

Abstract
Effective T‐cell surveillance of antigen‐presenting cells is dependent on the expression of an array of antigenic peptides bound to major histocompatibility complex (MHC) class I (MHC‐I) or class II (MHC‐II) molecules. Pathogens co‐evolving with their hosts exploit crucial translational regulatory mechanisms in order to evade host immune recognition and thereby sustain their infection. Evasion strategies that downregulate viral protein synthesis and thereby restrict antigen presentation to cytotoxic T‐cells through the endogenous MHC‐I pathway have been implicated in the pathogenesis of viral‐associated malignancies. An understanding of the mechanisms by which messenger RNA (mRNA) structure modulates both viral mRNA translation and the antigen processing machinery to escape immune surveillance, will stimulate the development of alternative therapeutic strategies focused on RNA‐directed drugs designed to enhance immune responses against infected cells. In this review, we discuss regulatory aspects of the MHC‐I pathway and summarize current knowledge of the role attributed by mRNA structure and other translational regulatory mechanisms in immune evasion. In particular we highlight the impact of recently identified G‐quadruplex structures within virally encoded transcripts as unique regulatory signals for translational control and antigen presentation. WIREs RNA 2015, 6:157–171. doi: 10.1002/wrna.1262
This article is categorized under:
-
1
RNA Structure and Dynamics > Influence of RNA Structure in Biological Systems
-
2
Translation > Translation Regulation
INTRODUCTION
To eliminate pathogens of viral or bacterial origin the adaptive immune system utilizes major histocompatibility complex class I (MHC‐I) molecules to bind and present epitopes from intracellular and extracellular antigens to CD8+ cytotoxic T lymphocytes (CTLs). These effector T cells scan the surface of virus‐infected cells to detect MHC‐I bound peptides (pMHC‐I) and destroy the cells by either direct lysis or through secretion of cytokines and chemokines.1, 2 The highly polymorphic nature of the MHC‐I molecules allows them to present a large repertoire of peptides representing the concealed intracellular antigens of infected or transformed cells. Generating and loading these peptides into cognate MHC‐1 binding grooves for antigen presentation defines the various steps of the MHC‐1 antigen presentation pathway. To counter this process, viruses have evolved various strategies to circumvent what is usually a highly effective immune surveillance system in order to limit the endogenous processing and presentation of viral peptides.3, 4, 5 Indeed, members of several viral families including Herpesviridae and Retroviridae, use sophisticated mechanisms to interfere at key steps of the MHC‐I antigen presentation pathway in order to evade immune recognition and as a result establish lifelong persistence in their host.6 An alternative mechanism utilized by viruses to limit the supply of viral peptides for T cell recognition is the downregulation of viral protein synthesis.7, 8, 9, 10, 11 Viruses that downregulate their protein synthesis at levels below the threshold required to initiate an immune response by host virus‐specific T cells may escape immune detection. While there has been steady progress over recent years in characterizing the successive steps underlying the antigen processing and presentation pathway, and in unraveling specialized functions of the components of the MHC‐1 presentation pathway,2, 12 our knowledge of the impact of translational regulatory mechanisms on MHC‐1 mediated antigen presentation remains limited.
In this review, we focus on translational regulatory mechanisms and their impact on antigen presentation, a critical step in the T cell‐mediated control of intracellular pathogens. While there have been numerous studies demonstrating an impact of messenger RNA (mRNA) structure on translation, we are only now gaining insights into how some viruses exploit mRNA structure to regulate the supply of viral antigens into the MHC‐I antigen presentation pathway. In parallel we discuss alternative translational mechanisms that are used by viruses to alter the nature of antigenic peptides. Characterization of the mechanisms by which viruses modulate their protein synthesis will not only help us in treating virus‐induced pathology, but also enable the design of safer and more immunogenic viral vectors such as vaccines or gene delivery systems. Furthermore, these insights will stimulate development of novel therapeutics based on antisense strategies or small molecules that target viral mRNA structure for overriding evasion mechanisms of immune detection.
MODULATION OF MHC‐I ANTIGEN PRESENTATION BY HUMAN VIRUSES
Viral replication and therefore its persistence rely on the host's protein synthesis machinery to generate numerous viral and cellular proteins within the infected cell. A high proportion of these viral cytosolic proteins are degraded by the proteasome to generate antigenic peptides for presentation to CTLs by MHC‐1 molecules (Figure 1). The 20S/26S proteasome is the main cytoplasmic processing complex responsible for generating peptides for the MHC‐I antigen presentation pathway.13 Proteolysis is regulated in an ATP‐dependent manner by a catalytic 20S core particle that is capped at both ends by 19S regulatory particles.14 This proteolytic chamber recognizes and unfolds ubiquitylated proteins for degradation. A designated transporter, referred to as the transporter associated with antigen processing (TAP), translocates the degraded peptides into the endoplasmic reticulum (ER) where resident chaperones facilitate their binding to newly synthesized MHC‐I molecules for vesicular migration through the golgi to the cell surface2 (Figure 1). Since the MHC‐I antigen presentation pathway plays a critical role in exposing virally infected cells to CD8+ T cells, it is not surprising that viruses have evolved immunomodulatory proteins to interfere with key steps of the presentation pathway. These evasive mechanisms include inhibiting peptide transport into the ER3 or inhibiting proteasomal degradation15 (Table 1). For example, herpes viruses including human cytomegalovirus (HCMV) and herpes simplex virus (HVS) have developed specific strategies to restrict antigen presentation through MHC‐I molecules by binding specific viral proteins to TAP, blocking peptide translocation into the ER.6, 30 HCMV protein US6 blocks ATP binding to prevent peptide transport through the TAP pore24, 25 while HVS protein ICP47 blocks peptide binding to TAP.22, 23 The adenoviral protein E3‐19K18, 19 and the HCMV protein US316, 17 both act to retain MHC‐I molecules in the ER, in addition to their inhibitory effects on the TAP‐associated glycoprotein, tapasin.19, 28, 29 It has also been shown that two HCMV proteins, US11 and US2, induce ER‐associated degradation of MHC‐1 molecules when bound to the ER binding immunoglobulin protein (BiP) chaperone (Table 1).20
Figure 1.
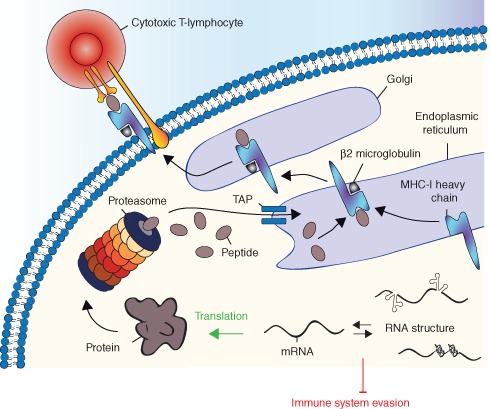
The MHC class I antigen presentation pathway. Once transcribed, viral mRNAs are translated into proteins that constitute the pool of MHC‐I restricted epitopes resulting from proteasomal degradation of nascent proteins and DRiPs. Degraded peptides are translocated into the endoplasmic reticulum (ER) by the transporter associated with antigen processing (TAP) where chaperones facilitate their binding to newly synthesized MHC‐I molecules for vesicular migration through the golgi to the cell surface. Cytotoxic T lymphocytes recognize viral antigenic peptides and initiate an immune response. Viruses that evade the immune system have evolved to interfere with key steps of the MHC‐I presentation pathway. Strategies that downregulate viral protein synthesis through translational control mechanisms can lead to immune evasion and subsequent latent infection.
Table 1.
