

Abstract
Background
Diet has a significant relationship with the risk of coronary heart disease (CHD). Traditionally the effect of diet on CHD was measured with the biomarker for low-density lipoprotein (LDL) cholesterol. However, LDL is not the only or even the most important biomarker for CHD risk. A suitably integrated view of the mechanism by which diet influences the detailed CHD pathogenetic pathways is therefore needed in order to better understand CHD risk factors and help with better holistic CHD prevention and treatment decisions.
Methods
A systematic review of the existing literature was conducted. From this an integrated CHD pathogenetic pathway system was constructed. CHD biomarkers, which are found on these pathways, are the only measurable data to link diet with these CHD pathways. They were thus used to simplify the link between diet and the CHD mechanism. Data were systematically analysed from 294 cohort studies of CHD biomarkers constituting 1 187 350 patients.
Results and discussion
The resulting integrated analysis provides insight into the higher-order interactions underlying CHD and high-glycemic load (HGL) diets. A novel “connection graph” illustrates the measurable relationship between HGL diets and the relative risks attributed to the important CHD serological biomarkers.
The “connection graph” vividly shows that HGL diets not only influence the lipid and metabolic biomarkers, but also the inflammation, coagulation and vascular function biomarkers in an important way.
Conclusion
A focus primarily on the low density lipoprotein cholesterol biomarker for CHD risk has led to the traditional guidelines of CHD dietary recommendations. This has however inadvertently led to HGL diets. The influence of HGL diets on the other CHD biomarkers is not always fully appreciated. Thus, new diets or other interventions which address the full integrated CHD impact, as shown in this paper, are required.
Keywords: High glycemic load, Coronary heart disease, Biomarkers
Background
Coronary heart disease (CHD) is the largest cause of death globally [1]. Cholesterol is commonly assumed to be a crucial element of CHD [2]. Therefore dietary recommendations have traditionally focused on the reduction of saturated fatty acids [3]. This has led to the adoption of low-fat, high carbohydrate diets [4]. However, such high-glycemic load (HGL) diets have been shown to increase the relative risk for CHD [3,5].
Forty percent of CHD deaths occur in men and women who have cholesterol levels lower than the average for the general population [6]. The focus on a single biomarker may thus be oversimplified.
But how does a HGL diet influence all the CHD pathogenetic pathways? The authors could not find a study which integrated all the CHD pathways activated by a HGL diet in order to give insight at a glance. This paper thus investigates the interconnectivity of the effects of HGL diets with CHD pathogenetic pathways. We then use CHD biomarkers, which measure the CHD risk of a pathway, to simplify the integrative CHD model.
We can thus investigate all the effects of a HGL diet on CHD risk, as opposed to only the effect of the pathways quantified by one biomarker, namely low-density lipoprotein (LDL) cholesterol.
Methods
Search criteria
We searched PubMed, Science Direct, Ebsco Host, and Google Scholar for publications with “coronary heart disease“ or “coronary artery disease” or “cardiovascular disease” or “CHD” as a keyword and combinations with “high glycemic load diets”, “relative risk prediction”, “network analysis”, “pathway analysis”, “interconnections”, “systems biology”, “pathogenesis”, “biomarkers”, “conventional biomarkers”, “drugs”, “therapeutics”, pharmacotherapeutics”, “hypercoagulability”, “hypercholesterolaemia”, “hyperglycaemia”, “hyperinsulinaemia”, “inflammation”, and “hypertension” in the title of the study.
We also searched all major relevant specialty journals in the areas of cardiology, nutrition, endocrinology, psychoneuroendocrinology, systems biology, physiology, CHD, the metabolic syndrome and diabetes, such as Circulation; Journal of the American College of Cardiology; Arteriosclerosis, Thrombosis and Vascular Biology; The Lancet; New England Journal of Medicine; American Journal of Medicine; Nature Medicine; Diabetes Care; Journal of Clinical Endocrinology and Metabolism; American Journal of Clinical Nutrition; Preventive Medicine; Molecular Psychology; and Journal of Physiology for similar or related articles.
Furthermore, we selected PubMed and Google Scholar for meta-analyses with keywords “coronary heart disease” or “coronary artery disease” or “cardiovascular disease” or “CHD”. We also reviewed articles referenced in primary sources and their relevant citations. However, unless cited more than 50 times, we included only articles published after 1998 as these contained the most relevant data.
Study selection
Only articles using the following risk measures were included: relative risk (RR), odds ratio (OR), or hazard ratio (HR). It was not the intention of this study to conduct individual meta-analyses of the individual biomarkers or lifestyle effects and thus the most recent meta-analysis of each biomarker was used for the risk data. Where no meta-analysis for CHD risk was available for a specific biomarker or lifestyle effect a single high quality representative study was used.
Only the trends from each meta-analysis that was adjusted for the most confounding variables was used and only where sufficient information was available on that trend. This was done so that the effects of most of the potential confounders could be adjusted for. This may, however, have increased the heterogeneity between studies, as not all studies adjusted for the same confounders.
CHD was classified as the incidence of atherosclerosis, coronary artery disease, or myocardial infarction. Where results were given for cardiovascular disease these were interpreted as CHD only in scenarios where the effect of stroke could be accounted for or results were presented separately. Biomarkers were only considered if they were associated with an increased or decreased risk of CHD.
In a general sense we characterised two different aspects that had an effect on CHD risk from the systems based view of CHD by using RR data. These aspects were the lifestyle effects and the risk associated with increased levels of certain biomarkers. The lifestyle effects were considered as effect versus control. In other words, the RR was calculated for the CHD incidence of a lifestyle versus a control or placebo group. For the biomarkers, however, a different approach had to be used due to the differing levels of markers which are possible in vivo.
The RR for HGL diet effects was retrieved from a meta-analysis based on prospective population based studies. The RR data for the biomarkers were also retrieved from meta-analyses based largely on prospective population based studies.
The RR for changes in biomarkers were, where possible, extracted from the most recent meta-analysis conducted on the specific biomarker. If no meta-analysis was available, a suitable high quality study was included. In order to limit errors in comparisons between biomarkers only RR given per increase of 1-standard deviation (SD) in the biomarker level was included. The standardisation of RR to RR per 1-SD prohibits the misrepresentation of risk due to the selection of extreme exposure contrasts [7].
Data extraction
The following data were extracted from the studies: journal citation; number of cases per lifestyle study for OR, RR and HR; total number of persons, including gender, per study; characterisation and severity of lifestyle; type/intensity of CHD; whether the risk was measured in RR, OR or HR; the risk per lifestyle study, and the 95% confidence intervals per lifestyle study.
Data analysis
Heterogeneity between studies was inevitable due to the large quantity of meta-analyses considered. Each underlying meta-analysis reported individually on the heterogeneity in their analysis. However, these effects were not so large as to discount the effects observed.
The individual meta-analyses also had detailed accounts of differences between studies and subgroup analyses. However, these aspects are not further elaborated on in this study as they were used as a measure of validity in the study inclusion process. The individual studies selected unfortunately represent only the risk associated with the cohort studied and cannot be accurately extrapolated to other populations without further research.
OR and HR were converted to RR using the approach outlined by Zou [8]. It must however be noted that some of the RR values in this article differ from convention. The need for this comes as a result of the visual scaling of the traditional relative risk. Traditionally, if one plots an RR = 3 and RR = 0.33, respectively, the one does not ‘look’ three times worse and the other three times better than the normal RR = 1. The reason is that the scales for the positive and negative effects are not numerically similar. A graph of ‘good’ and ‘bad’ RR can therefore be deceptive for the untrained person, e.g., a patient.
This article rather uses the method that the conventional RR = 3 is three times worse than the normal RR = 1. While the conventional RR = 0.33 means that the patient’s position is three times better than the normal RR = 1. Thus, in summary: a conventional RR = 3 is presented as per normal, as a 3-fold increase in risk and a conventional RR = 0.33 is presented as a 3-fold decrease in risk (1/0.33 = 3).
Results
Integrated model
The integrated model in Figure 1, which we developed, schematically illustrates the complexity of CHD. (A more detailed discussion of Figure 1 is given in Section “Pathogenetic effects of high glycemic load diets”). It is however important to realize that CHD involves inputs from hundreds of gene expressions and a number of tissues. Thus, analysing the individual components of the system would not be sufficient, as it is important to know how these components interact with each other [9]. For instance, genetic and lifestyle factors influence clinical traits by perturbing molecular networks [10]. A high-level systems-based view of CHD therefore has the potential to interrogate these molecular phenotypes and identify the patterns associated with the disease.
Figure 1.
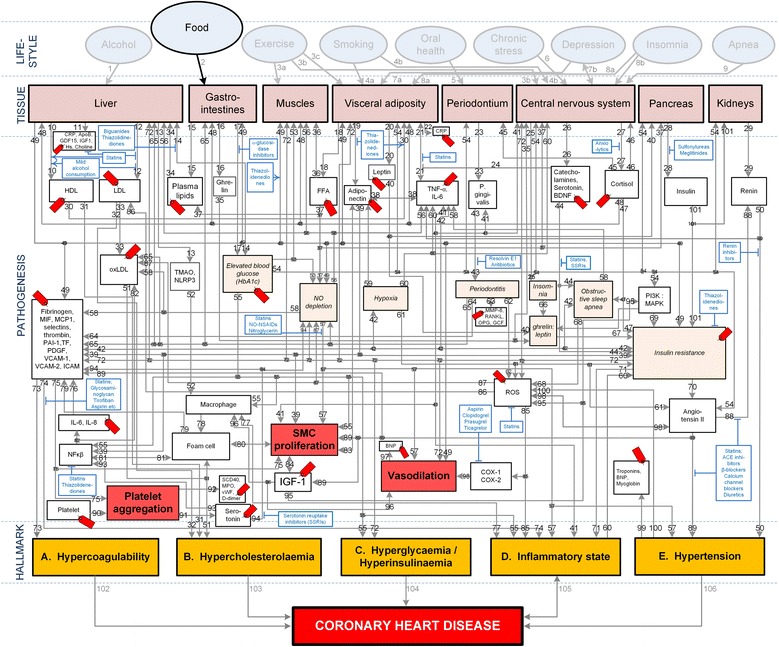
Conceptual model of general lifestyle effects, salient CHD pathogenetic pathways and CHD hallmarks. The affective pathway of pharmacotherapeutics, blue boxes, is shown in Figure 1, and salient serological biomarkers are indicated by the  icon. The blunted blue arrows denote antagonise or inhibit and pointed blue arrows denote up-regulate or facilitate. HDL denotes high-density lipoprotein; LDL, low-density lipoprotein; oxLDL, oxidised LDL; FFA, free fatty acids; TMAO, an oxidation product of trimethylamine (TMA); NLRP3, Inflammasome responsible for activation of inflammatory processes as well as epithelial cell regeneration and microflora; Hs, homocysteine; IGF-1, insulin-like growth factor-1; TNF-α , tumour necrosis factor-α; IL, interleukin; NO, nitric oxide; NO-NSAIDs, combinational NO-non-steroidal anti-inflammatory drug; SSRI, serotonin reuptake inhibitors; ROS, reactive oxygen species; NFκβ, nuclear factor-κβ; SMC, smooth muscle cell; HbA1c, glycosylated haemoglobin A1c; P. gingivalis, Porphyromonas gingivalis; vWF, von Willebrand factor; PDGF, platelet-derived growth factor; MIF, macrophage migration inhibitory factor; SCD-40, recombinant human sCD40 ligand; MPO, myeloperoxidase; MMP, matrix metalloproteinase; VCAM, vascular cell adhesion molecule; ICAM, intracellular adhesion molecule; CRP, C-reactive protein; PAI, plasminogen activator inhibitor; TF, tissue factor, MCP, monocyte chemoattractant protein; BDNF, brain-derived neurotrophic factor; PI3K, phosphatidylinositol 3-kinase; MAPK, mitogen-activated protein (MAP) kinase; RANKL, receptor activator of nuclear factor kappa-beta ligand; OPG, osteoprotegerin; GCF, gingival crevicular fluid; D-dimer, fibrin degradation product D; BNP, B-type natriuretic peptide; ACE, angiotensin-converting-enzyme; COX, cyclooxygenase; β-blocker, beta-adrenergic antagonists.
