

Abstract
Gastric acid challenge of the rat and mouse stomach is signalled to the brainstem as revealed by expression of c-Fos. The molecular sensors relevant to the detection of gastric mucosal acidosis are not known. Since the acid-sensing ion channels ASIC2 and ASIC3 are expressed by primary afferent neurons, we examined whether knockout of the ASIC2 or ASIC3 gene modifies afferent signalling of a gastric acid insult in the normal and inflamed stomach. The stomach of conscious mice (C57BL/6) was challenged with intragastric HCl; two hours later the activation of neurons in the nucleus tractus solitarii (NTS) of the brainstem was visualized by c-Fos immunocytochemistry. Mild gastritis was induced by addition of iodoacetamide (0.1 %) to the drinking water for 7 days. Exposure of the gastric mucosa to HCl (0.25 M) caused a 3-fold increase in the number of c-Fos-positive neurons in the NTS. This afferent input to the NTS remained unchanged by ASIC3 knockout, whereas ASIC2 knockout augmented the c-Fos response to gastric HCl challenge by 33 % (P<0.01). Pretreatment of wild-type mice with iodoacetamide induced mild gastritis, as revealed by increased myeloperoxidase activity, and enhanced the number of NTS neurons responding to gastric HCl challenge by 41 % (P<0.01). This gastric acid hyperresponsiveness was absent in ASIC3 knockout mice but fully preserved in ASIC2 knockout mice. The current data indicate that ASIC3 plays a major role in the acid hyperresponsiveness associated with experimental gastritis. In contrast, ASIC2 appears to dampen acid-evoked input from the stomach to the NTS.
Keywords: Acid-sensing ion channels, gastric acid hyperresponsiveness, gastritis, expression of c-Fos, nucleus of the solitary tract, vagal afferent neurons
INTRODUCTION
Gastric hydrochloric acid (HCl) is a noxious stimulus that following penetration through the mucosal barrier contributes to pain arising from the oesophagus, stomach and upper small intestine. Exposure of the rodent stomach to excess HCl elicits a visceromotor response indicative of pain (Lamb et al. 2003) and causes many neurons in the nucleus tractus solitarii (NTS) of the brainstem to express c-Fos, a marker of neuronal excitation (Schuligoi et al. 1998; Danzer et al. 2004; Wultsch et al. 2005). Both the gastric HCl-evoked visceromotor reaction and medullary c-Fos response are suppressed by vagotomy, which attests to a significant role of vagal afferent neurons in gastric chemonociception (Schuligoi et al. 1998; Lamb et al. 2003).
Intramucosal acidosis may excite sensory neurons by activation of molecular acid sensors such as the acid-sensing ion channels (ASICs) which belong to the voltage-insensitive, amiloride-sensitive epithelial Na+ channel/degenerin family (Waldmann and Lazdunski 1998; Kellenberger and Schild 2002; Holzer 2003; Kress and Waldmann 2006; Wemmie et al. 2006). The proton-gated members of the ASIC family are encoded by 3 different genes: ASIC1, ASIC2 and ASIC3, with ASIC1 and ASIC2 each having alternative splice variants termed ASIC1a, ASIC1b, ASIC2a and ASIC2b. Functional channels are made up of different ASIC subunits, most of which are expressed by vagal and spinal afferent neurons, although to different degrees (Alvarez de la Rosa et al. 2002; Benson et al. 2002; Page et al. 2004,2005; Schicho et al. 2004; Sugiura et al. 2005; Kress and Waldmann 2006).
ASIC1, ASIC2 and ASIC3 are gated by a drop in the external pH. While gene knockout studies have shown that mechanotransduction in colonic afferent neurons is altered in ASIC1, ASIC2 and ASIC3 null mice (Page et al. 2004,2005; Roza et al. 2004; Jones et al. 2005), the involvement of ASIC channels in gastric acid sensing has not yet been explored. Focusing on the role of ASIC2 and ASIC3, the first aim of this study was to examine whether gastric HCl-evoked afferent input to the brainstem as visualized by expression of c-Fos in the NTS is altered in ASIC2 knockout (ASIC2−/−) and ASIC3 knockout (ASIC3−/−) mice. The effect of gastric HCl challenge on gastric fluid retention and gross mucosal lesion formation was also investigated.
