

Abstract
The transcription factor E2F1 activates gene targets required for G1-S phase progression and for apoptosis, and exhibits increased expression levels in neurons in several CNS diseases including HIV encephalitis, Alzheimer disease, and Parkinson Disease. While E2F1 is known to regulate cell viability through activation of caspases, here we present evidence supporting the involvement of E2F1 in NMDA receptor-dependent, HIV-induced neuronal death mediated by calpains. Using an in vitro model of HIV-induced neurotoxicity that is dependent on NMDA receptor and calpain activation, we have shown that cortical neurons lacking functional E2F1 are less susceptible to neuronal death. Additionally, we report that neuronal E2F1 is cleaved by calpain to a stable 55-kiloDalton fragment following NR2B-dependent NMDA receptor stimulation. This cleavage of E2F1 is protein conformation-dependent and involves at least two cleavage events, one at each terminus of the protein. Intriguingly, the stabilized E2F1 cleavage product is produced in postmitotic neurons of all ages, but fails to be stabilized in cycling cells. Finally, we show that a matching E2F1 cleavage product is produced in human fetal neurons, suggesting that calpain cleavage of E2F1 may be produced in human cortical tissue. These results suggest neuronal E2F1 is processed in a novel manner in response to NMDA receptor-mediated toxicity, a mechanism implicated in HAND pathogenesis as well as several other diseases of the CNS.
Keywords: excitotoxicity, neuron, cell cycle, HAND, neurodegeneration, protease, calpain
Introduction
E2F1 is a highly conserved member of the E2F family, a group of nine transcription factors that coordinate a transcription program underlying cell cycle progression. E2F1-directed transcription is mediated by coordinated activity of the ‘winged helix’ DNA binding domain and C-terminal transactivation domain, which recruit basal transcription machinery to target gene promoters (Lang et al. 2001, Ross et al. 1999, Trouche et al. 1996). Embedded within the transactivation domain is the pocket binding motif for tumor suppressor Retinoblastoma protein (pRb), which binds E2F1 during quiescence and G1 to repress E2F1 activity (Helin et al. 1993, Lees et al. 1993). Hyperphosphorylation of pRb prior to S phase leads to dissociation of the pRb-E2F1 repressive complex allowing E2F1 to transactivate target genes involved in DNA replication such as DNA polymerase-α, dihydrofloate reductase, and proliferating cell nuclear antigen (DeGregori & Johnson 2006).
Aside from promoting cell proliferation, E2F1 functions as a potent inducer of apoptosis both in response to cytotoxic events such as irreparable DNA damage and during normal physiological processes like T-cell maturation (DeGregori & Johnson 2006). In these cases, E2F1 is stabilized by phosphorylation and protected from degradation through an interaction with the protein 14–3–3τ (Iaquinta & Lees 2007, Wang et al. 2004). As E2F1 accumulates, it can initiate apoptosis through transactivation of apoptotic targets such as Bax, caspase 3, 7–9, and p14ARF. Alternatively, E2F1 can trigger cell death through multiple transcription-independent mechanisms, by inhibiting activation of anti-apoptotic signals such as NFkappa-B, reducing levels of TRAF2, and inducing activation of calcium-sensitive cysteine protease calpain, which leads to proteolytic degradation of the Mdm2 homolog and p53 inhibitor, MDMx (Hou et al. 2001, Phillips et al. 1999, Strachan et al. 2005).
E2F1 regulation and function have been studied primarily in cycling cells. Little is known about the role of E2F1 in post-mitotic neurons. E2F1 is present in neurons during brain development and increases in expression through neuronal maturation (Kusek et al. 2001). This upregulation is also observed in differentiation of neural cell lines and cultured primary neurons (Kusek et al. 2001, Ting et al. 2014). In contrast to the nuclear localization of E2F1 in proliferating cells, neuronal E2F1 is predominantly cytoplasmic (Wang et al. 2010). Although neuronal E2F1 does retain some properties observed in mitotic cells, including its ability to induce apoptosis, these observations suggest E2F1 may have a novel function in neurons.
E2F1 deletion in vitro attenuates neuronal death in a number of toxicity models including potassium deprivation and Aβ peptide toxicity, while upregulation of E2F1 has been observed in post-mortem brain tissue of patients with Alzheimer Disease (AD), Parkinson Disease (PD) and Amyotrophic Lateral Sclerosis (ALS), supporting a link between E2F1, neuronal viability and neurodegeneration (Giovanni et al. 2000, Hoglinger et al. 2007, Jordan-Sciutto et al. 2001, O’Hare et al. 2000, Ranganathan & Bowser 2003). HIV-associated neurocognitive disorder (HAND) is a neurologic syndrome consisting of a spectrum of cognitive, motor and behavioral deficits. Although neurons themselves are not directly infected by HIV, neuropathological hallmarks of the disease include dendritic damage, synaptic loss and neuronal loss (Masliah et al. 1992, Masliah et al. 1997). Infiltration of HIV-infected macrophages into the central nervous system precedes neuronal damage. Such macrophages and subsequently activated resident microglia secrete inflammatory factors that alter the extracellular environment (Giulian et al. 1996, Gonzalez-Scarano & Martin-Garcia 2005). The neuronal response to the altered environment involves aberrant activation of the cell cycle regulatory machinery, including upregulation of E2F1 (Akay et al. 2011, Wang et al. 2007). Post-mortem tissue from patients with HIV encephalitis (HIVE), the pathological correlate of advanced disease, shows elevated levels of E2F1 in neurons from basal ganglia, hippocampus and prefrontal cortex, the brain regions most affected in HAND (Jordan-Sciutto et al. 2002), although prototypical E2F1 target genes remain unchanged (Wang et al. 2010). Interestingly, the E2F1 observed in tissue from patients with HIVE is also primarily cytoplasmic. Similar results were observed in cortical samples from SIV-infected encephalitic macaques (Jordan-Sciutto et al. 2000). These findings suggest that E2F1 correlates with HAND but likely fulfills a different function from its classical role as a nuclear transcription factor,
Our lab has previously shown that E2F1 is processed by calpain in dividing cells and overexpression of a cytoplasmic and transcriptionally-inactive E2F1 protein in dividing cells leads to calpain activation and calpain-dependent toxicity (Strachan et al. 2005). Although calpain is ubiquitously expressed, it is particularly abundant in the CNS. Calpain has also been implicated in synaptic potentiation (Khoutorsky & Spira 2009, Zadran et al. 2009), transcriptional regulation (Abe & Takeichi 2007, Khoutorsky & Spira 2009, Lynch & Gleichman 2007, Zadran et al. 2009) and in both acute and chronic neurodegeneration (Esteves et al. 2009, Gafni & Ellerby 2002, Gafni et al. 2004, Gascon et al. 2008, Kelly et al. 2005, Saito et al. 1993). Calpain has two prototypical isoforms, μ-calpain and m-calpain, that differ in their in vitro requirements for Ca+2 (μM for μ-calpain, mM for m-calpain). Although they display nearly identical substrate specificity, μ-calpain activation is associated with neuroprotective processes and m-calpain activation may contribute to pathological states (Wang et al. 2013). Sustained dysregulation of calcium homeostasis during many neurodegenerative conditions, specifically those involving excitotoxicity, leads to calpain cleavage of a wide range of substrates (Vosler et al. 2008). These cleaved substrates are either rendered inactive or display an altered function that may contribute to disease pathogenesis. Given these findings, we assessed the interplay of E2F1 and calpain in a calpain-dependent model of neurotoxicity utilizing an in vitro model of HIV-mediated neuronal damage.
Materials and Methods
Animals
E2F1tm1 transgenic mutant and wild-type mice are C57BL6/SV129 hybrids obtained from Jackson Labs (Bar Harbor, ME). Mice were housed under a 12-hr light/dark cycle and were allowed access to food and water ad libitum. Experiments were approved by the Institutional Animal Use and Care Committee and in accordance with the ARRIVE guidelines.
Chemicals and reagents
Antibodies used include: Actin (A2066), MAT (M6693) [Sigma], Akt1 (#2938), phospho-Akt1 (#4060), Erk1/2 (#4695), phospho-Erk1/2 (#9101), FLAG (#2368), Lamin A/C (#2032) [Cell Signaling], E2F1 (KH95), GAPDH (6C5) [Santa Cruz], MAP2 (SMI52) [Covance]. Calpain-cleaved aII-spectrin (A38) described previously (Roberts-Lewis et al. 1994) was a generous gift from Dr. Robert Siman (University of Pennsylvania). Other chemicals include N-methyl-d-Aspartate (NMDA), NMDA inhibitor MK801 (Tocris), calpain inhibitor MDL28170 (Tocris), recombinant calpain-1 (208712, Calbiochem), Nicotinamide (Sigma), Trichostatin A (Sigma), Lambda protein phosphatase (New England Biolabs), Conantokin G (Bachem), and Ifenprodil (Tocris), Goat anti-mouse-beta-lactamase TEM-1-conjugated secondary antibody and Fluorocillin™ Green reagent were from Invitrogen.