Immune evasion mechanisms by immune‐modulatory proteins involving key steps of the MHC‐I antigen presentation pathway
| Evasion Function | Gene/Protein | Virus | Evasion Mechanism | Refs |
|---|---|---|---|---|
| Inhibit MHC‐I transport | US3 | HCMV | Binding and retention of class I molecules in the ER | 16, 17 |
| E3/19K | Adenovirus | Binding and retention of class I molecules in the ER | 18, 19 | |
| US2, US11 | HCMV | Target MHC‐I molecules to ER for proteasomal degradation | 16, 17, 20 | |
| mK3 | MHV68 | Target MHC‐I molecules to ER for proteasomal degradation | 16, 17 | |
| Nef | HIV‐1 | Divert trafficking MHC‐I molecules from Golgi to lysosomes | 16, 17 | |
| kK3, kK5 | KSHV | Induces rapid endocytosis of cell surface MHC‐I molecules | 21 | |
| Inhibit transporter associated with antigen processing (TAP) | ICP‐47 | HSV | Blocks peptide binding to TAP in cytosol | 22, 23 |
| US6 | HCMV | Blocks ATP binding to prevents peptide transport through TAP pore | 24, 25 | |
| UL49.5 | BHV‐1 | Inhibit TAP‐mediated peptide transport | 26 | |
| BNLF2a | EBV | Inhibit TAP‐mediated peptide transport | 26 | |
| Effect on antigen processing | EBNA1 | EBV | Escapes proteasomal processing and limits EBNA1 self‐synthesis | 11, 15 |
| LANA1 | KSHV | Escapes proteasomal processing | 27 | |
| Effect on Tapasin | US3 | HCMV | Inhibit peptide optimization of tapasin | 28 |
| E3‐19K | Adenovirus | Inhibit recruiting function of tapasin | 19, 29 |
Viral proteins exploiting different mechanisms that impact on immune evasion target the MHC‐I antigen presentation pathway.
The MHC‐I antigen presentation pathway presented in Figure 1 highlights the importance of translation as the initial step in a long cascade of biological processes that ultimately result in the presentation of antigenic peptides. Consequently, viruses that have developed mechanisms to self‐regulate their protein synthesis at levels below the threshold required to initiate an immune response by host virus‐specific T cells may escape immune recognition.
RNA G‐quadruplexes in the EBV‐Nuclear Antigen 1 open Reading Frame Impact on Antigen Presentation
The double‐stranded DNA Epstein‐Barr virus (EBV), which infects over 90% of the world's population, is associated with a number of human malignancies including Burkitt's lymphoma, nasopharyngeal carcinoma, and Hodgkin's lymphoma. While the majority of EBV latent proteins elicit robust immune responses, EBV‐encoded nuclear antigen 1 (EBNA1) a genome maintenance protein (GMP) expressed in all EBV‐associated malignancies, self‐regulates its synthesis at levels sufficient to maintain viral infection but below the level required to initiate an immune response by host virus‐specific T cells.8, 11, 31 The glycine‐alanine repeat domain (GAr) of EBNA1 was initially thought to be responsible for limiting MHC‐I‐restricted presentation of EBNA1 epitopes to CTLs due to the GAr inhibiting its own proteasomal degradation and therefore presentation of EBNA1 peptides.15, 32 Indeed, deleting the GAr was shown to alleviate the inhibitory constraint on antigen presentation.31, 33 More recent investigations have demonstrated that reduced EBNA1 MHC‐I peptide presentation is due to downregulated EBNA1 synthesis9, 11, 31 and questioned whether the GAr has an impact on EBNA1 turnover.34 Moreover, studies by several groups have demonstrated that EBNA1‐specific epitopes are derived from newly synthesized protein and have shown both in vitro and in vivo, that the GAr inhibits mRNA translation of EBNA1 in cis. 31 These results support Yewdell's defective ribosomal products (DRiPs) hypothesis35 where DRiPs, a subset of rapidly degraded polypeptides (RDPs), supply peptides for MHC‐I molecules, thereby linking viral immune surveillance to translational control mechanisms and ensuring the immune system rapidly detects and eliminates virally infected cells. While the pendulum now seems to be swinging back in favor of mature functional proteins rather than DRiPs as the predominant source of MHC class I presented peptides,36, 37 recent investigations from the Khanna and Shastri group's demonstrated that EBNA1 peptide presentation is dependent on the proportion of generated DRiPs.8, 38
Interestingly, it was demonstrated that the inhibition of EBNA1 self‐synthesis and resultant decrease in antigen presentation is attributed to purine loading of the EBNA1 mRNA.9 Studies by Cristillo et al.39 who analyzed viral genome base compositions, suggested that many viruses purine‐load their RNAs to resemble host RNAs thereby avoiding triggering of the host cell's double‐stranded RNA (dsRNA) surveillance mechanism. They observed a similar ‘stealth’ strategy is exploited by EBNA1, which is uniquely expressed in the most basic form of EBV latency (latency I).40 It was proposed that the rightward transcribed (BKRF1) gene encoding EBNA1 has been under evolutionary pressure to accept mutations that increase the purine content of the top (mRNA‐synonymous) strand41 so that during latent infection EBNA1 evades recognition by CTLs by virtue of its purine‐biased GAr mRNA preventing interactions with host RNAs. Reduction of this purine‐bias within the GAr mRNA through codon‐modification dramatically reversed the inhibitory effect on EBNA1 self‐synthesis and antigen presentation.9 In addition, altering the peptide sequence of the native EBNA1 GAr using alternative reading frames did not alleviate the inhibition of both EBNA1 synthesis and epitope presentation,10 suggesting that regulatory structural elements within the EBNA1 GAr mRNA itself may regulate mRNA translation and antigen presentation. A subsequent analysis of the EBNA1 GAr encoding mRNA revealed that clustering of unusual RNA secondary structures, G‐quadruplexes, within the EBNA1 GAr encoding mRNA are involved in the cis‐acting translational regulation of the EBNA1 mRNA.7
G‐quadruplexes are noncanonical structures of nucleic acids formed within G‐rich DNA or RNA sequences. These structures are stabilized by the stacking of guanine‐tetrads that are formed by the coplanar arrangement of four G‐bases interacting by Hoogsteen hydrogen‐bonding42 (Figure 2(a)). G‐quadruplex motifs, which can act as transcriptional and translational repressors have previously been identified in telomeres, promoters, and gene bodies where they perform important regulatory roles in diverse biological processes including replication and gene expression.43, 44, 45 Using a combination of biophysical and immunological techniques in order to assess the role played by the EBNA1 GAr in reducing EBNA1 synthesis and limiting EBNA1‐restricted antigenic peptides, it was recently demonstrated that: (1) potassium‐dependent G‐quadruplexes are the major structural components of the native purine‐rich EBNA1 GAr mRNA; (2) the presence of G‐quadruplex motifs within the open reading frame (ORF) of EBNA1 act as ‘steric blocks’ to impede ribosome transit by inducing ribosome stalling and/or dissociation, and (3) destabilization of G‐quadruplex structures following codon‐modification of the EBNA1 mRNA encoding the GAr, to introduce stem‐loop structures, results in enhanced translation and recognition by antigen‐specific T cells.7 Overall the study demonstrated that EBV‐infected cells harboring G‐quadruplexes within the EBNA1 GAr mRNA are less efficiently recognized by virus‐specific T cells compared to EBV‐infected cells synthesizing a codon‐modified EBNA1 GAr mRNA which forms dsRNA structures due to altered base pairing following purine reduction. These results highlight the importance of G‐quadruplexes within virally encoded transcripts as unique regulatory signals for translational control and immune evasion.7
Figure 2.