Gastritis and gastric ulceration are associated with gastric hypersensitivity, and iodoacetamide-induced gastritis in rats has been found to enhance the visceromotor response to gastric distension and HCl challenge (Ozaki et al. 2002; Lamb et al. 2003). Further analysis has shown that gastric ulceration alters the kinetics of acid-induced currents in nodose ganglion neurons (Sugiura et al. 2005). Likewise, neurons in the cat brainstem exhibit hyperresponsiveness to oesophageal distension after acute exposure to HCl plus pepsin (Medda et al. 2005). Since the roles of ASIC2 and ASIC3 in gastritis-induced changes of gastric chemonociception have not yet been addressed, the second aim of this study was to test the hypothesis that ASIC2 and ASIC3 contribute to the gastritis-evoked hyperresponsiveness of the stomach – NTS system to gastric HCl.
METHODS
Experimental animals
All experiments were approved by an ethical committee at the Federal Ministry of Education, Science and Culture of the Republic of Austria and conducted according to the guidelines of the Committee for Research and Ethical Issues of the International Association for the Study of Pain and of the Directive of the European Communities Council of 24 November 1986 (86/609/EEC). The experiments were designed in such a way that the number of animals used and their suffering was minimized according to the directives of the ethical committee.
The experiments were carried out with germline ASIC2−/− and ASIC3−/− mice and wild-type mice of the same genetic background (C57BL/6). The mice were generated and bred at the CNRS-IPMC of the University of Nice-Sophia Antipolis in France, while the experiments were performed at the Medical University of Graz in Austria.
The generation of the ASIC2−/− mice has been reported elsewhere (Ettaiche et al. 2004; Roza et al. 2004). These mice are deficient in both known splice variants of ASIC2: ASIC2a and ASIC2b.
For the generation of ASIC3−/− mice, mouse genomic 129SvJ bacterial artificial chromosome (BAC) grids (Genome Systems, St. Louis, MO, USA) were hybridized with the ASIC3 coding sequence, and positive BACs were analyzed. The targeting vector was constructed as outlined in Figure 1. Homologous recombination at the 5′ end was verified by polymerase chain reaction with the oligonucleotides ACAGACACAGTGCCAGTAAGA and TCGCAGCGCATCGCCTTCTAT. For the 3′ end, homologous recombination was confirmed by Southern blot after BclI digestion with the BclI/AccI fragment indicated in Figure 1 as a probe. With the wild-type allele a fragment of 6.0 kb was labelled, whereas for the targeted allele a fragment of 6.8 kb hybridized with the probe.
Figure 1. Generation of ASIC3 null mice.
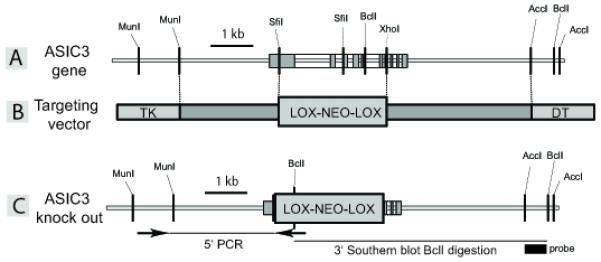
(A) ASIC3 gene and flanking sequences. ASIC3 exons are indicated by gray boxes. (B) In the ASIC3 targeting vector a SfiI / XhoI fragment of the ASIC3 gene was replaced by a neomycin resistance expression cassette flanked by two loxp sites. (C) Targeted ASIC3 allele which codes for a 43 amino acid protein truncated before the first transmembrane domain.
The initially obtained 129Sv/C57BL/6 mice were backcrossed for ten generations with C57BL/6 mice. The ASIC2 and ASIC3 null mice were viable, fertile and did not exhibit any obvious morphological or behavioral abnormalities. C57BL/6 mice were used as wild-type controls.
The experiments were carried out with female mice of all 3 genotypes, aged 2-4 months and weighing 20-30 g. The animals were housed in groups of 3-4 per cage under controlled temperature (21 °C) conditions and a 12 h light/dark cycle (lights on at 6:00 AM, lights off at 6:00 PM).
Iodoacetamide-induced gastritis
To induce gastritis, iodoacetamide (Sigma, Vienna, Austria) was added to the drinking water at a concentration of 0.1 % (w/w) for 7 days (Ozaki et al. 2002; Piqueras et al. 2003). Since iodoacetamide is light-sensitive, the iodoacetamide-containing drinking water was made up fresh every day. The control animals received normal drinking water. The gastric HCl challenge experiments were performed on day 8 of iodoacetamide pretreatment.
