

Abstract
Background
Plasma contains cell-derived extracellular vesicles (EVs), which participate in physiopathological processes and have potential applications as disease biomarker. However, the enumeration of EVs faces major problems, due to their sub-micrometer size and to intrinsic limitations in methods of characterization, mainly flow cytometry (FCM).
Objectives
Our objective is to enumerate EVs in plasma, by taking as the prototype the population of phosphatidylserine (PS)-exposing EVs, which constitute one of the major EV populations and are responsible for thrombotic disorders.
Methods
The concentration of PS-exposing EVs in platelet-free plasma (PFP) of healthy subjects was measured by FCM using either light scattering or fluorescence as the trigger and fluorescent Annexin-5 (Anx5) as the specific label. In addition, PS-exposing EVs were enumerated by electron microscopy (EM) after labeling with Anx5 gold nanoparticles and sedimentation on EM grids.
Results
We show that about 50× more Anx5-positive EVs are detected by FCM when detection is triggered on fluorescence as compared with light scattering. By fluorescence triggering, concentrations of 22 000–30 000 Anx5-positive EVs per μL PFP were determined, using two different flow cytometers. The limit of detection of the fluorescence triggering method was estimated at about 1000–2500 Anx5 molecules. Results from EM suggest that EVs down to 100–150 nm diameter are detected by fluorescence triggering.
Conclusion
This study presents a simple method for enumerating EVs. We believe that this method is applicable in a general context and will improve our understanding of the roles of EVs in pathophysiological situations, which will open avenues for the development of EV-based diagnosis assays.
Keywords: blood plasma, cell-derived microparticles, electron microscopy, flow cytometry, phosphatidylserines
To cite this article:Arraud N, Gounou C, Linares R, Brisson AR. A simple flow cytometry method improves the detection of phosphatidylserine-exposing extracellular vesicles. J Thromb Haemost 2015; 13: 237–47.
Introduction
Blood and other body fluids contain cell-derived extracellular vesicles (EVs) that consist of small pieces of cytoplasm surrounded by a lipid membrane. EVs are commonly classified according to their formation mechanism and their size [1,2]. EVs called microparticles or microvesicles are sub-micrometer vesicles that are shed from the cell plasma membrane, while exosomes are 50–100-nm vesicles that are secreted by cells via exocytosis of multivesicular bodies. In this paper, the term EV will be used to designate all types of cell-derived vesicles that are found in plasma [3,4].
Considered initially as cellular debris or waste particles [5,6], there is now increasing evidence that EVs are involved in numerous functions [7,8]. In blood, EVs participate in physiological processes of coagulation, inflammation or intercellular communication, while elevated EV levels have been reported in numerous diseases [9–11], including cardiovascular diseases, cancer, sepsis and autoimmune diseases [12–15]. EVs exposing the procoagulant lipid phosphatidylserine (PS) have attracted major interest for several reasons. According to the classical theory of EV formation at cells' plasma membranes, the exposure of PS molecules on the outer membrane leaflet constitutes an early step of cell activation processes, which precedes membrane blebbing and EV shedding [16–18]. Most studies on plasmatic EVs are in keeping with this theory and consider that PS-exposing EVs represent the majority, or even the totality of EVs [19–23]. In addition, PS and tissue factor-exposing EVs have been implicated in thrombotic events associated with various pathologies [11,24,25]. Furthermore, the physiological importance of PS-exposing EVs in hemostasis is underlined by the fact that two bleeding disorders, Scott syndrome and Castaman's defect [26,27], are characterized by an impairment in generating PS-exposing EVs.
However, despite intense research, current knowledge on EVs is still limited. This is mainly due to the small size of EVs, most of them being smaller than 500 nm [20,21,28], and to intrinsic limitations of methods applied for their characterization. Over the last two decades, flow cytometry (FCM) has been the main method used for characterizing EVs [1,2,29]. In the classical FCM approach, referred to hereafter as conventional FCM, objects are detected in a two-step process, first on the basis of their light scattering intensity, which must exceed a threshold value, and second on the basis of their specific labeling with fluorescent ligands. This approach has established the existence of EVs from different cellular origins in various body fluids [1,2,4,30–32]. However, major issues have emerged concerning the actual size and amount of EVs detected by FCM. In particular, it is now well established that polymer particles initially used as size calibrators do not constitute proper references for sizing EVs, due to their different light scattering properties [33,34]. Recent theoretical studies have reported that 500-nm polymer particles scatter light similarly to 800-nm EVs on a standard flow cytometer [35]. Also, we have recently shown experimentally that only one to a few % of the PS-exposing EVs observed by quantitative electron microscopy (EM) were detected by conventional FCM [28].
Other methods have been applied to the detection and quantification of EVs, including EM, atomic force microscopy (AFM), dynamic light scattering (DLS), nanoparticle tracking analysis (NTA) or resistive pulse sensing (RPS). These methods present specific advantages, as recently reviewed in [36]. However, FCM remains the method of choice for phenotyping large numbers of samples, as required in a biomedical context. It is worth noting that the detection of small objects is not a new issue in FCM. More than 30 years ago, dedicated flow cytometers were developed that were able to detect 100-nm viruses based on their light scattering properties [37,38]. More recently, Marie et al. [39,40] pioneered an alternative strategy that consisted of triggering detection of viruses on a fluorescence parameter instead of a light scattering parameter. In the EV field, this approach has been rarely applied [12,41], yet its power was recently illustrated with the detection of cell culture EVs using a high-end flow cytometer and an original labeling strategy in which a lipophilic fluorophore was incorporated into EV membranes [42].
Here, we present a simple method for detecting PS-exposing EVs from plasma samples by fluorescence (FL) triggering. In addition, we compare results obtained by this method and by a quantitative EM approach recently introduced for enumerating EVs [28].
Methods
Materials
Phe-Pro-Arg chloromethyl ketone (PPACK) was from Haematologic Technologies (Cryopep, Montpellier, France). Fluorescein-5-maleimide (Fluo-Mal) and cyanine-5-maleimide (Cy5-Mal) were from Invitrogen (Saint Aubin, France) and GE Healthcare (Velizy-Villacoublay, France), respectively. Size calibration fluorescent particles Estapor® F-XC100 (1-μm) and F-XC050 (500-nm) were from Merck Chemicals (Fontenay-sous-Bois, France). Fluorescence calibration particles Quantum™ fluorescein isothiocyanate (FITC)-5 molecule equivalent soluble fluorophore (MESF) were from Bangs Laboratories (Polysciences, Eppelheim, Germany). Sphero™ 1-μm Ultra Rainbow, 240-nm yellow fluorescent particles (YFP 240) and 2-μm AccuCount particles were from Spherotech (Interchim, Montluçon, France).