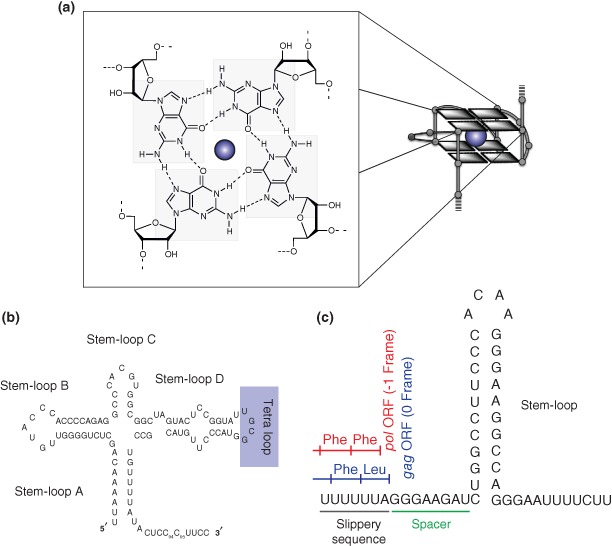
mRNA structures that affect MHC‐I mediated antigen presentation. (a) RNA G‐quadruplexes within gammaherpes viral ORFs negatively impact antigen presentation. G‐quadruplex structures are formed by guanine‐rich sequences and are stabilized by the stacking of guanine‐tetrads that are formed by the coplanar arrangement of four guanines interacting by Hoogsteen hydrogen‐bonding and stabilized by monovalent cations (usually K+). (b) Cloverleaf structures identified in the 5'‐untranslated regions of poliovirus. Cloverleaf structures in enteroviruses control the replication and translation of the viral genomes through the formation of ribonucleoprotein complexes. (c) HIV‐1 programmed ribosomal frameshift signal secondary structure. The formation of the stem‐loop structure downstream of a slippery sequence leads to the translocating ribosome to stall and induce a ‐1 frameshift that insures the synthesis of pol and gag which are encoded by the same ORF.
Identification of G‐quadruplex Motifs in Other Viral Maintenance Gene mRNAs
Intriguingly, in several other members of the gammaherpesviral family, similar purine‐biases are observed in the repetitive sequence‐encoding regions within genes, some of which have been demonstrated to be latency‐associated and known to regulate their self‐synthesis (Table 2). These include the ORF encoding latency‐associated nuclear antigen (LANA1), the GMP of human herpesvirus 8 (HHV8)46 and the ORF 73 of Ateline herpesvirus 3.10 Similar to EBNA1, clusters of putative G‐quadruplex motifs (PQSs) identified within the conserved purine‐rich sequences of these other gammaherpesviral maintenance gene mRNAs were analyzed by CD and 1H NMR spectroscopy and demonstrated to exhibit spectral signatures consistent with parallel G‐quadruplex formation.7 Several of the examined GMPs have been implicated in immune evasion, suggesting gammaherpesviruses exploit RNA G‐quadruplexes as structural regulatory elements impacting on antigen presentation.
Table 2.
RNA Quadruplex Motifs Are a Hallmark of Gammaherpes Viral ORFs
| Gammaherpesviruses | Viral Maintenance Proteins | Internal mRNA Repeat Size (bp) | Guanine Content of mRNA Repeats (%) | G‐Quadruplex‐Forming Sequences (5′→3′) | Occurrence Throughout the mRNA Repeat | % of Repeat Covered by Quadruplexes |
|---|---|---|---|---|---|---|
| Lymphocryptoviruses | ||||||
| Human herpesvirus 4 | EBNA1 | 711 | 62 | GGGGCAGGAGCAGGAGGA | 31 | 74 |
| Callitrichine herpesvirus 3 | ORF39 | 150 | 58 | GGGGUGGCAGAGGCGG | 5 | 60 |
| Macacine herpesvirus 4 | rhEBNA1 | 141 | 51 | GGUGCAGGGGGAAGCGG | 4 | 48 |
| Papiine herpesvirus 4 | baEBNA1 | 147 | 52 | GGUGCAGGAGCAGGAGG | 7 | 81 |
| Rhadinoviruses | ||||||
| Human herpesvirus 8 | LANA1 | 297 | 50 | seq1 – GGAGGAGGACGAGG | 11 | 58 |
| 501 | 48 | seq2 – GGAGGUGGAAGAGCAGG | 5 | 15 | ||
| Ateline herpesvirus 3 | ORF73 | 513 | 62 | GGGGGAGACGGGGGAGACGGGGGAGACGGGGGAGAC | 22 | 63 |
| Macaviruses | ||||||
| Alcelaphine herpesvirus 1 | ORF73 | 1278 | 41 | GGGGCCCGAGGGACCAGGGGGG | 5 | 10 |
| Ovine herpesvirus 2 | ORF73 | 984 | 45 | GGAGAAGGACCUGGAGGG | 15 | 35 |
Eight gammaherpes viral maintenance proteins, involved in immune evasion, are encoded by mRNAs possessing guanine‐rich repeats that possess the ability to form G‐quadruplex motifs.
The bolded guanines highlight the Gs most likely to be involved in the formation of guanine tetrads.
Furthermore, the effect of RNA G‐quadruplexes on viral mRNA translation and antigen presentation may have broader implications following the identification of G‐quadruplex motifs in the highly pyrimidine‐loaded retrovirus, Human T‐cell Leukemia (Lymphotropic) Virus‐1 (HTLV‐1) (unpublished data). Like EBV, HTLV‐1 is a latent, GC‐rich virus, which is able to persist in its human host asymptomatically. Larocca and colleagues reported an antisense transcript that like EBNA1 is heavily R‐loaded (purine‐loaded).39, 47 The HTLV‐1 antisense transcript translates into a basic leucine zipper protein (HBZ) that is poorly immunogenic.48 HBZ, a likely GMP for HTLV‐1 is increasingly seen as playing a major role in evasion of host immunosurveillance.48
RECOGNITION OF RNA STRUCTURES BY CELLULAR AND VIRAL PROTEINS
RNA binding proteins contain various domains that recognize structures within mRNAs in order to transport and localize mRNA, and control key steps of translation such as initiation, splicing, and termination. Viruses have evolved to select specific RNA structures in order to recruit cellular and viral proteins needed to initiate or regulate the maintenance of their genomes. It is noteworthy that EBNA1, whose synthesis is modulated through the formation of G‐quadruplex motifs within the ORF of its mRNA (see previous section), binds to G‐rich RNAs that are predicted to form G‐quadruplex structures.49 These observations suggest that EBNA1 may also control its synthesis through a feedback loop mechanism. Thus, EBV may regulate the pool of MHC‐I epitopes during the latency step of chronic infection by exploiting the complexity associated with EBNA1 mRNA translation.
In RNA positive‐stranded viruses, the genomic RNA serves as template for both translation and replication. The two pathways are exclusive and these viruses employ both viral and cellular factors to initiate one or the other. Most viral cis‐acting RNA elements are located in the 5′‐ and 3′‐untranslated regions of their genomes. The 5′‐noncoding regions of Entoroviral genomes are highly structured and contain two functional domains, which are involved in translation and RNA replication.50 The first domain comprises a cloverleaf (CL) structure (Figure 2(b)) that carries signals to control both translation and RNA replication. The poliovirus (PV) CL (88 nt) contains four stem‐loops (Stem‐loop A to D) that are involved in the formation of a ribonucleoprotein (RNP) complex. A second domain comprises an internal ribosomal entry site (IRES) that promotes translation. It has been shown that the formation of a RNP complex within the 5′‐UTR region of the PV genome, consisting of the CL structure, the cellular protein poly(rC)‐binding protein (PCBP) and the uncleaved viral proteinase 3CD is required to initiate viral RNA replication and translation.51 The cellular factor PCBP was shown to bind to the CL stem‐loop B domain while the viral protein 3CD interacts with the stem‐loop‐D domain. Mutational analysis has shown that the interaction of the CL structure with the cellular factor PCBP upregulates viral translation, while the binding of the viral protein 3CD represses translation and promotes negative‐strand RNA synthesis.52 Consequently, it has been proposed that the interaction of 3CD with the CL structure controls whether the genomic RNA is translated or replicated.