icon. The blunted blue arrows denote antagonise or inhibit and pointed blue arrows denote up-regulate or facilitate. HDL denotes high-density lipoprotein; LDL, low-density lipoprotein; oxLDL, oxidised LDL; FFA, free fatty acids; TMAO, an oxidation product of trimethylamine (TMA); NLRP3, Inflammasome responsible for activation of inflammatory processes as well as epithelial cell regeneration and microflora; Hs, homocysteine; IGF-1, insulin-like growth factor-1; TNF-α , tumour necrosis factor-α; IL, interleukin; NO, nitric oxide; NO-NSAIDs, combinational NO-non-steroidal anti-inflammatory drug; SSRI, serotonin reuptake inhibitors; ROS, reactive oxygen species; NFκβ, nuclear factor-κβ; SMC, smooth muscle cell; HbA1c, glycosylated haemoglobin A1c; P. gingivalis, Porphyromonas gingivalis; vWF, von Willebrand factor; PDGF, platelet-derived growth factor; MIF, macrophage migration inhibitory factor; SCD-40, recombinant human sCD40 ligand; MPO, myeloperoxidase; MMP, matrix metalloproteinase; VCAM, vascular cell adhesion molecule; ICAM, intracellular adhesion molecule; CRP, C-reactive protein; PAI, plasminogen activator inhibitor; TF, tissue factor, MCP, monocyte chemoattractant protein; BDNF, brain-derived neurotrophic factor; PI3K, phosphatidylinositol 3-kinase; MAPK, mitogen-activated protein (MAP) kinase; RANKL, receptor activator of nuclear factor kappa-beta ligand; OPG, osteoprotegerin; GCF, gingival crevicular fluid; D-dimer, fibrin degradation product D; BNP, B-type natriuretic peptide; ACE, angiotensin-converting-enzyme; COX, cyclooxygenase; β-blocker, beta-adrenergic antagonists.
Pathways can be tracked from a chosen lifestyle effect to a hallmark of CHD if the two states are connected by the pathogenesis of the disorder. The pathways are therefore a visual representation of previously published knowledge integrated here.
The pathogenetic pathways of interest for this review were only those between HGL diets (“Food”) and CHD. The effects of other lifestyle effects (e.g. moderate alcohol consumption, moderate intensity exercise, smoking, oral health, chronic stress, depression, insomnia and sleep apnoea) are not considered here.
The lifestyle effect of “Food” (Figure 1) was regarded as HGL diets (daily mean GL ≥ 142). “Tissue” in Figure 1 indicates the organ or type of tissue which is affected by a pathogenetic pathway or trait. “Pathogenesis” in Figure 1 indicates the pathological pathways of the disorder.
Salient serological biomarkers (shown in Figure 1 as  ) and pharmacotherapeutics (shown in Figure 1 as
) and pharmacotherapeutics (shown in Figure 1 as  ) that act on the pathways are also indicated in Figure 1. These pathogenetic pathways also lead to certain traits (e.g. insulin resistance) that lead to five pathophysiological end-states, which we designate as “hallmarks of CHD”, namely hypercoagulability, hypercholesterolaemia, hyperglycaemia/hyperinsulinaemia, an inflammatory state, and hypertension.
) that act on the pathways are also indicated in Figure 1. These pathogenetic pathways also lead to certain traits (e.g. insulin resistance) that lead to five pathophysiological end-states, which we designate as “hallmarks of CHD”, namely hypercoagulability, hypercholesterolaemia, hyperglycaemia/hyperinsulinaemia, an inflammatory state, and hypertension.
The formulation of this conceptual model required the consultation of numerous publications. The journal references which were used to describe the main pathogenetic pathways in the model are given in Table 1. It is however not the purpose of this review to describe in detail all these pathways. The aim is merely to simplify Figure 1 to show only the pathways relevant to HGL diets.
Table 1.
Pathogenetic pathways (in Figure 1 ) and cited works
| Pathway | Refs. | Pathway | Refs. | Pathway | Refs. | Pathway | Refs. | Pathway | Refs. | Pathway | Refs. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | [11,12] | 2 | [13-17] | 3 a,b,c | [18-20] | 4 a,b | [21-23] | 5 | [24-26] | 6 | [27-29] |
| 7 a,b | [30-35] | 8 a,b | [36-38] | 9 | [39] | 10 | [40,41-44] | 11 | [44,45] | 12 | [44] |
| 13 | [15-17] | 14 | [45-53] | 15 | [52-54] | 16 | [36-38] | 17 | [46-53] | 18 | [23,55-57] |
| 19 | [54,55] | 20 | [36-38] | 21 | [45,57-63] | 22 | [57] | 23 | [64-68] | 24 | [69-71] |
| 25 | [36-38] | 26 | [69-74] | 27 | [28,29,75-87] | 28 | [88-92] | 29 | [44,93] | 30 | [40,41-45] |
| 31 | [40,41-45] | 32 | [44] | 33 | [44] | 34 | [45,54-57] | 35 | [36-38] | 36 | [45,54-57] |
| 37 | [45,54-57] | 38 | [61,86,94-98] | 39 | [54,55] | 40 | [36-38] | 41 | [60,61,98] | 42 | [60,92] |
| 43 | [24,60,61,64-68] | 44 | [69-71] | 45 | [27,79,81] | 46 | [27,79,81] | 47 | [27,79,81] | 48 | [27,79,81] |
| 49 | [88-90,99] | 50 | [44,95,100] | 51 | [9,10,44,45,93,94,100-104] | 52 | [15,16] | 53 | [45-53] | 54 | [45-53] |
| 55 | [45-53,105-110] | 56 | [45,54-57] | 57 | [45,54-57,93,111-114] | 58 | [45,54-57,93] | 59 | [82-85] | 60 | [82-85] |
| 61 | [82-85] | 62 | [24,65] | 63 | [64-67] | 64 | [24,25] | 65 | [24,25,66] | 66 | [36-38] |
| 67 | [36-38] | 68 | [54-57] | 69 | [88] | 70 | [88-90] | 71 | [44,88-90,93,115,116] | 72 | [44,88-90,93,115,116] |
| 73 | [40,45,92] | 74 | [40,45,92] | 75 | [40,63,92,105,112] | 76 | [45,60,61] | 77 | [60,105] | 78 | [60,105] |
| 79 | [40,45,60,105] | 80 | [40,45,60,105] | 81 | [40,60,105] | 82 | [40,88,94] | 83 | [107-110] | 84 | [60] |
| 85 | [45,94,101,113,114] | 86 | [45,94] | 87 | [94] | 88 | [44,94,112,115,116] | 89 | [44,94,112,115,116] | 90 | [40,105,112] |
| 91 | [69-71] | 92 | [40,44,102] | 93 | [69,70] | 94 | [117-120] | 95 | [121-124] | 96 | [121-124] |
| 97 | [44] | 98 | [40,60,94,105] | 99 | [44] | 100 | [94] | 101 | [88-90] | 102 | [44,46,49,88,94,102] |
| 103 | [44,45,60,62] | 104 | [44,45,94,102,112] | 105 | [44,45,60,62,125,126] | 106 | [44,45,94,102,112] |
a,b,c denotes multiple pathways between lifestyle effects and CHD pathogenesis.
Despite the rich body of existing knowledge pertaining to CHD pathogenesis, lifestyle effects, and pharmacotherapeutics [9,10,45,101], a suitably integrated high-level conceptual model of CHD could not be found. A high-level model that consolidates the effects of HGL diets on relative risk of CHD and CHD biomarkers was therefore developed. This model could thus help elucidate the higher-order interactions underlying CHD [9] and provide new insights into dietary interventions.
Pathogenetic effects of high-glycemic load diets
Figure 1 indicates all possible pathogenetic pathways between the various lifestyle effects and CHD. In the present review only the CHD effects of HGL diets are appraised. The pathogenetic pathways which are activated by HGL diets are elucidated in Table 2. It is important to note that not all the pathogenetic pathways indicated in Figure 1 will be relevant in all patients, and all the pathways may not be active simultaneously.
Table 2.
Putative effects of high glycemic load diets and salient CHD pathogenetic pathways
| Lifestyle | Pathways, and pathway numbers corresponding to those in Figure 2 | Refs. |
|---|---|---|
| High- GL diets | a. 2-↑17-14-↑ blood glucose-55-↑ hyperglycaemia | a. [13,57,103] |
| b. 2-↑17-14-↑ blood glucose-54-19-↓ adiponectin-38-↑ TNFα-56-12-↑ LDL-33-↑ oxLDL-51-↑ hypercholesterolaemia | b. [13,57,103] | |
| c. 2-↑17-14-↑blood glucose-54-↑ PI3K:MAPK-69-↑ insulin resistance-70-↑ angiotensin II-89-↑ hypertension-100-↑ROS-85-↑ inflammatory state | c. [13,57,95,105] | |
| d. 2-↑17-14-↑ blood glucose-54-↑ PI3K:MAPK-69-↑ insulin resistance-70-↑ angiotensin II-88-50-↑ TNFα-41-↑ inflammatory state | d. [101] | |
| e. 2-↑17-14-↑ blood glucose-54-↑ PI3K:MAPK-69-↑ insulin resistance-70-↑ angiotensin II-89-↑ SMC proliferation | e. [62] | |
| f. 2-↑17-14-↑ blood glucose-54-↑ PI3K:MAPK-69-↑ insulin resistance-70-↑ angiotensin II-89-↓ IGF1-84-↑ SMC proliferation | f. [121-123] | |
| g. 2-↑17-14-↑ blood glucose-54-↑ PI3K:MAPK-69-↑ insulin resistance-70-↑ angiotensin II-89-↑ VCAM1/MCP1-73-↑ hypercoagulability | g. [60] | |
| h. 2-↑17-14-↑ blood glucose-54-↑ PI3K:MAPK-69-↑ insulin resistance-72-↑ platelet factors-73-↑ hypercoagulability | h. [40,48] | |
| i. 2-↑17-14-↑ blood glucose-54-19-↓ adiponectin-38-↑ TNFα-41-↑ P. gingivalis-43-↑ periodontitis-64-↑ platelet factors-73-↑ hypercoagulability | i. [27,40,48,66] | |
| j. 2-↑17-14-↑ blood glucose-54-19-↓ adiponectin-39-↑ insulin resistance | j. [101] | |
| k. 2-↑17-14-↑ blood glucose-54-19-↓ adiponectin-39-↑ SMC proliferation | k. [94] | |
| l. 2-↑17-14-↑ blood glucose-54-↑ PI3K:MAPK-69-↑ insulin resistance-72-↑ hyperglycaemia | l. [88,89] | |
| m. 2-↑17-14-↑ blood glucose-55-↑ SMC proliferation | m. [103] | |
| n. 2-↑17-14-↑ blood glucose-53-↑ NO depletion-57-↑ SMC proliferation | n. [28,89,105,111] | |
| o. 2-↑17-14-↑ blood glucose-53-↑ NO depletion-57-↓ vasodilation | o. [28,97,103,105] | |
| p. 2-↑17-14-↑ blood glucose-54-60-↑insulin resistance-72-↓ vasodilation | p. [101,103] | |
| q. 2-↑17-14-↑ blood glucose-54-↑angiotensin II-89-↑hypertension-100-↑ROS-85-↑inflammatory state | q. [28,93,103] | |
| r. 2-↑15-34-12-↑ LDL-33-↑ oxLDL-51- ↑ hypercholesterolaemia | r. [14] | |
| s. 2-↑15-34-13-↑ TMAO/NLRP3-52-macrophage-78-foam cell-↑ SMC proliferation | s. [15-17] | |
| t. 2-↑15-34-13-↑ TMAO/NLRP3-52-macrophage-51-↑ hypercholesterolaemia | t. [15-17] | |
| u. 2-↑15-34-13-↑ TMAO/NLRP3-52-macrophage-77-↑ inflammatory state | u. [15-17] |
↑denotes upregulation/increase, ↓denotes downregulation/decrease, x-y-z indicates pathway connecting x to y to z. HDL, high-density lipoprotein; LDL, low-density lipoprotein; oxLDL, oxidised LDL; FFA, free fatty acids; TNFα, tumour necrosis factor-α; IL6, interleukin-6; NO, nitric oxide; ROS, reactive oxygen species; BDNF, brain-derived neurotrophic factor; OSA, obstructive sleep apnoea; SMC, smooth muscle cell; P. gingivalis, Porphyromonas gingivalis; PI3K, phosphatidylinositol 3-kinase; MAPK, mitogen-activated protein (MAP) kinase; PI3K:MAPK, ratio of PI3K to MAPK; IGF 1, insulin-like growth factor-1; VCAM 1, vascular cell adhesion molecule-1; MCP 1, monocyte chemoattractant protein-1.