Experimental protocols
In all experiments, the mice were fasted for 16 h to ensure that the stomach was empty, but had free access to water, before HCl (0.25 M) or physiological saline (0.15 M NaCl) was administered intragastrically (IG) at a volume of 0.02 ml/g through a soft infant feeding tube (outer diameter 1.5 mm; Rüsch, Montevideo, Uruguay). After the IG treatment the animals were no longer allowed to drink until tissue collection 0.5 or 2 h later. For this purpose, the mice were euthanized by intraperitoneal injection of an overdose of pentobarbital (100 mg/kg).
Five specific studies were carried out. Study 1 was carried out to test whether IG administration of HCl to C57BL/6 mice would cause a similar expression of c-Fos in the NTS as seen previously in Him:OF1 mice (Wultsch et al. 2005). For this purpose, C57BL/6 mice received an IG bolus of saline or HCl (0.25, 0.35 and 0.5 M) 2 h before collection of the brainstem. In study 2, wild-type, ASIC2−/− and ASIC3−/− mice were compared in their c-Fos expression in the NTS 2 h after IG administration of saline or HCl (0.25 M). Study 3 was carried out to determine gastric emptying and gross gastric lesion formation in wild-type, ASIC2−/− and ASIC3−/− mice 0.5 h after IG administration of HCl (0.25 M). Study 4 was performed with wild-type mice to establish that addition of iodoacetamide (0.1 %) to the drinking water for 7 days induced mild gastritis as assessed by gastric myeloperoxidase (MPO) activity, gross gastric lesion formation and body weight change. In study 5 wild-type, ASIC2−/− and ASIC3−/− mice were pretreated with iodoacetamide (0.1 %) added to the drinking water for 7 days or received normal drinking water. On day 8 they were treated IG with HCl (0.25 M) 2 h before collection of the brainstem.
c-Fos immunocytochemistry
Following euthanasia, the mice were transcardially perfused with 0.1 M phosphate-buffered saline (PBS) of pH 7.4 (25 ml), followed by 4 % buffered paraformaldehyde (45 ml). The brainstems were removed, processed for c-Fos immunocytochemistry and analyzed as described previously (Wultsch et al. 2005). The coordinates of the brainstem region under study were interaural −3.96 to −3.40 and bregma −7.76 to −7.20 according to Paxinos and Franklin (2001). The sections were coded such that the examiner did not know which treatment group or genotype they came from. Eight sections per animal were analyzed, and all c-Fos-positive cells were randomly counted on one side of the NTS (Wultsch et al. 2005). In order to avoid that the same cells were counted twice, only every second section was taken for analysis. All counts in each section of each animal were averaged to give the number of c-Fos-positive cells in the NTS of that animal. These average values from each animal were then used to calculate the mean number of c-Fos-positive cells per section in the unilateral NTS of each experimental group.
Myeloperoxidase activity
The enzyme activity of MPO (donor:H2O2 oxidoreductase, EC 1.11.1.7) was determined according to previously described spectrophotometric techniques (Suzuki et al. 1983; Krawisz et al. 1984; Graff et al. 1998). At autopsy, full-thickness pieces of the gastric corpus (120-160 mg) were excised, shock-frozen in liquid nitrogen and stored at −70 °C until assay. After homogenisation in potassium phosphate buffer (0.05 M) of pH 7.4 and centrifugation at 40,000 g at 4 °C for 20 min, the pellets were taken up in potassium phosphate buffer (0.05 M) of pH = 6.0 containing 3.72 g/l ethylenediaminetetraacetic acid (Roth, Karlsruhe, Germany) and 5 g/l hexadecyltrimethylammonium bromide (Sigma, Vienna, Austria) which releases MPO from the primary granules of the neutrophils (Krawisz et al. 1984). The enzyme assay was based on the MPO-catalyzed oxidation of tetramethylbenzidine (Merck, Darmstadt, Germany) in the presence of H2O2 (Suzuki et al. 1983). The absorbance of the samples was measured at 655 nm.
MPO activity was expressed relative to the MPO activity of human neutrophils. To this end, a sample of human neutrophils (5 million cells in 2 ml of 0.05 M potassium phosphate buffer of pH = 7.4) was extracted in an identical manner as the tissue samples and assayed as described above. A standard curve was constructed by measuring the absorbance of the reaction mixture with various dilutions of the neutrophil extract. Since the MPO activity in the gastric tissue samples was determined as an index of neutrophil infiltration, the MPO activity of the tissue extracts was expressed as human neutrophil equivalents/mg wet tissue.