The RNP complex controlling the state of the virus is stabilized by other viral factors such as proteins 3AB for PV or 3Cpro for rhinoviruses.50 It is noteworthy that both isolated PV 3A protein and PV infection can inhibit functional MHC‐I dependent antigen presentation.53 The study of the effect of PV 3A protein expression on the presentation of hepatitis C virus antigens in cultured chimpanzee cells revealed that protein 3A slows the rate of MHC‐I transport to the cell surface and protects cells from CTL‐mediated lysis. It has been suggested that protein 3Cpro induces fragmentation of the golgi compartment and blocks intra‐golgi transport, thereby reducing the expression of MHC‐I antigens and slowing the secretion of proinflammatory cytokines.54 Thus translational activation through RNP complex formation controls the synthesis of proteins involved in immune evasion in enteroviruses. Further research is necessary to determine how these RNA structures and/or viral/cellular protein interactions may modulate the supply of antigenic peptides for MHC‐I molecules and the replication of RNA positive‐stranded viruses.
IMPACT OF CRYPTIC TRANSLATIONAL CONTROL MECHANISMS ON MHC‐I MEDIATED ANTIGEN PRESENTATION
MHC‐I restricted epitopes are widely accepted to be derived from viral proteins encoded by primary ORFs. However, recent studies have demonstrated that viruses also exploit alternative reading frames in order to fully express their genomes. The importance of these alternatively synthesized smaller polypeptides resulting from ‘cryptic’ translational control events are starting to be recognized as a crucial source of antigenic peptides.55 We will discuss in this section the two main mechanisms, frameshifting and non‐AUG translation initiation, that are used by viruses to exploit the genomic information present in alternative reading frames and the impact of nonconventional peptides presented by MHC‐I molecules.
Programmed ribosomal frameshifting is one of the many translational recoding processes used by viruses in order to appropriately express their genomes. Frameshifting results from a change or shift in the reading frame by one or two nucleotides during mRNA translation. Frameshifting is widely employed by RNA viruses, such as HIV‐1, coronaviruses and also influenza viruses to increase the diversity of proteins expressed from their compact genomes. This process is programmed by the mRNA nucleotide sequence and is often initiated as a consequence of mRNA secondary and tertiary structures such as stem‐loops and pseudoknots. An example is the HIV‐1 programmed ribosomal frameshifting secondary structures, depicted in Figure 2(c), which induce a −1 frameshift that insure the synthesis of pol and gag that are encoded by the same ORF.56
Several cryptic MHC‐I peptides have now been reported and the mechanisms by which viruses generate such peptides are starting to be elucidated.55 Mammalian ribosomes carefully select the correct initiation codon of ORFs for the synthesis of functional proteins but they have also been shown to sometimes use alternative start codons to translate from other regions of the mRNAs irrespective of the ORF location for MHC‐I presentation. The synthesized cryptic peptides compete with other MHC‐I peptides for cell surface presentation and T cell activation. One of the first examples of a cryptic antigenic peptide was described following a study investigating murine AIDS, revealing stimulated CTL responses toward a peptide encoded in an alternative reading frame of the LP‐BM5 gag gene.57 In HIV‐infected mice, CTLs target a HLA‐B*07‐restricted alternative reading frame epitope translated from the (+2) ORF within the gag gene. Similarly, simian immunodeficiency virus (SIV) infected rhesus macaques displayed a strong CTL response toward an epitope that was translated from the (+2) reading frame relative to the env ORF.58 Interestingly, it has been shown that nearly one‐quarter of the anti‐SIV CTL responses in SIV‐infected rhesus macaques are directed toward cryptic epitopes generated from alternative reading frame translation.59
In order to assess the impact of cryptic epitopes on immune surveillance, bioinformatic screening was performed to identify nonconventional antigens in HIV‐infected individuals.60 This approach identified 64 HLA‐associated viral polymorphisms translated in alternative reading frames of the HIV gag, pol and nef mRNAs. Surprisingly, no upstream in‐frame AUG start codons were identified and a codon normally encoding for leucine (CUG) was proposed as a start site for cryptic translation. Biochemical analyses of ribosomal initiation complexes at CUG start codons revealed that infected cells use an elongator leucine‐bound transfer RNA to initiate translation at cryptic start codons.61 Translation initiation at CUG codons was found to be independent of the canonical AUG initiator tRNA pathway and instead relied on the expression of eukaryotic initiation factor 2A. The synthesis of cryptic antigenic peptides may provide an escape mechanism from the overloading of conventional peptides on the surface of the infected cell. For example, it has been reported that the inclusion of nonconventional epitopes, including cryptic epitopes from HIV‐1 in DNA vaccines can trigger a much stronger response in rhesus macaques.62
Influenza A viruses (IAV) are negative‐stranded RNA viruses that are responsible for the worldwide spread of human or avian influenza. IAVs expressing the alternative full‐length protein, PB1‐F2, were responsible for causing the human influenza pandemics of the 20th century.63 The PB1‐F2 protein has been shown to control viral pathogenicity and mortality. In the absence of this protein, the virus replicates to similar levels in mouse lungs but shows decreased proliferation. Its expression, on the other hand, results in delayed clearance of the virus by the host immune system.64 PB1‐F2 was identified in the course of a systematic search for alternative reading‐frame peptides encoded by the IAV that are recognized by CD8+ T cells.63 A CTL peptide, (PB1‐F262‐70), not corresponding to any of the known standard viral open reading frames was found. Further genomic analysis of the IAV genome revealed the PB1‐F262‐70 peptide was derived from the conserved 87‐amino‐acid‐long PB1‐F2 protein encoded by the (+1) reading frame within the PB1 gene. PB1‐F2 and some truncation products resulting from a premature stop codon in the PB1‐F2 ORF can be detected in human, mouse, and avian cells infected by IAV.63 Studies involving Kozak and mutational analyses have shown that RNA structures within PB1 strongly favor translation at the (+1) reading frame's predicted PB1‐F2 start codon and are likely to be initiated by ribosomal scanning as opposed to internal ribosomal entry or other mechanisms.63 RNA secondary structure predictions revealed a conserved structured region at the start of the internal PB1‐F2 ORF for six influenza genomes comprising the human, avian and swine strains.65 This protein contributes to influenza viral pathogenicity through several mechanisms. One described mechanism is a pro‐apoptotic function in which PB1‐F2 was found to localize to Hela cell mitochondria. PB1‐F2 disturbs the mitochondrial membrane potential resulting in a fluctuation of cytochrome c into the cytoplasm triggering apoptosis in these cells.63 The induction of apoptosis by PB1‐F2 is proposed to occur specifically in immune cells in a strain dependent manner suggesting that the protein has a modulatory role in the host response to IAV by hastening the death of immune cells.63, 66 In addition to pro‐apoptotic activity, it has been reported that PB1‐F2 also has proinflammatory properties. It was observed that an asparagine (N) to serine (S) substitution at position 66 in the PB1‐F2 protein dramatically increased immunopathology and mortality by the 1918 pandemic strain and highly pathogenic H5N1 viruses.67 Also, transcriptional profiling of mice infected with a PB1‐F2 N66S‐expressing virus revealed an early suppression of interferon‐stimulated genes (ISGs)68 and in vitro experiments demonstrated an anti‐interferon activity of PB1‐F2 in relation to the mitochondrial antiviral signaling (MAVS) adaptor protein.69 Of note, PB1‐F2 N66S inhibited IFN induction more efficiently than wild‐type PB1‐F2.69 The wild‐type PB1‐F2 protein was initially shown to be highly unstable and to undergo proteasomal dependent degradation,63 however, more recent experiments performed in murine and duck cells demonstrate that protein levels of the N66S mutant but not the wild‐type increase following exposure of the infected cells to the proteasome inhibitor, lactacystin.70
These examples highlight the impact of cryptic translation and the generation of alternative antigenic peptides on MHC‐I mediated immune responses. Aberrant translational control mechanisms appear to be actively used by viruses in order to modulate pathogenicity (Figure 3). With only a few examples of RNA structures that control both the translation of alternative ORFs and antigenic peptide presentation having been reported to date, further identification and characterization of such important RNA structural regulatory elements would facilitate our understanding of the mechanisms exploited by viruses seeking to establish chronic infection.