Figure 1, Pathway: 2-17-14-blood glucose-55-hyperglycaemia shows how HGL diets are connected to hyperglycaemia through the increase of blood glucose due to carbohydrate consumption [127]. The resulting state of hyperglycaemia and concomitant hyperinsulinaemia are both CHD hallmarks in non-diabetic patients [128]. (Figure 1, Pathway: 2-17-14-blood glucose-55-hyperglycaemia).
The hyperglycaemia that result from HGL diets can also lead to an increase in the PI3K-to-MAPK ratio, through inhibition of the phosphatidylinositol 3-kinase (PI3K) insulin signalling pathway or the stimulation of the MAPK pathway [89]. This in turn increases insulin resistance [129]. (Figure 1, Pathway: 2-17-14-blood glucose-54-PI3K:MAPK-69-insulin resistance).
Decreased insulin sensitivity, due to insulin resistance, has been associated with increases in the serum levels of platelet factors, such as fibrinogen [130] and von Willebrand factor [131], and thus increased potential for hypercoagulability which is a CHD hallmark [132,133]. (Figure 1, Pathway: 2-17-14- blood glucose-54-PI3K:MAPK-69-insulin resistance-72-platelet factors-73-hypercoagulability).
Further, decreased adiponectin levels can result from increased adipose tissue levels stemming from excessive dietary intake due to HGL diets [86]. Decreases in plasma adiponectin concentrations can also decrease insulin sensitivity by decreasing muscle fat oxidation [134] and subsequently cause increased vasodilation [86] which is a hallmark of CHD. Additionally, it is possible for decreases in adiponectin levels to increase those of intramyocelular triacylglycerol which are correlated to insulin resistance [135,136]. (Figure 1, Pathway: 2-17-blood glucose-54-19-adiponectin-39-insulin resistance-vasodilation).
Figure 1 also shows why an insulin-resistant state may be pro-inflammatory, with the expression of the inflammatory mediator TNF-α by adipose tissue being a core aspect associated with plasma insulin [134]. Additionally, adipose tissue has been shown to express other pro-inflammatory mediators, including C-reactive protein (CRP). Macrophages residing in the adipose tissue may also be a source of pro-inflammatory factors by modulating the secretory activities of adipocytes [137]. (Figure 1, Pathway: 2-15-34-13-TMAO/NLRP3-52-macrophage-77-inflammatory state).
HGL diets can also lead to the accumulation of visceral fat, reduced lipoprotein lipase activity and reduced clearance of triglycerides. This leads to increased LDL levels, decreased high-density lipoprotein (HDL) levels, and increased LDL-to-HDL ratios [138], and eventually to hypercholesterolaemia [128] which contributes significantly to atherogenecity, leading to CHD [139]. (Figure 1, Pathway: 2-15-34-12-LDL-33-oxLDL-51-hypercholesterolaemia).
The CHD hallmark hypertension is directly correlated with visceral fat mass [140]. Hypertension may also be mediated through increased vascular and sympathetic tone created by reduced bioavailability of nitric oxide (NO) because of oxidative stress, and increased expression of angiotensinogen by adipose tissue leading to an activation of the renin-angiotensin system [141,142]. (Figure 1, Pathway: 2-17-14-blood glucose-54-angiotensin II-89-hypertension).
From the above high-level model, it is apparent that HGL diets have multiple effects on the pathogenetic mechanism of CHD. Therefore, it can be seen that with greater activation of the pathways connected to the hallmarks of CHD, a patient’s risk of CHD is further amplified. Thus, an integrated multi-faceted approach to therapeutics and lifestyle factors is necessary.
Biomarkers of coronary heart disease
The integrated model that was developed is a high-level conceptual model, from which the interconnectedness of CHD is immediately apparent (Figure 1). The model is however complicated, thus a novel approach was used to simplify it with regards to the consumption of a HGL diet. In order to simplify the integrated model, serological biomarkers (which can be easily measured) were used to link the effect of HGL diet to the corresponding CHD pathways.
Biomarkers are used as indicators of an underlying disorder or pathogenetic pathway, such as systemic inflammation that is a known aggravating factor in the pathogenesis of CHD [60,61,143]. The measurement of specific biomarkers therefore enables the prediction of the relative risk for CHD associated with these biomarkers [44]. As it is possible to accurately measure certain serum biomarker levels, they can also be used as patient-specific links to pathogenetic, lifestyle (e.g. diet) or pharmacotherapeutic (e.g. α-glucosidase inhibitors) factors. In essence, the biomarkers can be used to indicate the activation of underlying pathogenetic pathways of the disorder. The biomarkers associated with different pathways are indicated in Figure 1 as  .
.
Important CHD biomarkers which have been noted to change with chronic consumption of HGL diets are hyperglycaemia as represented by changes in the glycated haemoglobin levels [144] and hyperinsulinaemia as represented by increased serum insulin levels [145]. Further, additional biomarkers of interest would be the traditional cholesterol levels of LDL and HDL, which have both been noted to be affected by excessive consumption of HGL diets [145].
The authors could however not find a published study where all the important serum biomarkers were compared in order to show their relative importance regarding CHD risk prediction in terms of relative risk. We therefore attempted this in Table 3 and the results thereof are presented graphically in Figure 2.
Table 3.
Salient serological and functional biomarkers of CHD, and prospective ones
| Biomarker (class and salient examples) | Prediction of CHD relative risk (95% CI) | Size of studies (N = number of trials, n = number of patients) | Ref. |
|---|---|---|---|
| Lipid-related markers: | |||
| Triglycerides | 0.99 (0.94-1.05) | (N = 68, n = 302 430) | [146] |
| LDL | 1.25 (1.18-1.33) | (N = 15, n = 233 455) | [147] |
| HDL | 0.78 (0.74-0.82) | (N = 68, n = 302 430) | [146] |
| ApoB | 1.43 (1.35-1.51) | (N = 15, n = 233 455) | [147] |
| Leptin | 1.04 (0.92-1.17) | (n = 1 832) | [148] |
| Inflammation markers: | |||
| hsCRP | 1.20 (1.18-1.22) | (N = 38, n = 166 596) | [149] |
| IL-6 | 1.25 (1.19-1.32) | (N = 25, n = 42 123) | [150] |
| TNF-α | 1.17 (1.09-1.25) | (N = 7, n = 6 107) | [150] |
| GDF-15 | 1.40 (1.10-1.80) | (n = 1 740) | [151] |
| OPG | 1.41 (1.33-1.57) | (n = 5 863) | [152] |
| Marker of oxidative stress: | |||
| MPO | 1.17 (1.06-1.30) | (n = 2 861) | [153] |
| Marker of vascular function and neurohormonal activity: | |||
| BNP | 1.42 (1.24-1.63) | (N = 40, n = 87 474) | [154] |
| Homocysteine | 1.15 (1.09-1.22) | (N = 20, n = 22 652) | [155,156] |
| Coagulation marker: | |||
| Fibrinogen | 1.15 (1.13-1.17) | (N = 40, n = 185 892) | [149] |
| Necrosis marker: | |||
| Troponins | 1.15 (1.04-1.27) | (n = 3 265) | [157] |
| Renal function marker: | |||
| Urinary ACR | 1.57 (1.26-1.95) | (n = 626) | [158] |
| Metabolic markers: | |||
| HbA1c | 1.42 (1.16-1.74) | (N = 2, n = 2 442) | [159] |
| IGF-1 | 0.76 (0.56-1.04) | (n = 3 967) | [160] |
| Adiponectin | 0.97 (0.86-1.09) | (N = 14, n =21 272) | [161] |
| Cortisol | 1.10 (0.97-1.25) | (n = 2 512) | [162,163] |
| BDNF | ? | N/A | [71,73,74] |
| Insulin resistance (HOMA) | 1.46 (1.26-1.69) | (N = 17, n = 51 161) | [164] |
Only recent and/or highly cited papers have been cited here. n denotes number of participants; N, number of trials; ?, a possible, though not currently quantified effect on CHD risk; HDL, high-density lipoprotein; BNP, B-type natriuretic peptide; ACR, albumin–to-creatinine ratio; GDF-15, growth-differentiation factor-15; LDL, low-density lipoprotein; HbA1c, glycosylated haemoglobin A1c; hsCRP, high-sensitivity C-reactive protein; IL-6, interleukin-6; TNF-α, tumour necrosis factor-α; ApoB, apolipoprotein-B; IGF-1, insulin-like growth factor-1; MPO, myeloperoxidase; RANKL or OPG, osteoprotegerin; BDNF, brain-derived neurotrophic factor; HOMA, homeostatic model assessment.
Figure 2.
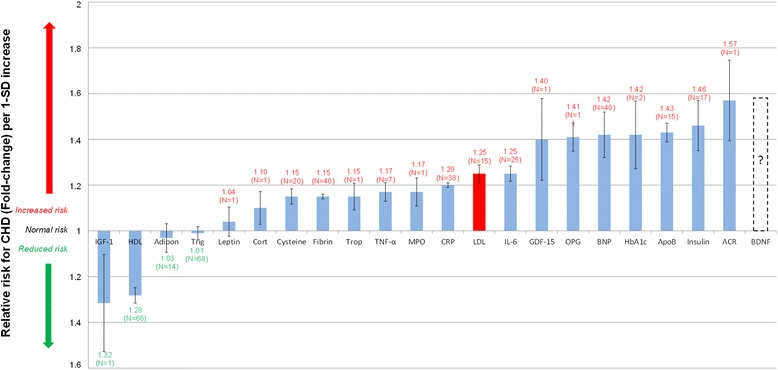
Normalised relative risks (fold-change) of salient current and potential biomarkers for CHD. Increased IGF-1 and HDL levels are associated with a moderately decreased CHD risk. (IGF-1 and HDL levels are significantly inversely correlated to relative risk for CHD.) N indicates number of trials; I, standard error; Adipo, adiponectin; HDL, high-density lipoprotein; BNP, B-type natriuretic peptide; ACR, albumin-to-creatinine ratio; GDF-15, growth-differentiation factor-15; Cysteine, Homocysteine; LDL, low-density lipoprotein; HbA1c, glycosylated haemoglobin A1c; Trop, troponins; Trigl, triglycerides; CRP, C-reactive protein; IL-6, interleukin-6; Fibrin, fibrinogen; Cort, cortisol; TNF-α, tumour necrosis factor-α; ApoB, apolipoprotein-B; IGF-1, insulin-like growth factor-1; MPO, myeloperoxidase; RANKL or OPG, osteoprotegerin; BDNF, brain-derived neurotrophic factor.
Table 3 presents the relative risk data from 294 cohort studies comprising 1 161 560 subjects. The results from the studies were thus interpreted and the averaged relative risks (with standard error (I) and study size (N)) were used to populate Figure 2. Figure 2 visually compares the RR associated with serological biomarkers per 1-standard deviation increase in said biomarker.