Preparation of primary neuronal cultures
Primary cortical cultures were prepared from E18 Sprague Dawley rat embryos or E16 murine embryos as described previously (Wang et al. 2007). Cells were plated on poly-L-lysine pre-coated 60mm dishes at a density of 2 × 106 cells per dish and maintained in neurobasal media with B27 supplement (Invitrogen) at 37°C, 5% CO2. Cultures were utilized at 21 days in vitro (DIV) unless stated otherwise.
Monocyte derived Macrophage (MDM) culture
HIVMDM supernatants were derived from monocyte-derived macrophage cultures infected with HIV-1 as described previously (Chen et al. 2002). HIV infection of macrophages was confirmed by serial quantification of p24 antigen levels in the culture supernatants over time and only HIVMDM supernatants exhibiting productive infection (p24 levels greater than 100 pg/ml of supernatant) were used to treat neuronal cultures. Supernatants from HIVMDM and non-infected macrophages (mock-infected MDM) were collected at selected time points after infection and stored at −80°C.
Western blotting
Whole cell extracts were prepared from primary rat cortical cultures. Cells were lysed for 20min with ice-cold lysis buffer consisting of 50mM Tris–HCl, 150mM NaCl, 0.1% SDS, 0.1% IGEPAL, 0.5% sodium deoxycholate, and a protease inhibitor cocktail (Sigma). Lysates were centrifuged at 16,100 g, at 4°C for 10 minutes and supernatants were collected. Protein concentrations were determined by Bradford assay, 30μg of protein was loaded in each lane of a 4–12% Bis-Tris gradient gel for protein separation and subsequently transferred onto PVDF membranes. Routine immunoblotting and autoradiography procedures were followed as described (Akay et al., 2011).
Subcellular fractionation
Cytoplasmic and nuclear protein extractions were performed using Panomics’ Nuclear Extraction Kit (Panomics Inc.) following the manufacturer’s protocol. Briefly, cells were lysed with ice cold lysis buffer (10 mM HEPES pH 7.9, 10mM KCl, 10 mM EDTA, 4% IGEPAL, 10mM DTT, protease inhibitor cocktail). Lysates were centrifuged at 16,100 g for 3 min and the supernatant (cytoplasmic fraction) was collected. The nuclear pellet was resuspended in buffer (20mM HEPES pH 7.9, 0.4M NaCl, 1mM EDTA, 10% glycerol, 10mM DTT, protease inhibitor cocktail) and incubated on a rocking platform at 4°C for 2 hours. Following centrifugation at 16,100 g for 5 min, the supernatant (nuclear extract) was collected. Protein (30μg) was resolved on a 4–12% gradient TBE gel and probed with antibodies against E2F1.
In vitro calpain cleavage assay
Primary rat cortical cultures were harvested using lysis buffer (137mM NaCl, 2mM EDTA, 10% Glycerol, 20mM Tris-HCl pH=8, 1% protease inhibitor cocktail). Resulting lysates were incubated with 2mM CaCl2 at 4°C for 6 hours in the presence or absence of 500nM of purified calpain-1 (EMD Millipore). The reactions were then terminated by SDS-PAGE sample buffer resolved on a 4–12% gradient Bis-Tris gel, followed by western blotting as described above. Calpain cleavage of affinity-purified FLAG/MAT-tagged E2F1 was performed using the above protocol with the following modifications: FLAG-transfected E2F1 protein was affinity-purified as indicated by manufacturer (Sigma). Prior to elution, FLAg-E2F1 was incubated at 4°C with 100ug neuronal protein lysate for 30 minutes followed by addition of calpain and CaCl2 as indicated above.
Neurotoxicity assessment
Neurotoxicity was quantified by a MAP2 cell-based ELISA as described previously (Wang et al. 2007). In parallel cultures, blinded-hand counts of Propidium Iodide-excluded MAP2-positive cells were performed to assess neuronal death as described (Akay et al. 2011).
Statistical analysis
Values are expressed as mean ± SEM. Paired results were analyzed by Student’s t-test. Data with multiple categories were analyzed by one-way ANOVA followed by the Newman–Keuls post-hoc test using Prism software (GraphPad Software). Values of p < 0.05 were considered significant.
Results
HIVMDM-induced neurotoxicity is attenuated in cortical neurons from E2F1 mutant mice
Increased E2F1 protein levels have previously been observed in post-mortem analysis from patients with HIVE (Jordan-Sciutto et al. 2002). We investigated the link between E2F1 and HIV-mediated neurodegeneration, employing an in vitro model of HIV-induced neurotoxicity in which primary rat cortical neuroglial cultures are treated at DIV 20 with conditioned media from HIV-infected human monocyte-derived macrophages (HIVMDM) (O’Donnell et al. 2006). In this model, neuronal death is dependent on NMDA receptor stimulation and calpain activation (O’Donnell et al. 2006). To assess the role of E2F1 in HIV-mediated neurotoxicity, we treated cortical cultures from E16 mouse embryos lacking functional E2F1 and from wildtype controls with either HIVMDM or Mock-infected macrophage supernatants (MOCK). Neuronal toxicity was assessed by cell-based ELISA for the dendritic marker MAP2 (Figure 1A) and confirmed through blinded hand-counts of MAP2-positive cells (Figure 1B). While HIVMDM supernatants cause dose-dependent neurotoxicity in wildtype cultures, E2F1 mutant cultures displayed lower levels of neurotoxicity relative to wildtype control cultures at all but the most concentrated HIVMDM dose (Figure 1A, 1B). The reduced toxicity in neurons lacking functional E2F1 suggests that loss of E2F1 confers partial protection against HIV-induced neuronal damage.
Figure 1. HIVMDM-induced neurotoxicity is attenuated in cortical neurons from E2F1 mutant mice.
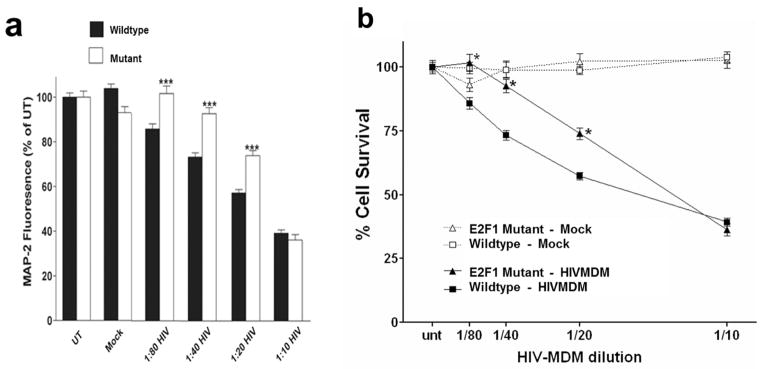
a. Cortical neurons from wild type (black bars) and E2F1 mutant mice (white bars) at DIV 20 were treated with HIVMDM supernatants or Mock-MDM supernatants at indicated dilutions for 20 hours. Resulting neuronal damage was measured by a cell-based MAP2 ELISA (*** denotes p<0.01) (White et al. 2011). b. Blinded hand counts of MAP2-positive, propidium iodide-negative cells from wildtype (square) and E2F1 mutant (triangle) cortical cultures treated with indicated dilutions of HIVMDM (black) and mock (white) supernatants for 20 hours (*denotes p<0.05).