Figure 3.
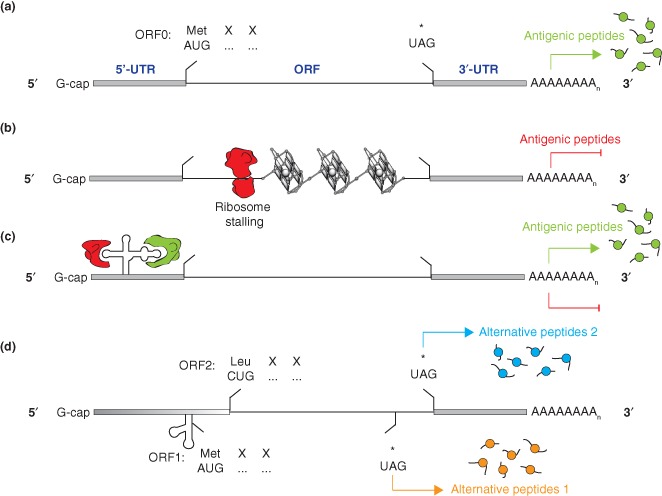
Schematic view of translational control mechanisms that regulate viral protein synthesis and antigen presentation. (a) Canonical translation of a viral mRNA is initiated at the AUG codon and leads to the synthesis of a viral protein that is the source of MHC‐I‐restricted antigenic peptides. (b) Formation of G‐quadruplex motifs within the ORF of gammaherpesviruses impede translation through ribosome stalling/dissociation and inhibit the presentation of MHC‐I‐restricted antigenic peptides. (c) RNA structures within the 5′‐untranslated region of enteroviruses control replication and translation of the viral genome by recruiting cellular and viral proteins. The ribonucleoprotein complex modulates the synthesis of viral proteins. (d) The translation of alternative reading frames induced by non‐AUG translation initiation or RNA structure mediated programmed frameshifting, is the source of alternative antigenic peptides that diversify the pool of MHC‐I‐restricted epitopes.
TARGETING RNA STRUCTURES TO CONTROL MHC‐I MEDIATED ANTIGEN PRESENTATION
We have described in the previous sections how secondary structures of RNA can impact on the translation of critical mRNAs encoding viral proteins that are the source of MHC‐I restricted epitopes. In this section we discuss novel opportunities of targeting viral RNA secondary structures and associated cis‐regulatory sequences or proteins in order to modulate the pool of antigenic determinants. Viruses often rely on a balance that necessitates the expression of viral proteins at levels sufficient to maintain viral replication but low enough to avoid stimulation of the immune surveillance system. By interfering with the translation of key mRNAs, one may anticipate to see dramatic changes in viral infectivity and survival particularly when stimulating or inhibiting the synthesis of both conventional and non‐conventional antigenic peptides.
As described previously, gammaherpes viruses comprise G‐quadruplex motifs within the ORF of the mRNAs encoding their GMPs. Within certain members of this viral family, G‐quadruplexes act as ribosomal roadblocks to prohibit a significant proportion of ribosomes from shuttling down the transcripts and in the process impede translation.7 Due to this decrease in translation, it was hypothesized that destabilization or stabilization of G‐quadruplex motifs may have a direct impact on the generation of antigen‐specific peptides. Reported observations that codon‐modification of the EBNA1 GAr mRNA destabilized G‐quadruplexes by inducing the formation of double‐stranded structures within the EBNA1 mRNA, prompted an assessment of the effect of potential exogenous factors such as antisense oligonucleotides to destabilize G‐quadruplex motifs. It was subsequently demonstrated that an antisense oligonucleotide complementary to the G‐quadruplex motif and selected for its ability to unwind G‐quadruplexes in vitro, was able to increase EBNA1 mRNA translation and enhance immune recognition by antigen‐specific T cells in EBV‐infected cells (Figure 4(a)).7 This result highlighted the ability of ribosomes to process RNA double‐stranded structure more efficiently than G‐quadruplex structure. In contrast, the addition of a G‐quadruplex stabilizing ligand, pyridostatin (Figure 4(a)), to EBNA1 in vitro translation assays and an in vivo transfection system showed the opposite effect: a more pronounced inhibition of EBNA1 mRNA translation and a resultant decrease in virus‐specific T‐cell recognition of EBV‐infected cells treated with the small molecule. G‐quadruplex destabilization, using the antisense oligonucleotide, or stabilization, using the small molecule G‐quadruplex ligand, led to a 25% increase or 40% decrease, respectively, in the activation of EBNA1‐specific T‐cells in treated EBV‐expressing HEK293 cells. The multifunctional roles of EBNA1 in initiating EBV‐genome replication and episomal maintenance, while also contributing to immune evasion, suggest that this protein is a promising target for immunotherapy. Stimulation of the immune recognition of EBV‐infected cells through G‐quadruplex destabilization or inhibition of replication of the EBV genome through G‐quadruplex stabilization may lead to potential elimination of viral latency.
Figure 4.
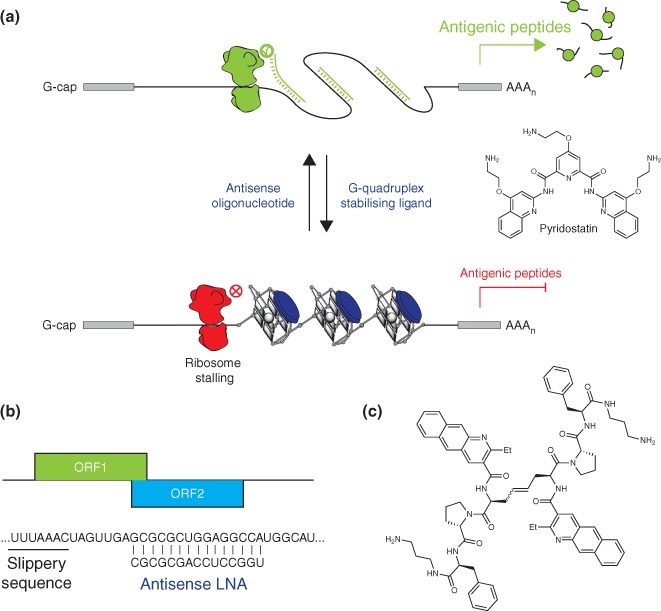
RNA structures within viral mRNAs are targets for translational control and antigen presentation. (a) Destabilization or stabilization of G‐quadruplexes using antisense oligonucleotides or small molecule G‐quadruplex ligands (Pyridostatin), respectively, stimulates or inhibits the synthesis of the EBV‐genome maintenance protein, EBNA1, and subsequent generation and presentation of MHC‐I‐restricted antigenic peptides. (b) The formation of mRNA secondary structures resulting from the presence of LNA downstream slippery sequences control ribosomal frameshift and the synthesis of alternative antigenic peptides. (c) Structure of a small molecule that binds and stabilizes the stem‐loop structure controlling the HIV‐1 gag‐pol programmed ribosomal frameshift. Stabilization of the RNA secondary structure enhances frameshifting in cells.