All other chemicals were of ultrapure grade (Sigma-Aldrich, Lyon, France). Ultrapure water with a resistivity of 18.2 MΩ.cm was produced by a RiOs5-Synergy system (Millipore, Molsheim, France).
Preparation of plasma samples
Blood was collected after written informed consent from five healthy male donors who had fasted for at least 12 h [43]. Blood was drawn from an antecubital vein using a 21-Gauge needle. A light tourniquet was applied during collection of the first tube, which was discarded, then the tourniquet was released and blood was collected in 4.5 mL BD Vacutainer® tubes containing 0.1 volume of 105 mm sodium citrate. The preparation of platelet-free plasma (PFP) was started less than 1 hour after blood collection and consisted of two cycles of centrifugation at 2500 × g for 15 min at 25 °C in an Eppendorf 5804 R centrifuge equipped with an A-4-44 swinging bucket rotor [23].
Production of fluorescently labeled Annexin-5 (Anx5-F*)
An Annexin-5 (Anx5) mutant containing a single cystein residue was produced and conjugated with Fluo-Mal or Cy5-Mal, as previously described [44].
Flow cytometry
PFP samples were diluted 10× in a buffer made of 150 mm NaCl, 2 mm NaN3, 10 μm PPACK, 10 mm HEPES, pH 7.4 (HBS buffer), then Anx5-F* was added at 2.8 nm final concentration, unless otherwise stated. Recalcification was achieved by adding 10 mm CaCl2. FCM was performed after at least 30-min incubation at ambient temperature. As negative controls, PFP samples were prepared as above in the absence of Ca2+.
The majority of FCM experiments were performed with a Beckman Coulter FC 500 (Villepinte, France) equipped with an additional solid phase 635 nm laser and running CXP Acquisition 2.2. A few experiments were performed with a Beckman Coulter Gallios in a 3 laser/10 color configuration and running Gallios cytometer 1.2.
A detailed description of the FCM operating conditions, data acquisition and analysis protocol is provided in Supplementary Methods.
Quantification of Anx5-positive-EVs by EM
Anx5-positive EVs were enumerated by EM after sedimentation on EM grids as previously described [28]. Briefly, PFP samples were labeled with Anx5-conjugated gold nanoparticles (Anx5-gold-NPs) [45] as follows: 100 μL PFP were mixed with 1 μL 1 mm PPACK, 1 μL Anx5-gold-NPs at 1-3 × 1016 NP/L, supplemented with 10 mm Ca2+, and incubated for 30 min at ambient temperature. Samples were then diluted 30× with 100 mm cacodylate buffer, pH 7.4, containing 2 mm Ca2+, and deposited into 4.5 mL polyallomer centrifuge tubes containing at the bottom a 12-mm diameter hemispherical epoxy resin support onto which four EM grids coated with a continuous carbon film had been fixed (Fig. S1). Samples were centrifuged in an Optima™ MAX-E Ultracentrifuge equipped with a MLS50 rotor at 100 000 × g for 1 h at 20 °C, after which the liquid above EM grids was discarded, and EM grids were recovered and air dried.
Grids were observed with a CM120 microscope (FEI, France) operated at 120 kV. Images were recorded with a USC1000-SSCCD camera (Gatan, Warrandale, PA, USA).
A stringent method was applied for counting Anx5-positive EVs, in which only objects that presented a high and homogenous gold-NP labeling with a well-defined shape were taken into account.
Results
Detection of Anx5-positive EVs in PFP samples by FS triggering
PS-exposing EVs were identified via specific labeling with fluorescent Anx5, a protein binding with high affinity to PS-exposing membranes in the presence of Ca2+ [46–48].
PFP samples were first analyzed by a conventional FCM approach, in which the trigger was set on the forward scatter (FS) parameter and the detection was limited to events labeled by Anx5-F* (Fig.1A,B). The concentration of Anx5-positive EVs detected by this approach was 457 ± 178 (n = 10) per μL pure PFP. This value is consistent with previous reports on EVs from normal PFP [20,23,28,30]. Displayed on a FS vs. side scatter (SS) color dot plot, most of the Anx5-positive EVs are superposed with the background from the buffer (Fig.1A,B). On an FL1 vs. SS plot, two populations of Anx5-positive EVs can be distinguished. About 30% of them (134 ± 91 per μL) form a distinct cluster of high fluorescence intensity events (colored blue in Fig.1B). As previously reported [28], these objects, which are characterized by a high fluorescence intensity and scattering properties similar to 500-nm polymer particles (Fig.1B left), correspond to empty erythrocytes and are called hereafter erythrocyte ghosts. The rest of the Anx5-positive EVs are colored red in Fig.1B.
Figure 1.
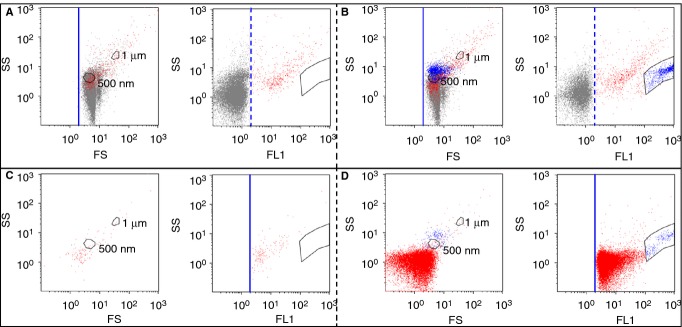
Flow cytometry (FCM) analysis of a platelet-free plasma (PFP) sample by FS triggering (A,B) and FL triggering (C,D), in the absence of Ca2+ (A, C) and in the presence of Ca2+ (B, D). (A-D). Each of the four panels presents on the left an FS vs. SS color dot plot and on the right an FL1 vs. SS color dot plot. Thresholds are represented by a solid blue line, while fluorescence positivity gates are represented by a dashed blue line. The positions of 500-nm and 1-μm polystyrene particles are indicated in each FS vs. SS plot (Fig. S2). (A,B) FS triggering analysis. The acquisition time was 10 min. Events labeled with Anx5-Fluo are separated into two groups: events of high fluorescence intensity forming a well-defined cluster are colored blue, and all the other Anx5-positive events are colored red. (C,D) FL triggering analysis. The acquisition time was 1 min, which explains why the cluster of erythrocyte ghosts (colored blue) contains less events than in (B).