HIVMDM-treated rat cortical cultures display lower molecular weight E2F1
We have previously shown that E2F1 expression is predominantly cytoplasmic in post-mitotic neurons and that mRNA levels of E2F1 transcriptional targets remain unchanged in neurons following HIVMDM treatment (Wang et al. 2010). To explore the role of E2F1 in HIV-mediated neurotoxicity, we treated primary neuroglial cultures from rat cortex at DIV21 with HIVMDM supernatants and assessed E2F1 levels in subcellular fractions via immunoblot. E2F1 was primarily in the GAPDH-positive cytosolic fraction of both untreated and HIVMDM-treated neuronal cultures and largely absent from the Lamin A/C-containing nuclear fractions (Figure 2B). While the subcellular distribution of E2F1 seemed unchanged following HIVMDM treatment, the cytosolic fraction from HIVMDM-treated cultures contained a lower molecular weight E2F1 immunoreactive band in place of the full length E2F1 found in untreated and mock-treated neuronal cultures (Figure 2B). This lower molecular weight E2F1 (LMW-E2F1) band migrates at an apparent molecular weight 5 kDa below the full sized band and displays a dose-dependent accumulation during HIVMDM treatment, showing increased production with decreasing dilutions of HIVMDM supernatant treatment (Figure 2C). Furthermore, the LMW-E2F1 is detectable as early as 6 hours following initial treatment and accumulates over time (Figure 2D). These results suggest that neuronal E2F1 is cytoplasmic and changes in size in response to HIV-associated neurotoxins.
Figure 2. HIVMDM-treated rat cortical cultures display lower molecular weight E2F1 (LMW-E2F1).

a,b. Immunoblot analysis of LaminA/C-positive nuclear fraction and GAPDH-positive cytoplasmic fraction from rat cortical cultures at DIV14 (a) and DIV21 (b) following 20-hour HIVMDM or Mock treatment. c. Immunoblot analysis of E2F1 from GAPDH-positive, LaminA/C-negative cytosolic fractions of rat cortical cultures (DIV21) treated with indicated dilutions of HIVMDM for 20 hours. Coomassie gel stain used for loading control (a, b, c). d. E2F1 immunoblot analysis of rat cortical cultures at DIV 21 following a time-course of HIVMDM or Mock treatment (Actin: loading control).
Production of LMW-E2F1 is mediated by NR2A/2B-containing NMDA Receptor stimulation
Since NMDA receptor subunit expression is developmentally regulated, HIVMDM does not induce toxicity in neuronal cultures until DIV 14–21 when NR2B-containing receptor maturation occurs (Wang et al. 2007). Interestingly, production of LMW-E2F1 by HIVMDM treatment is not detectable until after neuronal cultures reach 2 weeks of age (Figure 2A) suggesting that LMW-E2F1 may be dependent on NMDA receptor (NMDAR) stimulation. To investigate this possibility, we pretreated cortical cultures at DIV 21 with the NMDAR antagonist MK801 for 30 minutes prior to the addition of HIVMDM supernatant. Blocking NMDAR activation with MK801 eliminated the production of LMW-E2F1 following HIVMDM treatment (Figure 3A). Treatment of neurons with synthetic NMDAR agonist NMDA also led to the generation of LMW-E2F1, confirming that LMW-E2F1 production is NMDAR-dependent (Figure 4C). To determine whether LMW-E2F1 production was similarly mediated by NR2B containing receptors, we pretreated neuronal cultures with antagonists to different NMDA receptor subtypes prior to incubation with HIVMDM supernatants. While Zinc (NR1/2A) and Ifenprodil (NR1/2B) failed to block HIVMDM-induced formation of LMW-E2F1, Conantokin-G (all NR2B-containing) inhibited LMW-E2F1 production (Figure 3C). These results demonstrate that LMW-E2F1 production is selectively dependent on NR2A/NR2B-containing NMDAR stimulation.
Figure 3. Production of LMW-E2F1 is mediated by NR2A/2B-containing NMDAR stimulation.

a. Immunoblot of subcellular fractions from rat cortical cultures (DIV21) treated for 20 hours with HIVMDM or mock supernatants. For indicated samples, NMDAR antagonist MK801 (10μM) was added to cultures 30 minutes prior to supernatant treatment. Coomassie-stained gel serves as a loading control. b,c. Rat cortical cultures at DIV21 were pre-treated for 1 hour with different NMDAR subunit inhibitors (Table, c) followed by HIVMDM treatment. Conantokin G (CoG, 10μM), which blocks NR2A/2B-containing NMDARs prevented the production of LMW-E2F1, while Ifenprodil (Ifn, 10μM), which blocks NR2B/2B-containing NMDARs, and Zinc (Zn, 500nM) which blocks NR2A-containing NMDARs, had no effect on HIVMDM-induced formation of LMW-E2F1. Coomassie-stained gel serves as a loading control.
Figure 4. LMW-E2F1 is a calpain cleavage product.

a. Samples from primary rat cortical cultures at DIV21 were immunoblotted for calpain-cleaved aII-spectrin to measure calpain activation following HIVMDM treatment. Coomassie gel stain serves as a loading control. b. To block calpain activation, cortical cultures were pretreated with calpain inhibitor MDL28170 (20uM) for 30 minutes prior to HIVMDM. Coomassie gel stain serves as a loading control. c. In vitro calpain cleavage assay of neuronal lysates from untreated cultures at DIV 21 with calpain-1 (10ug/mL) and 2mM CaCl2. Reaction mixture was incubated at 4°C for 6 hours. Untreated lysate from untreated and NMDA-treated cultures serve as controls for full length and LMW-E2F1 (top and bottom arrowheads, respectively). d. In vitro calpain cleavage assay to test direct cleavage of E2F1 by calpain. Addition of MDL28170 to neuronal lysates prior to incubation with calpain-1 and CaCl2 blocked formation of LMW-E2F1 (pre-MDL). Following 4 hours of the 6-hour incubation of neuronal lysate with calpain-1, MDL28170 was added along with affinity-purified FLAG-E2F1 to the reaction (post-MDL and post-FLAG-E2F1, respectively) to determine whether exogenous E2F1 is protected by calpain inhibition following cleavage of endogenous targets.
LMW-E2F1 is a calpain cleavage product
Our in vitro model of HIV-induced neurotoxicity depends on NMDAR stimulation and also on activation of calpains, the calcium-sensitive cysteine proteases subsequently activated by NMDAR-dependent Ca2+ influx (O’Donnell et al. 2006, Wang et al. 2007, White et al. 2011). Calpain was activated in the present experiments as revealed by increased levels of calpain-cleaved αII-spectrin in HIVMDM-treated neurons (Figure 4A). We therefore hypothesized that NMDAR-dependent production of LMW-E2F1 results from proteolytic cleavage of E2F1 by calpain. To test the involvement of calpain, we pretreated neuronal cultures with the cell-permeable pharmacologic inhibitor MDL28170 (Calpain inhibitor III) prior to HIVMDM incubation and measured resulting LMW-E2F1 production by immunoblot. Pharmacologic inhibition of calpains prevented the production of LMW-E2F1 while preserving the full length form of E2F1 observed in untreated neuronal cultures, suggesting that LMW-E2F1 is dependent on calpain activation (Figure 4B). To validate E2F1 as a bona fide substrate of calpain, we performed an in vitro calpain cleavage assay on untreated neuronal protein lysates. Incubation of the lysates with calcium and calpain reproduced the LMW-E2F1 product observed in HIVMDM- and NMDA-treated neurons, (Figure 4C). To definitively show that E2F1 is cleaved directly by calpain rather than through an event downstream of calpain activity, we performed an in vitro cleavage assay on neuronal lysates as before, then inhibited calpain activity by adding MDL28170 to the cleavage reaction and subsequently incubated the reaction with recombinant FLAG-E2F1 protein. While endogenous E2F1 was cleaved by initial incubation with calpain, calpain inhibition blocked cleavage of the subsequently added FLAG-E2F1, again suggesting that the effect of calpain on E2F1 is through direct cleavage (Figure 4D). Finally, we incubated recombinant 35S-labeled E2F1 with μ-calpain or m-calpain in the presence of calcium. E2F1 was cleaved by both calpains in a similar time course (Figure S1). Together, these results demonstrate that E2F1 is cleaved by calpain to a smaller stable isoform following HIVMDM treatment and NMDAR stimulation.