Similarly, several groups have investigated the use of antisense oligonucleotides and derivatives to control programmed frameshifting events. This approach relies on the hypothesis that the formation of an RNA secondary structure downstream of a slippery sequence that is known to induce frameshifting will shift ribosomes into a new frame (Figure 4(b)). The efficiency of frameshifting using morphilino, 2′‐O‐methyl phosphorothioate, locked nucleic acid (LNA) and RNA antisense oligonucleotides can be as high as 40% and depends on the stability of the secondary structure rather than the three‐dimensional structure formed.71, 72, 73 The targeted slippery sequences comprise the HIV gag‐pol, UUUUUUA, MMTV gag‐pro, AAAAAAC and RSV gag‐pol, AAAUUA frameshift sites. This in vitro research investigation highlights the potential of synthetic oligonucleotides to interfere with the translation of key mRNAs to insure infectivity and survival of RNA viruses. Unfortunately, at present no results for an effect on the in vivo synthesis of related MHC‐I restricted epitopes using this approach exist, however, one would expect that interfering with translation would ultimately affect the pool of antigenic peptides. This approach may also be used to stimulate translation at non‐AUG start codons by creating RNA secondary structures in close proximity of tRNA binding sites. The use of an antisense microwalk assay may allow the identification of sequences and structures controlling the synthesis of cryptic epitopes.
The main goal of identifying RNA structures that impact on MHC‐I mediated antigen presentation is to define potential targets for modulating the pool of antigenic peptides. The use of small molecules for this purpose represents an alternative way to design vaccines in order to control viral infectivity. By targeting the EBNA1 repeat with a small molecule, a G‐quadruplex stabilizing ligand, allowed regulation of EBNA1 synthesis and the subsequent modulation of EBNA1 specific antigenic peptides. On a similar note, high‐throughput screening and combinatorial chemistry approaches have been successfully used in order to identify small molecules that interact with the HIV‐1 RNA stem‐loop (Figure 2(c)) frameshift stimulatory structure.71 The compound depicted in Figure 4(c) was shown to selectively bind this RNA, and enhanced frameshifting by greater than 50% in a dual‐luciferase assay in HEK293 cells and also strongly inhibited the infectivity of pseudotyped HIV‐1 virions.74
These examples highlight the relevance of stabilizing or creating structures in the vicinity of cis‐regulating sequences within viral mRNAs in order to control translation. The limited amount of data available in the literature supports this approach to control the presentation of MHC‐I restricted epitopes and the activation of virus‐specific T‐cells. The identification and characterization of exogenous agents able to stabilize or induce relevant RNA structures represents an opportunity to interfere with the MHC‐I antigen presentation pathway, which will lead to a better understanding of how viruses escape the immune system.
CONCLUSION AND PERSPECTIVES
Recent insights into viral immune evasion mechanisms have provided an important awareness of how some viruses exploit mRNA structure to modulate viral protein synthesis and antigen presentation in order to escape immune detection and establish a successful latent infection. In this review, we have described several mechanisms that link recently identified structural elements within viral mRNAs to protein translation and MHC‐I‐restricted antigen presentation. In particular, we have highlighted a novel function of RNA G‐quadruplexes within gammaherpesviral GMP transcripts through cis‐acting modulation of their mRNA translation and MHC‐I antigen presentation. Altering the translational regulatory effects of the G‐quadruplex motifs and their resultant impact on antigen presentation has the potential to enhance immune recognition of virally infected cells or inhibit replication of the viral genome, leading to elimination of viral latency. With no available vaccines to prevent EBV and other gammaherpesviral‐associated malignancies, the identification of new therapeutic targets based on RNA G‐quadruplex structures within viral GMP mRNAs will promote the advancement of RNA‐directed drug design to reduce the burden of associated viral malignancies. While recent demonstrations that an antisense oligonucleotide and a small molecule G‐quadruplex binding ligand can selectively target EBNA1 G‐quadruplexes to impact on EBNA1 synthesis, the next challenge will require rigorous assessment of the mechanistic impact and specificity of these exogenous agents in vivo.
We also described RNP complexes and cryptic translational regulatory mechanisms that potentially impact on the supply of antigenic peptides to MHC‐I molecules. Although RNA structures within the 5′‐UTR of RNA viruses, that control the translation and replication of viral genomes, have been characterized, further effort is needed to fully appreciate their impact on antigen presentation. Only limited data are available regarding the impact of these RNA structures on immune evasion. Chemical tools could be used to inhibit the formation of RNP complexes needed to initiate translation and to unravel the mechanisms used by RNA viruses to regulate the pool of antigenic determinants. Further research is also required to identify the relevant RNA structures controlling the generation of antigenic peptides resulting from frameshift events and non‐AUG translation initiation. The characterization of such structures will allow deciphering of the mechanism of cryptic translation and open the way to new approaches to modulate CTL activation.
ACKNOWLEDGMENTS
We thank C. Mayer, R. Khanna and R. Tellam for valuable discussions and proofreading of the manuscript. P. M. is supported by a program grant funded by Cancer Research UK. J.T. was supported by a NHMRC Career Development Award Fellowship (#496712) and NHMRC Project Grants (#496684 and APP1005091).
Conflict of interest: The authors have declared no conflicts of interest for this article.
REFERENCES
- 1. Burrows SR, Moss DJ, Khanna R. Understanding human T‐cell‐mediated immunoregulation through herpesviruses. Immunol Cell Biol 2011, 89:352–358. [DOI] [PubMed] [Google Scholar]
- 2. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol 2013, 31:443–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ploegh HL. Viral strategies of immune evasion. Science 1998, 280:248–253. [DOI] [PubMed] [Google Scholar]
- 4. Elliott T, Williams A. The optimization of peptide cargo bound to MHC class I molecules by the peptide‐loading complex. Immunol Rev 2005, 207:89–99. [DOI] [PubMed] [Google Scholar]
- 5. Hansen TH, Bouvier M. MHC class I antigen presentation: learning from viral evasion strategies. Nat Rev Immunol 2009, 9:503–513. [DOI] [PubMed] [Google Scholar]
- 6. Khanna R, Burrows SR. Role of cytotoxic T lymphocytes in Epstein‐Barr virus‐associated diseases. Annu Rev Microbiol 2000, 54:19–48. [DOI] [PubMed] [Google Scholar]
