.121
Miller et al.122 prepared multiblock hybrid copolymers of PEG (3400 Da) and matrix metalloproteinase (MMP)-sensitive peptide via Michael-type addition of thiol and acrylate. Acrylate functionalized macromers were prepared using slight access of PEGDA and the molecular weight of these macromers is controlled by the stoichiometry of the reaction, with a high proportion of resultant macromer species greater than 500 kDa. These macromers were combined with cell-adhesive acrylate-PEG-CGRGDS and PEGDA for the preparation of synthetic matrices that support the sprouting of endothelial cells. Because of the inaccessibility of the acrylate ends groups of the high molecular weight macromer, it is highly likely that low molecular weight species contributed to the gelation whereas the high molecular weight components contribute to the soluble fraction of the hybrid hydrogel.
Alternatively, a bifunctional RAFT chain transfer agent that contains an enzymatically degradable oligopeptide sequence was used for the synthesis of telechelic poly(HPMA) or HPMA copolymer-doxorubicin (DOX) conjugates. Post-polymerization aminolysis revealed the thiol moieties at both ends, through which chain extension was initiated with a bis-maleimide (Michael addition) to afford a linear high molecular weight multiblock HPMA copolymer conjugate. These polymers are enzymatically degradable; in addition to releasing the drug (DOX), the degradation of the polymer backbone resulted in products with molecular weights similar to the starting material and below the renal threshold. The new multiblock HPMA copolymers hold potential as new carriers of anticancer drugs.123
4. Modular Synthesis of Hybrid Hyperbranched or Dendritic Polymers
Hyperbranched polymers124 and dendrimers125 are highly branched macromolecules with a three-dimensional dentritic architecture. While hyperbranched polymers are less defined and irregularly shaped, dendrimers are monodisperse and have a globular structure. Due to their unique structures and abundant functional groups, both types of polymers have found applications in areas such as cancer therapy, biosensors for diagnostics and light harvesting scaffolds. While hyperbranched polymers can be conveniently synthesized via one-pot polymerization of appropriately designed monomers (e.g. ABn or A2 + Bn, where n is 2 or greater), dendrimers are prepared step by step via convergent and divergent approaches. Capitalizing on the unique features of bioorthognal chemistries, researchers have devised efficient synthesis of sophisticated dendrimers in a minimum number of chemical steps by maximizing the number of reactive chain ends while minimizing the number of reaction steps required in the process.126–128 Here we highlight examples on the orthogonal construction of hyperbranched polymers and dendrimers containing biological motifs.
Hybrid hyperbranched polymers or dendrimers have been synthesized for drug/gene delivery purposes. Employing a straightforward approach, Chen et al synthesized cationic hyperbranched glycoconjugated polymer via Michael-addition polymerization of gentamycin and N,N'-methylenebisacrylamide. Owing to the presence of various primary, secondary, and tertiary amines in the polymers, these hyperbranched glycoconjugated polymers showed high buffering capacity and strong DNA condensation ability, resulting in the high transfection efficiency. Additionally, the hybrid polymer was found to inhibit the growth of cancer cells and bacteria efficiently owing to the repetitive presentation of natural aminoglycosides on the polymeric backbone.129 Enzyme-sensitive, amphiphilic peptide dendrimer drug conjugate, mPEGylated dendron-GFLG-DOX (Figure 5),130 was synthesized via two-step highly efficient CuAAC reaction. The conjugate self-assembled into compact nanoparticles with a negatively charged surface. The nanoparticles with 9.62 wt% of DOX showed enzyme-sensitive property by drug release tests. The nanoparticles were found to accumulate and retain within the tumor for a long time and exhibit enhanced antitumor efficacy compared to the free DOX.
Figure 5.

Synthesis of amphiphilic mPEGylated dendron-GFLG-DOX conjugate. Nanoparticles were assembled from the DOX-containing conjugate.130
Hybrid dendrimers are frequently synthesized for the multivalent presentation of cell/bacteria binding motifs. An orthogonal coupling strategy was developed by combining thiol–ene and SN2 reactions, which was subsequently applied to the accelerated synthesis of multifunctional dendrimers using carbohydrate building blocks. The β-D-galactopyranoside-coated dendrimer exhibited nanomolar binding affinity with the bacterial lectin.131 Grandjean et al. described successive hydrazone/thioether orthogonal ligations of a dendrimeric lysine core with antigenic peptides and mannoside derivatives. This method provides a flexible one-pot synthesis for artificial conjugates of clustered carbohydrate receptor ligands bearing antigens.23, 132:
Alkene and alkyne functional polyester-based dendrimers of generation 1 to 4 have been prepared and reacted under free-radical conditions with 2-acetamido-2-deoxy-1-thio--D-glucose (GlcNAc-SH). As the alkene-dendrimers underwent the addition of one thiyl radical per ene group whereas each yne group of alkyne-dendrimers was saturated by two thiyl radicals, a collection of glycodendrimers with glycan density ranging from six to ninety-six GlcNAc per dendrimer were obtained The glycodendrimers were found to inhibit wheat germ agglutinin at nanomolar concentrations.23 Using thiol–ene or thiol–yne, esterification, and azide–alkyne chemistry, Sharma et al. generated dendrimers having chemically heterogeneous layers (Figure 6). The applicability of this novel approach towards the construction of biologically active glycodendrimers having dense surface sugar residues within low dendrimer generations was fully demonstrated using Erythrina cristagalli, a leguminous lectin known to bind natural killer cells through its galactoside recognition ability. The dendrimer's surface was decorated with an azido derivative of N-acetyllactosamine using click chemistry which led to new glycodendrimers having high affinities as compared to the corresponding monovalent analog.133
Figure 6.

Molecular structures of lactose-decorated glycodendrimers synthesized via a combination of thiol–ene or thiol–yne, esterification, and azide–alkyne chemistries.133
5. Modular Synthesis of Hybrid Hydrogel Networks
Orthogonal coupling of multifunctional building blocks with complementary reactivity in a step-wise fashion beyond the gel point gives rise to crosslinked networks with tissue-like properties. Compared to networks synthesized by convention chemistries, bioorthognally constructed hydrogels are theoretically more homogeneous, exhibit less interference from other functionalities and are more biocompatible to cells and biomacromolecules. Because the reaction rate can be readily adjusted, gelation kinetics and consequently network properties can be easily manipulated. Using biocompatible polymers and bioactive peptides/proteins as the modular building blocks and employing bioorthogonal reactions, various groups are working on the development of cell-instructive synthetic ECM to interrogate complex processes of tissue formation and regeneration.134
Covalent hydrogel networks are not necessarily static.135 Over time, the soluble polymers entrapped in the network can slowly diffuse out. If chemically or enzymatically degradable linkages are present in the network, hydrogels will erode and become softer as a result of matrix degradation.136–138 Certain chemistries are reversible; thus hydrogels prepared with these chemistries can be dynamic or erodible.139 If responsive signals or motifs are incorporated, hydrogel properties can be altered in response to a specific signal input.134, 140–144 Bioorthogonal reactions are uniquely suitable for time-dependent modulation of hydrogel properties.145 For example, hydrogels could become stiffened or softened over time, or could present or erase a biological signal post-gelation in the presence of cells. A cytocompatible covalent adaptable hydrogel capable of mimicking the modulus and stress relaxation properties of many complex biological tissues might be conducive to cell growth.