Stabilized calpain-cleaved E2F1 is generated preferentially in neurons
To determine whether calpain cleavage may play a role in other cell or tissue types, we performed in vitro calpain cleavage assays on lysates from several different types of cultured cells and determined how efficiently the stabilized E2F1 cleavage product accumulated. Interestingly, the E2F1 cleavage product was produced in a number of different neuronal cell types including primary rat cortical neurons, primary rat hippocampal neurons, and retinoic acid-differentiated SHSY5y neuroblastoma cells, but did not stably accumulate in non-neuronal cells such as HEK293 cells and undifferentiated SHSY5y cells (Figure 5A). We found similar results in mouse tissue homogenates, where cleaved E2F1 was stably produced in brain samples but not those of other organs (data not shown). These findings suggest that calpain cleavage of E2F1 into a stable isoform occurs preferentially in neurons. Given that undifferentiated and differentiated SHSY5y cells exhibited different capacities for generating cleaved E2F1 and that cleaved E2F1 production was only detected in HIVMDM-treated neuronal cultures at DIV 14 or later, we suspected that formation of the stabilized cleavage product may be influenced by neuronal maturation. To test this possibility, we performed in vitro calpain cleavage assays on protein lysates from rat primary cortical neurons at different ages in culture. Incubation of protein lysates with activated calpain produced the stabilized E2F1 cleavage product at all culture ages tested (Figure 5B), suggesting that both immature and mature neurons have the capacity to generate the calpain cleavage product. Together, these results suggest that production and stabilization of calpain-cleaved E2F1 occurs preferentially in cells of neuronal lineage.
Figure 5. Stabilized calpain-cleaved E2F1 is generated preferentially in neurons.

a. In vitro calpain cleavage assays were performed on protein lysates from indicated cell types to assess the influence of cell type on production of stabilized calpain-cleaved E2F1. Lysates were collected from primary rat cortical and hippocampal cultures at DIV21, and from differentiated SHSY5y neuroblastoma cells following 10-day retinoic acid treatment. b. In vitro calpain cleavage assay of lysates from primary rat cortical neurons at indicated ages in culture.
Stabilization of cleaved E2F1 is protein conformation-dependent
To determine what factors may be responsible for stabilizing cleaved E2F1 in neurons, we first tested whether heat denaturation of the neuronal protein lysate affected the ability of activated calpain to produce the stabilized product. In vitro calpain cleavage assays were performed on boiled and unboiled neuronal protein lysates and assessed by immunoblot for presence of the stabilized E2F1 cleavage product. We found that cleaved E2F1 production was disrupted by heat denaturation of the lysate, even when purified, natively folded E2F1 was added to the boiled lysate prior to calpain incubation (Figure 6A). This result suggests that native conformation of neuronal proteins is necessary for stabilizing the cleaved product, providing a potential explanation for how calpain-cleaved E2F1 is preferentially stabilized in neurons.
Figure 6. Stabilization of cleaved E2F1 is protein conformation-dependent.

a. In vitro calpain cleavage assays were performed on rat cortical neuronal lysates from cultures at DIV21 that were boiled or unboiled. Purified FLAG-E2F1 protein that was added to the boiled lysate failed to show stable accumulation of cleaved E2F1 when incubated with calpain-1. FLAG-MAPK served as a control to demonstrate calpain substrate specificity in the assay. b. To determine the effect of protein phosphorylation on the stabilization of calpain-cleaved E2F1, neuronal lysate (05μg/μL) was treated with λ protein phosphatase (λPP, 600 units) for 1 hour at 25°C prior to incubation with calpain-1 and CaCl2 at 4°C. Phospho-Akt levels normalized to total Akt levels were measured to confirm effect of λPP treatment. c,d. Rat cortical cultures were treated for 24 hours with either 1μM Trichostatin A (TSA) to inhibit class I and II HDACs (c) or Nicotinamide (low=1μM, high=25μM) to inhibit class III HDACs (d). Cultures were harvested and total protein lysates were subjected to in vitro calpain cleavage assay. HDAC inhibition was confirmed by increased levels of acetylated α-tubulin over total α-tubulin and decreased levels of phospho-Erk1/2 over total ERK.
A growing number of studies have demonstrated that the post-translational modifications, particularly phosphorylation, of calpain substrates or their binding partners can influence their ability to be cleaved by calpain (Bi et al. 2000, Huang et al. 1997, Nicolas et al. 2002, Qin et al. 2010). As E2F1 is both acetylated and phosphorylated in mitotic cells, we disrupted phosphorylation and acetylation of the neuronal protein lysates to determine whether these modifications might alter calpain processing of E2F1. To dephosphorylate neuronal proteins, lysates were incubated with lambda protein phosphatase, a pan phosphatase with activity toward phosphorylated serine, threonine, and tyrosine residues, prior to incubation with activated calpain. While dephosphorylation activity was confirmed through a decrease in phospho-Akt levels, stabilized calpain-cleaved E2F1 was still produced in our in vitro calpain cleavage assay following phosphatase treatment (Figure 6B). We similarly disrupted global acetylation levels in the neuronal lysates through inhibition of histone deacetylases (HDACs) with Trichostatin A, which inhibits class I and II HDACs. Despite observing decreased acetylated alpha-tubulin from Trichostatin A treatment, production of calpain-cleaved E2F1 appeared unaffected (Figure 6C). We also inhibited class III HDACs (sirtuins) using Nicotinamide treatment. While Nicotinamide has effects on other targets, Nicontinamide-mediated sirtuin inhibition has frequently been confirmed through decreased Erk1/2 signaling (Lee et al. 2011, Li et al. 2008, Zhao et al. 2012). We did observe decreased levels of phospho-Erk1/2 following Nicotinamide treatment; however, as with trichostatin A treatment, we observed no significant effect on the stable production of cleaved E2F1 (Figure 6D). Similar results were also observed following deglycosylation of the neuronal lysates (data not shown). Together, these results suggest that post-translational modifications of the surrounding neuronal proteins may not be important for stabilizing the calpain-cleaved E2F1 product.
Calpain cleaves E2F1 at the N- and C-terminus
To better understand the kinetics of calpain cleavage of E2F1, we assessed E2F1 products produced by addition of calpain over time. By performing a time course of our in vitro calpain cleavage assay, we were able to identify an intermediate E2F1 cleavage product that was produced early on during calpain incubation (Figure 7A). This intermediate product was further processed to the final stabilized cleavage product observed in neurons following HIVMDM treatment, suggesting that calpain cleavage of E2F1 involves at least two distinct cleavage events. Arresting calpain activity during this time course prevented further conversion of full-length E2F1 to the intermediate product and blocked production of the stabilized final product (Figure 7A′), further demonstrating that both the intermediate and final E2F1 products are directly produced by calpain cleavage. To identify where the E2F1 protein is cleaved by calpain, we made use of a construct encoding a dually epitope-tagged E2F1 protein with the FLAG-tag at the N-terminus and a metal affinity (MAT)-tag at the C-terminus. We affinity-purified the E2F1 protein from transfected HEK293 cells and with the protein still bound by the affinity resin, we then incubated the purified protein with neuronal protein lysate to reproduce the environment in which production of stabilized calpain-cleaved E2F1 is observed. Following incubation with activated calpain, we assessed which epitope tags were lost and retained in the eluted fraction by immunoblot analysis. We found that both FLAG and MAT tags were lost following calpain incubation (Figure 7B) supporting the concept of two distinct cleavage events at each terminus of E2F1.
Figure 7. Calpain cleaves E2F1 at N- and C-terminus.

a. Time course of in vitro calpain cleavage assay was performed on lysates from rat cortical cultures for indicated times. Samples were run on a 10% Bis-Tris polyacrylamide gel to better resolve molecular weight differences of E2F1 bands due to calpain processing. Samples from early time points show generation of an intermediate product (middle arrow) before complete conversion to the stabilized calpain-cleaved E2F1 product (bottom arrow). Lysate from NMDA-treated cultures served as a positive control for calpain-cleaved E2F1. Calpain-cleaved αII-spectrin shows progressive calpain activity over time course. a′. Time course of calpain cleavage assay was performed as in (a) but with addition of MDL28170 (20uM) beginning at t=20 minutes to inhibit calpain activity at all subsequent time points. b. Differentially epitope-tagged FLAG-E2F1-MAT overexpressed in HEK293 cells was affinity purified with either FLAG antibody or Nickel resin. Purified protein was incubated with neuronal lysate (50ug) for 30 minutes while still resin-bound prior to performing an in vitro calpain cleavage assay and subsequent elution. Eluted protein was assessed for presence of N-terminal FLAG tag and C-terminal MAT tag by immunoblot analysis. FLAG-MAPK-MAT served as a control to demonstrate E2F1-specific calpain processing and as a positive control along with FLAG-BAP for successful purification.