Detection of Anx5-positive EVs by FL triggering
Figure1(C,D) presents the results obtained when the same PFP sample was analyzed with the trigger set on the FL1 parameter, in the absence of Ca2+ (Fig.1C) and in the presence of Ca2+ (Fig.1D). After recalcification, a large number of Anx5-positive EVs were detected, namely 22 326 ± 206/μL PFP for this sample. Most of these Anx5-positive EVs formed a compact cluster, with a minimal fluorescence intensity of 2 arbitrary units (colored red in Fig.1D). The erythrocyte ghosts, identified by their high fluorescence intensity, formed a minor group of objects (colored blue in Fig.1D). Their number, namely 249 ± 208 (n = 10) per μL PFP, was larger than the number measured by FS triggering, which is likely to be due to the weak scattering intensity of some of them.
Using this approach, referred to hereafter as FL triggering, we determined an average concentration of 22 245 ± 3664 (n = 10) Anx5-positive EVs per μL PFP. Similar values were obtained with either Anx5-Fluo or Anx5-Cy5 (Table S1), hence Anx5-Fluo and Anx5-Cy5 were used interchangeably in the rest of this study. It is important to note that the numbers of Anx5-positive EVs measured by FL triggering are highly reproducible, with less than 5% difference between replicates.
By comparing the concentrations of Anx5-positive EVs detected by FL and FS triggering in 10 PFP samples, we conclude that the FL triggering approach enables us to detect 55 ± 24 (n = 10) more EVs than the conventional FS triggering approach.
The values reported above were obtained from 10 PFP samples that were selected because they were prepared and analyzed in the same experimental conditions. During this study, 30 additional PFPs from healthy subjects were also analyzed, although with slightly different conditions of preparation or FCM analysis. The concentrations of Anx5-positive EVs detected by FL and FS triggering were determined for these 30 PFPs, indicating that FL triggering detects 51 ± 24 x more EVs than FS triggering.
Chelation of Ca2+ with EDTA resulted in a near total disappearance of the Anx5-positive EVs, as expected from the Ca2+-dependency of Anx5 binding to PS-exposing membranes (Fig. S3C) [47]. Similarly, a near total reversion of the signal was observed after addition of 0.5% Triton X-100 (Fig. S3D), confirming the lipidic nature of EVs. In addition, we found that after sedimentation of PFP samples at 20 000 × g, most of the Anx5-positive EVs disappeared from the supernatants (Fig. S3E) and were recovered in the resuspended pellets (Fig. S3F). This ensemble of data allows us to conclude that the Anx5-positive objects detected by FL triggering are genuine Anx5-positive EVs.
The Ca2+ concentration of 10 mm used for recalcifying PFP samples was selected because it corresponds to the plateau value of Ca2+ concentration curves (Fig. S4). Furthermore, we verified with synthetic large unilamellar vesicles (LUVs) that, in the conditions used here, Anx5 binding is specific to PS-exposing membranes, with no detectable binding to phosphatidylethanolamine-exposing liposomes (Fig. S5).
Influence of Anx5-F* concentration on the detection of Anx5-positive EVs
Next, we investigated the influence of Anx5-F* concentration on the detection of Anx5-positive EVs. For both FS and FL triggering, the number of Anx5-positive EVs detected was found to increase with increasing Anx5-F* concentration, until a plateau value was reached (Fig.2). In the conditions used here, namely 10 ×-diluted PFP, the plateau was reached at 0.8 nm Anx5-F*. Most experiments reported in this paper were performed with 2.8 nm Anx5-F*, therefore in saturating conditions.
Figure 2.
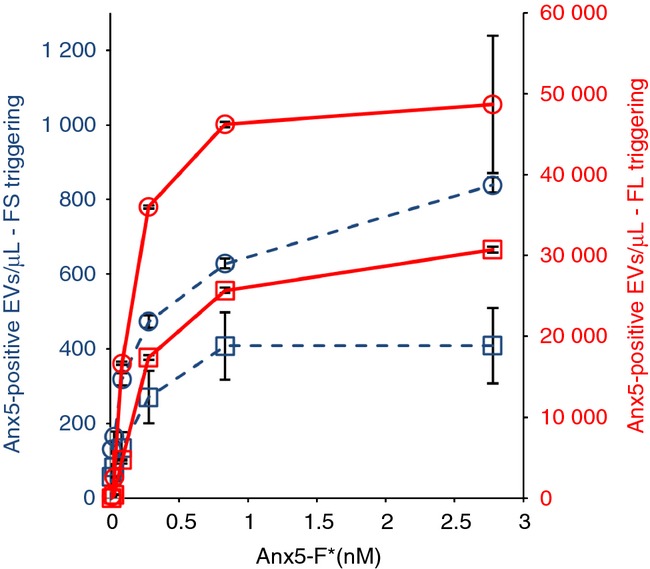
Influence of Anx5-F* concentration on the concentrations of Anx5-positive extracellular vesicles (EVs) detected by FS and fluorescence (FL) triggering. Curves representing the concentrations of Anx5-positive EVs (expressed per μL of pure platelet-free plasma) detected by FS (dashed lines) and FL triggering (plain lines) at various Anx5-F* concentrations, for two platelet-free plasma (PFP) samples (represented by squares and circles, respectively). The concentrations of Anx5-F* range from 0 to 2.8 nM. Each point represents the mean ± SD of two independent aliquots measured in duplicate.
For the two PFP samples analyzed in Fig.2, the concentrations of Anx5-positive EVs detected by FL triggering were, respectively, 58 (circles) and 75 (squares) × larger than the concentrations detected by FS triggering.
Influence of PFP dilution on the detection of Anx5-positive EVs
In order to evaluate the possible occurrence of coincidence effects on the detection of Anx5-positive EVs [36,49], we determined the concentration of Anx5-positive EVs for serial 2-fold dilutions of PFP samples, using a fixed concentration of 2.8 nm Anx5-F*.
Figure3 shows the results of two independent experiments in which Anx5-positive EVs were detected using either FS triggering (blue symbols) or FL triggering (red symbols), for dilution factors ranging from 4× to 128×. The concentration of Anx5-positive EVs in diluted PFP samples is observed to decrease linearly with the reciprocal dilution factor. In addition, the mean fluorescence intensity of EVs detected by FL triggering remains constant over the entire dilution range (Fig. S6). These results indicate the absence of coincidence effects on the detection of Anx5-positive EVs. Therefore, as the experiments reported here were performed with 10 ×-diluted PFP samples, we conclude that the 22 000 Anx5-positive EVs determined by FL triggering correspond to individual EVs.
Figure 3.