The main outcome from the relative risk comparison in Figure 2 is that it allows one to compare the relative risk of CHD associated with changes in certain biomarkers. From the figure, it is clear that adverse changes in certain biomarkers, such as ApoB, present a much greater risk than the generally considered LDL cholesterol (Shown in Figure 2 in red). Additionally, glycated haemoglobin A1c (HbA1c), an easy-to-measure biomarker that is well correlated with HGL diets [144], is associated with a large increased risk. This type of consideration thus alludes to biomarkers such as ApoB and insulin resistance that are potentially more important for lifestyle and pharmaceutical interventions.
Although the numerical values of relative risk presented in this study are based on large, clustered clinical trials, and thus give a good idea of average effects, it is acknowledged that individual patients will have very specific CHD profiles. However, Figure 1 is still relevant to everyone and should thus provide general insight into relevant risk factors. Therefore, Figure 1 could inter alia reveal further pathways still available for biomarker and drug discovery.
Effects of high-glycemic load diets
The pathogenesis of different lifestyle effects are illustrated in Figure 1 and the specific paths regulated by HGL diets are detailed in Table 2. It is therefore possible to quantify the effects of HGL diets on the RR of CHD using Figure 1 as a model for the pathogenesis of CHD. By considering the pathogenesis of HGL diets, the pathways activated thereby are elucidated in Figure 1. Certain pathways might be quantified by the measurement of specific biomarkers (shown as  in Figure 1).
in Figure 1).
The effects of HGL diets on CHD are further characterised by the ‘connection graph’ in Figure 3. The ‘connection graph’ is a simplification of the pathogenesis of CHD presented in Figure 1. Within this graph none of the underlying pathogenesis is neglected, but only the CHD biomarkers affected by HGL diets are indicated. The pathways, from Figure 1, through which the consumption of HGL diets effect the biomarkers are shown on the connections.
Figure 3.
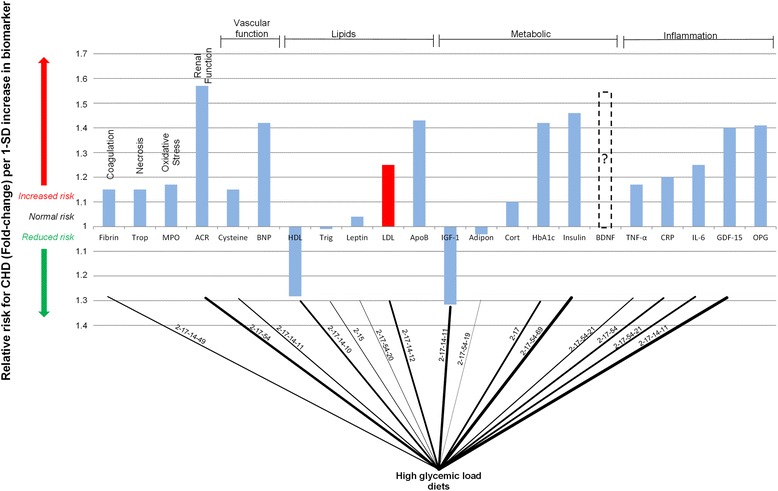
Interconnection of relative risk effects of high glycemic load diets and serological biomarkers for CHD. “ACR” denotes albumin-to-creatinine ratio; Trop, troponins; Fibrin, fibrinogen; MPO, myeloperoxidase; BNP, B-type natriuretic peptide; Cysteine, Homocysteine; HDL, high-density lipoprotein; LDL, low-density lipoprotein; Trigl, triglycerides; ApoB, Apolipoprotein-B; Adipon, adiponectin; HbA1c, glycosylated haemoglobin A1c; Cort, cortisol; IGF-1, insulin-like growth factor-1; BDNF, brain-derived neurotrophic factor; GDF-15, growth-differentiation factor-15; CRP, C-reactive protein; IL-6, interleukin-6; TNF-α, tumour necrosis factor-α; RANKL or OPG, osteoprotegerin.
To make further deductions from the ‘connection graph’ the biomarkers have been sorted into classes in terms of their clinical effect. The classes are renal function, necrosis, coagulation, oxidative stress, vascular function, lipids, metabolic function and inflammation. The ‘connection graph’ therefore allows easy visual recognition of the effects of different lifestyle factors, in this case HGL diets on the biomarkers of CHD.
The pathogenetic pathways (from Figure 1) are superimposed on the connecting lines in Figure 3. Therefore, increasing line thickness indicates a connection with greater pathogenetic effect (as quantified by the biomarker’s relative risk prediction of CHD). For example, the relative risk of CHD is relatively low when considering leptin, thus the connection line between HGL diets and leptin is thin.
From the connection graph, it is clear that there are many connections between HGL diets and the biomarkers of CHD. Firstly, it is rather evident that chronic consumption of a HGL diet would serve to induce chronic hyperglycaemia [165]. This chronic hyperglycaemia will be evident in increased HbA1c levels [166] which predicts an increased RR of CHD [159].
Since hyperglycaemia stimulates insulin secretion [167], chronic hyperglycaemia could also serve to increase insulin resistance, by the over-production of insulin [131]. Insulin resistance, which predicts an increased RR of CHD [164], is associated with hyperinsulinaemia [168].
The metabolic marker adiponectin (Figure 3) is also linked to HGL diets, through increased obesity and visceral adiposity possible from HGL diets [169] which are known to reduce the plasma levels of adiponectin [170].
Increased fibrinogen levels, a coagulation biomarker in Figure 3, are postulated to be caused by increased insulin resistance [130], however this pathogenesis is not fully understood. It is however clear that there is some causal relationship between increased serum insulin levels and increased fibrinogen levels [130,131,171] and a possible state of hypercoagulation. Therefore HGL diet induced insulin resistance may have an effect on coagulation, which is a hallmark of CHD.
It has been found that high carbohydrate diets can affect changes in lipid profile, regardless of the cholesterol, protein or fat content [172,173]. Similar trends are observed in HGL diets which have been found to provide reductions in HDL levels and increased LDL and triacylglycerol levels [55,174] as shown in Figure 3. These results suggest that HGL diets have an attributable effect on the traditional CHD biomarkers HDL and LDL.
Therefore, it can be seen that HGL diets affect all of the aforementioned serological biomarkers in such a manner that the risk for CHD would be increased. The negative effects of HGL diets on a patient’s risk for CHD can thus be quantified in a general sense through the consideration of the connection graph in Figure 3. Furthermore, it is possible to consider patient-specific reactions to HGL diets by measuring said patients biomarker levels.
It is thus evident that two of the major aspects of HGL diets which serve to increase the relative risk for CHD would be the hyperglycaemia and hyperinsulinaemia that may result from these diets. Both these factors are also associated with a greatly increased risk for CHD.
Further potential mediation of CHD risk may also be due to increased fibrinogen levels as a result of hyperinsulinaemia. HGL diets also have adverse impacts on lipids levels through decreased levels of HDL and increased levels of LDL, both conditions of which serve to increase the risk of CHD.
In general, based on a recent meta-analysis of eight studies where modest heterogeneity was present [175], HGL diets are associated with an increased RR of 1.36 (95% confidence interval 1.13 to 1.63). This smaller-than-expected RR effect can be somewhat explained by the heterogeneity of the study, i.e. the difference in risk between men and women. In general, women have been found to have a higher relative risk for CHD in association with HGL diets [3,175].
Heterogeneity is to be expected in the combined risk for CHD as some studies have found that there is no increased risk due to HGL diets in men [176], while other studies have found no increased risk association with women [177].
Discussion
As can be seen from the preceding discussion, the adoption of HGL diets can have negative impacts on the pathogenesis of CHD which is evident through the modification of several CHD biomarkers. The implication from this is that an increased risk for CHD is observed with the consumption of HGL diets. It is therefore the opinion of the authors that modern dietary guidelines for patients at risk of CHD should reflect this as there is an inadvertent danger of consuming a HGL diet based on current dietary guidelines.
The latest AHA dietary guidelines have attempted to focus on overall diet quality, rather than on specific macronutrient content. Some emphasis was placed on restricting or increasing the consumption of certain types of foods, such as increasing high-fibre foods and decreasing high-trans-fat foods [4]. However, these and previous guidelines have inadvertently caused the adoption of high-carbohydrate diets in order to increase fibre intake and reduce trans-fats [173,178,179] which may lead to HGL diets.
It is acknowledged that the intent of the AHA guidelines was never to increase carbohydrate intake, but instead to increase the intake of fibre through high-fibre carbohydrates and to decrease the consumption of saturated fats. Unfortunately, many patients opt for foods that do not meet the required fibre consumption guidelines [178] which results in the inherent carbohydrates imparting a greater GL, which has been negatively associated with CHD risk in this paper and others [180].
Much of the problems with the dietary recommendations as described by the AHA is the eventual use of high-carbohydrate content foods. It has been proven that high- carbohydrate diets have adverse effects on many of the risk factors which are targeted by the AHA guidelines, including lipid profiles and blood glucose levels [172,178]. A comparison of three different diets by McAuley and co-workers showed that the use of the traditional AHA guideline diet proved to be the worst of the three diets for mediating the risk factors for CHD [178].
Dietary recommendations have long been focused on the type of ingested food [4,181]. However, it has recently become more evident that the type of food ingested is less important than the overall amount of calories ingested [173,178]. Therefore, adherence to any low calorie diet is more important than the specific type of diet [182].
Thus an easy-to-follow and understand diet is obviously required in order to adequately address the issue of “heart healthy” diets and CHD. It is clear from Figure 1 that there is an abundance of links between the hallmarks of CHD and hyperglycaemia and insulin resistance from HGL diets. This was highlighted in the discussion of the pathways that are activated by HGL diets.
The importance of hyperglycaemia and insulin resistance is further highlighted by the increased risks associated with each prospective biomarker [159,164]. As the effects of HGL diets are largely dependent on carbohydrate absorption into the blood stream [127], it may be interesting to consider the effect of inhibiting this absorption. In the integrated system, in Figure 1, the pathway representing carbohydrate absorption is pathway-17, which as indicated can be regulated with the use of α-glucosidase inhibitors [183].
The α-glucosidase inhibitors thus give some insight into the effect of reduced carbohydrate consumption, as would be possible to achieve with a low GL diet. The α-glucosidase inhibitor acarbose has been successfully employed to counteract the effects of carbohydrates in diabetic patients [184,185].
The use of α-glucosidase inhibitors serves to delay the breakdown of carbohydrates in the gut, which slows down the absorption of sugars [183]. This reduces plasma glucose levels, which in turn reduces the requirement of plasma insulin, both risk factors for CHD (Figure 2).
If one then considers that the use of acarbose in diabetic patients resulted in a much lower incidence of CHD according to a meta-analysis of seven studies compromising 2180 patients. It was found that the RR for CHD was 0.36 (95% CI 0.16 to 0.80) in diabetic patients using acarbose compared to the control group [186]. This equates to a 2.78-fold reduction in CHD risk when using our notation.
This substantial relative risk reduction achieved with acarbose [186] accentuates the importance of the specific path on which this pharmacotherapeutic acts (Pathway 17). Through the inhibition of carbohydrate digestion in the stomach, α-glucosidase inhibitors reduce blood glucose levels (HbA1c) and reduce insulin levels, increasing insulin sensitivity. Therefore, if α-glucosidase inhibitors are effective to regulate blood glucose levels and insulin resistance, then much of the risk reduction can be explained by the combined effects of decreased blood glucose levels and increased insulin sensitivity [187].
It is important to note that the CHD risk reduction effects that have been observed from treatment with α-glucosidase inhibitors were found in studies on patients with type 2 diabetes mellitus [186]. It is thus conceivable that the reductions in CHD risk achieved could be greater than expected due to the increased risk for CHD associated with type 2 diabetes mellitus [188]. However the underlying effect of α-glucosidase inhibitors on blood glucose and insulin levels may retain it as a suitable candidate for treatment and prevention of CHD in non-diabetic patients.
The effectiveness of α-glucosidase inhibitors in reducing CHD risk in diabetic patients clearly elucidates the importance of the main pathways which they regulate with regards to CHD. This may therefore indicate the importance of regulating these pathways in non-diabetic patients to prevent CHD, such as through the adoption of low GL diets.