Presence of LMW-E2F1 in human fetal neurons
The data from our in vitro model of HIV-mediated neurotoxicity firmly establish that a calpain-cleaved E2F1 fragment accumulates in primary rodent neuronal cultures following HIVMDM treatment. To determine whether E2F1 in human neurons is similarly processed by calpain, we cultured human fetal neurons and performed an in vitro calpain cleavage assay on the protein lysates obtained from these cells. As observed with primary rat neuronal cultures, activated calpain processed E2F1 from human neurons to a smaller, stable product (Figure 8A). This product matched the size of the calpain-cleaved E2F1 isoform from primary rat neurons, suggesting that E2F1 in human neurons displays the same capability to be processed to the smaller product. A smaller E2F1 isoform was also observed in protein lysates from human cortical autopsy tissue by immunoblot. Interestingly, several of the samples from HIV+ patients with minor cognitive motor disorder (MCMD) and with HIV-associated dementia (HAD) contained a lower molecular weight E2F1 band that was not found in a sample from an HIV+ neurocognitively normal patient (Figure S2). Production of this lower band did not correlate with post-mortem interval (Pearson’s Correlation: r=−0.103, p=0.684) or diagnosis (Fisher’s Exact: p=0.571 for HAD, p=0.545 for MCMD, p=0.576 for HAD+MCMD). Together, these results provide evidence for the production of a calpain-cleaved E2F1 isoform in human neurons.
Figure 8. Lower molecular weight E2F1 is produced in in human fetal neurons.
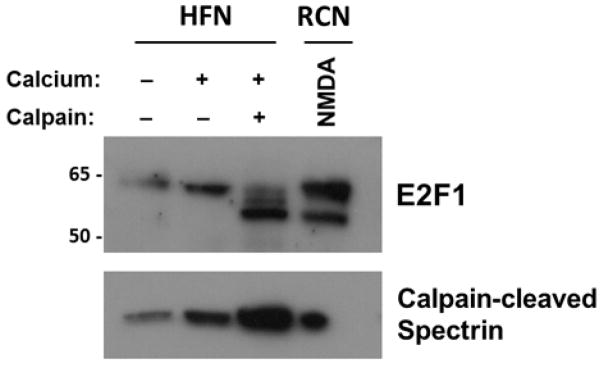
Protein lysates from cultured human fetal neurons (HFN) at DIV 17 were subjected to an in vitro calpain cleavage assay as described above. Immunoblot analysis of resulting samples assessed levels of E2F1 and calpain-cleaved αII-spectrin to measure calpain activation. Lysate from NMDA-treated rat cortical cultures (RCN) served as a positive control for calpain-cleaved E2F1.
Discussion
Unlike other proteolytic systems such as lysosomes and the ubiquitin-proteasome, calpains do not simply breakdown their substrates. In many cases, calpain proteolytic processing leads to modulatory effects on substrate structure and function. Here, we present evidence supporting the involvement of E2F1 in HIV-induced neuronal death mediated by calpains. Using an in vitro model of HIV-induced neurotoxicity that is dependent on calpain activation, we have shown that cortical neurons lacking functional E2F1 are less susceptible to neuronal death. Additionally, we report that neuronal E2F1 is cleaved by calpain to a stable 55 kiloDalton fragment following NR2B-dependent NMDA receptor stimulation. This cleavage of E2F1 is protein conformation-dependent and involves at least two cleavage events, one at each terminus of the protein. Intriguingly, the stabilized E2F1 cleavage product is produced in postmitotic neurons of all ages, but fails to be stabilized in cycling cells. We have previously reported that neuronal E2F1 displays the ability to shuttle between the cytosol and nucleus, a result also observed in differentiated keratinocytes (Ivanova et al. 2007, Strachan et al. 2005). Despite its nuclear-cytoplasmic shuttling capability, our results and previously published studies indicate that E2F1 is predominantly cytoplasmic in postmitotic neurons, similar to the localization recently reported for E2F1 in fully differentiated oligodendrocytes (Figure 2A, 2B, 3A)(Magri et al. 2014, Wang et al. 2010). Furthermore, neuronal E2F1 appears to be retained in the cytosol of HIVMDM-treated neurons following cleavage by calpain (Figure 2A, 3A), suggesting that the functional implications of E2F1 cleavage are distinct from those of Beta-catenin or Cdk5. This observation is further supported by the reported cytosolic E2F1 staining in neurons from post-mortem brain tissue of patients with HIVE, AD, ALS and PD (Hoglinger et al. 2007, Jordan-Sciutto et al. 2001, Ranganathan et al. 2001). Finally, our previous studies using this in vitro model have demonstrated that transcription of several E2F target genes remains unchanged in neurons following HIVMDM treatment (Wang et al. 2010). These observations all argue against a functional effect of E2F1 cleavage on its activity as a transcription factor. Future studies to define the E2F1 protein sequence that is preserved following cleavage may shed light on the role of this calpain-cleaved transcription factor when it is retained in the cytosol.
Determining cleavage sites within calpain substrates is often challenging given the poor understanding of how substrate specificity is achieved. While several studies have reported amino acid preferences for calpain activity based on analysis of published substrate cleavage sites, these preferences are often insufficient to correctly predict cleavage sites in novel calpain substrates (DuVerle et al. 2011, Tompa et al. 2004). Furthermore, calpain substrates are often cleaved in multiple places, compounding the difficulty of accurately defining the resulting fragment. For example, the microtubule-associated protein Tau is cleaved by calpain at a minimum of three internal sites, resulting in a stable 17-kilodalton fragment that has been reported to play a role in neurotoxicity (Liu et al. 2011, Park & Ferreira 2005). Similarly, voltage-gated sodium channel NaV1.2 is cleaved by calpain in two regions of the alpha-subunit, producing channel fragments that display altered channel properties (von Reyn et al. 2012, von Reyn et al. 2009). Our data suggest a similar pattern of calpain processing for E2F1 in neurons. At least two cleavage events, one at the N-terminus and one at the C-terminus, are responsible for producing the stabilized E2F1 cleavage product (Figure 7). E2F1 has a Cyclin A binding domain that begins nearly 70 amino acids into its N-terminus. In dividing cells, E2F1-Cyclin A binding through this domain helps regulate E2F1-DNA binding. However, given the cytosolic localization of E2F1 in mature neurons, it is unlikely that the domain serves the same function in this cellular context. Similarly, the C-terminus of E2F1 contains the transactivation domain along with the pRB pocket binding domain, which has been studied exclusively in the context of E2F1-mediated transcription. While these sequences have well-characterized roles in cycling cells, it is unclear how they may function in the neuronal cytosol. To what extent either of these domains is affected by cleavage is also unknown. Based on our results, the E2F1 cleavage product displays a 5–10 kDa reduction in apparent molecular weight, suggesting a loss of 45–90 amino acids. Future efforts to define the calpain cleavage sites are necessary to determine the consequence of disrupting the N- and C-terminal domains.
Despite a growing list of calpain substrates that have recently been identified, the rules surrounding substrate recognition and sequence cleavage are yet to be fully elucidated. In this study, we identified a stabilized E2F1 isoform produced by calpain processing. Intriguingly, we were unable to observe this stabilized product in cells other than neurons (Figure 4). To our knowledge, this is the first example of a calpain substrate exhibiting distinct processing patterns in different cell types. To understand how this could be achieved, we first tested whether the stabilized cleavage product was dependent on protein conformation (Figure 6A). We found that protein conformation was indeed critical for the stabilized accumulation of cleaved E2F1, suggesting that neuronal E2F1 may contain unique binding partners that protect regions of the protein from further degradation. Interestingly, the E2F1 protein sequence has a number of P-X-X-P motifs known to bind Src homology 3 (SH3) domains (Alexandropoulos et al. 1995). We have previously reported that neuronal E2F1 is enriched in synaptosomes and co-localizes with the synaptic scaffolding protein PSD-95, which contains a C-terminal SH3 domain (Ting et al. 2014). Exploring whether PSD-95 or other synaptic proteins may bind E2F1 through SH3 binding domains may provide further clarity on the dependence of calpain-cleaved E2F1 on protein conformation. In addition to examining native protein structure, we also tested whether post-translational modifications such as phosphorylation and acetylation may play a role in modifying calpain activity toward E2F1 in different cellular environments (Figure 6B–6D). There are multiple examples showing calpain preferentially cleaving substrates based on their phosphorylation state. Notably, the tumor suppressor p53 is preferentially cleaved by calpain when phosphorylated at one of its serine residues in developing neurons (Qin et al. 2010). A separate study focusing on calpain-mediated cleavage of NMDA receptors found that tyrosine phosphorylation of NR2 subunits protected their C-terminal domains from calpain processing (Bi et al. 2000). Finally, αII-spectrin, one of the prototypical calpain substrates, also shows decreased sensitivity to calpain cleavage when phosphorylated at one of its tyrosine residues (Nicolas et al. 2002). While post-translational modifications to E2F1 have not been explored in neurons, we thought they may provide a basis for calpain to differentially process neuronal E2F1. Given the numerous instances when E2F1 is phosphorylated and acetylated in dividing cells, we attempted to pharmacologically disrupt both of these modification systems in neuronal lysates to determine whether calpain cleavage of E2F1 is altered. However, neither phosphatase treatment nor inhibition of HDACs or Sirtuins significantly affected the formation of the stabilized E2F1 calpain cleavage product (Figure 6). Whether other post-translational modifications are involved in directing calpain processing of E2F1 is still a possibility. A comprehensive understanding of neuronal E2F1 post-translational modifications will likely be necessary to fully explore this issue.