In this section, we highlight the synthesis of hydrogels using modular building blocks and employing step growth orthogonal coupling. Owing to its cytocompatible and bio-inert nature, PEG is widely employed for the preparation of hydrogel networks, thereby serving as a blank slate for the incorporation of bioactive signals. Because PEG does not contain abundant functional groups along the polymer backbone, frequently 4 and 8-armed PEG are used. To overcome these limitations, anionic copolymerization of ethylene oxide with glycidyl ether derivatives afforded PEG based copolymers with diverse backbone functionality.146–148 Alternatively, hyperbranched poly(glycerol) (hPG) was used in place of PEG.149 Hyaluronic acid (HA) is a linear polysaccharide with disaccharide repeats of D-glucuronic acid and N-acetyl-D-glucosamine. Unlike PEG, HA is abundantly expressed in the natural ECM, interacting with various proteins or proteoglycans to organize the ECM, to activate various signaling pathways, to maintain tissue homeostasis and to facilitate tumor metastasis.150, 151 Biomimetic synthetic extracellular matrices have been produced using HA derivatives carrying orthogonal functional groups in combination with synthetic polymers or peptides.152, 153 Finally, a new class of de novo saccharide-peptide copolymers has been used in the synthesis of hybrid hydrogels.154–159
5.1. Hydrogels synthesized by oxime/hydrazone chemistry
We have synthesized HA-based hydrogels160 employing hydrazone ligation using hydrazide-modified HA (HA-ADH) and an HA derivative carrying aldehyde groups (HA-ALD). The orthogonal nature of the hydrazone chemistry, combined with the rapid gelation kinetics, permits in situ cell encapsulation and subsequent 3D culture for the creation of physiologically relevant prostate cancer models.161–163 The hydrazone ligation permits facile incorporation of therapeutic molecules for local release purposes.160 Structural proteins can also be integrated in the network without compromising their assembly properties and bioactivities. Vocal fold fibroblasts encapsulated in the composite matrix adopted a fibroblastic morphology, proliferated readily, expressed genes encoding important vocal fold ECM proteins and actively modulated the viscoelasticity of the constructs through a cell-mediated remodeling process.164 The same hydrazone chemistry, when restricted in the inverse emulsion droplets resulted in nanoporous HA microgels.165 The resultant microgels contain reactive handles that can be used for bioconjugation166 or crosslinking purposes.165, 167 Simple mixing of these functional microgels with an aqueous solution of HA-ADH, HA-ALD or PEG-dialdehyde results in a hierachically structured, elastic hydrogel within 5 minutes. This type of network (referred to as doubly crosslinked network, DXN) contains highly crosslinked HA microgels in a loosely crosslinked secondary HA network. The viscoelastic properties of the matrix can be readily modulated by varying the particle size, surface functional group, inter-particle and intra-particle crosslinking.168 When appropriately functionalized with collagen like polypeptide169 or gelatin,170 the HA DXNs facilitate integrin mediated attachment of MSCs and matrix mediated osteogenic differentiation (Figure 7). Separately, Patenaude and Hoare applied the same chemistry to the preparation of injectable HA/poly(N-isopropylacrylamide) hydrogels.171
Figure 7.


(A): Chemical structures of HA derivatives used for hydrogel synthesis by the Jia Group. (B): SEM (i) and cryoSEM (ii–iv) images of HA microgels synthesized by inverse emulsion crosslinking using HA-ADH and HA-ALD (i), HA bulk gel synthesized by HA-SH and HA-AM (ii), HA DXN by mixing aldehyde functionalized HA microgels with HA-ADH and collagen integrated HA-ADH/HA-ALD gel. (C): Confocal images of cells cultured in HA gels. Cytoskeleton and nuclei staining of C4-2B prostate cancer cells (i), LNCaP prostate cancer cells (ii), vocal fold fibroblasts (iii) and MSCs (iv) (i–iii): F-actin and nuclei were stained green and blue respectively; (iv) F-actin, vinculin and nuclei were stained red, green and blue, respectively.160, 161, 163–165, 167, 185
The reversible nature of the hydrazone chemistry, combined with the tunability of the reaction kinetics, has led to the discovery of novel hydrogel properties. Hydrolysis of the hydrazone linkage recreates the respective hydrazide and aldehyde groups. If respective partners are located in close proximity, local network integrity can be restored via the reformation of new hydrazone bonds.172 Hydrazone-based hydrogel networks are reported to be dynamically adaptable and self-healing. McKinnon et al.173 described PEG hydrogels formed through the ligation of an aliphatic hydrazine-terminated multiarm PEG macromer with aldehyde-terminated multi-arm PEG macromer. Rapid gelation occurred under physiological conditions via the formation of bis-aliphatic hydrazone bond without the need for analine catalysis and not surprisingly, the hydrolysis rate is pH-dependent. The modular nature of hydrogel construction and the large number of easily tuned variables provide access to gels with a wide range of modulus and stress relaxation characteristics. These covalently adaptable hydrogels, when incorporated with RGD, also through hydrazone bonds, allow for the development of physiologically relevant morphologies for mouse myoblasts, whereas static, non-adaptable gels prevent cytoskeletal rearrangement and extension. Taken together, these studies show that hydrazone linked hydrogels offer unique advantages in terms of dynamic tunability and should serve as a valuable complement to existing hydrogel technologies.174
5.2. Hydrogels synthesized by Michael addition or thiol-ene chemistry
Michael-type addition reaction is a versatile orthogonal reaction for the formation of hydrogel networks. Frequently, acrylate, maleimide or acrylamide-based double bonds are used. Cross-reaction with native proteins is not an issue since most cysteine thiols in proteins exist in the oxidized S-S form and lysine amines react with these enes at a much lower rate. Using thiolated HA (HA-SH), prepared using a dihydrazide reagent containing an internal disulfide bond175–177 synthetic ECM has been developed using PEG diacrylate (PEGDA) as the Michael donor.152, 178 When thiol is used in a stoichiometric excess to acrylate, the fast Michael addition reaction contributes to the initial network formation and the slow disulfide bond formation gradually increases gel stiffness.179, 180 Depending on the molecular weight of HA and PEG, percent functional group incorporation in HA, concentrations of HA and PEG and thiol/acrylate ratio, hydrogels with elastic modulus varying from 11 Pa to 3500 Pa have been prepared.181 Co-crosslinking thiolated HA with other thiolated biomacromolecules (heparin or gelatin) creates a more complex network containing immobilized biological cues for growth factor sequestration or for integrin engagement.182 This type of hydrogel system has been commercialized and widely used in cell therapy, growth factor delivery and the regeneration of healthy bladder, bone, cartilage, sinus, spinal cord and vocal fold tissues and the creation of disease models.152
Although the incorporation of thiolated gelatin in HA gels facilitates the attachment and spreading of fibroblasts on the hydrogel surface, cells remain rounded inside the gel in 3D.183 In order for cells of mesenchymal origin to spread in 3D hydrogel networks, both matrix metalloproteinase (MMP) degradable crosslinker and RGD signals must be incorporated and cell spreading is more significant in gels with higher RGD density.184 These observations reconfirm the early observations from the Hubbell group134, 140 on vinyl sulfone-terminated multiarm PEG macromers and a cysteine-terminated MMP substrate as the crosslinkers that both RGD and MMP signals for 3D attachment and migration of cells of mesenchymal origin.
The hydrolysis of the ester linkages at the crosslinking points in the above HA/PEG gels may compromise the overall gel stability and complicate the interpretation of cellular enzymatic processes. To improve the hydrolytic stability of HA gels, acrylate groups were introduced to HA via an amide linkage (HA-AM). Thus, HA gels are formed within 30 minutes of mixing of HA-SH and HA-AM and slow gel stiffening occurs overnight, reaching a plateau modulus of 234±30 Pa. The orthogonal reaction permits direct encapsulation of prostate cancer cells as well as HA microgels containing sequestered growth factors.185 Prostate cancer cells entrapped in HA matrices formed distinct multicellular aggregates which grew and merged to form spherical aggregates, expressing E-cadherin, and showing cortical organization of F-actin. Compared to 2D-cultured cells, the engineered tumoroids increased the expression of pro-angiogenic factors and multidrug resistant proteins. The engineered models (Figure 7) were utilized to assess the treatment efficacy of nanoparticle-based drug delivery systems.163, 185, 186
Guan and coworkers synthesized copolymers containing peptide-saccharide along the backbone.157 Chemically crosslinked hydrogels (Figure 8) were prepared via Michael addition by mixing hybrid polymers with cysteine or vinyl sulfone groups. Intriguingly, a single amino acid mutation (valine (V), cysteine (C), tyrosine (Y)) in the polymer backbone has profound effects on the gel properties and the behaviors of encapsulated chondrocytes.154 The synthetic saccharide/peptide hydrogels have also proven useful for islet transplantation.156
Figure 8.