Conclusions
The authors were intrigued by the possible negative effects of HGL diets on a patient’s risk for CHD as well as the over emphasis of LDL cholesterol. As LDL is not the only or even the most important biomarker for CHD risk, a more detailed integrated view of diet and the CHD mechanism as well as its biomarkers were attempted.
The integrative view highlights the increased potential CHD risk that is associated with HGL diets. This potential risk is clearly elucidated in the wide range of CHD pathogenetic pathways which are mediated by HGL diets and the large array of CHD biomarkers which are affected as vividly shown in the simplified “connection graph”. HGL diets do not only influence the lipid and metabolic biomarkers, but also coagulation and vascular function biomarkers.
The use of α-glucosidase inhibitors is also found as substantially beneficial in CHD prevention efforts in diabetic patients by controlling important pathways shown in the integrated view of CHD. This further emphasises the importance of blood glucose and insulin levels in the prevention of CHD in diabetic patients. The array of biomarkers affected by these pharmacotherapeutic interventions would also indicate that these conditions could be of importance to non-diabetic patients.
Acknowledgements
The angel investor was Dr Arnold van Dyk. TEMM International (Pty) Ltd funded this study. We also acknowledge the fact that the integrated view is relevant to other lifestyle issues and for full comprehension will have to be replicated again in other articles describing these.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All of the authors have been involved in the writing of this manuscript and have read and approved the final text.
Contributor Information
Marc J Mathews, Email: mjmathews@rems2.com.
Leon Liebenberg, Email: lliebenberg@researchtoolbox.com.
Edward H Mathews, Email: ehmathews@researchtoolbox.com.
References
- 1.Mathers CD, Boerma T, Fat DM. Global and regional causes of death. Br Med Bull. 2009;92:7–32. doi: 10.1093/bmb/ldp028. [DOI] [PubMed] [Google Scholar]
- 2.Goff DC, Lloyd-Jones DM, Bennett G, Coady S, D’Agostino RB, Gibbons R, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association task force on practice guidelines. J Am Coll Cardiol. 2014;129:49S–73. doi: 10.1016/j.jacc.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mirrahimi A, de Souza RJ, Chiavaroli L, Sievenpiper JL, Beyene J, Hanley AJ, et al. Associations of glycemic index and load with coronary heart disease events: a systematic review and meta-analysis of prospective cohorts. J Am Heart Assoc. 2012;1:e000752. doi: 10.1161/JAHA.112.000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lichtenstein AH, Appel LJ, Brands M, Carnethon M, Daniels S, Franch HA, et al. Diet and lifestyle recommendations revision 2006: a scientific statement from the American Heart Association nutrition committee. Circulation. 2006;114:82–96. doi: 10.1161/CIRCULATIONAHA.106.176158. [DOI] [PubMed] [Google Scholar]
- 5.Fan J, Song Y, Wang Y, Hui R, Zhang W. Dietary glycemic index, glycemic load, and risk of coronary heart disease, stroke, and stroke mortality: a systematic review with meta-analysis. PloS one. 2012;7:e52182. doi: 10.1371/journal.pone.0052182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith SC., Jr Current and future directions of cardiovascular risk prediction. Am J Cardiol. 2006;97:28–32. doi: 10.1016/j.amjcard.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 7.Kavvoura FK, Liberopoulos G, Ioannidis JP. Selection in reported epidemiological risks: an empirical assessment. PLoS Med. 2007;4:e79. doi: 10.1371/journal.pmed.0040079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zou G. A modified poisson regression approach to prospective studies with binary data. Am J Epidemiol. 2004;159:702–6. doi: 10.1093/aje/kwh090. [DOI] [PubMed] [Google Scholar]
- 9.Lusis AJ, Weiss JN. Cardiovascular networks systems-based approaches to cardiovascular disease. Circulation. 2010;121:157–70. doi: 10.1161/CIRCULATIONAHA.108.847699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lusis AJ, Attie AD, Reue K. Metabolic syndrome: from epidemiology to systems biology. Nat Rev Genet. 2008;9:819–30. doi: 10.1038/nrg2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bollen M, Keppens S, Stalmans W. Specific features of glycogen metabolism in the liver. Biochem J. 1998;336:19–31. doi: 10.1042/bj3360019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rimm EB, Williams P, Fosher K, Criqui M, Stampfer MJ. Moderate alcohol intake and lower risk of coronary heart disease: meta-analysis of effects on lipids and haemostatic factors. BMJ. 1999;319:1523–8. doi: 10.1136/bmj.319.7224.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uchiki T, Weikel KA, Jiao W, Shang F, Caceres A, Pawlak D, et al. Glycation‐altered proteolysis as a pathobiologic mechanism that links dietary glycemic index, aging, and age‐related disease (in nondiabetics) Aging Cell. 2012;11:1–13. doi: 10.1111/j.1474-9726.2011.00752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waqar AB, Koike T, Yu Y, Inoue T, Aoki T, Liu E, et al. High-fat diet without excess calories induces metabolic disorders and enhances atherosclerosis in rabbits. Atherosclerosis. 2010;213:148–55. doi: 10.1016/j.atherosclerosis.2010.07.051. [DOI] [PubMed] [Google Scholar]
- 15.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, DuGar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–86. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 17.Shanahan F. The gut microbiota-a clinical perspective on lessons learned. Nat Rev Gastroentero. 2012;9:609–14. doi: 10.1038/nrgastro.2012.145. [DOI] [PubMed] [Google Scholar]
- 18.Golbidi S, Laher I. Exercise and the cardiovascular system. Cardiol Res Pract. 2012;2012:e210852. doi: 10.1155/2012/210852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Macera CA, Hootman JM, Sniezek JE. Major public health benefits of physical activity. Arthritis Care Res. 2003;49:122–8. doi: 10.1002/art.10907. [DOI] [PubMed] [Google Scholar]
- 20.Thompson PD. Exercise and physical activity in the prevention and treatment of atherosclerotic cardiovascular disease. Arterioscler Thromb Vasc Biol. 2003;23:1319–21. doi: 10.1161/01.ATV.0000087143.33998.F2. [DOI] [PubMed] [Google Scholar]
- 21.Badrick E, Kirschbaum C, Kumari M. The relationship between smoking status and cortisol secretion. J Clin Endocrinol Metab. 2007;92:819–24. doi: 10.1210/jc.2006-2155. [DOI] [PubMed] [Google Scholar]
- 22.Reaven G, Tsao PS. Insulin resistance and compensatory hyperinsulinemia. The key player between cigarette smoking and cardiovascular disease? J Am Coll Cardiol. 2003;41:1044–7. doi: 10.1016/s0735-1097(02)02982-0. [DOI] [PubMed] [Google Scholar]
- 23.Efstathiou SP, Skeva II, Dimas C, Panagiotou A, Parisi K, Tzanoumis L, et al. Smoking cessation increases serum adiponectin levels in an apparently healthy Greek population. Atherosclerosis. 2009;205:632–6. doi: 10.1016/j.atherosclerosis.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 24.Granados-Principal S, El-Azem N, Quiles JL, Perez-Lopez P, Gonzalez A, Ramirez-Tortosa M. Relationship between cardiovascular risk factors and periodontal disease: current knowledge. In: Gasparyan AY, editor. Cardiovascular Risk Factors. Shanghai: InTech; 2012. pp. 193–216. [Google Scholar]
- 25.Meurman JH, Sanz M, Janket S-J. Oral health, atherosclerosis, and cardiovascular disease. Crit Rev Oral Biol Med. 2004;15:403–13. doi: 10.1177/154411130401500606. [DOI] [PubMed] [Google Scholar]
- 26.Fisher MA, Borgnakke WS, Taylor GW. Periodontal disease as a risk marker in coronary heart disease and chronic kidney disease. Curr Opin Nephrol Hypertens. 2010;19:519–26. doi: 10.1097/MNH.0b013e32833eda38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walker BR. Glucocorticoids and cardiovascular disease. Eur J Endocrinol. 2007;157:545–59. doi: 10.1530/EJE-07-0455. [DOI] [PubMed] [Google Scholar]
- 28.Costa R, Sanches A, Cunha TS, Moura MJCS, Tanno AP, Casarini DE. Dyslipidemia - From Prevention to Treatment. Shanghai: InTech; 2011. Dyslipidemia induced by stress; pp. 367–90. [Google Scholar]
- 29.McEwen BS. Central effects of stress hormones in health and disease: understanding the protective and damaging effects of stress and stress mediators. Eur J Pharmacol. 2008;583:174–85. doi: 10.1016/j.ejphar.2007.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Musselman DL, Evans DL, Nemeroff CB. The relationship of depression to cardiovascular disease: epidemiology, biology, and treatment. Arch Gen Psychiatry. 1998;55:580–92. doi: 10.1001/archpsyc.55.7.580. [DOI] [PubMed] [Google Scholar]
- 31.Celano CM, Huffman JC. Depression and cardiac disease: a review. Cardiol Rev. 2011;19:130–42. doi: 10.1097/CRD.0b013e31820e8106. [DOI] [PubMed] [Google Scholar]
- 32.von Känel R. Psychosocial stress and cardiovascular risk: current opinion. Swiss Med Wkly. 2012;142:w13502. doi: 10.4414/smw.2012.13502. [DOI] [PubMed] [Google Scholar]
- 33.Sher Y, Lolak S, Maldonado JR. The impact of depression in heart disease. Curr Psychiatry Rep. 2010;12:255–64. doi: 10.1007/s11920-010-0116-8. [DOI] [PubMed] [Google Scholar]
- 34.Everson-Rose SA, Lewis TT, Karavolos K, Dugan SA, Wesley D, Powell LH. Depressive symptoms and increased visceral fat in middle-aged women. Psychosom Med. 2009;71:410–6. doi: 10.1097/PSY.0b013e3181a20c9c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weber-Hamann B, Werner M, Hentschel F, Bindeballe N, Lederbogen F, Deuschle M, et al. Metabolic changes in elderly patients with major depression: evidence for increased accumulation of visceral fat at follow-up. Psychoneuroendocrino. 2006;31:347–54. doi: 10.1016/j.psyneuen.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 36.Spiegel K, Tasali E, Leproult R, Van Cauter E. Effects of poor and short sleep on glucose metabolism and obesity risk. Nat Rev Endocrinol. 2009;5:253–61. doi: 10.1038/nrendo.2009.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spiegel K, Knutson K, Leproult R, Tasali E, Van Cauter E. Sleep loss: a novel risk factor for insulin resistance and Type 2 diabetes. J Appl Physiol. 2005;99:2008–19. doi: 10.1152/japplphysiol.00660.2005. [DOI] [PubMed] [Google Scholar]
- 38.Knutson KL, Spiegel K, Penev P, Van Cauter E. The metabolic consequences of sleep deprivation. Sleep Med Rev. 2007;11:163–78. doi: 10.1016/j.smrv.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shamsuzzaman AS, Gersh BJ, Somers VK. Obstructive sleep apneaimplications for cardiac and vascular disease. JAMA. 2003;290:1906–14. doi: 10.1001/jama.290.14.1906. [DOI] [PubMed] [Google Scholar]
- 40.Jackson SP. Arterial thrombosis-insidious, unpredictable and deadly. Nat Med. 2011;17:1423–36. doi: 10.1038/nm.2515. [DOI] [PubMed] [Google Scholar]
- 41.Vykoukal D, Davies MG. Vascular biology of metabolic syndrome. J Vasc Surg. 2011;54:819–31. doi: 10.1016/j.jvs.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heinecke JW. The not-so-simple HDL story: a new era for quantifying HDL and cardiovascular risk? Nat Med. 2012;18:1346–7. doi: 10.1038/nm.2930. [DOI] [PubMed] [Google Scholar]