Our findings suggest that calpain cleavage of E2F1 occurs during a neurotoxic event and contributes to this process as we have observed reduced neurotoxicity in HIVMDM-treated cortical neurons lacking functional E2F1 (Figure 1). Interestingly, we were able to completely block calpain cleavage of E2F1 using the NR2B subunit inhibitor Conantokin G, suggesting that cleaved E2F1 requires NR2B-NMDAR stimulation (Figure 3B). A recent study detailed how synaptic NMDAR stimulation leads to μ-Calpain activation and subsequent neuroprotection, while extrasynaptic NMDAR stimulation leads to m-Calpain activation that promotes neurotoxicity (Wang et al. 2013). A study that deleted both μ-Calpain and m-Calpain in the CNS of mice define roles for these proteases in both excitotoxic death and synaptic plasticity (Amini et al. 2013). Given that NR2B subunits are typically found at extrasynaptic locations, the NR2B-dependent nature of E2F1 processing by calpain suggests that it requires extrasynaptic NMDAR activity and subsequent neurotoxic activation of m-calpain. Although the NR2B-dependent nature of E2F1 processing suggests that it is part of a neurotoxic pathway, it is important to note that both m-calpain and μ-calpain are able to proteolytically cleave E2F1 (Figure S1), and that calpain-cleaved E2F1 may fulfill a neuroprotective function either in our model of HIV-induced neurotoxicity or possibly in other neuronal contexts such as development. Indeed, we have shown that calpain-cleaved E2F1 can be produced in immature neurons that have not yet expressed NR2B subunits (Figure 5B), suggesting the possibility of a role in synaptic development. It should also be noted that the attenuated HIVMDM-induced neurotoxicity observed in E2F1 mutant neurons could be due to a loss of physiologic E2F1 function rather than absence of a gain-of-toxic function in wildtype cultures. While it seems unlikely that E2F1 in the cytosol could promote neurotoxicity by activating transcription of apoptotic genes, it is quite possible that it could influence calcium homeostasis. Previous work from our lab has shown that overexpression of a transcriptionally inactive N-terminally truncated E2F1 protein in HEK293 cells causes calpain-dependent cell death (Strachan et al. 2005). Interestingly, this truncated E2F1 product is cytoplasmic, much like the neuronal E2F1 protein that we detect in primary neuronal cultures. While these results were obtained from cell lines, they provide an intriguing comparator to calpain-cleaved neuronal E2F1 and suggest the possibility that the E2F1 cleavage product may serve to amplify calpain activation. In fact, a number of calpain substrates have been reported to perform this function, promoting further cytosolic calcium increases upon cleavage. For instance, the Type 1 IP3 receptor, which functions as an ER calcium channel, disrupts intracellular Ca2+ buffering when cleaved by calpain (Kopil et al. 2012). Similarly, L-type Calcium channels in cultured neurons are cleaved by calpain, leading to C-terminal truncation of their α1 pore-forming subunit and enhanced Ca2+ influx from the L-type channels (Hell et al. 1996, Klockner et al. 1995). Cleavage of these substrates and others help create a positive feed-forward pathway that further increases intracellular calcium levels, thereby amplifying calpain activation. Our results showing the presence of a lower molecular weight E2F1 band in a subset of subcortical tissue samples from patients with HAND may similarly be due to intracellular calcium dysregulation (Figure S2), and additional studies with greater statistical power are warranted. Interestingly, neuronal E2F1 has been reported to bind Necdin, a factor involved in maintaining the terminally differentiated identity of postmitotic neurons (Kuwako et al. 2004). Further, Necdin has been linked to intracellular calcium regulation through its interaction with NEFA, a calcium-binding protein located on the ER cisternae which regulates ER calcium release (Taniguchi et al. 2000). As Necdin interacts with the C-terminal domain of E2F1, further work to determine if calpain-cleaved E2F1 could participate in calcium regulation through changing its interaction with Necdin and NEFA would provide further insight into this pathway.
Our findings indicate that E2F1 exhibits novel regulation by calpain cleavage in neurons. Further, cleavage of E2F1 by calpain is independent of known phosphorylation and acetylation events and appears to be dependent on an interacting protein partner. Finally, an isoform of E2F1 with similar size to calpain-cleaved E2F1 is apparent in cortex from HIV-infected patients with cognitive deficits while it is absent in cognitively normal patients. Understanding the role of this stabilized calpain cleavage project will provide novel therapeutic implications for treating patients with cognitive decline linked to excititoxic injury including HAND, AD, ALS and PD.
Supplementary Material
Acknowledgments
We thank Dr. Harry Ischiropoulos, Dr. Judy Grinspan, Dr. Robert Neumaur, and Dr. Cagla Akay for assistance with experimental design and preparation of the manuscript. We would also like to thank Dr. David Park and Dr. Olimpia Meuicci for providing reagents and E2F1 mutant mice. We also thank the CNAC Tissue processing Core at Temple University, particularly Dr. Dianne Langford, for providing primary human neurons, the National NeuroAIDS Tissue Consortium for providing tissue from HIV-positive patients and Margaret Maronski for her help in preparing primary rat neuronal cultures. This work was supported by the NIH.
Abbreviations
- DIV
days in vitro
- HAD
HIV-associated dementia
- HAND
HIV-associated neurocognitive disorders
- HIVMDM
HIV-infected Monocyte-derived-Macrophage
- MAP2
microtubule associated protein-2
- NMDAR
N-methyl-D-aspartate receptor
- NR2A
NMDA receptor subunit 2A
- NR2B
NMDA receptor subunit 2B
- PSD-95
postsynaptic density protein 95
References
- Abe K, Takeichi M. NMDA-receptor activation induces calpain-mediated beta-catenin cleavages for triggering gene expression. Neuron. 2007;53:387–397. doi: 10.1016/j.neuron.2007.01.016. [DOI] [PubMed] [Google Scholar]
- Akay C, Lindl KA, Shyam N, Nabet B, Goenaga-Vazquez Y, Ruzbarsky J, Wang Y, Kolson DL, Jordan-Sciutto KL. Activation status of integrated stress response pathways in neurones and astrocytes of HIV-associated neurocognitive disorders (HAND) cortex. Neuropathology and applied neurobiology. 2012;38:175–200. doi: 10.1111/j.1365-2990.2011.01215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akay C, Lindl KA, Wang Y, White MG, Isaacman-Beck J, Kolson DL, Jordan-Sciutto KL. Site-specific hyperphosphorylation of pRb in HIV-induced neurotoxicity. Molecular and cellular neurosciences. 2011;47:154–165. doi: 10.1016/j.mcn.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandropoulos K, Cheng G, Baltimore D. Proline-rich sequences that bind to Src homology 3 domains with individual specificities. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:3110–3114. doi: 10.1073/pnas.92.8.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amini M, Ma CL, Farazifard R, et al. Conditional disruption of calpain in the CNS alters dendrite morphology, impairs LTP, and promotes neuronal survival following injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:5773–5784. doi: 10.1523/JNEUROSCI.4247-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi R, Rong Y, Bernard A, Khrestchatisky M, Baudry M. Src-mediated tyrosine phosphorylation of NR2 subunits of N-methyl-D-aspartate receptors protects from calpain-mediated truncation of their C-terminal domains. The Journal of biological chemistry. 2000;275:26477–26483. doi: 10.1074/jbc.M003763200. [DOI] [PubMed] [Google Scholar]