(A): De novo designed saccharide-peptide hybrid copolymer, when crosslinked, creates a synthetic matrix suitable for cell culture purposes. (B): Cysteine or vinyl sulfone containing saccharide-peptide copolymers were mixed to produce hydrogels via Michael Addition. Hydrogel properties can be tuned by varying the hydrophobicity/hydrophilicity ratio.154, 157
Micrometer-sized hydrogel particles containing living cells were fabricated with an exquisite control through the use of droplet-based microfluidics and PEG and hyperbranched polyglycerol (hPG). Gelation was achieved via the nucleophilic Michael addition of dithiolated PEG macro-cross-linkers to acrylated hPG building blocks. Microgel properties were varied through the use of PEG linkers with different molecular weights along with different concentrations of macromonomers. Fibroblasts and lymphoblasts were successfully encapsulated in the microgel with high viability.187
Thiol-ene photochemistry has been extensively explored for hydrogel synthesis because this photoinitiated polymerization occurs at neutral pH and can be controlled both spatially and temporally.188–190 Proteins and cells have been entrapped in these gels for the controlled release of protein therapeutics and 3D culture purposes. For example, the bioactivity of lysozyme was maintained above 90% following the exposure to thiol-ene photopolymerization conditions.191 Bovine chondrocytes were encapsulated in a similar gel with high viability and synthesized ECM resembling that of a hylain cartilage. Contrarily, when encapsulated in a radically crosslinked gel, lysozyme exhibits a compromised activity (50%) and chondrocytes adopt a hypertrophic phenotype, thus pointing to the advantages of step growth mechanism for hydrogen preparation.192
Cell-adhesive, MMP-degradable PEG hydrogels were prepared using thiol-ene photopolymerization. MSCs entrapped in the resultant matrices undergo classical trilineage differentiation in the presence of differentiation media. Pancreatic ductal epithelial cells (transformed cell line) formed ductal cyst-like structures in 3D and cellular aggregation was dependent on the immobilized laminin-derived peptide signals as well as MMP-mediated matrix degradation.193, 194 Similarly, norbornene-functionalized HA (HA-Norb) was combined with di-thiols to create non-toxic hydrogels with a wide range of mechanical properties. By limiting the initial extent of crosslinking, HA-Norb gels were synthesized with remaining pendent norbornene groups that could be reacted with thiol-containing molecules in the presence of light and an initiator, including with spatial control. Secondary reactions with a di-thiol crosslinker changed mechanical properties, whereas reaction with mono-thiol peptides had no influence on the gel elastic modulus. This orthogonal chemistry was used sequentially to pattern multiple peptides into a single hydrogel, demonstrating the robustness of this system for the formation of complex hydrogels195
Application of thiol-ene chemistry in a confined space or at an interface has resulted in the production of microgels or multilayered hydrogels. For example, taking advantages of the ability of eosin-Y to generate radicals upon visible light exposure and its high diffusivity, hydrogels with multilayer structures were prepared by step growth thiol-ene photochemical reactions. The initial light exposure resulted in the formation of the core hydrogels, through which the residual eosin-Y diffuses outwards to initiate further crosslinking. The thickness of the thiol-ene gel coating could be easily controlled by adjusting visible light exposure time, eosin-Y concentration initially loaded in the core gel, or macromer concentration in the coating solution.196, 197
5.3. Hydrogels synthesized by cycloaddition reactions
Diels–Alder click reaction was applied to the synthesis of HA hydrogels by reacting furan-modified HA with bis-maleimide-PEG. Biomolecules were photopatterned into the hydrogel by two-photon laser processing, resulting in spatially defined growth factor gradients. The Young’s modulus was controlled by either changing the hydrogel concentration or the furan substitution on the HA backbone, thereby decoupling the hydrogel concentration from mechanical properties. Porosity was prepared by cryogelation, and the addition of galactose further influenced the porosity, pore size, and Young’s modulus of the cryogels. These HA-based hydrogels offer a tunable platform with a diversity of properties for directing cell function, with applications in tissue engineering and regenerative medicine.198–202
CuAAC was applied to hydrogel synthesis as early as 2006.203 The goal was to make a more perfect hydrogel network with robust mechanical properties. However, an efficient chemistry does not guarantee perfect networks. In fact, carbon black, a form of paracrystalline carbon, has to be incorporated in the gel to improve the mechanical properties. CuAAC of tetrakis(2-propynyloxymethyl)-methane (TMOP), diazide end-functionalized triblock copolymers of poly(β-caprolactone) with PEG (N3-PCL-PEG-PCL-N3) afforded amphiphilic co-networks whose properties can be tuned by varying the hydrophilic/hydrophobic ratio. Both hydrophilic and hydrophobic drugs can be encapsulated in the resultant networks and the drug release is attributable to the well-defined molecular structure and tunable hydrophobic/hydrophilic composition of the hydrogels.204
The toxicity issues associated with Cu motivated researchers to explore the utility of other orthogonal reactions for hydrogel synthesis. For example, the inverse-electron-demand D-A reaction between norbornene and tetrazine has been utilized for gelation purposes. Mixing of multifunctional PEG-tetrazine macromer with a dinorbornene peptide resulted in hydrogel formation within minutes. MSCs were encapsulated in such gels with high post-encapsulation viability. The specificity of the tetrazine norbornene reaction allows for sequential modification of the network via thiol-ene photochemistry.78 pH-Cleavable cell-laden microgels (Figure 9) were fabricated in microfluidic channels by SPAAC employing PEG dicyclooctyne and dendritic poly(glycerol) displaying azide groups through an acid-labile benzacetal linker. Cells were encapsulated in the microgel with a high viability and were subsequently selectively released by varying the pH without compromising the overall cell viability and cell spreading. The capture and release microgel platform allows cells to be studied and manipulated during the encapsulation and then be isolated and harvested by decomposition of the microgel scaffolds.205
Figure 9.

Cell encapsulation and release in microgels synthesized via SPAAC.205
The Jia/Fox groups have successfully demonstrated the application of tetrazine ligation to create hydrogel materials via a diffusion-controlled interfacial gelation (Figure 10) at the gel-liquid interface.80 Syringe delivery of tetrazine modified HA (HA-Tz, pink, viscosity molecular weight: 218 kDa) to a bath of bisTCO crosslinker (colorless, MW=1,253 Da) resulted in the instantaneous formation of a crosslinked shell (colorless), through which bisTCO diffused inwards to introduce further crosslinking. Alternatively, injection of bisTCO into a bath of HA-Tz allowed instant formation of a crosslinked shell, through which bisTCO diffused outwards to create water-filled hydrogel channels. The interfacial polymerization process permitted the creation of HA hydrogels with pre-determined spatial distribution of TCO-tagged Alexa Fluor 647 (Alexa-TCO) without external triggers or templates by timed alteration of the TCO bath composition. For example, microspheres with a multilayer structure were created by simply alternating the presence/absence of Alexa-TCO in the TCO bath during crosslinking. Similarly, microspheres with a radial gradient of the fluorescent tag (Figure 10) were readily created by continuously varying the Alexa-TCO concentration (0–0.47 µM) during the 2-h crosslinking process. This interfacial gelation is projected to find utility in the fabrication of cell-instructive matrices for the growth of in vitro tissue models.
Figure 10.

Interfacial bioorthogonal crosslinking. Gel (colorless) interface forms when HA-Tz (pink) contacts a solution of bisTCO. The interfacial kinetics allowed simultaneous introduction of tagging groups (red star in A and B). Spatio-temporal control can be achieved by simply varying the relative concentration of the tagging group as a function of time. (C, D): Confocal images of HA microspheres containing spatially tagged Alexa-TCO (red) in an onion-like layered structure (C) or in a radial gradient (D).80
5.4. Hydrogels synthesized by other orthogonal chemistries
Other orthogonal chemistries are less explored for hydrogel synthesis due to the complication in synthesis and/or slow kinetics. Staudinger ligation has been explored for the covalent stablization of ionically crosslinked alginate hydrogels using azide functionalized alginate and 1-methyl-2-diphenylphosphinoterephthalate-terminated, telechelic PEG.206 Native chemical ligation has been explored to create covalently crosslinked hydrogels using macromonomers of four-armed PEG with either thioester or N-terminal cysteine peptides.207 A similar strategy has been applied to prepare anti-inflammatory hydrogels supporting islet cell survival in the presence of diffusible pro-inflammatory cytokines.208, 209
Enzyme-catalyzed reactions, although highly specific and naturally biocompatible, are underexplored for the synthesis of bioactive hydrogels. Ehrbar et al. used activated coagulation transglutaminase factor XIIIa (FXIIIa) for gelation and site-specific coupling of cell adhesion ligands and engineered growth factor proteins to PEG-based proteolytically degradable hydrogels. Primary stromal cells can invade and proteolytically remodel these networks both in an in vitro and in vivo setting. These hybrid networks can potentially serve as alternatives for fibrin as provisional drug delivery platforms in tissue engineering.210 Using a photocaged FXIIIa substrate, Mosiewicz et al created PEG-based hydrogels with masked peptide. Subsequent laser-scanning lithography afforded highly localized biomolecule tethering. This approach for the 3D manipulation of cells within gels should open up avenues for the study and manipulation of cell signaling.211
Mosiewicz et al.212 employed phosphopantetheinyl transferase (PPTase) to catalyze covalent cross-linking of PEG-based hydrogels (Figure 11). Gels were formed within minutes under physiological conditions by mixing two aqueous precursors containing multiarm PEG macromers end-functionalized with the PPTase substrate Coenzyme A (CoA) and a genetically engineered dimer of a carrier protein. Bioactive hydrogels were produced by covalent incorporation of a CoA-functionalized cell adhesion peptide (RGDS), resulting in specific adhesion of primary fibroblasts on the hydrogel surfaces. 3D encapsulation of cells resulted in high cell viability (ca. 95%) and single cell migration over long distances within RGDS-modified gels. In general, enzyme-catalyzed gelation reactions usually result in heterogeneous hydrogels with low mechanical strength due to the low conversion imposed by the inability of enzymes to diffuse readily during the gelation process.213
Figure 11.