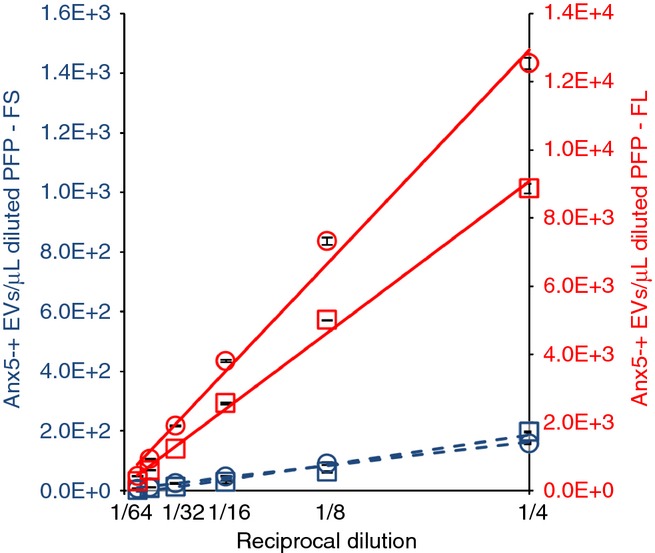
Influence of platelet-free plasma (PFP) dilution on the detection of Anx5-positive extracellular vesicles (EVs). Curves representing the concentrations of Anx5-positive EVs in diluted PFP samples detected either by FS (dashed lines) or fluorescence (FL) triggering (plain lines) vs. the reciprocal dilution factor, for two PFP samples (represented by squares and circles, respectively). The calculated linear regression lines are presented. R² values higher than 0.96 were obtained for all regression lines. By extrapolating the regression lines to pure PFP (dilution factor 1 x), Anx5-positive EVs concentrations of 35 666 (squares) and 50 239 (circles) are obtained. Each point represents the mean ± SD of two independent aliquots measured in duplicate.
In the two independent experiments presented in Fig.3, the numbers of Anx5-positive EVs detected by FL triggering were respectively 45 (squares) and 79 (circles) × larger than the values measured by FS triggering.
Determination of the number of Anx5-F* molecules required for detecting single Anx5-positive EVs by FL triggering
Next, we focused on the determination of the sensitivity of the FL triggering approach, which can be defined as the minimal number of Anx5-F* molecules needed for detecting one single Anx5-positive EV. The fluorescence intensities of a mixture of MESF-FITC calibration particles containing from 0 to 756 000 MESF were measured by FS triggering (Fig.4A,B), from which a calibration curve relating the number of fluorophores of the particles to their fluorescence intensity was drawn (Fig.4C).
Figure 4.

Determination of the number of Anx5-F* molecules needed to detect individual extracellular vesicles (EVs) by fluorescence (FL) triggering. (A-C) FS triggering analysis of a mixture of Quantum™ fluorescein isothiocyanate (FITC)-5 MESF particles made of six particle populations containing, respectively, 0, 1484, 9828, 47 640, 188 438 and 756 101 MESF. (A) The 7.7-μm fluorescent particles form a homogeneous cluster, located in the upper right corner of the FS vs. SS color dot plot. (B) Histogram of fluorescence intensities, showing four resolved peaks (A,B,C,D) corresponding to particles containing 9828 fluorophores and above, while the 1484 MESF particles form a peak (E) overlapping the blank particles (F), which present some auto-fluorescence. (C) Calibration curve correlating the MESF-FITC of the particles to their mean fluorescence intensity, taken from (B). (D) Enlarged view from the low-fluorescence values of the calibration curve (C). The minimal fluorescence intensity of 2 arbitrary units measured by FL triggering is indicated (horizontal arrow), together with its corresponding MESF value, namely 2500 MESF (vertical arrow). (E) FL1 triggering analysis of the Quantum™ FITC-5 MESF particles. Four peaks (A,B,C,D) corresponding to particles containing 9828 MESF and above are detected, while the population of 1484 MESF particles is not detected. The threshold is represented by a solid blue line.
Based on this calibration curve, the minimal fluorescence intensity measured by FL triggering, which is equal to 2 fluorescence arbitratry units, is found to correspond to about 2500 MESF. As the Anx5-F* molecules used in this study bear one single fluorophore per protein [44], we conclude that the sensitivity of the FL triggering approach corresponds to 2500 Anx5-F* (Fig.4D).
This result was further confirmed by analyzing the mixture of MESF-FITC calibration particles by FL triggering (Fig.4E). Indeed, only four populations of particles were detected, those containing 9828 fluorophores and above. The 1484 MESF particles, of 1.34 mean fluorescence intensity, were not detected by FL triggering.
Enumeration of Anx5-positive EVs by EM
In order to determine the minimal size of Anx5-positive EVs detected by FL triggering, we analyzed the same PFP samples by EM. As we reported recently [28], Anx5-positive EVs can be enumerated reliably by EM after labeling with Anx5-gold-NPs and high-speed sedimentation on EM grids (Fig. S1).
A gallery of EV images obtained by this procedure is presented in Fig.5. Most EVs (Fig.5A–D) present a near-circular shape, as expected from spherical EVs projected onto a flat support. Some EVs present a tubular morphology (Fig.5C,D), while a minor number consists of large fragments and erythrocyte ghosts (Fig.5E).
Figure 5.

Gallery of Anx5-positive extracellular vesicles (EVs) observed by EM. (A-E) Representative images of Anx5-positive EVs observed on EM grids after sedimentation. Most EVs present a near-circular shape. Very rare gold-nanoparticles (NPs) are present in the background, illustrating the high specificity of Anx5-gold-NP labeling. Some EVs devoid of Anx5-gold-NPs are labeled with arrows, including 50-nm spherical EVs in (B) and a 2-μm tubular EV in (C). The size of the Anx5-positive EVs is indicated on the images (in nm). Considering that a spherical EV, of radius R and area 4πR², transforms upon sedimentation into a flattened circular pancake, of radius R' and area 2πR'², the radius of the flattened pancake is related to the radius of the original sphere by the relationship R = R'√2. The size indicated in the images for the circular EVs is the diameter of the equivalent spheres. Scale bars: (A-D) 200 nm; (E) 2 μm. (F) Size histogram of Anx5-positive EVs. About 80% Anx5-positive EVs are comprised between 100 and 400 nm. Histogram determined over 500 images of EVs recorded on three EM grids. Error bars correspond to SD between the three individual datasets [28].