- Chen W, Sulcove J, Frank I, Jaffer S, Ozdener H, Kolson DL. Development of a human neuronal cell model for human immunodeficiency virus (HIV)-infected macrophage-induced neurotoxicity: apoptosis induced by HIV type 1 primary isolates and evidence for involvement of the Bcl-2/Bcl-xL-sensitive intrinsic apoptosis pathway. J Virol. 2002;76:9407–9419. doi: 10.1128/JVI.76.18.9407-9419.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregori J, Johnson DG. Distinct and Overlapping Roles for E2F Family Members in Transcription, Proliferation and Apoptosis. Current molecular medicine. 2006;6:739–748. doi: 10.2174/1566524010606070739. [DOI] [PubMed] [Google Scholar]
- DuVerle DA, Ono Y, Sorimachi H, Mamitsuka H. Calpain cleavage prediction using multiple kernel learning. PloS one. 2011;6:e19035. doi: 10.1371/journal.pone.0019035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteves AR, Arduino DM, Swerdlow RH, Oliveira CR, Cardoso SM. Dysfunctional mitochondria uphold calpain activation: Contribution to Parkinson’s disease pathology. Neurobiol Dis. 2009 doi: 10.1016/j.nbd.2009.12.011. [DOI] [PubMed] [Google Scholar]
- Gafni J, Ellerby LM. Calpain activation in Huntington’s disease. J Neurosci. 2002;22:4842–4849. doi: 10.1523/JNEUROSCI.22-12-04842.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafni J, Hermel E, Young JE, Wellington CL, Hayden MR, Ellerby LM. Inhibition of calpain cleavage of huntingtin reduces toxicity: accumulation of calpain/caspase fragments in the nucleus. J Biol Chem. 2004;279:20211–20220. doi: 10.1074/jbc.M401267200. [DOI] [PubMed] [Google Scholar]
- Gascon S, Sobrado M, Roda JM, Rodriguez-Pena A, Diaz-Guerra M. Excitotoxicity and focal cerebral ischemia induce truncation of the NR2A and NR2B subunits of the NMDA receptor and cleavage of the scaffolding protein PSD-95. Mol Psychiatry. 2008;13:99–114. doi: 10.1038/sj.mp.4002017. [DOI] [PubMed] [Google Scholar]
- Giovanni A, Keramaris E, Morris EJ, Hou ST, O’Hare M, Dyson N, Robertson GS, Slack RS, Park DS. E2F1 mediates death of B-amyloid-treated cortical neurons in a manner independent of p53 and dependent on Bax and caspase 3. The Journal of biological chemistry. 2000;275:11553–11560. doi: 10.1074/jbc.275.16.11553. [DOI] [PubMed] [Google Scholar]
- Giulian D, Yu J, Li X, Tom D, Li J, Wendt E, Lin SN, Schwarcz R, Noonan C. Study of receptor-mediated neurotoxins released by HIV-1-infected mononuclear phagocytes found in human brain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1996;16:3139–3153. doi: 10.1523/JNEUROSCI.16-10-03139.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nature reviews Immunology. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Helin K, Harlow E, Fattaey A. Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Mol Cell Biol. 1993;13:6501–6508. doi: 10.1128/mcb.13.10.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Breeze LJ, Wang KK, Chavkin C, Catterall WA. N-methyl-D-aspartate receptor-induced proteolytic conversion of postsynaptic class C L-type calcium channels in hippocampal neurons. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:3362–3367. doi: 10.1073/pnas.93.8.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoglinger GU, Breunig JJ, Depboylu C, et al. The pRb/E2F cell-cycle pathway mediates cell death in Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:3585–3590. doi: 10.1073/pnas.0611671104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou ST, Cowan E, Walker T, Ohan N, Dove M, Rasqinha I, MacManus JP. The transcription factor E2F1 promotes dopamine-evoked neuronal apoptosis by a mechanism independent of transcriptional activation. J Neurochem. 2001;78:287–297. doi: 10.1046/j.1471-4159.2001.00402.x. [DOI] [PubMed] [Google Scholar]
- Huang C, Tandon NN, Greco NJ, Ni Y, Wang T, Zhan X. Proteolysis of platelet cortactin by calpain. The Journal of biological chemistry. 1997;272:19248–19252. doi: 10.1074/jbc.272.31.19248. [DOI] [PubMed] [Google Scholar]
- Iaquinta PJ, Lees JA. Life and death decisions by the E2F transcription factors. Current opinion in cell biology. 2007;19:649–657. doi: 10.1016/j.ceb.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova IA, Vespa A, Dagnino L. A novel mechanism of E2F1 regulation via nucleocytoplasmic shuttling: determinants of nuclear import and export. Cell cycle (Georgetown, Tex) 2007;6:2186–2195. doi: 10.4161/cc.6.17.4650. [DOI] [PubMed] [Google Scholar]
- Jordan-Sciutto K, Rhodes J, Bowser R. Altered subcellular distribution of transcriptional regulators in response to Abeta peptide and during Alzheimer’s disease. Mechanisms of ageing and development. 2001;123:11–20. doi: 10.1016/s0047-6374(01)00334-7. [DOI] [PubMed] [Google Scholar]
- Jordan-Sciutto KL, Wang G, Murphey-Corb M, Wiley CA. Cell cycle proteins exhibit altered expression patterns in lentiviral-associated encephalitis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22:2185–2195. doi: 10.1523/JNEUROSCI.22-06-02185.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan-Sciutto KL, Wang G, Murphy-Corb M, Wiley CA. Induction of cell-cycle regulators in simian immunodeficiency virus encephalitis. The American journal of pathology. 2000;157:497–507. doi: 10.1016/S0002-9440(10)64561-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly BL, Vassar R, Ferreira A. Beta-amyloid-induced dynamin 1 depletion in hippocampal neurons. A potential mechanism for early cognitive decline in Alzheimer disease. J Biol Chem. 2005;280:31746–31753. doi: 10.1074/jbc.M503259200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoutorsky A, Spira ME. Activity-dependent calpain activation plays a critical role in synaptic facilitation and post-tetanic potentiation. Learn Mem. 2009;16:129–141. doi: 10.1101/lm.1275709. [DOI] [PubMed] [Google Scholar]
- Klockner U, Mikala G, Varadi M, Varadi G, Schwartz A. Involvement of the carboxyl-terminal region of the alpha 1 subunit in voltage-dependent inactivation of cardiac calcium channels. The Journal of biological chemistry. 1995;270:17306–17310. doi: 10.1074/jbc.270.29.17306. [DOI] [PubMed] [Google Scholar]
- Kopil CM, Siebert AP, Foskett JK, Neumar RW. Calpain-cleaved type 1 inositol 1,4,5-trisphosphate receptor impairs ER Ca(2+) buffering and causes neurodegeneration in primary cortical neurons. Journal of neurochemistry. 2012;123:147–158. doi: 10.1111/j.1471-4159.2012.07859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusek JC, Greene RM, Pisano MM. Expression of the E2F and retinoblastoma families of proteins during neural differentiation. Brain research bulletin. 2001;54:187–198. doi: 10.1016/s0361-9230(00)00447-0. [DOI] [PubMed] [Google Scholar]
- Kuwako K, Taniura H, Yoshikawa K. Necdin-related MAGE proteins differentially interact with the E2F1 transcription factor and the p75 neurotrophin receptor. The Journal of biological chemistry. 2004;279:1703–1712. doi: 10.1074/jbc.M308454200. [DOI] [PubMed] [Google Scholar]
- Lang SE, McMahon SB, Cole MD, Hearing P. E2F transcriptional activation requires TRRAP and GCN5 cofactors. J Biol Chem. 2001;276:32627–32634. doi: 10.1074/jbc.M102067200. [DOI] [PubMed] [Google Scholar]
- Lee YM, Shin SI, Shin KS, Lee YR, Park BH, Kim EC. The role of sirtuin 1 in osteoblastic differentiation in human periodontal ligament cells. Journal of periodontal research. 2011;46:712–721. doi: 10.1111/j.1600-0765.2011.01394.x. [DOI] [PubMed] [Google Scholar]
- Lees JA, Saito M, Vidal M, Valentine M, Look T, Harlow E, Dyson N, Helin K. The retinoblastoma protein binds to a family of E2F transcription factors. Mol Cell Biol. 1993;13:7813–7825. doi: 10.1128/mcb.13.12.7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xu W, McBurney MW, Longo VD. SirT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2 signaling and protects neurons. Cell metabolism. 2008;8:38–48. doi: 10.1016/j.cmet.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MC, Kobeissy F, Zheng W, Zhang Z, Hayes RL, Wang KK. Dual vulnerability of tau to calpains and caspase-3 proteolysis under neurotoxic and neurodegenerative conditions. ASN neuro. 2011;3:e00051. doi: 10.1042/AN20100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch DR, Gleichman AJ. Picking up the pieces: the roles of functional remnants of calpain-mediated proteolysis. Neuron. 2007;53:317–319. doi: 10.1016/j.neuron.2007.01.014. [DOI] [PubMed] [Google Scholar]