(A) General mechanism of phosphopantetheinyl transferase (PPTase)-catalyzed transfer of phosphopantethein prostetic group of CoA to the serine residue of ACP; (B) Hydrogel formed between branched PEG-CoA and fluorescent ACP fusion protein.212
5.5 Manipulation of hydrogel properties using multiple chemistries
Bioorthogonal reactions are frequently combined to generate complex hydrogels with dynamic properties. In some cases, certain functional groups participate in multiple types of reactions, some of which may not necessarily be orthogonal in nature. In other cases, two independent chemistries are used to sculpt the hydrogels. Burdick and coworkers devised a step-wise approach to fabricate hydrogels that stiffen over time in the presence of cells. The initial network was established by Michael addition between thiol and methacrylate or maleimide and the additional crosslinking was achieved by radical chain polymerization. Time dependent gel stiffening was found to selectively differentiate MSCs,214 which is directed by the generation of degradation-mediated cellular traction independently of cell morphology or matrix mechanics. Moreover, switching the permissive hydrogel to a restrictive state through delayed secondary crosslinking reduced further hydrogel degradation, suppressed traction, and caused a switch from osteogenesis to adipogenesis in the absence of changes to the extended cellular morphology.215
Photoinitiated thiol-ene chemistry is frequently combined with other orthogonal chemistries for light-directed spatial patterning purposes in the presence or absence of cells. For example, thiol-ene photochemistry was combined with aniline-catalyzed oxime ligation or CuAAC for 3D patterning of peptides in PEG gels post gelation.216 Using copper-free click chemistry (Figure 12), DeForest et al. directly encapsulated cells in PEG-based hydrogels. Subsequently, thiol–ene photocoupling chemistry is introduced that enables patterning of biological functionalities within the gel in real time and with micrometer-scale resolution. This material system enables researchers to tailor independently the biophysical and biochemical properties of the cell culture microenvironments in situ.145 This synthetic approach uniquely allows for the direct fabrication of biologically functionalized gels with ideal structures that can be photopatterned, and all in the presence of cells.188
Figure 12.

Two independent orthogonal chemistries were applied to the preparation of the PEG/peptide network. The initial gelation was induced by SPAAC and subsequent spatial patterning was achieved using thiol-ene photocoupling chemistry.145
6. Conclusion and Perspective
In this article, we discussed modular approaches for the synthesis of hybrid polymeric biomaterials using diverse building blocks and employing orthogonal reactions. These highly efficient and selective reactions have enabled facile control over the composition, structure and properties of polymeric biomaterials, considerably expanding the design options. Covalent integration of bioactive motifs in linear, hyperbranched or dendritic polymers has given rise to bioconjugates with unique biological functions. Bioorthogonal chemistry is particularly attractive for the fabrication of hydrogels in the presence of living cells to produce biologically relevant 3D constructs with desired mechanical properties and spatial presentation of biological signals. Proteins can also be entrapped in the matrix with high bioactivity and be released in a controlled manner. Light-triggerable chemistries and diffusion controlled ligation mechanism are particularly attractive for spatial patterning purposes.
Although orthogonal chemistries have become an enabling technology for the synthesis of hybrid biomaterials, it is important to note that a “good chemistry” does not necessarily lead to a “good material”. Polymers and networks synthesized by modular coupling inevitably follow the characteristics of step growth polymerization in that the molecular weight of the polymers or the average molecular weight between crosslinks exhibit high polydispersity. Orthogonal reactions when inappropriately applied defeat the purpose of the modular design. For example, ultrafast tetrazine/TCO ligation,63 when applied to solution polymerization or crosslinking, results in oligomeric cyclic products or highly heterogeneous, ill-defined hydrogels.80 The application of this chemistry at the interface enabled the diffusion controlled process that opens up a wide range of biomaterials applications. Furthermore, hydrogel properties are not solely dependent on the crosslinking chemistry. Phase separation and network defects217 may negatively affect network properties. Multiarm PEGs are frequently used for hydrogel synthesis. The high functionality facilitates rapid gelation but inevitably leads to more network defects. An attractive approach is the combination of orthogonal chemistry with orthogonal supramolecular assembly.218
Overall, orthogonal chemistries have provided researchers with the unprecedented ability to link two entities in high yield without interference from other functional groups. These chemistries have become powerful and enabling tools for polymer and materials synthesis. As organic chemists continue to expand the chemistry toolbox, further advancement in biomaterials synthesis and fabrication is anticipated. With improved biological understanding of biological design principles, more sophisticated and biologically relevant materials with controlled spatial and temporal properties will lead to further advances in the biomedical field.
Acknowledgements
Work in the authors’ laboratories has been funded by grants from the National Science Foundation (DMR: 0643226 and 1206310), National Institutes of Health (R01 DC008965; R01 DE022969, P20 GM103541-10). We acknowledge the Delaware COBRE program (NIGMS: P30 GM110758-01) for instrumentation support. KTD thanks Robert W. Gore for the Gore Fellowship. The authors wish to acknowledge Genzyme for generously providing HA.
References
- 1.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. New York: Garland Science; 2002. [Google Scholar]
- 2.Hounsell EF, Davies MJ, Renouf DV. Glycoconjugate J. 1996;13:19–26. doi: 10.1007/BF01049675. [DOI] [PubMed] [Google Scholar]
- 3.Kiessling LL, Gestwicki JE, Strong LE. Curr. Opin. Chem. Biol. 2000;4:696–703. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 4.Vrhovski B, Weiss AS. Eur. J. Biochem. 1998;258:1–18. doi: 10.1046/j.1432-1327.1998.2580001.x. [DOI] [PubMed] [Google Scholar]
- 5.Friedl P, Brocker EB. Cell. Mol. Life Sci. 2000;57:41–64. doi: 10.1007/s000180050498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gosline J, Lillie M, Carrington E, Guerette P, Ortlepp C, Savage K. Phil. Trans. R. Soc. Lond. B. 2002;357:121–132. doi: 10.1098/rstb.2001.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosso F, Giordano A, Barbarisi M, Barbarisi A. J. Cell. Physiol. 2004;199:174–180. doi: 10.1002/jcp.10471. [DOI] [PubMed] [Google Scholar]
- 8.Barker TH. Biomaterials. 2011;32:4211–4214. doi: 10.1016/j.biomaterials.2011.02.027. [DOI] [PubMed] [Google Scholar]
- 9.Vandermeulen GWM, Klok HA. Macromol. Biosci. 2004;4:383–398. doi: 10.1002/mabi.200300079. [DOI] [PubMed] [Google Scholar]