- 43.Rader DJ, Tall AR. The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis? Nat Med. 2012;18:1344–6. doi: 10.1038/nm.2937. [DOI] [PubMed] [Google Scholar]
- 44.Vasan RS. Biomarkers of cardiovascular disease molecular basis and practical considerations. Circulation. 2006;113:2335–62. doi: 10.1161/CIRCULATIONAHA.104.482570. [DOI] [PubMed] [Google Scholar]
- 45.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–25. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 46.Aronson D, Rayfield EJ. How hyperglycemia promotes atherosclerosis: molecular mechanisms. Cardiovasc Diabetol. 2002;1:1–10. doi: 10.1186/1475-2840-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beckman JA, Creager MA, Libby P. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA. 2002;287:2570–81. doi: 10.1001/jama.287.19.2570. [DOI] [PubMed] [Google Scholar]
- 48.Rydén L, Standl E, Bartnik M, Van den Berghe G, Betteridge J, De Boer M-J, et al. Guidelines on diabetes, pre-diabetes, and cardiovascular diseases: executive summary The Task Force on Diabetes and Cardiovascular Diseases of the European Society of Cardiology (ESC) and of the European Association for the Study of Diabetes (EASD) Eur Heart J. 2007;28:88–136. doi: 10.1093/eurheartj/ehl260. [DOI] [PubMed] [Google Scholar]
- 49.Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365:1333–46. doi: 10.1016/S0140-6736(05)61032-X. [DOI] [PubMed] [Google Scholar]
- 50.Gerstein HC, Miller ME, Genuth S, Ismail-Beigi F, Buse JB, Goff DC, Jr, et al. Long-term effects of intensive glucose lowering on cardiovascular outcomes. N Engl J Med. 2011;364:818–28. doi: 10.1056/NEJMoa1006524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khardori R, Nguyen DD. Glucose control and cardiovascular outcomes: reorienting approach. Front Endocrinol (Lausanne) 2012;3:1–5. doi: 10.3389/fendo.2012.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gorgojo Martínez JJ. Glucocentricity or adipocentricity: a critical view of consensus and clinical guidelines for the treatment of type 2 diabetes mellitus. Endocrinol Nutr. 2011;58:541–9. doi: 10.1016/j.endonu.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 53.Zoungas S, Chalmers J, Ninomiya T, Li Q, Cooper M, Colagiuri S, et al. Association of HbA1c levels with vascular complications and death in patients with type 2 diabetes: evidence of glycaemic thresholds. Diabetologia. 2012;55:636–43. doi: 10.1007/s00125-011-2404-1. [DOI] [PubMed] [Google Scholar]
- 54.Leibundgut G, Arai K, Orsoni A, Yin H, Scipione C, Miller ER, et al. Oxidized phospholipids are present on plasminogen, affect fibrinolysis, and increase following acute myocardial infarction. J Am Coll Cardiol. 2012;59:1426–37. doi: 10.1016/j.jacc.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tsimikas S, Hall JL. Lipoprotein (a) as a potential causal genetic risk factor of cardiovascular disease: a rationale for increased efforts to understand its pathophysiology and develop targeted therapies. J Am Coll Cardiol. 2012;60:716–21. doi: 10.1016/j.jacc.2012.04.038. [DOI] [PubMed] [Google Scholar]
- 56.Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–17. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levitan EB, Cook NR, Stampfer MJ, Ridker PM, Rexrode KM, Buring JE, et al. Dietary glycemic index, dietary glycemic load, blood lipids, and C-reactive protein. Metabolism. 2008;57:437–43. doi: 10.1016/j.metabol.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takefuji S, Yatsuya H, Tamakoshi K, Otsuka R, Wada K, Matsushita K, et al. Smoking status and adiponectin in healthy Japanese men and women. Prev Med. 2007;45:471–5. doi: 10.1016/j.ypmed.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 59.Tsai J-S, Guo F-R, Chen S-C, Lue B-H, Chiu T-Y, Chen C-Y, et al. Smokers show reduced circulating adiponectin levels and adiponectin mRNA expression in peripheral blood mononuclear cells. Atherosclerosis. 2011;218:168–73. doi: 10.1016/j.atherosclerosis.2011.04.025. [DOI] [PubMed] [Google Scholar]
- 60.Libby P. Atherosclerosis in inflammation. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 61.Packard RR, Libby P. Inflammation in atherosclerosis: from vascular biology to biomarker discovery and risk prediction. Clin Chem. 2008;54:24–38. doi: 10.1373/clinchem.2007.097360. [DOI] [PubMed] [Google Scholar]
- 62.Epstein FH, Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340:115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 63.Trepels T, Zeiher AM, Fichtlscherer S. The endothelium and inflammation. Endothelium. 2006;13:423–9. doi: 10.1080/10623320601061862. [DOI] [PubMed] [Google Scholar]
- 64.Paquette DW, Brodala N, Nichols TC. Cardiovascular disease, inflammation, and periodontal infection. Periodontol 2000. 2007;44:113–26. doi: 10.1111/j.1600-0757.2006.00196.x. [DOI] [PubMed] [Google Scholar]
- 65.Persson GR, Persson RE. Cardiovascular disease and periodontitis: an update on the associations and risk. J Clin Periodontol. 2008;35:362–79. doi: 10.1111/j.1600-051X.2008.01281.x. [DOI] [PubMed] [Google Scholar]
- 66.Kebschull M, Demmer R, Papapanou P. “Gum bug, leave my heart alone!”-epidemiologic and mechanistic evidence linking periodontal infections and atherosclerosis. J Dent Res. 2010;89:879–902. doi: 10.1177/0022034510375281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Machuca G, Segura-Egea JJ, Jiménez-Beato G, Lacalle JR, Bullón P. Clinical indicators of periodontal disease in patients with coronary heart disease: A 10 years longitudinal study. Med Oral Patol Oral Cir Bucal. 2012;17:e569. doi: 10.4317/medoral.17848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li X, Kolltveit KM, Tronstad L, Olsen I. Systemic diseases caused by oral infection. Clin Microbiol Rev. 2000;13:547–58. doi: 10.1128/cmr.13.4.547-558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27:24–31. doi: 10.1016/j.it.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Noble EE, Billington CJ, Kotz CM, Wang C. The lighter side of BDNF. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1053. doi: 10.1152/ajpregu.00776.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Feder A, Nestler EJ, Charney DS. Psychobiology and molecular genetics of resilience. Nat Rev Neurosci. 2009;10:446–57. doi: 10.1038/nrn2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Karatsoreos IN, McEwen BS. Psychobiological allostasis: resistance, resilience and vulnerability. Trends Cogn Sci. 2011;15:576–84. doi: 10.1016/j.tics.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 74.Calabrese F, Molteni R, Racagni G, Riva MA. Neuronal plasticity: a link between stress and mood disorders. Psychoneuroendocrino. 2009;34:S208–16. doi: 10.1016/j.psyneuen.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 75.Liebenberg L, Mathews EH. A practical quantification of blood glucose production due to high-level chronic stress. Stress Health. 2012;28:327–32. doi: 10.1002/smi.2415. [DOI] [PubMed] [Google Scholar]
- 76.Kubzansky LD, Kawachi I, Spiro A, Weiss ST, Vokonas PS, Sparrow D. Is worrying bad for your heart? A prospective study of worry and coronary heart disease in the Normative Aging Study. Circulation. 1997;95:818–24. doi: 10.1161/01.cir.95.4.818. [DOI] [PubMed] [Google Scholar]
- 77.Einvik G, Ekeberg Ø, Klemsdal TO, Sandvik L, Hjerkinn EM. Physical distress is associated with cardiovascular events in a high risk population of elderly men. BMC Cardiovasc Disord. 2009;9:14. doi: 10.1186/1471-2261-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vogelzangs N, Seldenrijk A, Beekman AT, van Hout HP, de Jonge P, Penninx BW. Cardiovascular disease in persons with depressive and anxiety disorders. J Affect Disord. 2010;125:241–8. doi: 10.1016/j.jad.2010.02.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dungan KM, Braithwaite SS, Preiser J-C. Stress hyperglycaemia. Lancet. 2009;373:1798–807. doi: 10.1016/S0140-6736(09)60553-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dallman MF, Akana SF, Laugero KD, Gomez F, Manalo S, Bell M, et al. A spoonful of sugar: feedback signals of energy stores and corticosterone regulate responses to chronic stress. Physiol Behav. 2003;79:3–12. doi: 10.1016/s0031-9384(03)00100-8. [DOI] [PubMed] [Google Scholar]
- 81.Weissman C. The metabolic response to stress: an overview and update. Anesthesiology. 1990;73:308–27. doi: 10.1097/00000542-199008000-00020. [DOI] [PubMed] [Google Scholar]
- 82.Kohler M, Stradling JR. Mechanisms of vascular damage in obstructive sleep apnea. Nat Rev Cardiol. 2010;7:677–85. doi: 10.1038/nrcardio.2010.145. [DOI] [PubMed] [Google Scholar]
- 83.Levy P, Bonsignore M, Eckel J. Sleep, sleep-disordered breathing and metabolic consequences. Eur Respir J. 2009;34:243–60. doi: 10.1183/09031936.00166808. [DOI] [PubMed] [Google Scholar]
- 84.Punjabi NM, Polotsky VY. Disorders of glucose metabolism in sleep apnea. J Appl Physiol. 2005;99:1998–2007. doi: 10.1152/japplphysiol.00695.2005. [DOI] [PubMed] [Google Scholar]
- 85.Besedovsky L, Lange T, Born J. Sleep and immune function. Pflügers Arch Eur J Physiol. 2012;463:121–37. doi: 10.1007/s00424-011-1044-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oliveira CS, Giuffrida F, Crispim F, Saddi-Rosa P, Reis AF. ADIPOQ and adiponectin: the common ground of hyperglycemia and coronary artery disease? Arq Bras Endocrinol Metabol. 2011;55:446–54. doi: 10.1590/s0004-27302011000700003. [DOI] [PubMed] [Google Scholar]
- 87.Ip MS, Lam B, Ng MM, Lam WK, Tsang KW, Lam KS. Obstructive sleep apnea is independently associated with insulin resistance. Am J Respir Crit Care Med. 2002;165:670–6. doi: 10.1164/ajrccm.165.5.2103001. [DOI] [PubMed] [Google Scholar]
- 88.Bornfeldt KE, Tabas I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell. 2011;14:575–85. doi: 10.1016/j.cmet.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocr Rev. 2007;28:463–91. doi: 10.1210/er.2007-0006. [DOI] [PubMed] [Google Scholar]
- 90.Nigro J, Osman N, Dart AM, Little PJ. Insulin resistance and atherosclerosis. Endocr Rev. 2006;27:242–59. doi: 10.1210/er.2005-0007. [DOI] [PubMed] [Google Scholar]
- 91.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 92.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–71. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stocker R, Keaney JF. Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 94.Van Gaal LF, Mertens IL, Christophe E. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–80. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- 95.Vettore M, Leao A, Monteiro Da Silva A, Quintanilha R, Lamarca G. The relationship of stress and anxiety with chronic periodontitis. J Clin Periodontol. 2003;30:394–402. doi: 10.1034/j.1600-051x.2003.10271.x. [DOI] [PubMed] [Google Scholar]
- 96.Ng SK, Keung Leung W. A community study on the relationship between stress, coping, affective dispositions and periodontal attachment loss. Community Dent Oral Epidemiol. 2006;34:252–66. doi: 10.1111/j.1600-0528.2006.00282.x. [DOI] [PubMed] [Google Scholar]