- Magri L, Swiss VA, Jablonska B, et al. E2F1 Coregulates Cell Cycle Genes and Chromatin Components during the Transition of Oligodendrocyte Progenitors from Proliferation to Differentiation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:1481–1493. doi: 10.1523/JNEUROSCI.2840-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Achim CL, Ge N, DeTeresa R, Terry RD, Wiley CA. Spectrum of human immunodeficiency virus-associated neocortical damage. Annals of neurology. 1992;32:321–329. doi: 10.1002/ana.410320304. [DOI] [PubMed] [Google Scholar]
- Masliah E, Heaton RK, Marcotte TD, et al. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Annals of neurology. 1997;42:963–972. doi: 10.1002/ana.410420618. [DOI] [PubMed] [Google Scholar]
- Morgello S, Gelman BB, Kozlowski PB, et al. The National NeuroAIDS Tissue Consortium: a new paradigm in brain banking with an emphasis on infectious disease. Neuropathol Appl Neurobiol. 2001;27:326–335. doi: 10.1046/j.0305-1846.2001.00334.x. [DOI] [PubMed] [Google Scholar]
- Nicolas G, Fournier CM, Galand C, et al. Tyrosine phosphorylation regulates alpha II spectrin cleavage by calpain. Mol Cell Biol. 2002;22:3527–3536. doi: 10.1128/MCB.22.10.3527-3536.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell LA, Agrawal A, Jordan-Sciutto KL, Dichter MA, Lynch DR, Kolson DL. Human immunodeficiency virus (HIV)-induced neurotoxicity: roles for the NMDA receptor subtypes. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:981–990. doi: 10.1523/JNEUROSCI.4617-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hare MJ, Hou ST, Morris EJ, Cregan SP, Xu Q, Slack RS, Park DS. Induction and modulation of cerebellar granule neuron death by E2F-1. The Journal of biological chemistry. 2000;275:25358–25364. doi: 10.1074/jbc.M001725200. [DOI] [PubMed] [Google Scholar]
- Park SY, Ferreira A. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:5365–5375. doi: 10.1523/JNEUROSCI.1125-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips AC, Ernst MK, Bates S, Rice NR, Vousden KH. E2F-1 potentiates cell death by blocking antiapoptotic signaling pathways. Molecular cell. 1999;4:771–781. doi: 10.1016/s1097-2765(00)80387-1. [DOI] [PubMed] [Google Scholar]
- Qin Q, Liao G, Baudry M, Bi X. Role of calpain-mediated p53 truncation in semaphorin 3A-induced axonal growth regulation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13883–13887. doi: 10.1073/pnas.1008652107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan S, Bowser R. Alterations in G(1) to S phase cell-cycle regulators during amyotrophic lateral sclerosis. Am J Pathol. 2003;162:823–835. doi: 10.1016/S0002-9440(10)63879-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan S, Scudiere S, Bowser R. Hyperphosphorylation of the retinoblastoma gene product and altered subcellular distribution of E2F-1 during Alzheimer’s disease and amyotrophic lateral sclerosis. Journal of Alzheimer’s disease : JAD. 2001;3:377–385. doi: 10.3233/jad-2001-3403. [DOI] [PubMed] [Google Scholar]
- Roberts-Lewis JM, Savage MJ, Marcy VR, Pinsker LR, Siman R. Immunolocalization of calpain I-mediated spectrin degradation to vulnerable neurons in the ischemic gerbil brain. J Neurosci. 1994;14:3934–3944. doi: 10.1523/JNEUROSCI.14-06-03934.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross JF, Liu X, Dynlacht BD. Mechanism of transcriptional repression of E2F by the retinoblastoma tumor suppressor protein. Mol Cell. 1999;3:195–205. doi: 10.1016/s1097-2765(00)80310-x. [DOI] [PubMed] [Google Scholar]
- Saito K, Elce JS, Hamos JE, Nixon RA. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci U S A. 1993;90:2628–2632. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachan GD, Koike MA, Siman R, Hall DJ, Jordan-Sciutto KL. E2F1 induces cell death, calpain activation, and MDMX degradation in a transcription independent manner implicating a novel role for E2F1 in neuronal loss in SIV encephalitis. Journal of cellular biochemistry. 2005;96:728–740. doi: 10.1002/jcb.20574. [DOI] [PubMed] [Google Scholar]
- Taniguchi N, Taniura H, Niinobe M, Takayama C, Tominaga-Yoshino K, Ogura A, Yoshikawa K. The postmitotic growth suppressor necdin interacts with a calcium-binding protein (NEFA) in neuronal cytoplasm. The Journal of biological chemistry. 2000;275:31674–31681. doi: 10.1074/jbc.M005103200. [DOI] [PubMed] [Google Scholar]
- Ting JH, Marks DR, Schleidt SS, Wu JN, Zyskind JW, Lindl KA, Blendy JA, Pierce RC, Jordan-Sciutto KL. Targeted gene mutation of E2F1 evokes age-dependent synaptic disruption and behavioral deficits. Journal of neurochemistry. 2014 doi: 10.1111/jnc.12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P, Buzder-Lantos P, Tantos A, Farkas A, Szilagyi A, Banoczi Z, Hudecz F, Friedrich P. On the sequential determinants of calpain cleavage. The Journal of biological chemistry. 2004;279:20775–20785. doi: 10.1074/jbc.M313873200. [DOI] [PubMed] [Google Scholar]
- Trouche D, Cook A, Kouzarides T. The CBP co-activator stimulates E2F1/DP1 activity. Nucleic Acids Res. 1996;24:4139–4145. doi: 10.1093/nar/24.21.4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Reyn CR, Mott RE, Siman R, Smith DH, Meaney DF. Mechanisms of calpain mediated proteolysis of voltage gated sodium channel alpha-subunits following in vitro dynamic stretch injury. Journal of neurochemistry. 2012;121:793–805. doi: 10.1111/j.1471-4159.2012.07735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Reyn CR, Spaethling JM, Mesfin MN, Ma M, Neumar RW, Smith DH, Siman R, Meaney DF. Calpain mediates proteolysis of the voltage-gated sodium channel alpha-subunit. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:10350–10356. doi: 10.1523/JNEUROSCI.2339-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosler PS, Brennan CS, Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Molecular neurobiology. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Liu K, Lin FT, Lin WC. A role for 14-3-3 tau in E2F1 stabilization and DNA damage-induced apoptosis. The Journal of biological chemistry. 2004;279:54140–54152. doi: 10.1074/jbc.M410493200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Briz V, Chishti A, Bi X, Baudry M. Distinct roles for mu-calpain and m-calpain in synaptic NMDAR-mediated neuroprotection and extrasynaptic NMDAR-mediated neurodegeneration. J Neurosci. 2013;33:18880–18892. doi: 10.1523/JNEUROSCI.3293-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Shyam N, Ting JH, Akay C, Lindl KA, Jordan-Sciutto KL. E2F1 localizes predominantly to neuronal cytoplasm and fails to induce expression of its transcriptional targets in human immunodeficiency virus-induced neuronal damage. Neuroscience letters. 2010;479:97–101. doi: 10.1016/j.neulet.2010.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, White MG, Akay C, et al. Activation of cyclin-dependent kinase 5 by calpains contributes to human immunodeficiency virus-induced neurotoxicity. Journal of neurochemistry. 2007;103:439–455. doi: 10.1111/j.1471-4159.2007.04746.x. [DOI] [PubMed] [Google Scholar]
- White MG, Wang Y, Akay C, Lindl KA, Kolson DL, Jordan-Sciutto KL. Parallel high throughput neuronal toxicity assays demonstrate uncoupling between loss of mitochondrial membrane potential and neuronal damage in a model of HIV-induced neurodegeneration. Neuroscience research. 2011;70:220–229. doi: 10.1016/j.neures.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadran S, Qin Q, Bi X, Zadran H, Kim Y, Foy MR, Thompson R, Baudry M. 17-Beta-estradiol increases neuronal excitability through MAP kinase-induced calpain activation. Proc Natl Acad Sci U S A. 2009;106:21936–21941. doi: 10.1073/pnas.0912558106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Luo P, Guo Q, et al. Interactions between SIRT1 and MAPK/ERK regulate neuronal apoptosis induced by traumatic brain injury in vitro and in vivo. Experimental neurology. 2012;237:489–498. doi: 10.1016/j.expneurol.2012.07.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.