- 10.Lutolf MP, Hubbell JA. Nat. Biotechnol. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 11.Jia X, Kiick KL. Macromol Biosci. 2009;9:140–156. doi: 10.1002/mabi.200800284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Azagarsamy MA, Anseth KS. ACS Macro Lett. 2013;2:5–9. doi: 10.1021/mz300585q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iha RK, Wooley KL, Nystrom AM, Burke DJ, Kade MJ, Hawker CJ. Chem. Rev. 2009;109:5620–5686. doi: 10.1021/cr900138t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hawker CJ, Wooley KL. Science. 2005;309:1200–1205. doi: 10.1126/science.1109778. [DOI] [PubMed] [Google Scholar]
- 15.Ramil CP, Lin Q. Chem. Commun. 2013;49:11007–11022. doi: 10.1039/c3cc44272a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.King M, Wagner A. Bioconjugate Chem. 2014;25:825–839. doi: 10.1021/bc500028d. [DOI] [PubMed] [Google Scholar]
- 17.McKay CS, Finn MG. Chem. Biol. 2014;21:1075–1101. doi: 10.1016/j.chembiol.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shih HW, Kamber DN, Prescher JA. Curr. Opin. Chem. Biol. 2014;21:103–111. doi: 10.1016/j.cbpa.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 19.Dirksen A, Dawson PE. Bioconjugate Chem. 2008;19:2543–2548. doi: 10.1021/bc800310p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ulrich S, Boturyn D, Marra A, Renaudet O, Dumy P. Chem.-Eur. J. 2014;20:34–41. doi: 10.1002/chem.201302426. [DOI] [PubMed] [Google Scholar]
- 21.Mather BD, Viswanathan K, Miller KM, Long TE. Prog. Polym. Sci. 2006;31:487–531. [Google Scholar]
- 22.Hoyle CE, Bowman CN. Angew. Chem.-Int. Edit. 2010;49:1540–1573. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 23.Ghirardello M, Oberg K, Staderini S, Renaudet O, Berthet N, Dumy P, Hed Y, Marra A, Malkoch M, Dondoni A. J. Polym. Sci. Pol. Chem. 2014;52:2422–2433. [Google Scholar]
- 24.Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G. Angew. Chem.-Int. Edit. 2002;41:1668–1698. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 25.Sanyal A. Macromol. Chem. Phys. 2010;211:1417–1425. [Google Scholar]
- 26.Kolb HC, Finn MG, Sharpless KB. Angew. Chem.-Int. Edit. 2001;40 doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. 2004-+. [DOI] [PubMed] [Google Scholar]
- 27.Agard NJ, Prescher JA, Bertozzi CR. J. Am. Chem. Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 28.Sletten EM, Bertozzi CR. Accounts Chem. Res. 2011;44:666–676. doi: 10.1021/ar200148z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Devaraj NK, Weissleder R. Accounts Chem. Res. 2011;44:816–827. doi: 10.1021/ar200037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knall AC, Slugovc C. Chem. Soc. Rev. 2013;42:5131–5142. doi: 10.1039/c3cs60049a. [DOI] [PubMed] [Google Scholar]
- 31.Selvaraj R, Fox JM. Curr. Opin. Chem. Biol. 2013;17:753–760. doi: 10.1016/j.cbpa.2013.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agut W, Taton D, Lecommandoux S. Macromolecules. 2007;40:5653–5661. [Google Scholar]
- 33.Quadir MA, Martin M, Hammond PT. Chem. Mat. 2014;26:461–476. [Google Scholar]
- 34.Hermanson GT. Bioconjugate techniques. Amsterdam: Elsevier/Academic Press; 2008. [Google Scholar]
- 35.Dirksen A, Dirksen S, Hackeng TM, Dawson PE. J. Am. Chem. Soc. 2006;128:15602–15603. doi: 10.1021/ja067189k. [DOI] [PubMed] [Google Scholar]
- 36.Kool ET, Park DH, Crisalli P. J. Am. Chem. Soc. 2013;135:17663–17666. doi: 10.1021/ja407407h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmidt P, Zhou LN, Tishinov K, Zimmermann K, Gillingham D. Angew. Chem.-Int. Edit. 2014;53:10928–10931. doi: 10.1002/anie.201406132. [DOI] [PubMed] [Google Scholar]
- 38.Kalia J, Raines RT. Angew. Chem.-Int. Edit. 2008;47:7523–7526. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rizzi SC, Ehrbar M, Halstenberg S, Raeber GP, Schmoekel HG, Hagenmuller H, Muller R, Weber FE, Hubbell JA. Biomacromolecules. 2006;7:3019–3029. doi: 10.1021/bm060504a. [DOI] [PubMed] [Google Scholar]
- 40.Baldwin AD, Kiick KL. Bioconjugate Chem. 2011;22:1946–1953. doi: 10.1021/bc200148v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoyle CE, Lee TY, Roper T. J. Polym. Sci. Pol. Chem. 2004;42:5301–5338. [Google Scholar]
- 42.Lowe AB. Polym. Chem. 2014;5:4820–4870. [Google Scholar]
- 43.Northrop BH, Coffey RN. J. Am. Chem. Soc. 2012;134:13804–13817. doi: 10.1021/ja305441d. [DOI] [PubMed] [Google Scholar]
- 44.Stenzel MH. ACS Macro Lett. 2013;2:14–18. doi: 10.1021/mz3005814. [DOI] [PubMed] [Google Scholar]
- 45.Gandini A. Prog. Polym. Sci. 2013;38:1–29. [Google Scholar]
- 46.Chen XX, Dam MA, Ono K, Mal A, Shen HB, Nutt SR, Sheran K, Wudl F. Science. 2002;295:1698–1702. doi: 10.1126/science.1065879. [DOI] [PubMed] [Google Scholar]
- 47.Inglis AJ, Nebhani L, Altintas O, Schmidt FG, Barner-Kowollik C. Macromolecules. 2010;43:5515–5520. [Google Scholar]
- 48.Berg R, Straub BF. Beilstein J. Org. Chem. 2013;9:2715–2750. doi: 10.3762/bjoc.9.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Presolski SI, Hong V, Cho SH, Finn MG. J. Am. Chem. Soc. 2010;132:14570–14576. doi: 10.1021/ja105743g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Appukkuttan P, Mehta VP, Van der Eycken EV. Chem. Soc. Rev. 2010;39:1467–1477. doi: 10.1039/b815717k. [DOI] [PubMed] [Google Scholar]
- 51.Hong V, Presolski SI, Ma C, Finn MG. Angew. Chem.-Int. Edit. 2009;48:9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Organic Letters. 2004;6:2853–2855. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 53.del Amo DS, Wang W, Jiang H, Besanceney C, Yan AC, Levy M, Liu Y, Marlow FL, Wu P. J. Am. Chem. Soc. 2010;132:16893–16899. doi: 10.1021/ja106553e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Besanceney-Webler C, Jiang H, Zheng TQ, Feng L, del Amo DS, Wang W, Klivansky LM, Marlow FL, Liu Y, Wu P. Angew. Chem.-Int. Edit. 2011;50:8051–8056. doi: 10.1002/anie.201101817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Uttamapinant C, Tangpeerachaikul A, Grecian S, Clarke S, Singh U, Slade P, Gee KR, Ting AY. Angew. Chem.-Int. Edit. 2012;51:5852–5856. doi: 10.1002/anie.201108181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Uttamapinant C, Sanchez MI, Liu DS, Yao JZ, Ting AY. Nat. Protoc. 2013;8:1620–1634. doi: 10.1038/nprot.2013.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brewer GJ. Chem. Res. Toxicol. 2010;23:319–326. doi: 10.1021/tx900338d. [DOI] [PubMed] [Google Scholar]
- 58.Kay P, Wagner JR, Gagnon H, Day R, Klarskov K. Chem. Res. Toxicol. 2013;26:1333–1339. doi: 10.1021/tx400061e. [DOI] [PubMed] [Google Scholar]
- 59.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc Natl Acad Sci U S A. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jewett JC, Bertozzi CR. Chem. Soc. Rev. 2010;39:1272–1279. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blackman ML, Royzen M, Fox JM. J. Am. Chem. Soc. 2008;130 doi: 10.1021/ja8053805. 13518-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thalhammer F, Wallfahrer U, Sauer J. Tetrahedron Lett. 1990;31:6851–6854. [Google Scholar]
- 63.Taylor MT, Blackman ML, Dmitrenko O, Fox JM. J. Am. Chem. Soc. 2011;133:9646–9649. doi: 10.1021/ja201844c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lang K, Davis L, Wallace S, Mahesh M, Cox DJ, Blackman ML, Fox JM, Chin JW. J. Am. Chem. Soc. 2012;134:10317–10320. doi: 10.1021/ja302832g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Devaraj NK, Weissleder R, Hilderbrand SA. Bioconjugate Chem. 2008;19:2297–2299. doi: 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kamber DN, Nazarova LA, Liang Y, Lopez SA, Patterson DM, Shih HW, Houk KN, Prescher JA. J. Am. Chem. Soc. 2013;135:13680–13683. doi: 10.1021/ja407737d. [DOI] [PubMed] [Google Scholar]