The concentration of Anx5-positive EVs determined by EM was 30 615 ± 10 243 (n = 11) per μL PFP. Although we lack a proper control for evaluating the yield of the sedimentation procedure, two independent results suggest strongly that most of the EVs present in centrifugation tubes are recuperated on EM grids. First, when a PFP sample is centrifuged a first time and its supernatant is submitted to a second centrifugation, basically no EVs are observed on EM grids. Second, the sedimentation conditions are sufficient to sediment EVs of small size, as demonstrated by the observation of such EVs on EM grids (e.g. in Fig.5B which shows two unlabeled EVs of about 50 nm diameter marked with arrows). We consider therefore that the value of about 30 000 Anx5-positive EVs per μL PFP corresponds to the large majority of this EV population. Yet, due to the stringent method of EV counting and the possible deformation of small EVs upon sedimentation, we consider likely that EVs smaller than about 100–150 nm diameter are under-estimated in this analysis.
The number of Anx5-positive EVs observed by EM is larger than the number determined by FL triggering (about 22 000 per μL). As about one-third of Anx5-positive EVs measured by EM range from 100 to 200 nm (Fig.5F), we estimate that EVs down to about 200 nm diameter were detected by FL triggering with the flow cytometer settings used here. This point will be further discussed below.
Application of the FL triggering approach to another flow cytometer
At a later stage of this study, we applied the FL triggering approach to another flow cytometer, namely a Gallios instrument. The concentration of Anx5-positive EVs detected by FL triggering was measured in five PFP samples and found to be 31 975 ± 13 306 per μL PFP (n = 5). The absolute number of Anx5-positive EVs detected with the Gallios is therefore higher than with the FC 500, as expected from the respective sensitivity of these instruments [50] (Fig. S7).
We determined the sensitivity of the FL triggering approach with this flow cytometer, as described above, and found that about 1000 Anx5-F* were required to detect individual Anx5-positive EVs (Fig. S8).
As the number of Anx5-positive EVs detected by FL triggering on the Gallios is almost equal to the number observed by EM, we conclude that EVs down to about 100–150 nm diameter were detected by FL triggering on this instrument.
Phenotyping of Anx5-positive EVs
Finally, we attempted to phenotype the Anx5-positive EVs detected by FL triggering. We focused on the populations of EVs derived from platelets and erythrocytes, which were identified by labeling with anti-CD41-mAb-PE and anti-CD235a-mAb-PE, respectively. As shown in Fig.6, when PFP was double labeled with Anx5-Cy5 and either anti-CD41-PE (Fig.6B) or anti-CD235a-PE (Fig.6C), a population of events positive for both markers was observed in the FL6 vs. FL2 color dot plots. We found that, for this PFP sample, 23% and 8% of the Anx5-positive EVs detected by FL triggering derived from platelets and erythrocytes, respectively.
Figure 6.

Phenotyping of Anx5-positive extracellular vesicles (EVs) detected by fluorescence (FL) triggering. A platelet-free plasma (PFP) sample was double labeled with Anx5-Cy5 and either anti-mouse-IgG-PE (A), anti-CD41-mAb-PE (B) or anti-CD235a-mAb-PE (C). FL6 fluorescence was used as a trigger, which means that all the detected events are Anx5-positive EVs. Anti-mouse-IgG-PE was used as an isotypic antibody to create a PE positivity gate on the FL6 vs. FL2 color dot plots. Events positive for only Anx5 are colored blue, while events positive for both Anx5 and a PE-conjugated mAb are colored red. On the bottom row, events labeled with Anx5 and/or a PE-conjugated antibody are backgated and displayed on FS vs. SS color dot plots. (C, top row) A small population of erythrocyte ghosts (EG) is observed in the top right corner (colored green). EG are characterized by their high intensity for both Anx5 and anti-CD235a labeling.
These results provide a proof of principle of the phenotyping of PFP EVs by FL triggering. A complete study will be described elsewhere.
Discussion
In this paper, we present a simple FCM method that improves the detection of PS-exposing EVs in plasma samples. In this method, the detection of EVs is based on their fluorescence signal, instead of their scattering intensity as in conventional FCM. Although the detection of small objects by FCM based on their fluorescence is a strategy that was introduced more than 10 years ago [12,39–41], this strategy has only been applied recently in the EV field [42,49,51].
In this study, we focused on PS-exposing EVs, which are considered to constitute a major population of cell-derived EVs and have been the subject of most FCM studies on EVs [18–23]. The main result of our work is that about 50× more Anx5-positive EVs are detected by the FL triggering approach as compared with a conventional FS triggering approach. For several years it has been recognized that only a fraction of EVs were detected by FCM, yet their exact amount remained unclear. This study is therefore in agreement with our previous EM study, which concluded that conventional FCM detects only a few per cent of EVs, as further discussed below [28].
The FL triggering method presents several advantages: (i) its simplicity, as it can be performed with most commercial flow cytometers; (ii) its reproducibility, as intra-sample variation coefficients of less than 5% were observed; (iii) its speed, as it requires only several minutes per sample; (iv) its cost effectiveness, as it requires only about 100 nanograms Anx5-F* per sample, together with small sample volumes (e.g. less than 100 μL plasma); and (v) this method is applicable to unprocessed plasma samples, thus avoiding potential artifacts from filtration or sedimentation. In addition, the fact that Anx5 binding to PS-exposing membranes can be induced by simple addition of Ca2+ constitutes a further advantage, because highly reliable negative controls can be obtained by analyzing the same samples in the absence of Ca2+. In comparison, negative controls in FCM involve in general the use of isotypic antibodies, often of unknown fluorophore to antibody ratio, which may lead to unsatisfactory results.
Using two different flow cytometers, we determined concentrations of Anx5-positive EVs of about 22 000 and 30 000 per μL normal PFP. Both values are of the same order of magnitude when compared with values previously reported from FS triggering studies, namely around 500 EVs per μL [20,23,28,30]. The additional fact that the values obtained independently by EM are close to those from FL triggering constitutes a strong support for the validity of both methods.
It is logical to wonder which percentage of the whole population of Anx5-positive EVs are detected by FL triggering and what is the smallest size of EVs detected by this method. Two independent sets of results provide some answers to these questions. First, using fluorescence calibration particles, we determined the number of Anx5-F* molecules necessary for detecting individual Anx5-positive EVs, namely about 2500 and 1000 Anx5-F* molecules for the FC 500 and Gallios, respectively. Considering that an Anx5 molecule covers a surface of 30 nm² on synthetic PS-exposing membranes [52–54] and approximating EVs to spheres made of pure lipids, 1000 and 2500 Anx5 molecules would cover vesicles of 100 and 150-nm diameter, respectively. These values must be considered with caution, because EVs are not pure liposomes and it is most likely that the presence of membrane proteins reduces the membrane surface accessible for Anx5 binding. Second, we found that the concentrations of Anx5-positive EVs detected by EM and by FL triggering with the Gallios were almost identical. As EM detects Anx5-positive EVs down to 100–150 nm in diameter, we conclude that the Anx5-positive EVs detected with the Gallios have similar size, while slightly larger EVs are detected with the FC 500. These two independent sets of results lead therefore to the same conclusion, which is that FL triggering allows detecting EVs of 100–150 nm diameter.