Gastric fluid recovery and gastric lesion formation
After euthanasia the abdomen of the animals was quickly opened by a midline incision, and the stomach clamped at the lower oesophageal sphincter and pylorus. Then the fluid-filled stomach was weighed, opened along the greater curvature, blotted dry on tissue paper and re-weighed to determine the volume of the fluid present in the stomach. Gastric fluid recovery was calculated by expressing the weight of the fluid present in the stomach as a percentage of the weight of the fluid administered into the stomach (Wultsch et al. 2005). To determine gastric lesion formation, the stomach was pinned flat on a silicon elastomer-coated plate and covered with PBS. The stomach was photographed, the image transferred to a personal computer, and gross gastric injury assessed by computerized planimetry (Wultsch et al. 2005). The mucosal area covered by visible haemorrhagic damage was expressed as a percentage of the total area of the glandular mucosa (Wultsch et al. 2005).
Statistics
Statistical evaluation of the results was performed on SPSS 11.5 (SPSS Inc., Chicago, IL, USA) with Student’s t-test (two treatments groups), one-way analysis of variance (ANOVA) (more than two treatment groups) or two-way ANOVA (genotype and treatment as factors) followed by the Games-Howell test. All data are presented as means ± SEM, n referring to the number of mice in each group. Probability values of P < 0.05 were regarded as significant.
RESULTS
Concentration-dependent effect of gastric HCl to induce c-Fos in the NTS of wild-type mice
In a pilot study involving 8 wild-type mice it was tested whether IG administration of HCl to C57BL/6 wild-type mice would cause a similar expression of c-Fos in the NTS as that previously characterized in Him:OF1 mice (Wultsch et al. 2005). Relative to saline (0.15 M NaCl), IG administration of increasing concentrations of HCl (0.25, 0.35, 0.5 M) enhanced the number of c-Fos=-expressing cells in the NTS in a HCl concentration-dependent manner (Figure 2). Some neurons in the area postrema also exhibited c-Fos-like immunoreactivity (Figure 3A) but this response was not further analyzed. The concentration of 0.25 M HCl augmented the number of c-Fos-positive cells by a factor of 3.0, which compared well with the 2.7-fold rise previously seen in Him:OF1 mice (Wultsch et al. 2005). After gastric exposure to 0.35 M HCl, the number of c-Fos-positive neurons in the NTS rose by a factor of 4.6, which appeared to be the maximal effect attainable since there was no further increase in c-Fos expression after gastric challenge with 0.5 M HCl (Figure 2). All further experiments were carried out with 0.25 M HCl (pH = 0.70), because at this stimulus strength both increases and decreases in the c-Fos response could be demonstrated.
Figure 2. Effect of gastric acid challenge on c-Fos expression in the nucleus tractus solitarii (NTS) of wild-type mice.

The graph shows the concentration-related effect of IG administered HCl, relative to NaCl, to increase the expression of c-Fos in the unilateral NTS of wild-type mice. NaCl (0.15 M; depicted as 0 M HCl) and HCl (0.25, 0.35 and 0.5 M) were administered IG 2 h before immunocytochemistry. Since this experiment was a pilot experiment, each concentration of HCl was tested in 2 mice only. The values shown represent the results of the individual experiments (black dots) and their means (horizontal line).
Figure 3. Expression of c-Fos in the nucleus tractus solitarii (NTS) and area postrema (AP) of wild-type (A,B), ASIC2−/− (C,D) and ASIC3−/− (E,F) mice.

The animals were treated IG with 0.25 M HCl 2 h before immunocytochemical visualization of c-Fos-positive cells in the brainstem. The animals had either received normal drinking water (A,C,E) or drinking water containing 0.1 % iodoacetamide (B,D,F) for 7 days before the gastric acid challenge experiment. HCl induced many cells in the medial and subpostremal nuclei of the NTS and some cells in the AP to express c-Fos. CC, central canal. Calibration bar: 0.1 mm.
The specificity of c-Fos immunoreactivity was proved by the absence of any immunoreactive signal when the c-Fos blocking peptide had been added to the primary antibody dilution.
Gastric HCl-evoked expression of c-Fos in the NTS of wild-type, ASIC2−/− and ASIC3−/− mice
When saline was administered IG to wild-type, ASIC2−/− and ASIC3−/− mice, the number of NTS neurons expressing c-Fos did not significantly differ between the 3 genotypes. Gastric exposure to HCl (0.25 M) increased the number of c-Fos-positive neurons in the NTS of all genotypes to a significant extent (Figure 4). Importantly, ASIC2−/− mice responded to gastric HCl challenge with a 33 % larger expression of c-Fos in the NTS than wild-type mice (P<0.01), whereas ASIC3−/− mice did not significantly differ in this respect from wild-type animals (Figures 3A,C,E and 4).