- 67.Patterson DM, Jones KA, Prescher JA. Mol. Biosyst. 2014;10:1693–1697. doi: 10.1039/c4mb00092g. [DOI] [PubMed] [Google Scholar]
- 68.Patterson DM, Nazarova LA, Xie B, Kamber DN, Prescher JA. J. Am. Chem. Soc. 2012;134:18638–18643. doi: 10.1021/ja3060436. [DOI] [PubMed] [Google Scholar]
- 69.Sachdeva A, Wang KH, Elliott T, Chin JW. J. Am. Chem. Soc. 2014;136:7785–7788. doi: 10.1021/ja4129789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seckute J, Yang J, Devaraj NK. Nucleic Acids Res. 2013;41:9. doi: 10.1093/nar/gkt540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang J, Seckute J, Cole CM, Devaraj NK. Angew. Chem.-Int. Edit. 2012;51:7476–7479. doi: 10.1002/anie.201202122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen WX, Wang DZ, Dai CF, Hamelberg D, Wang BH. Chem. Commun. 2012;48:1736–1738. doi: 10.1039/c2cc16716f. [DOI] [PubMed] [Google Scholar]
- 73.Karver MR, Weissleder R, Hilderbrand SA. Angew. Chem.-Int. Edit. 2012;51:920–922. doi: 10.1002/anie.201104389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Niederwieser A, Spate AK, Nguyen LD, Jungst C, Reutter W, Wittmann V. Angew. Chem.-Int. Edit. 2013;52:4265–4268. doi: 10.1002/anie.201208991. [DOI] [PubMed] [Google Scholar]
- 75.Haun JB, Devaraj NK, Hilderbrand SA, Lee H, Weissleder R. Nat. Nanotechnol. 2010;5:660–665. doi: 10.1038/nnano.2010.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Keliher EJ, Reiner T, Turetsky A, Hilderbrand SA, Weissleder R. Chem Med Chem. 2011;6:424–427. doi: 10.1002/cmdc.201000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hansell CF, Espeel P, Stamenovic MM, Barker IA, Dove AP, Du Prez FE, O'Reilly RK. J. Am. Chem. Soc. 2011;133:13828–13831. doi: 10.1021/ja203957h. [DOI] [PubMed] [Google Scholar]
- 78.Alge DL, Azagarsamy MA, Donohue DF, Anseth KS. Biomacromolecules. 2013;14:949–953. doi: 10.1021/bm4000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cok AM, Zhou H, Johnson JA. Macromol. Symp. 2013;329:108–112. [Google Scholar]
- 80.Zhang H, Dicker KT, Xu X, Jia X, Fox JM. ACS Macro Lett. 2014;3:727–731. doi: 10.1021/mz5002993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Saxon E, Bertozzi CR. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 82.Nilsson BL, Kiessling LL, Raines RT. Organic Letters. 2000;2:1939–1941. doi: 10.1021/ol0060174. [DOI] [PubMed] [Google Scholar]
- 83.Dawson PE, Muir TW, Clarklewis I, Kent SBH. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 84.Armstrong RW, Combs AP, Tempest PA, Brown SD, Keating TA. Accounts Chem. Res. 1996;29:123–131. [Google Scholar]
- 85.Kappe CO. Molecules. 1998;3:1–9. [Google Scholar]
- 86.Yang B, Zhao Y, Wang SQ, Zhang YL, Fu CK, Wei Y, Tao L. Macromolecules. 2014;47:5607–5612. [Google Scholar]
- 87.Koeller KM, Wong CH. Nature. 2001;409:232–240. doi: 10.1038/35051706. [DOI] [PubMed] [Google Scholar]
- 88.Odian G. Principles of Polymerization. Hoboken, NJ: John Wiley & Sons, Inc.; 2004. [Google Scholar]
- 89.Goldmann AS, Glassner M, Inglis AJ, Barner-Kowollik C. Macromol. Rapid Commun. 2013;34:810–849. doi: 10.1002/marc.201300017. [DOI] [PubMed] [Google Scholar]
- 90.Wong CH, Zimmerman SC. Chem. Commun. 2013;49:1679–1695. doi: 10.1039/c2cc37316e. [DOI] [PubMed] [Google Scholar]
- 91.Grieshaber SE, Farran AJ, Bai S, Kiick KL, Jia X. Biomacromolecules. 2012;13:1774–1786. doi: 10.1021/bm3002705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Grieshaber SE, Farran AJ, Lin-Gibson S, Kiick KL, Jia X. Macromolecules. 2009;42:2532–2541. doi: 10.1021/ma802791z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Grieshaber SE, Paik BA, Bai S, Kiick KL, Jia X. Soft matter. 2013;9:1589–1599. doi: 10.1039/C2SM27496E. PMID: 23976897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Termonia Y. Biomacromolecules. 2004;5:2404–2407. doi: 10.1021/bm049662x. [DOI] [PubMed] [Google Scholar]
- 95.Zhou CC, Leng BX, Yao JR, Qian J, Chen X, Zhou P, Knight DP, Shao ZZ. Biomacromolecules. 2006;7:2415–2419. doi: 10.1021/bm060199t. [DOI] [PubMed] [Google Scholar]
- 96.Sahin E, Kiick KL. Biomacromolecules. 2009;10:2740–2749. doi: 10.1021/bm900474k. [DOI] [PubMed] [Google Scholar]
- 97.Hamley IW. Biomacromolecules. 2014;15:1543–1559. doi: 10.1021/bm500246w. [DOI] [PubMed] [Google Scholar]
- 98.Joo JH, Ko DY, Moon HJ, Shinde UP, Park MH, Jeong B. Biomacromolecules. 2014;15:3664–3670. doi: 10.1021/bm500942p. [DOI] [PubMed] [Google Scholar]
- 99.Tsarevsky NV, Sumerlin BS, Matyjaszewski K. Macromolecules. 2005;38:3558–3561. [Google Scholar]
- 100.Qin AJ, Lam JWY, Tang BZ. Chem. Soc. Rev. 2010;39:2522–2544. doi: 10.1039/b909064a. [DOI] [PubMed] [Google Scholar]
- 101.Nagao Y, Takasu A. J. Polym. Sci. Pol. Chem. 2010;48:4207–4218. [Google Scholar]
- 102.Berthet MA, Zarafshani Z, Pfeifer S, Lutz JF. Macromolecules. 2010;43:44–50. [Google Scholar]
- 103.Schwartz E, Breitenkamp K, Fokin VV. Macromolecules. 2011;44:4735–4741. [Google Scholar]
- 104.Wang Y, Zhang R, Xu N, Du FS, Wang YL, Tan YX, Ji SP, Liang DH, Li ZC. Biomacromolecules. 2011;12:66–74. doi: 10.1021/bm101005j. [DOI] [PubMed] [Google Scholar]
- 105.Guo JS, Wei Y, Zhou DF, Cai PQ, Jing XB, Chen XS, Huang YB. Biomacromolecules. 2011;12:737–746. doi: 10.1021/bm1013662. [DOI] [PubMed] [Google Scholar]
- 106.Sheng X, Rock DM, Mauldin TC, Kessler MR. Polymer. 2011;52:4435–4441. [Google Scholar]
- 107.Guo JS, Meng FB, Li XY, Wang MZ, Wu YJ, Jing XB, Huang YB. Macromol. Biosci. 2012;12:533–546. doi: 10.1002/mabi.201100394. [DOI] [PubMed] [Google Scholar]
- 108.June SM, Bissel P, Long TE. J. Polym. Sci. Pol. Chem. 2012;50:3797–3805. [Google Scholar]
- 109.Miladi IA, Mudraboyina BP, Oueslati A, Drockenmuller E, Ben Romdhane H. J. Polym. Sci. Pol. Chem. 2014;52:223–231. [Google Scholar]
- 110.Molina-Pinilla I, Bueno-Martinez M, Hakkou K, Galbis JA. J. Polym. Sci. Pol. Chem. 2014;52:629–638. [Google Scholar]
- 111.Roux R, Sallet L, Alcouffe P, Chambert S, Sintes-Zydowicz N, Fleury E, Bernard J. ACS Macro Lett. 2012;1:1074–1078. doi: 10.1021/mz300281u. [DOI] [PubMed] [Google Scholar]
- 112.Greene AC, Zhu JH, Pochan DJ, Jia XQ, Kiick KL. Macromolecules. 2011;44:1942–1951. doi: 10.1021/ma102869y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Grieshaber SE, Nie T, Yan C, Zhong S, Teller SS, Clifton RJ, Pochan DJ, Kiick KL, Jia X. Macromol Chem Phys. 2011;212:229–239. doi: 10.1002/macp.201000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Luo K, Yang JY, Kopeckova P, Kopecek J. Macromolecules. 2011;44:2481–2488. doi: 10.1021/ma102574e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Averick S, Karacsony O, Mohin J, Yong X, Moellers NM, Woodman BF, Zhu WP, Mehl RA, Balazs AC, Kowalewski T, Matyjaszewski K. Angew. Chem.-Int. Edit. 2014;53:8050–8055. doi: 10.1002/anie.201402827. [DOI] [PubMed] [Google Scholar]
- 116.Zhou JW, Guimard NK, Inglis AJ, Namazian M, Lin CY, Coote ML, Spyrou E, Hilf S, Schmidt FG, Barner-Kowollik C. Polym. Chem. 2012;3:628–639. [Google Scholar]
- 117.Liu S, Zhang H, Fox JM, Jia X. Adv Mater. 2014 submitted. [Google Scholar]
- 118.Goldberg M, Mahon K, Anderson D. Adv. Drug Deliv. Rev. 2008;60:971–978. doi: 10.1016/j.addr.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zaquen N, Wenn B, Ranieri K, Vandenbergh J, Junkers T. J. Polym. Sci. Pol. Chem. 2014;52:178–187. [Google Scholar]
- 120.Walker CN, Sarapas JM, Kung V, Hall AL, Tew GN. ACS Macro Lett. 2014;3:453–457. doi: 10.1021/mz5001288. [DOI] [PubMed] [Google Scholar]