In principle, the FL triggering approach should be applicable to the quantification of EVs of other phenotypes. The prerequisites are that EVs present a sufficient number of receptors, that bright fluorescent ligands are available, and that reliable negative controls are designed.
This study presents the proof of concept of the quantification of EVs by the FL triggering method, demonstrated here in the case of PS-exposing EVs from plasma. As shown here, the use of different flow cytometers leads to different results. Therefore, in order to be able to compare results from different laboratories, standardized procedures will have to be established, including the use of fluorescent calibration particles, the use of well-controlled fluorescent proteins, and sample preparation methods [23].
We foresee that applying the FL triggering detection strategy on flow cytometers with improved optics and detector sensitivity [34,42,50,55] should allow an efficient identification and enumeration of all EVs, including the smallest ones. This is mandatory for a better understanding of the diversity of EVs' functions, and also for the development of EV-based biomarker assays.
Addendum
N. Arraud supervised and performed the FCM studies; C. Gounou produced the Anx5 proteins and performed part of the FCM studies; R. Linares performed the EM experiments and synthesized the Anx5-conjugated gold nanoparticles; A. R. Brisson coordinated the entire project.
Acknowledgments
We thank Ms M. Guillonet and Drs. G. Le Provost, F. Chaléat and F. Tarascon (Laboratoire Mutualiste d'Analyse Médicales de Pessac) for their help with the collection of blood samples. This study was supported by ANR (grants EMPB-MP-NPAuA5-2007-021-01 and 11-BSV1-03501-PlacentA5 to AB).
Disclosure of Conflict of Interests
A. R. Brisson reports a patent WO/2007/122259 pending. All other authors state that they have conflicts of interests.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Supplementary Methods
Table S1. Comparison of the concentrations of Anx5-positive EVs detected by FS and FL triggering with either Anx5-Fluo or Anx5-Cy5 labeling.
Fig. S1. Scheme of the procedure of sedimentation of EVs on EM grids.
Fig. S2. Characterization of the FC 500 (A,B) and the Gallios (C,D) flow cytometers used in this study by means of size calibration fluorescent particles.
Fig. S3. Effect of Ca2+ chelation, Triton X-100 and centrifugation on the detection of Anx5-positive EVs by FL triggering.
Fig. S4. Influence of the Ca2+ concentration on the number of Anx5-EVs detected by FL triggering.
Fig. S5. Binding of Anx5-F* to 200-nm liposomes, by FL triggering.
Fig. S6. Effect of PFP dilution on the mean fluorescence intensity of Anx5-positive EVs.
Fig. S7. Comparative analysis of a PFP sample with either a FC 500 or a Gallios flow cytometer, by FS and FL triggering.
Fig. S8. Determination of the limit of detection of the Gallios flow cytometer by FL triggering, using fluorescence calibration particles.
References
- 1.György B, Szabó TG, Pásztói M, Pál Z, Misják P, Aradi B, László V, Pállinger É, Pap E, Kittel Á, Nagy G, Falus A, Buzás EI. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci. 2011;68:2667–88. doi: 10.1007/s00018-011-0689-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Pol E, Böing AN, Harrison P, Sturk A, Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev. 2012;64:676–705. doi: 10.1124/pr.112.005983. [DOI] [PubMed] [Google Scholar]
- 3.Gould SJ, Raposo G. As we wait: coping with an imperfect nomenclature for extracellular vesicles. J Extracell Vesicles. 2013;2:20389. doi: 10.3402/jev.v2i0.20389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Witwer KW, Buzás EI, Bemis LT, Bora A, Lässer C, Lötvall J, Nolte-'t Hoen EN, Piper MG, Sivaraman S, Skog J, Théry C, Wauben MH, Hochberg F. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles. 2013;2:20360. doi: 10.3402/jev.v2i0.20360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolf P. The nature and significance of platelet products in human plasma. Br J Haematol. 1967;13:269–88. doi: 10.1111/j.1365-2141.1967.tb08741.x. [DOI] [PubMed] [Google Scholar]
- 6.Pan BT, Teng K, Wu C, Adam M, Johnstone RM. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J Cell Biol. 1985;101:942–8. doi: 10.1083/jcb.101.3.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–9. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 8.Burger D, Schock S, Thompson CS, Montezano AC, Hakim AM, Touyz RM. Microparticles: biomarkers and beyond. Clin Sci. 2013;124:423–41. doi: 10.1042/CS20120309. [DOI] [PubMed] [Google Scholar]
- 9.Horstman LL, Ahn YS. Platelet microparticles: a wide-angle perspective. Crit Rev Oncol Hematol. 1999;30:111–42. doi: 10.1016/s1040-8428(98)00044-4. [DOI] [PubMed] [Google Scholar]
- 10.vanWijk MJ, van Bavel E, Sturk A, Nieuwland R. Microparticles in cardiovascular diseases. Cardiovasc Res. 2003;59:277–87. doi: 10.1016/s0008-6363(03)00367-5. [DOI] [PubMed] [Google Scholar]
- 11.Morel O, Toti F, Hugel B, Bakouboula B, Camoin-Jau L, Dignat-George F, Freyssinet J-M. Procoagulant microparticles disrupting the vascular homeostasis equation? Arterioscler Thromb Vasc Biol. 2006;26:2594–604. doi: 10.1161/01.ATV.0000246775.14471.26. [DOI] [PubMed] [Google Scholar]
- 12.Wiedmer T, Hall SE, Ortel TL, Kane WH, Rosse WF, Sims PJ. Complement-induced vesiculation and exposure of membrane prothrombinase sites in platelets of paroxysmal nocturnal hemoglobinuria. Blood. 1993;82:1192–6. [PubMed] [Google Scholar]