Figure 4. Expression of c-Fos in the nucleus tractus solitarii (NTS) of wild-type, ASIC2−/− and ASIC3−/− mice without gastritis.
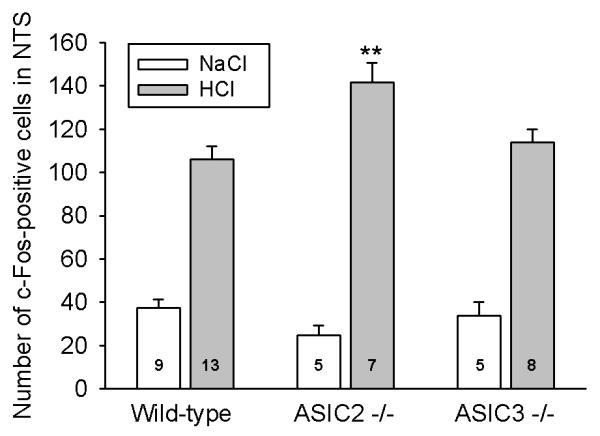
Expression of c-Fos in the unilateral NTS was visualized 2 h after IG administration of NaCl (0.15 M) and HCl (0.25 M). The values represent means ± SEM, n as indicated. **P < 0.01 versus HCl-treated wild-type animals (two-way ANOVA followed by Games-Howell test).
Gastric volume recovery and mucosal damage after gastric HCl exposure of wild-type, ASIC2−/− and ASIC3−/− mice
Gastric fluid recovery and gross mucosal damage were examined 30 min after IG administration of HCl (0.25 M), at a time point when these parameters have been shown to be maximal (Wultsch et al. 2005). While 30 min after IG administration of saline only 24 % of the injected volume is recovered from the stomach (Wultsch et al. 2005), 95-111 % of the administered volume was regained after IG administration of HCl (Table 1). These rates of gastric volume recovery did not significantly differ between wild-type, ASIC2−/− and ASIC3−/− mice subjected to IG HCl challenge.
Table 1. Gastric volume recovery and gross mucosal lesion formation after gastric HCl challenge in wild-type, ASIC2−/− and ASIC3−/− mice.
| Test parameter | Wild-type | ASIC2−/− | ASIC3−/− |
|---|---|---|---|
| Volume recovery (%) | 111 ± 10.4 (11) | 94.9 ± 6.4 (6) | 107 ± 7.6 (7) |
| Gastric lesions (%) | 0.49 ± 0.08 (7) | 0.72 ± 0.31 (6) | 1.1 ± 0.22 (7) |
Wild-type, ASIC2−/− and ASIC3−/− mice were treated IG with 0.25 M HCl 30 min before gastric volume recovery and macroscopic gastric injury were quantified. The gastric volume recovery is expressed as a percentage of the volume administered into the stomach, and the extent of the gastric lesions is given as a percentage of the area of the total glandular mucosa. The values represent means ± SEM, n as indicated in brackets. There was no significant difference between the 3 genotypes (F2,29=0.631, F2,17=2.340, one-way ANOVA followed by Games-Howell test).
The macroscopic injury of the gastric corpus mucosa observed after IG administration of HCl (0.25 M) was minor and, as reported previously (Wultsch et al. 2005), was not significantly different from that observed after IG treatment with NaCl (0.15 M). The visible lesions consisted of petechiae and a few small streaks of haemorrhage. As shown in Table 1, the formation of gastric haemorrhagic damage following gastric HCl challenge did not significantly differ between wild-type, ASIC2−/− and ASIC3−/− mice.
Effect of iodoacetamide pretreatment in wild-type mice
Relative to normal water consumption, addition of iodoacetamide (0.1 %) to the drinking water for 7 days increased MPO activity in the gastric corpus wall of wild-type mice (Table 2). Visual inspection of the gastric mucosa did not reveal any macroscopically visible damage. The iodoacetamide-consuming mice did not exhibit any behavioral signs of illness, and their weight gain during iodoacetamide pretreatment was not significantly smaller than that seen in animals drinking normal water (Table 2).
Table 2. Effect of chronic iodoacetamide pretreatment on gastric MPO activity and body weight gain of wild-type mice.
| Test parameter | Control | Iodoacetamide |
|---|---|---|
| MPO activity (neutrophil equivalents/mg) | 3,134 ± 644 (7) | 9,997 ± 1,701 (7)* |
| Body weight gain (g) | 0.83 ± 0.10 (6) | 0.61 ± 0.17 (6) |
Iodoacetamide (0.1 %) was added to the drinking water for 7 days, while control mice drank normal water. MPO activity in the gastric corpus wall was determined on the 8th day of pretreatment and expressed as neutrophil equivalents per mg tissue. The body weight gain was calculated as the difference in body weight before and after the 7 day pretreatment period. The values represent means ± SEM, n as indicated in brackets.