- 121.Turunc O, Meier MAR. J. Polym. Sci. Pol. Chem. 2012;50:1689–1695. [Google Scholar]
- 122.Miller JS, Shen CJ, Legant WR, Baranski JD, Blakely BL, Chen CS. Biomaterials. 2010;31:3736–3743. doi: 10.1016/j.biomaterials.2010.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pan HZ, Yang JY, Kopeckova P, Kopecek J. Biomacromolecules. 2011;12:247–252. doi: 10.1021/bm101254e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gao C, Yan D. Prog. Polym. Sci. 2004;29:183–275. [Google Scholar]
- 125.Walter MV, Malkoch M. Chem. Soc. Rev. 2012;41:4593–4609. doi: 10.1039/c2cs35062a. [DOI] [PubMed] [Google Scholar]
- 126.Antoni P, Robb MJ, Campos L, Montanez M, Hult A, Malmstrom E, Malkoch M, Hawker CJ. Macromolecules. 2010;43:6625–6631. [Google Scholar]
- 127.Killops KL, Campos LM, Hawker CJ. J. Am. Chem. Soc. 2008;130 doi: 10.1021/ja8006325. 5062-+. [DOI] [PubMed] [Google Scholar]
- 128.Semsarilar M, Ladmiral V, Perrier S. Macromolecules. 2010;43:1438–1443. [Google Scholar]
- 129.Chen MS, Hu M, Wang DL, Wang GJ, Zhu XY, Yan DY, Sun J. Bioconjugate Chem. 2012;23:1189–1199. doi: 10.1021/bc300016b. [DOI] [PubMed] [Google Scholar]
- 130.Li N, Yi QY, Luo K, Guo CH, Pan DY, Gu ZW. Biomaterials. 2014;35:9529–9545. doi: 10.1016/j.biomaterials.2014.07.059. [DOI] [PubMed] [Google Scholar]
- 131.Kottari N, Chabre YM, Shiao TC, Rej R, Roy R. Chem. Commun. 2014;50:1983–1985. doi: 10.1039/c3cc46633g. [DOI] [PubMed] [Google Scholar]
- 132.Grandjean C, Rommens C, Gras-Masse H, Melnyk O. Angew. Chem.-Int. Edit. 2000;39 1068-+. [Google Scholar]
- 133.Sharma R, Naresh K, Chabre YM, Rej R, Saadeh NK, Roy R. Polym. Chem. 2014;5:4321–4331. [Google Scholar]
- 134.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Proc Nat Acad Sci. 2003;100:5413–5418. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kloxin CJ, Bowman CN. Chem. Soc. Rev. 2013;42:7161–7173. doi: 10.1039/c3cs60046g. [DOI] [PubMed] [Google Scholar]
- 136.Ulbrich K, Subr V, Podperova P, Buresova M. J. Control. Release. 1995;34:155–165. [Google Scholar]
- 137.Sahoo S, Chung C, Khetan S, Burdick JA. Biomacromolecules. 2008;9:1088–1092. doi: 10.1021/bm800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Science. 2009;324:59–63. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kharkar PM, Kiick KL, Kloxin AM. Chem. Soc. Rev. 2013;42:7335–7372. doi: 10.1039/c3cs60040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lutolf MP, Raeber GP, Zisch AH, Tirelli N, Hubbell JA. Adv. Mater. 2003;15 888-+. [Google Scholar]
- 141.Xiao LX, Zhu JH, Londono JD, Pochan DJ, Jia XQ. Soft matter. 2012;8:10233–10237. doi: 10.1039/C2SM26566D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hu JM, Zhang GQ, Liu SY. Chem. Soc. Rev. 2012;41:5933–5949. doi: 10.1039/c2cs35103j. [DOI] [PubMed] [Google Scholar]
- 143.Higuchi A, Ling QD, Kumar SS, Chang Y, Kao TC, Munusamy MA, Alarfaj AA, Hsu ST, Umezawa A. Prog. Polym. Sci. 2014;39:1585–1613. [Google Scholar]
- 144.Kharkar PM, Kloxin AM, Kiick KL. J. Mat. Chem. B. 2014;2:5511–5521. doi: 10.1039/c4tb00496e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.DeForest CA, Polizzotti BD, Anseth KS. Nat. Mater. 2009;8:659–664. doi: 10.1038/nmat2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mangold C, Wurm F, Frey H. Polym. Chem. 2012;3:1714–1721. [Google Scholar]
- 147.Schulte B, Walther A, Keul H, Moller M. Macromolecules. 2014;47:1633–1645. [Google Scholar]
- 148.Thomas A, Muller SS, Frey H. Biomacromolecules. 2014;15:1935–1954. doi: 10.1021/bm5002608. [DOI] [PubMed] [Google Scholar]
- 149.Jin HB, Huang W, Zhu XY, Zhou YF, Yan DY. Chem. Soc. Rev. 2012;41:5986–5997. doi: 10.1039/c2cs35130g. [DOI] [PubMed] [Google Scholar]
- 150.Serban MA, Prestwich GD. Methods. 2008;45:93–98. doi: 10.1016/j.ymeth.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dicker KT, Gurski LA, Pradhan-Bhatt S, Witt RL, Farach-Carson MC, Jia X. Acta Biomater. 2014;10:1558–1570. doi: 10.1016/j.actbio.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Burdick JA, Prestwich GD. Adv. Mater. 2011;23:H41–H56. doi: 10.1002/adma.201003963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Xu X, Jha AK, Harrington DA, Farach-Carson MC, Jia X. Soft matter. 2012;8:3280–3294. doi: 10.1039/C2SM06463D. PMID: 2241994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chawla K, Yu TB, Stutts L, Yen M, Guan ZB. Biomaterials. 2012;33:6052–6060. doi: 10.1016/j.biomaterials.2012.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kushner AM, Guan ZB. Angew. Chem.-Int. Edit. 2011;50:9026–9057. doi: 10.1002/anie.201006496. [DOI] [PubMed] [Google Scholar]
- 156.Liao SW, Rawson J, Omori K, Ishiyama K, Mozhdehi D, Oancea AR, Ito T, Guan ZB, Mullen Y. Biomaterials. 2013;34:3984–3991. doi: 10.1016/j.biomaterials.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Liao SW, Yu TB, Guan ZB. J. Am. Chem. Soc. 2009;131:17638–17646. doi: 10.1021/ja907097t. [DOI] [PubMed] [Google Scholar]
- 158.Metzke M, O'Connor N, Maiti S, Nelson E, Guan ZB. Angew. Chem.-Int. Edit. 2005;44:6529–6533. doi: 10.1002/anie.200501944. [DOI] [PubMed] [Google Scholar]
- 159.Roland JT, Guan Z. J Am Chem Soc. 2004;126:14328–14329. doi: 10.1021/ja0448871. [DOI] [PubMed] [Google Scholar]
- 160.Jia X, Colombo G, Padera R, Langer R, Kohane DS. Biomaterials. 2004;25:4797–4804. doi: 10.1016/j.biomaterials.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 161.Gurski LA, Jha AK, Zhang C, Jia X, Farach-Carson MC. Biomaterials. 2009;30:6076–6085. doi: 10.1016/j.biomaterials.2009.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gurski LA, Xu X, Labrada LN, Nguyen NT, Xiao L, van Golen KL, Jia X, Farach-Carson MC. PLoS One. 2012;7:16. doi: 10.1371/journal.pone.0050075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Xu X, Farach-Carson MC, Jia X. Biotechnology advances. 2014;32:1256–1268. doi: 10.1016/j.biotechadv.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Farran AJ, Teller SS, Jha AK, Jiao T, Hule RA, Clifton RJ, Pochan DP, Duncan RL, Jia X. Tissue Eng Part A. 2010;16:1247–1261. doi: 10.1089/ten.tea.2009.0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jia X, Yeo Y, Clifton RJ, Jiao T, Kohane DS, Kobler JB, Zeitels SM, Langer R. Biomacromolecules. 2006;7:3336–3344. doi: 10.1021/bm0604956. [DOI] [PubMed] [Google Scholar]
- 166.Jha AK, Yang W, Kirn-Safran CB, Farach-Carson MC, Jia X. Biomaterials. 2009;30:6964–6975. doi: 10.1016/j.biomaterials.2009.09.009. PMID: 19775743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jha AK, Hule RA, Jiao T, Teller SS, Clifton RJ, Duncan RL, Pochan DJ, Jia X. Macromolecules. 2009;42:537–546. doi: 10.1021/ma8019442. PMID: 20046226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jha AK, Malik MS, Farach-Carson MC, Duncan RL, Jia X. Soft matter. 2010;6:5045–5055. doi: 10.1039/C0SM00101E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Krishna OD, Jha AK, Jia X, Kiick KL. Biomaterials. 2011;32:6412–6424. doi: 10.1016/j.biomaterials.2011.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Jha AK, Xu X, Duncan RL, Jia X. Biomaterials. 2011;32:2466–2478. doi: 10.1016/j.biomaterials.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Patenaude M, Hoare T. Biomacromolecules. 2012;13:369–378. doi: 10.1021/bm2013982. [DOI] [PubMed] [Google Scholar]
- 172.Lee KY, Bouhadir KH, Mooney DJ. Macromolecules. 2000;33:97–101. [Google Scholar]