- 13.Manly DA, Wang J, Glover SL, Kasthuri R, Liebman HA, Key NS, Mackman N. Increased microparticle tissue factor activity in cancer patients with Venous Thromboembolism. Thromb Res. 2010;125:511–2. doi: 10.1016/j.thromres.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boilard E, Nigrovic PA, Larabee K, Watts GFM, Coblyn JS, Weinblatt ME, Massarotti EM, Remold-O'Donnell E, Farndale RW, Ware J, Lee DM. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 2010;327:580–3. doi: 10.1126/science.1181928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delabranche X, Boisramé-Helms J, Asfar P, Berger A, Mootien Y, Lavigne T, Grunebaum L, Lanza F, Gachet C, Freyssinet J-M, Toti F, Meziani F. Microparticles are new biomarkers of septic shock-induced disseminated intravascular coagulopathy. Intensive Care Med. 2013;39:1695–703. doi: 10.1007/s00134-013-2993-x. [DOI] [PubMed] [Google Scholar]
- 16.Bevers EM, Comfurius P, Zwaal RFA. Changes in membrane phospholipid distribution during platelet activation. Biochim Biophys Acta. 1983;736:57–66. doi: 10.1016/0005-2736(83)90169-4. [DOI] [PubMed] [Google Scholar]
- 17.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–16. [PubMed] [Google Scholar]
- 18.Zwaal RFA, Schroit AJ. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood. 1997;89:1121–32. [PubMed] [Google Scholar]
- 19.Biró E, Sturk-Maquelin KN, Vogel GMT, Meuleman DG, Smit MJ, Hack CE, Sturk A, Nieuwland R. Human cell-derived microparticles promote thrombus formation in vivo in a tissue factor-dependent manner. J Thromb Haemost. 2003;1:2561–8. doi: 10.1046/j.1538-7836.2003.00456.x. [DOI] [PubMed] [Google Scholar]
- 20.Aras O, Shet A, Bach RR, Hysjulien JL, Slungaard A, Hebbel RP, Escolar G, Jilma B, Key NS. Induction of microparticle- and cell-associated intravascular tissue factor in human endotoxemia. Blood. 2004;103:4545–53. doi: 10.1182/blood-2003-03-0713. [DOI] [PubMed] [Google Scholar]
- 21.György B, Módos K, Pállinger É, Pálóczi K, Pásztói M, Misják P, Deli MA, Sipos Á, Szalai A, Voszka I, Polgár A, Tóth K, Csete M, Nagy G, Gay S, Falus A, Kittel Á, Buzás EI. Detection and isolation of cell-derived microparticles are compromised by protein complexes resulting from shared biophysical parameters. Blood. 2011;117:e39–48. doi: 10.1182/blood-2010-09-307595. [DOI] [PubMed] [Google Scholar]
- 22.Morel O, Jesel L, Freyssinet J-M, Toti F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler Thromb Vasc Biol. 2011;31:15–26. doi: 10.1161/ATVBAHA.109.200956. [DOI] [PubMed] [Google Scholar]
- 23.Lacroix R, Judicone C, Poncelet P, Robert S, Arnaud L, Sampol J, Dignat-George F. Impact of pre-analytical parameters on the measurement of circulating microparticles: towards standardization of protocol. J Thromb Haemost. 2012;10:437–46. doi: 10.1111/j.1538-7836.2011.04610.x. [DOI] [PubMed] [Google Scholar]
- 24.Owens AP, 3rd, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res. 2011;108:1284–97. doi: 10.1161/CIRCRESAHA.110.233056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubin O, Crettaz D, Canellini G, Tissot J-D, Lion N. Microparticles in stored red blood cells: an approach using flow cytometry and proteomic tools. Vox Sang. 2008;95:288–97. doi: 10.1111/j.1423-0410.2008.01101.x. [DOI] [PubMed] [Google Scholar]
- 26.Zwaal RFA, Comfurius P, Bevers EM. Scott syndrome, a bleeding disorder caused by defective scrambling of membrane phospholipids. Biochim Biophys Acta. 2004;1636:119–28. doi: 10.1016/j.bbalip.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 27.Castaman G, Yu-Feng L, Battistin E, Rodeghiero F. Characterization of a novel bleeding disorder with isolated prolonged bleeding time and deficiency of platelet microvesicle generation. Br J Haematol. 1997;96:458–63. doi: 10.1046/j.1365-2141.1997.d01-2072.x. [DOI] [PubMed] [Google Scholar]
- 28.Arraud N, Linares R, Tan S, Gounou C, Pasquet J-M, Mornet S, Brisson AR. Extracellular vesicles from blood plasma: determination of their morphology, size, phenotype and concentration. J Thromb Haemost. 2014;12:614–27. doi: 10.1111/jth.12554. [DOI] [PubMed] [Google Scholar]
- 29.Jy W, Horstman LL, Jimenez JJ, Ahn YS. Measuring circulating cell-derived microparticles. J Thromb Haemost. 2004;2:1842–3. doi: 10.1111/j.1538-7836.2004.00936.x. [DOI] [PubMed] [Google Scholar]
- 30.Ayers L, Kohler M, Harrison P, Sargent I, Dragovic R, Schaap M, Nieuwland R, Brooks SA, Ferry B. Measurement of circulating cell-derived microparticles by flow cytometry: sources of variability within the assay. Thromb Res. 2011;127:370–7. doi: 10.1016/j.thromres.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 31.Horstman LL, Jy W, Minagar A, Bidot CJ, Jimenez JJ, Alexander JS, Ahn YS. Cell-derived microparticles and exosomes in neuroinflammatory disorders. Int Rev Neurobiol. 2007;79:227–68. doi: 10.1016/S0074-7742(07)79010-4. [DOI] [PubMed] [Google Scholar]
- 32.Chironi GN, Boulanger CM, Simon A, Dignat-George F, Freyssinet J-M, Tedgui A. Endothelial microparticles in diseases. Cell Tissue Res. 2009;335:143–51. doi: 10.1007/s00441-008-0710-9. [DOI] [PubMed] [Google Scholar]
- 33.van der Pol E, Hoekstra AG, Sturk A, Otto C, van Leeuwen TG, Nieuwland R. Optical and non-optical methods for detection and characterization of microparticles and exosomes. J Thromb Haemost. 2010;8:2596–607. doi: 10.1111/j.1538-7836.2010.04074.x. [DOI] [PubMed] [Google Scholar]
- 34.Chandler WL, Yeung W, Tait JF. A new microparticle size calibration standard for use in measuring smaller microparticles using a new flow cytometer. J Thromb Haemost. 2011;9:1216–24. doi: 10.1111/j.1538-7836.2011.04283.x. [DOI] [PubMed] [Google Scholar]
- 35.van der Pol E, van Gemert MJC, Sturk A, Nieuwland R, van Leeuwen TG. Single vs. swarm detection of microparticles and exosomes by flow cytometry. J Thromb Haemost. 2012;10:919–30. doi: 10.1111/j.1538-7836.2012.04683.x. [DOI] [PubMed] [Google Scholar]
- 36.van der Pol E, Coumans F, Varga Z, Krumrey M, Nieuwland R. Innovation in detection of microparticles and exosomes. J Thromb Haemost. 2013;11:36–45. doi: 10.1111/jth.12254. [DOI] [PubMed] [Google Scholar]