- 7. Murat P, Zhong J, Lekieffre L, Cowieson NP, Clancy JL, Preiss T, Balasubramanian S, Khanna R, Tellam J. G‐quadruplexes regulate Epstein‐Barr virus‐encoded nuclear antigen 1 mRNA translation. Nat Chem Biol 2014, 10:358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tellam J, Fogg MH, Rist M, Connolly G, Tscharke D, Webb N, Heslop L, Wang F, Khanna R. Influence of translation efficiency of homologous viral proteins on the endogenous presentation of CD8+ T cell epitopes. J Exp Med 2007, 204:525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tellam J, Smith C, Rist M, Webb N, Cooper L, Vuocolo T, Connolly G, Tscharke DC, Devoy MP, Khanna R. Regulation of protein translation through mRNA structure influences MHC class I loading and T cell recognition. Proc Natl Acad Sci USA 2008, 105:9319–9324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tellam JT, Lekieffre L, Zhong J, Lynn DJ, Khanna R. Messenger RNA Sequence Rather than Protein Sequence Determines the Level of Self‐synthesis and Antigen Presentation of the EBV‐encoded Antigen, EBNA1. PLoS Pathog 2012, 8:e1003112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yin Y, Manoury B, Fahraeus R. Self‐inhibition of synthesis and antigen presentation by Epstein‐Barr virus‐encoded EBNA1. Science 2003, 301:1371–1374. [DOI] [PubMed] [Google Scholar]
- 12. Cresswell P, Bangia N, Dick T, Diedrich G. The nature of the MHC class I peptide loading complex. Immunol Rev 1999, 172:21–28. [DOI] [PubMed] [Google Scholar]
- 13. Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 1994, 78:761–771. [DOI] [PubMed] [Google Scholar]
- 14. Baumeister W, Walz J, Zuhl F, Seemuller E. The proteasome: paradigm of a self‐compartmentalizing protease. Cell 1998, 92:367–380. [DOI] [PubMed] [Google Scholar]
- 15. Levitskaya J, Sharipo A, Leonchiks A, Ciechanover A, Masucci MG. Inhibition of ubiquitin/proteasome‐dependent protein degradation by the Gly‐Ala repeat domain of the Epstein‐Barr virus nuclear antigen 1. Proc Natl Acad Sci USA 1997, 94:12616–12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Farrell HE, Davis‐Poynter NJ. From sabotage to camouflage: viral evasion of cytotoxic T lymphocyte and natural killer cell‐mediated immunity. Semin Cell Dev Biol 1998, 9:369–378. [DOI] [PubMed] [Google Scholar]
- 17. Fruh K, Gruhler A, Krishna RM, Schoenhals GJ. A comparison of viral immune escape strategies targeting the MHC class I assembly pathway. Immunol Rev 1999, 168:157–166. [DOI] [PubMed] [Google Scholar]
- 18. Andersson M, Paabo S, Nilsson T, Peterson PA. Impaired intracellular transport of class I MHC antigens as a possible means for adenoviruses to evade immune surveillance. Cell 1985, 43:215–222. [DOI] [PubMed] [Google Scholar]
- 19. Li L, Muzahim Y, Bouvier M. Crystal structure of adenovirus E3‐19K bound to HLA‐A2 reveals mechanism for immunomodulation. Nat struct Mol Biol 2012, 19:1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hegde NR, Chevalier MS, Wisner TW, Denton MC, Shire K, Frappier L, Johnson DC. The role of BiP in endoplasmic reticulum‐associated degradation of major histocompatibility complex class I heavy chain induced by cytomegalovirus proteins. J Biol Chem 2006, 281:20910–20919. [DOI] [PubMed] [Google Scholar]
- 21. Coscoy L, Ganem D. Kaposi's sarcoma‐associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc Natl Acad Sci USA 2000, 97:8051–8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fruh K, Ahn K, Djaballah H, Sempe P, van Endert PM, Tampe R, Peterson PA, Yang Y. A viral inhibitor of peptide transporters for antigen presentation. Nature 1995, 375:415–418. [DOI] [PubMed] [Google Scholar]
- 23. Hill A, Jugovic P, York I, Russ G, Bennink J, Yewdell J, Ploegh H, Johnson D. Herpes simplex virus turns off the TAP to evade host immunity. Nature 1995, 375:411–415. [DOI] [PubMed] [Google Scholar]
- 24. Ahn K, Gruhler A, Galocha B, Jones TR, Wiertz EJ, Ploegh HL, Peterson PA, Yang Y, Fruh K. The ER‐luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity 1997, 6:613–621. [DOI] [PubMed] [Google Scholar]
- 25. Lehner PJ, Karttunen JT, Wilkinson GW, Cresswell P. The human cytomegalovirus US6 glycoprotein inhibits transporter associated with antigen processing‐dependent peptide translocation. Proc Natl Acad Sci USA 1997, 94:6904–6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hislop AD, Ressing ME, van Leeuwen D, Pudney VA, Horst D, Koppers‐Lalic D, Croft NP, Neefjes JJ, Rickinson AB, Wiertz EJ. A CD8+ T cell immune evasion protein specific to Epstein‐Barr virus and its close relatives in Old World primates. J Exp Med 2007, 204:1863–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zaldumbide A, Ossevoort M, Wiertz EJ, Hoeben RC. In cis inhibition of antigen processing by the latency‐associated nuclear antigen I of Kaposi sarcoma herpes virus. Mol Immunol 2007, 44:1352–1360. [DOI] [PubMed] [Google Scholar]
- 28. Sadasivan B, Lehner PJ, Ortmann B, Spies T, Cresswell P. Roles for calreticulin and a novel glycoprotein, tapasin, in the interaction of MHC class I molecules with TAP. Immunity 1996, 5:103–114. [DOI] [PubMed] [Google Scholar]
- 29. Bennett EM, Bennink JR, Yewdell JW, Brodsky FM. Cutting edge: adenovirus E19 has two mechanisms for affecting class I MHC expression. J Immunol 1999, 162:5049–5052. [PubMed] [Google Scholar]
- 30. Khanna R, Moss D, Gandhi M. Technology insight: applications of emerging immunotherapeutic strategies for Epstein‐Barr virus‐associated malignancies. Nat Clin Pract Oncol 2005, 2:138–149. [DOI] [PubMed] [Google Scholar]
- 31. Blake N. Immune evasion by gammaherpesvirus genome maintenance proteins. J Gen Virol 2010, 91:829–846. [DOI] [PubMed] [Google Scholar]
- 32. Sharipo A, Imreh M, Leonchiks A, Imreh S, Masucci MG. A minimal glycine‐alanine repeat prevents the interaction of ubiquitinated I kappaB α with the proteasome: a new mechanism for selective inhibition of proteolysis. Nat Med 1998, 4:939–944. [DOI] [PubMed] [Google Scholar]
- 33. Levitskaya J, Coram M, Levitsky V, Imreh S, Steigerwald‐Mullen PM, Klein G, Kurilla MG, Masucci MG. Inhibition of antigen processing by the internal repeat region of the Epstein‐Barr virus nuclear antigen‐1. Nature 1995, 375:685–688. [DOI] [PubMed] [Google Scholar]
- 34. Daskalogianni C, Apcher S, Candeias MM, Naski N, Calvo F, Fahraeus R. Gly‐Ala repeats induce position‐ and substrate‐specific regulation of 26 S proteasome‐dependent partial processing. J Biol Chem 2008, 283:30090–30100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yewdell JW, Reits E, Neefjes J. Making sense of mass destruction: quantitating MHC class I antigen presentation. Nat Rev Immunol 2003, 3:952–961. [DOI] [PubMed] [Google Scholar]
- 36. Colbert JD, Farfan‐Arribas DJ, Rock KL. Substrate‐induced protein stabilization reveals a predominant contribution from mature proteins to peptides presented on MHC class I. J Immunol 2013, 191:5410–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rock KL, Farfan‐Arribas DJ, Colbert JD, Goldberg AL. Re‐examining class‐I presentation and the DRiP hypothesis. Trends Immunol 2014, 35:144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cardinaud S, Starck SR, Chandra P, Shastri N. The synthesis of truncated polypeptides for immune surveillance and viral evasion. PLoS One 2010, 5:e8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cristillo AD, Mortimer JR, Barrette IH, Lillicrap TP, Forsdyke DR. Double‐stranded RNA as a not‐self alarm signal: to evade, most viruses purine‐load their RNAs, but some (HTLV‐1, Epstein‐Barr) pyrimidine‐load. J Theor Biol 2001, 208:475–491. [DOI] [PubMed] [Google Scholar]
- 40. Thorley‐Lawson DA, Miyashita EM, Khan G. Epstein‐Barr virus and the B cell: that's all it takes. Trends Microbiol 1996, 4:204–208. [DOI] [PubMed] [Google Scholar]
- 41. Karlin S, Blaisdell BE, Mocarski ES, Brendel V. A method to identify distinctive charge configurations in protein sequences, with application to human herpesvirus polypeptides. J Mol Biol 1988, 205:165–177. [DOI] [PubMed] [Google Scholar]