P < 0.01 versus control (Student’s t test).
Relative to normal water consumption, iodoacetamide pretreatment of wild-type mice enhanced the number of NTS neurons expressing c-Fos in response to gastric HCl (0.25 M) challenge by 41 % (Figures 3A,B and 5).
Figure 5. Expression of c-Fos in the nucleus tractus solitarii (NTS) of wild-type, ASIC2−/− and ASIC3−/− mice with gastritis.
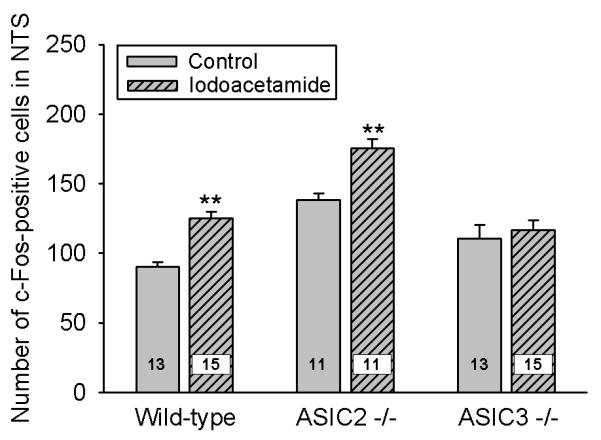
Expression of c-Fos in the unilateral NTS was visualized 2 h after IG administration of HCl (0.25 M) to wild-type, ASIC2−/− and ASIC3−/− mice that had been pretreated with iodoacetamide (0.1%) added to the drinking water for 7 days or were allowed to drink normal water (control). The values represent means ± SEM, n as indicated. ** P < 0.01 versus control animals of the same genotype (two-way ANOVA followed by Games-Howell test). ASIC2−/− control mice, but not ASIC3−/− control mice, were significantly (P < 0.01) different from wild-type control mice.
Effect of iodoacetamide pretreatment on gastric HCl-evoked expression of c-Fos in the NTS of ASIC2−/− and ASIC3−/− mice
Although ASIC2−/− mice drinking normal water exhibited a significantly higher number of c-Fos-immunoreactive NTS neurons following gastric exposure to HCl (0.25 M) than wild-type mice (Figures 4 and 5), the ability of iodoacetamide (0.1 %) pretreatment to augment the gastric HCl-evoked c-Fos response in the NTS was preserved in ASIC2−/− mice (Figure 5). Thus, the NTS of both wild-type and ASIC2−/− mice pretreated with iodoacetamide contained on average 35 and 37 more neurons expressing c-Fos in response to gastric HCl challenge than that of wild-type and ASIC2−/− mice drinking normal water, respectively (Figure 5).
In contrast, the ability of iodoacetamide pretreatment to significantly boost the NTS response to gastric HCl challenge was lost in ASIC3−/− mice (Figure 5). In some of these animals gastric MPO was determined 2 h after IG administration of HCl, i.e., at the time when the brainstem was removed for c-Fos immunocytochemistry. The MPO activity in the stomach of ASIC3−/− mice drinking normal water amounted to 4,371 ± 1,418 and that in ASIC3−/− mice pretreated with iodoacetamide to 9,184 ± 1,256 neutrophil equivalents/mg (n = 3, P = 0.064, Student’s t test). The latter value was very similar to that measured in iodoacetamide-pretreated wild-type mice (Table 2).
DISCUSSION
This study tested the hypothesis whether the H+-gated ion channels ASIC2 and ASIC3 contribute to the acid sensitivity of the afferent stomach – NTS pathway in the normal and inflamed stomach. Exposure of the rat gastric mucosa to excess HCl is signalled to the NTS, but not spinal cord (Schuligoi et al. 1998), as visualized by immunocytochemical expression of c-Fos, a marker of neuronal excitation (Hughes and Dragunow 1995; Munglani and Hunt 1995). Vagotomy suppresses the medullary c-Fos response to gastric HCl challenge (Schuligoi et al. 1998; Michl et al. 2001), which is consistent with the NTS being the major central projection area of vagal afferents (Norgren and Smith 1988; Altschuler et al. 1989). In analogy to this situation in rats, we suppose that the major pathway which in mice signals a gastric HCl insult to the brainstem is vagal afferent in nature. It need be considered, however, that the NTS receives visceral input not only via vagal afferents but also via a spinosolitary pathway (Menetrey and Basbaum 1987; Gamboa-Esteves et al. 2001).