- 97.Michel T, Vanhoutte PM. Cellular signaling and NO production. Pflügers Arch Eur J Physiol. 2010;459:807–16. doi: 10.1007/s00424-009-0765-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Biondi-Zoccai GG, Abbate A, Liuzzo G, Biasucci LM. Atherothrombosis, inflammation, and diabetes. J Am Coll Cardiol. 2003;41:1071–7. doi: 10.1016/s0735-1097(03)00088-3. [DOI] [PubMed] [Google Scholar]
- 99.Schadt EE. Molecular networks as sensors and drivers of common human diseases. Nature. 2009;461:218–23. doi: 10.1038/nature08454. [DOI] [PubMed] [Google Scholar]
- 100.Kitano H, Oda K, Kimura T, Matsuoka Y, Csete M, Doyle J, et al. Metabolic syndrome and robustness tradeoffs. Diabetes. 2004;53:S6–15. doi: 10.2337/diabetes.53.suppl_3.s6. [DOI] [PubMed] [Google Scholar]
- 101.Wang JC, Bennett M. Aging and atherosclerosis mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. 2012;111:245–59. doi: 10.1161/CIRCRESAHA.111.261388. [DOI] [PubMed] [Google Scholar]
- 102.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–28. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 103.Mazzone T, Chait A, Plutzky J. Cardiovascular disease risk in type 2 diabetes mellitus: insights from mechanistic studies. Lancet. 2008;371:1800–9. doi: 10.1016/S0140-6736(08)60768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Diez D, Wheelock ÅM, Goto S, Haeggström JZ, Paulsson-Berne G, Hansson GK, et al. The use of network analyses for elucidating mechanisms in cardiovascular disease. Mol Biosyst. 2010;6:289–304. doi: 10.1039/b912078e. [DOI] [PubMed] [Google Scholar]
- 105.Libby P, DiCarli M, Weissleder R. The vascular biology of atherosclerosis and imaging targets. J Nucl Med. 2010;51:33S–7. doi: 10.2967/jnumed.109.069633. [DOI] [PubMed] [Google Scholar]
- 106.Mougios V. Exercise biochemistry. Champaign, IL: Human Kinetics; 2006. [Google Scholar]
- 107.Wykrzykowska J, Lehman S, Williams G, Parker JA, Palmer MR, Varkey S, et al. Imaging of inflamed and vulnerable plaque in coronary arteries with 18F-FDG PET/CT in patients with suppression of myocardial uptake using a low-carbohydrate, high-fat preparation. J Nucl Med. 2009;50:563–8. doi: 10.2967/jnumed.108.055616. [DOI] [PubMed] [Google Scholar]
- 108.Sanz J, Fayad ZA. Imaging of atherosclerotic cardiovascular disease. Nature. 2008;451:953–7. doi: 10.1038/nature06803. [DOI] [PubMed] [Google Scholar]
- 109.Rudd JH, Hyafil F, Fayad ZA. Inflammation imaging in atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:1009–16. doi: 10.1161/ATVBAHA.108.165563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Christen T, Sheikine Y, Rocha VZ, Hurwitz S, Goldfine AB, Di Carli M, et al. Increased glucose uptake in visceral versus subcutaneous adipose tissue revealed by PET imaging. JACC Cardiovasc Imaging. 2010;3:843–51. doi: 10.1016/j.jcmg.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest. 2001;108:1341–8. doi: 10.1172/JCI11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vanhoutte P, Shimokawa H, Tang E, Feletou M. Endothelial dysfunction and vascular disease. Acta Physiol Scand. 2009;196:193–222. doi: 10.1111/j.1748-1716.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- 113.Rader DJ, Daugherty A. Translating molecular discoveries into new therapies for atherosclerosis. Nature. 2008;451:904–13. doi: 10.1038/nature06796. [DOI] [PubMed] [Google Scholar]
- 114.Pountos I, Georgouli T, Bird H, Giannoudis PV. Nonsteroidal anti-inflammatory drugs: prostaglandins, indications, and side effects. Int J Infereron Cytokine Mediator Res. 2011;3:19–27. [Google Scholar]
- 115.Chan D, Ng LL. Biomarkers in acute myocardial infarction. BMC Med. 2010;8:34. doi: 10.1186/1741-7015-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Singh V, Tiwari RL, Dikshit M, Barthwal MK. Models to study atherosclerosis: a mechanistic insight. Curr Vasc Pharmacol. 2009;7:75–109. doi: 10.2174/157016109787354097. [DOI] [PubMed] [Google Scholar]
- 117.Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS. Nuclear factor-κB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci U S A. 2010;107:2669–74. doi: 10.1073/pnas.0910658107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D, et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci U S A. 2003;100:1920–5. doi: 10.1073/pnas.0438019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.von Känel R, Mills PJ, Fainman C, Dimsdale JE. Effects of psychological stress and psychiatric disorders on blood coagulation and fibrinolysis: a biobehavioral pathway to coronary artery disease? Psychosom Med. 2001;63:531–44. doi: 10.1097/00006842-200107000-00003. [DOI] [PubMed] [Google Scholar]
- 120.Haroon E, Raison CL, Miller AH. Psychoneuroimmunology meets neuropsychopharmacology: translational implications of the impact of inflammation on behavior. Neuropsychopharmacol. 2012;37:137–62. doi: 10.1038/npp.2011.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.von der Thüsen JH, Borensztajn KS, Moimas S, van Heiningen S, Teeling P, van Berkel TJ, et al. IGF-1 has plaque-stabilizing effects in atherosclerosis by altering vascular smooth muscle cell phenotype. Am J Pathol. 2011;178:924–34. doi: 10.1016/j.ajpath.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shai S-Y, Sukhanov S, Higashi Y, Vaughn C, Rosen CJ, Delafontaine P. Low circulating insulin-like growth factor I increases atherosclerosis in ApoE-deficient mice. Am J Physiol Heart Circ Physiol. 2011;300:H1898. doi: 10.1152/ajpheart.01081.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ruidavets J, Luc G, Machez E, Genoux A, Kee F, Arveiler D, et al. Effects of insulin-like growth factor 1 in preventing acute coronary syndromes: The PRIME study. Atherosclerosis. 2011;218:464–9. doi: 10.1016/j.atherosclerosis.2011.05.034. [DOI] [PubMed] [Google Scholar]
- 124.Higashi Y, Sukhanov S, Anwar A, Shai S-Y, Delafontaine P. Aging, atherosclerosis, and IGF-1. J Gerontol A Biol Sci Med Sci. 2012;67:626–39. doi: 10.1093/gerona/gls102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Krishnadas R, Cavanagh J. Depression: an inflammatory illness? J Neurol Neurosurg Psychiatry. 2012;83:495–502. doi: 10.1136/jnnp-2011-301779. [DOI] [PubMed] [Google Scholar]
- 126.Gardner A, Boles RG. Beyond the serotonin hypothesis: mitochondria, inflammation and neurodegeneration in major depression and affective spectrum disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:730–43. doi: 10.1016/j.pnpbp.2010.07.030. [DOI] [PubMed] [Google Scholar]
- 127.Ludwig DS. The glycemic index: physiological mechanisms relating to obesity, diabetes, and cardiovascular disease. JAMA. 2002;287:2414–23. doi: 10.1001/jama.287.18.2414. [DOI] [PubMed] [Google Scholar]
- 128.Strasser B. Physical activity in obesity and metabolic syndrome. Ann N Y Acad Sci. 2013;1281:141–59. doi: 10.1111/j.1749-6632.2012.06785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rose AJ, Richter EA. Skeletal muscle glucose uptake during exercise: how is it regulated? Physiology. 2005;20:260–70. doi: 10.1152/physiol.00012.2005. [DOI] [PubMed] [Google Scholar]
- 130.Raynaud E, Pérez-Martin A, Brun J-F, Aissa-Benhaddad A, Fédou C, Mercier J. Relationships between fibrinogen and insulin resistance. Atherosclerosis. 2000;150:365–70. doi: 10.1016/s0021-9150(99)00373-1. [DOI] [PubMed] [Google Scholar]
- 131.Meigs JB, Mittleman MA, Nathan DM, Tofler GH, Singer DE, Murphy-Sheehy PM, et al. Hyperinsulinemia, hyperglycemia, and impaired hemostasis: the Framingham Offspring study. JAMA. 2000;283:221–8. doi: 10.1001/jama.283.2.221. [DOI] [PubMed] [Google Scholar]
- 132.Cimenti C, Schlagenhauf A, Leschnik B, Schretter M, Tschakert G, Gröschl W, et al. Low endogenous thrombin potential in trained subjects. Thromb Res. 2013;131:e281–5. doi: 10.1016/j.thromres.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 133.Rauramaa R, Salonen JT, Seppänen K, Salonen R, Venäläinen J, Ihanainen M, et al. Inhibition of platelet aggregability by moderate-intensity physical exercise: a randomized clinical trial in overweight men. Circulation. 1986;74:939–44. doi: 10.1161/01.cir.74.5.939. [DOI] [PubMed] [Google Scholar]
- 134.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 135.Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–6. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- 136.Guo Z. Intramyocellular lipid kinetics and insulin resistance. Lipids Health Dis. 2007;6:421. doi: 10.1186/1476-511X-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–30. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Warburton DE, Nicol CW, Bredin SS. Health benefits of physical activity: the evidence. Can Med Assoc J. 2006;174:801–9. doi: 10.1503/cmaj.051351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Reaven GM. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 140.Klein S, Burke LE, Bray GA, Blair S, Allison DB, Pi-Sunyer X, et al. Clinical implications of obesity with specific focus on cardiovascular disease. A statement for professionals from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism: endorsed by the American College of Cardiology Foundation. Circulation. 2004;110:2952–67. doi: 10.1161/01.CIR.0000145546.97738.1E. [DOI] [PubMed] [Google Scholar]
- 141.Brown NJ, Agirbasli MA, Williams GH, Litchfield WR, Vaughan DE. Effect of activation and inhibition of the renin-angiotensin system on plasma PAI-1. Hypertension. 1998;32:965–71. doi: 10.1161/01.hyp.32.6.965. [DOI] [PubMed] [Google Scholar]
- 142.Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL, et al. Seventh report of the joint National Committee on Prevention, detection, evaluation, and treatment of high blood pressure. Hypertension. 2003;42:1206–52. doi: 10.1161/01.HYP.0000107251.49515.c2. [DOI] [PubMed] [Google Scholar]
- 143.Becker AE, de Boer OJ, van der Wal AC. The role of inflammation and infection in coronary artery disease. Annu Rev Med. 2001;52:289–97. doi: 10.1146/annurev.med.52.1.289. [DOI] [PubMed] [Google Scholar]
- 144.Liu S, Willett WC, Stampfer MJ, Hu FB, Franz M, Sampson L, et al. A prospective study of dietary glycemic load, carbohydrate intake, and risk of coronary heart disease in US women. Am J Clin Nutr. 2000;71:1455–61. doi: 10.1093/ajcn/71.6.1455. [DOI] [PubMed] [Google Scholar]
- 145.Denova-Gutiérrez E, Huitrón-Bravo G, Talavera JO, Castañón S, Gallegos-Carrillo K, Flores Y, et al. Dietary glycemic index, dietary glycemic load, blood lipids, and coronary heart disease. J Nutr Metab. 2010;2010:e170680. doi: 10.1155/2010/170680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000. doi: 10.1001/jama.2009.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sniderman AD, Williams K, Contois JH, Monroe HM, McQueen MJ, de Graaf J, et al. A meta-analysis of low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk. Circ Cardiovasc Qual Outcomes. 2011;4:337–45. doi: 10.1161/CIRCOUTCOMES.110.959247. [DOI] [PubMed] [Google Scholar]