- 42. Parkinson GN, Lee MP, Neidle S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 2002, 417:876–880. [DOI] [PubMed] [Google Scholar]
- 43. Balasubramanian S, Hurley LH, Neidle S. Targeting G‐quadruplexes in gene promoters: a novel anticancer strategy? Nat Rev Drug Discov 2011, 10:261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Neidle S, Parkinson G. Telomere maintenance as a target for anticancer drug discovery. Nat Rev Drug Discov 2002, 1:383–393. [DOI] [PubMed] [Google Scholar]
- 45. Rodriguez R, Miller KM, Forment JV, Bradshaw CR, Nikan M, Britton S, Oelschlaegel T, Xhemalce B, Balasubramanian S, Jackson SP. Small‐molecule‐induced DNA damage identifies alternative DNA structures in human genes. Nat Chem Biol 2012, 8:301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rainbow L, Platt GM, Simpson GR, Sarid R, Gao SJ, Stoiber H, Herrington CS, Moore PS, Schulz TF. The 222‐ to 234‐kilodalton latent nuclear protein (LNA) of Kaposi's sarcoma‐associated herpesvirus (human herpesvirus 8) is encoded by orf73 and is a component of the latency‐associated nuclear antigen. J Virol 1997, 71:5915–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Larocca D, Chao LA, Seto MH, Brunck TK. Human T‐cell leukemia virus minus strand transcription in infected T‐cells. Biochem Biophys Res Commun 1989, 163:1006–1013. [DOI] [PubMed] [Google Scholar]
- 48. Cook LB, Elemans M, Rowan AG, Asquith B. HTLV‐1: persistence and pathogenesis. Virology 2013, 435:131–140. [DOI] [PubMed] [Google Scholar]
- 49. Norseen J, Johnson FB, Lieberman PM. Role for G‐quadruplex RNA binding by Epstein‐Barr virus nuclear antigen 1 in DNA replication and metaphase chromosome attachment. J Virol 2009, 83:10336–10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu Y, Wimmer E, Paul AV. Cis‐acting RNA elements in human and animal plus‐strand RNA viruses. Biochim Biophys Acta 2009, 1789:495–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Andino R, Rieckhof GE, Baltimore D. A functional ribonucleoprotein complex forms around the 5' end of poliovirus RNA. Cell 1990, 63:369–380. [DOI] [PubMed] [Google Scholar]
- 52. Andino R, Rieckhof GE, Achacoso PL, Baltimore D. Poliovirus RNA synthesis utilizes an RNP complex formed around the 5′‐end of viral RNA. EMBO J 1993, 12:3587–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Deitz SB, Dodd DA, Cooper S, Parham P, Kirkegaard K. MHC I‐dependent antigen presentation is inhibited by poliovirus protein 3A. Proc Natl Acad Sci USA 2000, 97:13790–13795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhou Z, Mogensen MM, Powell PP, Curry S, Wileman T. Foot‐and‐mouth disease virus 3C protease induces fragmentation of the Golgi compartment and blocks intra‐Golgi transport. J Virol 2013, 87:11721–11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Starck SR, Shastri N. Non‐conventional sources of peptides presented by MHC class I. Cell Mol Life Sci 2011, 68:1471–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ, Varmus HE. Characterization of ribosomal frameshifting in HIV‐1 gag‐pol expression. Nature 1988, 331:280–283. [DOI] [PubMed] [Google Scholar]
- 57. Mayrand SM, Schwarz DA, Green WR. An alternative translational reading frame encodes an immunodominant retroviral CTL determinant expressed by an immunodeficiency‐causing retrovirus. J Immunol 1998, 160:39–50. [PubMed] [Google Scholar]
- 58. Maness NJ, Valentine LE, May GE, Reed J, Piaskowski SM, Soma T, Furlott J, Rakasz EG, Friedrich TC, Price DA, et al. AIDS virus specific CD8+ T lymphocytes against an immunodominant cryptic epitope select for viral escape. J Exp Med 2007, 204:2505–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Maness NJ, Walsh AD, Piaskowski SM, Furlott J, Kolar HL, Bean AT, Wilson NA, Watkins DI. CD8+ T cell recognition of cryptic epitopes is a ubiquitous feature of AIDS virus infection. J Virol 2010, 84:11569–11574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Berger CT, Carlson JM, Brumme CJ, Hartman KL, Brumme ZL, Henry LM, Rosato PC, Piechocka‐Trocha A, Brockman MA, Harrigan PR, et al. Viral adaptation to immune selection pressure by HLA class I‐restricted CTL responses targeting epitopes in HIV frameshift sequences. J Exp Med 2010, 207:61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Starck SR, Jiang VV, Pavon‐Eternod M, Prasad S, McCarthy B, Pan T, Shastri N. Leucine‐tRNA initiates at CUG start codons for protein synthesis and presentation by MHC class I. Science 2012, 336:1719–1723. [DOI] [PubMed] [Google Scholar]
- 62. Maness NJ, Wilson NA, Reed JS, Piaskowski SM, Sacha JB, Walsh AD, Thoryk E, Heidecker GJ, Citron MP, Liang X, et al. Robust, vaccine‐induced CD8(+) T lymphocyte response against an out‐of‐frame epitope. J Immunol 2010, 184:67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, Basta S, O'Neill R, Schickli J, Palese P, et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat Med 2001, 7:1306–1312. [DOI] [PubMed] [Google Scholar]
- 64. Zamarin D, Ortigoza MB, Palese P. Influenza A virus PB1‐F2 protein contributes to viral pathogenesis in mice. J Virol 2006, 80:7976–7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Moss WN, Priore SF, Turner DH. Identification of potential conserved RNA secondary structure throughout influenza A coding regions. RNA 2011, 17:991–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. McAuley JL, Chipuk JE, Boyd KL, Van De Velde N, Green DR, McCullers JA. PB1‐F2 proteins from H5N1 and 20 century pandemic influenza viruses cause immunopathology. PLoS Pathog 2010, 6:e1001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Conenello GM, Zamarin D, Perrone LA, Tumpey T, Palese P. A single mutation in the PB1‐F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog 2007, 3:1414–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Conenello GM, Tisoncik JR, Rosenzweig E, Varga ZT, Palese P, Katze MG. A single N66S mutation in the PB1‐F2 protein of influenza A virus increases virulence by inhibiting the early interferon response in vivo. J Virol 2011, 85:652–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Varga ZT, Ramos I, Hai R, Schmolke M, Garcia‐Sastre A, Fernandez‐Sesma A, Palese P. The influenza virus protein PB1‐F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog 2011, 7:e1002067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schmolke M, Manicassamy B, Pena L, Sutton T, Hai R, Varga ZT, Hale BG, Steel J, Perez DR, Garcia‐Sastre A. Differential contribution of PB1‐F2 to the virulence of highly pathogenic H5N1 influenza A virus in mammalian and avian species. PLoS Pathog 2011, 7:e1002186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Brakier‐Gingras L, Charbonneau J, Butcher SE. Targeting frameshifting in the human immunodeficiency virus. Expert Opin Ther Targets 2012, 16:249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Howard MT, Gesteland RF, Atkins JF. Efficient stimulation of site‐specific ribosome frameshifting by antisense oligonucleotides. RNA 2004, 10:1653–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yu CH, Noteborn MH, Olsthoorn RC. Stimulation of ribosomal frameshifting by antisense LNA. Nucleic Acids Res 2010, 38:8277–8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ofori LO, Hilimire TA, Bennett RP, Brown NW Jr, Smith HC, Miller BL. High‐affinity recognition of HIV‐1 frameshift‐stimulating RNA alters frameshifting in vitro and interferes with HIV‐1 infectivity. J Med Chem 2014, 57:723–732. [DOI] [PMC free article] [PubMed] [Google Scholar]