The gastric HCl concentrations (0.15 - 0.35 M) that induced c-Fos in the NTS of Him:OF1 (Wultsch et al. 2005) and C57BL/6 (present study) mice were supraphysiological but did not cause visible gastric damage. Gastric injury and hyperosmolarity of the HCl stimulus have previously been ruled out as major factors contributing to the gastric HCl-evoked expression of c-Fos in the NTS (Michl et al. 2001; Danzer et al. 2004). Consequently, the afferent signals are thought to result from intramucosal acidosis (Danzer et al. 2004; Wultsch et al. 2005) caused by the excess transmucosal H+ gradient that drives luminal H+ ions across the gastric mucosal barrier into the lamina propria where they excite afferent neurons either directly or indirectly via neuroactive factors released in the tissue.
In addressing the implication of ASIC2 and ASIC3 as gastric acid sensors we found that knockout of the ASIC2 or ASIC3 gene had no significant effect on c-Fos expression in the NTS following IG administration of saline. This result indicates that baseline activity in the afferent stomach – NTS axis and any response to the mechanical stimulation caused by gastric gavage were not altered in ASIC2−/− and ASIC3−/− mice. In contrast, the NTS response to gastric HCl challenge was significantly enhanced by ASIC2 gene knockout. This observation is reminiscent of the increase in the mechanosensitivity of gastro-oesophageal mucosal nerve endings seen in ASIC2−/− mice, whereas the mechanosensitivity of other populations of afferent nerve fibres in the gut is increased or decreased to various degrees (Roza et al. 2004; Page et al. 2005). Our observation implies that ASIC2 dampens afferent signalling of a gastric HCl insult and that hence knockout of ASIC2 results in disinhibition of afferent input to the NTS. It remains to be shown which mechanisms underlie the inhibitory effect of ASIC2 on afferent signalling from the acid-threatened stomach and whether this effect takes place at the level of primary afferent or NTS neurons.
Unlike ASIC2 gene deletion, knockout of ASIC3 did not alter the gastric HCl-evoked input to the NTS. It would appear that ASIC3 does not contribute to the acid sensitivity of afferent neurons in the normal stomach. Although the expression of ASIC1 and ASIC2 is not altered in ASIC3−/− mice (Price et al. 2001), it need be considered that compensatory changes in other acid sensors may have masked the functional implications of ASIC2 and ASIC3 in our germline knockout mice. This argument is consistent with the expression of multiple acid sensors on afferent neurons, including the transient receptor potential vanilloid 1 ion channel which can be activated by HCl in the absence of inflammation (Holzer 2003; Jones et al. 2005; Sugiura et al. 2005).
Gastric fluid retention was assessed to examine whether the medullary c-Fos response was influenced by variations in the duration of gastric HCl exposure (Danzer et al. 2004; Wultsch et al. 2005). As shown before in the rat and mouse, 95 % or more of the IG administered volume of HCl (0.25 M) is recovered from the stomach 30 min later, compared with a 15-24 % recovery rate when saline is administered IG (Holzer et al. 2003; Danzer et al. 2004; Wultsch et al. 2005). This retention of gastric fluid reflects inhibition of gastric emptying and secretion of gastric fluid. Since gastric fluid recovery remained unaltered in ASIC2−/− and ASIC3−/− mice it follows that the processes underlying HCl-induced gastric fluid retention do not depend on these proton-gated ion channels. In addition, the minor gastric injury due to HCl was not significantly altered in ASIC2−/− and ASIC3−/− mice. However, the tendency towards increased lesion formation in ASIC3−/− mice deserves further analysis as it suggests that ASIC3 is relevant to mucosal protection from HCl-induced injury.
The second aim was to explore whether ASIC2 and ASIC3 contribute to the acid hyperresponsiveness of the inflamed stomach. Exposure of the stomach to iodoacetamide for one week was found to significantly increase the medullary c-Fos response to gastric HCl challenge, which is in keeping with the ability of iodoacetamide-induced gastritis to augment the visceromotor response to gastric distension or a HCl insult (Ozaki et al. 2002; Lamb et al. 2003). The induction of low-grade inflammation was verified by the significantly enhanced MPO activity in the gastric corpus in the absence of a significant weight loss or other signs of illness. Increased levels of MPO reflect inflammation-associated infiltration of neutrophils and monocytes into the tissue (Krawisz et al. 1984), and the 3.2-fold increase in the MPO activity seen here compares well with the limited increase in MPO activity caused by iodoacetamide in the rat stomach (Ozaki et al. 2002).