- 37.Hercher M, Mueller W, Shapiro HM. Detection and discrimination of individual viruses by flow cytometry. J Histochem Cytochem. 1979;27:350–2. doi: 10.1177/27.1.374599. [DOI] [PubMed] [Google Scholar]
- 38.Steen HB. Flow cytometer for measurement of the light scattering of viral and other submicroscopic particles. Cytometry. 2004;57A:94–9. doi: 10.1002/cyto.a.10115. [DOI] [PubMed] [Google Scholar]
- 39.Marie D, Brussaard CPD, Thyrhaug R, Bratbak G, Vaulot D. Enumeration of marine viruses in culture and natural samples by flow cytometry. Appl Environ Microbiol. 1999;65:45–52. doi: 10.1128/aem.65.1.45-52.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brussaard CPD, Marie D, Bratbak G. Flow cytometric detection of viruses. J Virol Methods. 2000;85:175–82. doi: 10.1016/s0166-0934(99)00167-6. [DOI] [PubMed] [Google Scholar]
- 41.Bevers EM, Wiedmer T, Comfurius P, Shattil SJ, Weiss HJ, Zwaal RFA, Sims PJ. Defective Ca2 + -induced microvesiculation and deficient expression of procoagulant activity in erythrocytes from a patient with a bleeding disorder: a study of the red blood cells of Scott syndrome. Blood. 1992;79:380–8. [PubMed] [Google Scholar]
- 42.van der Vlist EJ, Nolte-'t Hoen ENM, Stoorvogel W, Arkesteijn GJA, Wauben MHM. Fluorescent labeling of nano-sized vesicles released by cells and subsequent quantitative and qualitative analysis by high-resolution flow cytometry. Nat Protoc. 2012;7:1311–26. doi: 10.1038/nprot.2012.065. [DOI] [PubMed] [Google Scholar]
- 43.Cantero M, Conejo J, Parra T, Jiménez A, Carballo F, de Arriba G. Interference of chylomicrons in analysis of platelets by flow cytometry. Thromb Res. 1998;91:49–52. doi: 10.1016/s0049-3848(98)00044-9. [DOI] [PubMed] [Google Scholar]
- 44.Bouter A, Gounou C, Bérat R, Tan S, Gallois B, Granier T, d'Estaintot BL, Pöschl E, Brachvogel B, Brisson AR. Annexin-A5 assembled into two-dimensional arrays promotes cell membrane repair. Nat Commun. 2011;2:270. doi: 10.1038/ncomms1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brisson A, Mornet S. 2007. Functionalization of Gold Nanoparticles with Oriented Proteins. Application to the High-Density Labelling of Cell Membranes Patent WO/2007/122259.
- 46.Andree HA, Reutelingsperger CP, Hauptmann R, Hemker HC, Hermens WT, Willems GM. Binding of vascular anticoagulant alpha (VAC α) to planar phospholipid bilayers. J Biol Chem. 1990;265:4923–8. [PubMed] [Google Scholar]
- 47.Tait JF, Gibson DF, Smith C. Measurement of the affinity and cooperativity of annexin V–membrane binding under conditions of low membrane occupancy. Anal Biochem. 2004;329:112–9. doi: 10.1016/j.ab.2004.02.043. [DOI] [PubMed] [Google Scholar]
- 48.Richter RP, Lai Kee Him J, Tessier B, Tessier C, Brisson AR. On the kinetics of adsorption and two-dimensional self-assembly of annexin a5 on supported lipid bilayers. Biophys J. 2005;89:3372–85. doi: 10.1529/biophysj.105.064337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nolan JP, Stoner SA. A trigger channel threshold artifact in nanoparticle analysis. Cytometry A. 2013;83A:301–5. doi: 10.1002/cyto.a.22255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robert S, Lacroix R, Poncelet P, Harhouri K, Bouriche T, Judicone C, Wischhusen J, Arnaud L, Dignat-George F. High-sensitivity flow cytometry provides access to standardized measurement of small-size microparticles—brief report. Arterioscler Thromb Vasc Biol. 2012;32:1054–8. doi: 10.1161/ATVBAHA.111.244616. [DOI] [PubMed] [Google Scholar]
- 51.Stoner S, Nolan J. 2013. Flow Cytometric Analysis of Single Lipid Membrane Vesicles. CYTO 2013 San Diego;; Abstract no. 85.
- 52.Pigault C, Follenius-Wund A, Schmutz M, Freyssinet J-M, Brisson A. Formation of two-dimensional arrays of annexin V on phosphatidylserine-containing liposomes. J Mol Biol. 1994;236:199–208. doi: 10.1006/jmbi.1994.1129. [DOI] [PubMed] [Google Scholar]
- 53.Reviakine I, Bergsma-Schutter W, Mazères-Dubut C, Govorukhina N, Brisson A. Surface topography of the p3 and p6 Annexin V crystal forms determined by atomic force microscopy. J Struct Biol. 2000;131:234–9. doi: 10.1006/jsbi.2000.4286. [DOI] [PubMed] [Google Scholar]
- 54.Oling F, Bergsma-Schutter W, Brisson A. Trimers, dimers of trimers, and trimers of trimers are common building blocks of Annexin A5 two-dimensional crystals. J Struct Biol. 2001;133:55–63. doi: 10.1006/jsbi.2000.4337. [DOI] [PubMed] [Google Scholar]
- 55.Hjuler Nielsen M, Beck-Nielsen H, Nørgaard Andersen M, Handberg A. A flow cytometric method for characterization of circulating cell-derived microparticles in plasma. J Extracell Vesicles. 2014;3:20795–807. doi: 10.3402/jev.v3.20795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Comparison of the concentrations of Anx5-positive EVs detected by FS and FL triggering with either Anx5-Fluo or Anx5-Cy5 labeling.
Fig. S1. Scheme of the procedure of sedimentation of EVs on EM grids.
Fig. S2. Characterization of the FC 500 (A,B) and the Gallios (C,D) flow cytometers used in this study by means of size calibration fluorescent particles.
Fig. S3. Effect of Ca2+ chelation, Triton X-100 and centrifugation on the detection of Anx5-positive EVs by FL triggering.
Fig. S4. Influence of the Ca2+ concentration on the number of Anx5-EVs detected by FL triggering.
Fig. S5. Binding of Anx5-F* to 200-nm liposomes, by FL triggering.
Fig. S6. Effect of PFP dilution on the mean fluorescence intensity of Anx5-positive EVs.
Fig. S7. Comparative analysis of a PFP sample with either a FC 500 or a Gallios flow cytometer, by FS and FL triggering.
Fig. S8. Determination of the limit of detection of the Gallios flow cytometer by FL triggering, using fluorescence calibration particles.