- 173.McKinnon DD, Domaille DW, Cha JN, Anseth KS. Chem. Mat. 2014;26:2382–2387. doi: 10.1002/adma.201303680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.McKinnon DD, Domaille DW, Cha JN, Anseth KS. Adv. Mater. 2014;26:865–872. doi: 10.1002/adma.201303680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Shu XZ, Liu YC, Palumbo F, Prestwich GD. Biomaterials. 2003;24:3825–3834. doi: 10.1016/s0142-9612(03)00267-9. [DOI] [PubMed] [Google Scholar]
- 176.Shu XZ, Liu YC, Palumbo FS, Lu Y, Prestwich GD. Biomaterials. 2004;25:1339–1348. doi: 10.1016/j.biomaterials.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 177.Shu XZ, Prestwich GD. In: Chemistry and biology of hyaluronan. Garg HG, Hales CA, editors. Oxford: Elsevier Ltd.; 2004. pp. 475–504. [Google Scholar]
- 178.Prestwich GD, Kuo JW. Curr Pharm Biotechnol. 2008;9:242–245. doi: 10.2174/138920108785161523. [DOI] [PubMed] [Google Scholar]
- 179.Ghosh K, Shu XZ, Mou R, Lombardi J, Prestwich GD, Rafailovich MH, Clark RAF. Biomacromolecules. 2005;6:2857–2865. doi: 10.1021/bm050361c. [DOI] [PubMed] [Google Scholar]
- 180.Seelbach RJ, Fransen P, Peroglio M, Pulido D, Lopez-Chicon P, Duttenhoefer F, Sauerbier S, Freiman T, Niemeyer P, Semino C, Albericio F, Alini M, Royo M, Mata A, Eglin D. Acta Biomater. 2014;10:4340–4350. doi: 10.1016/j.actbio.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 181.Vanderhooft JL, Alcoutlabi M, Magda JJ, Prestwich GD. Macromol. Biosci. 2009;9:20–28. doi: 10.1002/mabi.200800141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hosack LW, Firpo MA, Scott JA, Prestwich GD, Peattie RA. Biomaterials. 2008;29:2336–2347. doi: 10.1016/j.biomaterials.2008.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Chen X, Thibeault SL. Tissue Eng. Part A. 2012;18:2528–2538. doi: 10.1089/ten.tea.2012.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lei YG, Gojgini S, Lam J, Segura T. Biomaterials. 2011;32:39–47. doi: 10.1016/j.biomaterials.2010.08.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Xu X, Gurski LA, Zhang C, Harrington DA, Farach-Carson MC, Jia X. Biomaterials. 2012;33:9049–9060. doi: 10.1016/j.biomaterials.2012.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Xu X, Sabanayagam CR, Harrington DA, Farach-Carson MC, Jia X. Biomaterials. 2014;35:3319–3330. doi: 10.1016/j.biomaterials.2013.12.080. PMID: 24447463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rossow T, Heyman JA, Ehrlicher AJ, Langhoff A, Weitz DA, Haag R, Seiffert S. J. Am. Chem. Soc. 2012;134:4983–4989. doi: 10.1021/ja300460p. [DOI] [PubMed] [Google Scholar]
- 188.Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. Adv. Mater. 2009;21 doi: 10.1002/adma.200901808. 5005-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Aimetti AA, Machen AJ, Anseth KS. Biomaterials. 2009;30:6048–6054. doi: 10.1016/j.biomaterials.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lin CC, Ki CS, Shih H. J. Appl. Polym. Sci. 2015;132:11. doi: 10.1002/app.41563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.McCall JD, Anseth KS. Biomacromolecules. 2012;13:2410–2417. doi: 10.1021/bm300671s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Roberts JJ, Bryant SJ. Biomaterials. 2013;34:9969–9979. doi: 10.1016/j.biomaterials.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Anderson SB, Lin CC, Kuntzler DV, Anseth KS. Biomaterials. 2011;32:3564–3574. doi: 10.1016/j.biomaterials.2011.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Raza A, Ki CS, Lin CC. Biomaterials. 2013;34:5117–5127. doi: 10.1016/j.biomaterials.2013.03.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Gramlich WM, Kim IL, Burdick JA. Biomaterials. 2013;34:9803–9811. doi: 10.1016/j.biomaterials.2013.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Shih H, Fraser AK, Lin CC. ACS applied materials & interfaces. 2013;5:1673–1680. doi: 10.1021/am302690t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Shih H, Lin CC. Macromol. Rapid Commun. 2013;34:269–273. doi: 10.1002/marc.201200605. [DOI] [PubMed] [Google Scholar]
- 198.Adzima BJ, Aguirre HA, Kloxin CJ, Scott TF, Bowman CN. Macromolecules. 2008;41:9112–9117. doi: 10.1021/ma801863d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Adzima BJ, Kloxin CJ, DeForest CA, Anseth KS, Bowman CN. Macromol. Rapid Commun. 2012;33:2092–2096. doi: 10.1002/marc.201200599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Koehler KC, Alge DL, Anseth KS, Bowman CN. Biomaterials. 2013;34:4150–4158. doi: 10.1016/j.biomaterials.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Koehler KC, Anseth KS, Bowman CN. Biomacromolecules. 2013;14:538–547. doi: 10.1021/bm301789d. [DOI] [PubMed] [Google Scholar]
- 202.Owen SC, Fisher SA, Tam RY, Nimmo CM, Shoichet MS. Langmuir. 2013;29:7393–7400. doi: 10.1021/la305000w. [DOI] [PubMed] [Google Scholar]
- 203.Malkoch M, Vestberg R, Gupta N, Mespouille L, Dubois P, Mason AF, Hedrick JL, Liao Q, Frank CW, Kingsbury K, Hawker CJ. Chem. Commun. 2006:2774–2776. doi: 10.1039/b603438a. [DOI] [PubMed] [Google Scholar]
- 204.Yuan Y, Zhang AK, Ling J, Yin LH, Chen Y, Fu GD. Soft matter. 2013;9:6309–6318. [Google Scholar]
- 205.Steinhilber D, Rossow T, Wedepohl S, Paulus F, Seiffert S, Haag R. Angew. Chem.-Int. Edit. 2013;52:13538–13543. doi: 10.1002/anie.201308005. [DOI] [PubMed] [Google Scholar]
- 206.Gattas-Asfura KM, Stabler CL. Biomacromolecules. 2009;10:3122–3129. doi: 10.1021/bm900789a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hu BH, Su J, Messersmith PB. Biomacromolecules. 2009;10:2194–2200. doi: 10.1021/bm900366e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Su J, Hu BH, Lowe WL, Kaufman DB, Messersmith PB. Biomaterials. 2010;31:308–314. doi: 10.1016/j.biomaterials.2009.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Strehin I, Gourevitch D, Zhang Y, Heber-Katz E, Messersmith PB. Biomater. Sci. 2013;1:603–613. doi: 10.1039/C3BM00201B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ehrbar M, Rizzi SC, Hlushchuk R, Djonov V, Zisch AH, Hubbell JA, Weber FE, Lutolf MP. Biomaterials. 2007;28:3856–3866. doi: 10.1016/j.biomaterials.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 211.Mosiewicz KA, Kolb L, van der Vlies AJ, Martino MM, Lienemann PS, Hubbell JA, Ehrbar M, Lutolf MP. Nat. Mater. 2013;12:1071–1077. doi: 10.1038/nmat3766. [DOI] [PubMed] [Google Scholar]
- 212.Mosiewicz KA, Johnsson K, Lutolf MP. J. Am. Chem. Soc. 2010;132 doi: 10.1021/ja9098164. 5972-+. [DOI] [PubMed] [Google Scholar]
- 213.Grieshaber SE, Jha AK, farran AJE, Jia X. In: Biomaterials for Tissue Engineering: A Review of the Past and Future Trends. Burdick JA, Mauck RL, editors. New York: Springer; 2011. pp. 9–46. [Google Scholar]
- 214.Guvendiren M, Burdick JA. Nat. Commun. 2012;3:9. doi: 10.1038/ncomms1792. [DOI] [PubMed] [Google Scholar]
- 215.Khetan S, Guvendiren M, Legant WR, Cohen DM, Chen CS, Burdick JA. Nat. Mater. 2013;12:458–465. doi: 10.1038/nmat3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Lin F, Yu JY, Tang W, Zheng JK, Defante A, Guo K, Wesdemiotis C, Becker ML. Biomacromolecules. 2013;14:3749–3758. doi: 10.1021/bm401133r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Zhou HX, Woo J, Cok AM, Wang MZ, Olsen BD, Johnson JA. Proc. Natl. Acad. Sci. U. S. A. 2012;109:19119–19124. doi: 10.1073/pnas.1213169109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Elacqua E, Lye DS, Weck M. Acc Chem Res. 2014;47:2405–2416. doi: 10.1021/ar500128w. [DOI] [PubMed] [Google Scholar]
- 219.Chen YX, Triola G, Waldmann H. Accounts Chem. Res. 2011;44:762–773. doi: 10.1021/ar200046h. [DOI] [PubMed] [Google Scholar]