- 148.Luc G, Empana J, Morange P, Juhan-Vague I, Arveiler D, Ferrieres J, et al. Adipocytokines and the risk of coronary heart disease in healthy middle aged men: the PRIME Study. Int J Obes. 2009;34:118–26. doi: 10.1038/ijo.2009.204. [DOI] [PubMed] [Google Scholar]
- 149.Kaptoge S, Di Angelantonio E, Pennells L, Wood AM, White IR, Gao P, et al. C-reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med. 2012;367:1310–20. doi: 10.1056/NEJMoa1107477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kaptoge S, Seshasai SRK, Gao P, Freitag DF, Butterworth AS, Borglykke A, et al. Inflammatory cytokines and risk of coronary heart disease: new prospective study and updated meta-analysis. Eur Heart J. 2014;35:578–89. doi: 10.1093/eurheartj/eht367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Daniels LB, Clopton P, Laughlin GA, Maisel AS, Barrett-Connor E. Growth-differentiation factor-15 is a robust, independent predictor of 11-year mortality risk in community-dwelling older adults the Rancho Bernardo Study. Circulation. 2011;123:2101–10. doi: 10.1161/CIRCULATIONAHA.110.979740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mogelvang R, Pedersen SH, Flyvbjerg A, Bjerre M, Iversen AZ, Galatius S, et al. Comparison of osteoprotegerin to traditional atherosclerotic risk factors and high-sensitivity C-reactive protein for diagnosis of atherosclerosis. Am J Cardiol. 2012;109:515–20. doi: 10.1016/j.amjcard.2011.09.043. [DOI] [PubMed] [Google Scholar]
- 153.Rana JS, Arsenault BJ, Després J-P, Côté M, Talmud PJ, Ninio E, et al. Inflammatory biomarkers, physical activity, waist circumference, and risk of future coronary heart disease in healthy men and women. Eur Heart J. 2009;32:336–44. doi: 10.1093/eurheartj/ehp010. [DOI] [PubMed] [Google Scholar]
- 154.Di Angelantonio E, Chowdhury R, Sarwar N, Ray KK, Gobin R, Saleheen D, et al. B-type natriuretic peptides and cardiovascular risk systematic review and meta-analysis of 40 prospective studies. Circulation. 2009;120:2177–87. doi: 10.1161/CIRCULATIONAHA.109.884866. [DOI] [PubMed] [Google Scholar]
- 155.Humphrey LL, Fu R, Rogers K, Freeman M, Helfand M. Homocysteine level and coronary heart disease incidence: a systematic review and meta-analysis. Mayo Clin Proc. 2008;83:1203–12. doi: 10.4065/83.11.1203. [DOI] [PubMed] [Google Scholar]
- 156.Homocysteine Studies Collaboration Homocysteine and risk of ischemic heart disease and stroke: a meta-analysis. JAMA. 2002;288:2015–22. doi: 10.1001/jama.288.16.2015. [DOI] [PubMed] [Google Scholar]
- 157.Wang TJ, Wollert KC, Larson MG, Coglianese E, McCabe EL, Cheng S, et al. Prognostic utility of novel biomarkers of cardiovascular stress: the Framingham Heart Study. Circulation. 2012;126:1596–604. doi: 10.1161/CIRCULATIONAHA.112.129437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kistorp C, Raymond I, Pedersen F, Gustafsson F, Faber J, Hildebrandt P. N-terminal pro-brain natriuretic peptide, c-reactive protein, and urinary albumin levels as predictors of mortality and cardiovascular events in older adults. JAMA. 2005;293:1609–16. doi: 10.1001/jama.293.13.1609. [DOI] [PubMed] [Google Scholar]
- 159.Pai JK, Cahill LE, Hu FB, Rexrode KM, Manson JE, Rimm EB. Hemoglobin A1c is associated with increased risk of incident coronary heart disease among apparently healthy, nondiabetic men and women. J Am Heart Assoc. 2013;2:e000077. doi: 10.1161/JAHA.112.000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Schneider HJ, Wallaschofski H, Volzke H, Markus MR, Doerr M, Felix SB, et al. Incremental effects of endocrine and metabolic biomarkers and abdominal obesity on cardiovascular mortality prediction. PloS one. 2012;7:e33084. doi: 10.1371/journal.pone.0033084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kanhai D, Kranendonk M, Uiterwaal C, Graaf Y, Kappelle L, Visseren F. Adiponectin and incident coronary heart disease and stroke. A systematic review and meta‐analysis of prospective studies. Obes Rev. 2013;14:555–67. doi: 10.1111/obr.12027. [DOI] [PubMed] [Google Scholar]
- 162.Smith GD, Ben-Shlomo Y, Beswick A, Yarnell J, Lightman S, Elwood P. Cortisol, testosterone, and coronary heart disease: prospective evidence from the Caerphilly study. Circulation. 2005;112:332–40. doi: 10.1161/CIRCULATIONAHA.104.489088. [DOI] [PubMed] [Google Scholar]
- 163.Hamer M, Endrighi R, Venuraju SM, Lahiri A, Steptoe A. Cortisol responses to mental stress and the progression of coronary artery calcification in healthy men and women. PloS One. 2012;7:e31356. doi: 10.1371/journal.pone.0031356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gast KB, Tjeerdema N, Stijnen T, Smit JW, Dekkers OM. Insulin resistance and risk of incident cardiovascular events in adults without diabetes: meta-analysis. PloS one. 2012;7:e52036. doi: 10.1371/journal.pone.0052036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Brand-Miller JC, Holt SH, Pawlak DB, McMillan J. Glycemic index and obesity. Am J Clin Nutr. 2002;76:281S–5. doi: 10.1093/ajcn/76/1.281S. [DOI] [PubMed] [Google Scholar]
- 166.Kohnert K-D, Augstein P, Heinke P, Zander E, Peterson K, Freyse E-J, et al. Chronic hyperglycemia but not glucose variability determines HbA1c levels in well-controlled patients with type 2 diabetes. Diabetes Res Clin Pract. 2007;77:420–6. doi: 10.1016/j.diabres.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 167.Henquin J-C. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49:1751–60. doi: 10.2337/diabetes.49.11.1751. [DOI] [PubMed] [Google Scholar]
- 168.Laakso M. How good a marker is insulin level for insulin resistance? Am J Epidemiol. 1993;137:959–65. doi: 10.1093/oxfordjournals.aje.a116768. [DOI] [PubMed] [Google Scholar]
- 169.Willett W, Manson J, Liu S. Glycemic index, glycemic load, and risk of type 2 diabetes. Am J Clin Nutr. 2002;76:274S–80. doi: 10.1093/ajcn/76/1.274S. [DOI] [PubMed] [Google Scholar]
- 170.Pischon T, Girman CJ, Hotamisligil GS, Rifai N, Hu FB, Rimm EB. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA. 2004;291:1730–7. doi: 10.1001/jama.291.14.1730. [DOI] [PubMed] [Google Scholar]
- 171.Válek J, Válková L, Vlasáková Z, Topinka V. Increased fibrinogen levels in the offspring of hypertensive men: relation with hyperinsulinemia and the metabolic syndrome. Arterioscler Thromb Vasc Biol. 1995;15:2229–33. doi: 10.1161/01.atv.15.12.2229. [DOI] [PubMed] [Google Scholar]
- 172.Jeppesen J, Schaaf P, Jones C, Zhou M, Chen Y, Reaven G. Effects of low-fat, high-carbohydrate diets on risk factors for ischemic heart disease in postmenopausal women. Am J Clin Nutr. 1997;65:1027–33. doi: 10.1093/ajcn/65.4.1027. [DOI] [PubMed] [Google Scholar]
- 173.Dumesnil JG, Turgeon J, Tremblay A, Poirier P, Gilbert M, Gagnon L, et al. Effect of a low-glycaemic index–low-fat–high protein diet on the atherogenic metabolic risk profile of abdominally obese men. Br J Nutr. 2001;86:557–68. doi: 10.1079/bjn2001427. [DOI] [PubMed] [Google Scholar]
- 174.Liu S, Manson JE, Stampfer MJ, Holmes MD, Hu FB, Hankinson SE, et al. Dietary glycemic load assessed by food-frequency questionnaire in relation to plasma high-density-lipoprotein cholesterol and fasting plasma triacylglycerols in postmenopausal women. Am J Clin Nutr. 2001;73:560–6. doi: 10.1093/ajcn/73.3.560. [DOI] [PubMed] [Google Scholar]
- 175.Dong J-Y, Zhang Y-H, Wang P, Qin L-Q. Meta-analysis of dietary glycemic load and glycemic index in relation to risk of coronary heart disease. Am J Cardiol. 2012;109:1608–13. doi: 10.1016/j.amjcard.2012.01.385. [DOI] [PubMed] [Google Scholar]
- 176.Sieri S, Krogh V, Berrino F, Evangelista A, Agnoli C, Brighenti F, et al. Dietary glycemic load and index and risk of coronary heart disease in a large italian cohort: the EPICOR study. Arch Intern Med. 2010;170:640–7. doi: 10.1001/archinternmed.2010.15. [DOI] [PubMed] [Google Scholar]
- 177.Burger KN, Beulens JW, Boer JM, Spijkerman AM. Dietary glycemic load and glycemic index and risk of coronary heart disease and stroke in Dutch men and women: the EPIC-MORGEN study. PloS One. 2011;6:e25955. doi: 10.1371/journal.pone.0025955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.McAuley K, Hopkins C, Smith K, McLay R, Williams S, Taylor R, et al. Comparison of high-fat and high-protein diets with a high-carbohydrate diet in insulin-resistant obese women. Diabetologia. 2005;48:8–16. doi: 10.1007/s00125-004-1603-4. [DOI] [PubMed] [Google Scholar]
- 179.De Souza RJ, Swain JF, Appel LJ, Sacks FM. Alternatives for macronutrient intake and chronic disease: a comparison of the OmniHeart diets with popular diets and with dietary recommendations. Am J Clin Nutr. 2008;88:1–11. doi: 10.1093/ajcn/88.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hardy DS, Hoelscher DM, Aragaki C, Stevens J, Steffen LM, Pankow JS, et al. Association of glycemic index and glycemic load with risk of incident coronary heart disease among Whites and African Americans with and without type 2 diabetes: the Atherosclerosis Risk in Communities study. Ann Epidemiol. 2010;20:610–6. doi: 10.1016/j.annepidem.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Krauss RM, Eckel RH, Howard B, Appel LJ, Daniels SR, Deckelbaum RJ, et al. AHA dietary guidelines revision 2000: a statement for healthcare professionals from the Nutrition Committee of the American Heart Association. Circulation. 2000;102:2284–99. doi: 10.1161/01.cir.102.18.2284. [DOI] [PubMed] [Google Scholar]
- 182.Dansinger ML, Gleason JA, Griffith JL, Selker HP, Schaefer EJ. Comparison of the Atkins, Ornish, Weight Watchers, and Zone diets for weight loss and heart disease risk reduction: a randomized trial. JAMA. 2005;293:43–53. doi: 10.1001/jama.293.1.43. [DOI] [PubMed] [Google Scholar]
- 183.Krentz AJ, Bailey CJ. Oral antidiabetic agents. Drugs. 2005;65:385–411. doi: 10.2165/00003495-200565030-00005. [DOI] [PubMed] [Google Scholar]
- 184.Mertes G. Safety and efficacy of acarbose in the treatment of type 2 diabetes: data from a 5-year surveillance study. Diabetes Res Clin Pract. 2001;52:193–204. doi: 10.1016/s0168-8227(01)00221-2. [DOI] [PubMed] [Google Scholar]
- 185.Hasche H, Mertes G, Bruns C, Englert R, Genthner P, Heim D, et al. Effects of acarbose treatment in Type 2 diabetic patients under dietary training: a multicentre, double-blind, placebo-controlled, 2-year study. Diabetes Nutr Metab. 1999;12:277–85. [PubMed] [Google Scholar]
- 186.Hanefeld M, Cagatay M, Petrowitsch T, Neuser D, Petzinna D, Rupp M. Acarbose reduces the risk for myocardial infarction in type 2 diabetic patients: meta-analysis of seven long-term studies. Eur Heart J. 2004;25:10–6. doi: 10.1016/s0195-668x(03)00468-8. [DOI] [PubMed] [Google Scholar]
- 187.Derosa G, Maffioli P, D’Angelo A, Fogari E, Bianchi L, Cicero AF. Acarbose on insulin resistance after an oral fat load: a double-blind, placebo controlled study. J Diabetes Complications. 2011;25:258–66. doi: 10.1016/j.jdiacomp.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 188.Kannel WB, McGee D. Diabetes and glucose tolerance as risk factors for cardiovascular disease: the Framingham study. Diabetes Care. 1979;2:120–6. doi: 10.2337/diacare.2.2.120. [DOI] [PubMed] [Google Scholar]