The major discovery of this work was that ASIC3, but not ASIC2, participates in the gastritis-evoked hyperresponsiveness to gastric acid. The failure of iodoacetamide-pretreated ASIC3−/− mice to develop gastric HCl hyperresponsiveness cannot be ascribed to a failure of iodoacetamide to induce mild gastritis in these mice, because their gastric MPO activity was very similar to that measured in iodoacetamide-pretreated wild-type mice. We suppose that the ASIC3-mediated hyperresponsiveness to gastric acid takes place at the level of primary afferent neurons, since in the afferent stomach – NTS axis ASIC3 is primarily associated with these neurons (Waldmann and Lazdunski 1998; Alvarez de la Rosa et al. 2002; Schicho et al. 2004; Page et al. 2005). It awaits to be examined whether ASIC3-mediated hyperresponsiveness to gastric HCl arises from an increase in channel expression or channel sensitivity to HCl. Because ASIC3 also contributes to the mechanical hyperresponsiveness of the inflamed colon (Jones et al. 2005), it would seem that ASIC3 is an important target for the therapeutic management of hyperalgesia associated with gastrointestinal inflammation. This conjecture is in keeping with the inflammation-evoked increase in ASIC3 transcription of sensory neurons (Voilley et al. 2001) and the upregulation of ASIC3 in the intestine of patients with Crohn’s disease (Yiangou et al. 2001).
Iodoacetamide pretreatment induced gastric acid hyperresponsiveness in ASIC2−/− mice much as it did in wild-type mice, despite the fact that ASIC2 (ASIC2a and ASIC2b) gene knockout itself enhanced the medullary c-Fos response to gastric HCl challenge. It would appear that this effect of ASIC2 gene knockout is unrelated to the ASIC3-dependent mechanism whereby mild gastritis leads to gastric acid hyperresponsiveness of the stomach – NTS axis, and it is conceivable that the effect of ASIC2 to dampen afferent input to the brainstem takes place primarily at the NTS level. Since ASIC2b is inactive as a homomultimeric channel (Kellenberger and Schild 2002; Kress and Waldmann 2006), we speculate that the ASIC2-mediated inhibition of afferent input from the acid-threatened stomach to the brainstem is mediated by ASIC2a. ASIC2b forms functional heteromultimers with other ASIC subunits, particularly ASIC3, and the currents generated by ASIC2b/ASIC3 heteromultimers closely reflect acid-induced currents in afferent neurons (Benson et al. 2002; Kress and Waldmann 2006). Deletion of ASIC2b is hence likely to alter acid-induced currents in sensory neurons, and the ASIC2 knockout phenotype is likely to be determined by the cellular distribution of ASIC2 and ASIC3 and their functional interaction along the afferent stomach – NTS axis.
In summary, the present findings attribute ASIC2 and ASIC3 diverse roles in the afferent signalling from the acid-threatened stomach to the brainstem. While ASIC2 has a depressant action, ASIC3 contributes to the development of gastritis-associated hyperresponsiveness to gastric acid. Since vagal afferents constitute the major afferent pathway from the stomach to the NTS and there is emerging evidence that vagal afferents and the NTS play a role in gastric chemonociception (Schuligoi et al. 1998; Gamboa-Esteves et al. 2001; Michl et al. 2001; Boscan et al. 2002; Lamb et al. 2003), our observations may have two important implications. Firstly, if ASIC3 expression or function is upregulated in gastric inflammation, ASIC3 could prove to be a biomarker for afferent hyperresponsiveness associated with inflammatory or functional disorders of the foregut. Secondly, ASIC3 could represent a novel target for treating gastric hyperalgesia, if specific ASIC3 blockers (Diochot et al. 2004; Dube et al. 2005) are able to reverse inflammation-induced afferent hyperresponsiveness.
ACKNOWLEDGEMENTS
The authors are grateful to Dr. Nicolas Guy (Valbonne, France) for his help in the generation of the knockout mice and to AstraZeneca (Södertälje, Sweden) for ES cell transfections and blastocyst injections. This study was supported by the Jubilee Funds of the Austrian National Bank (grant 9858), Zukunftsfonds Steiermark (grant 262), Austrian Scientific Research Funds (FWF grant L25-B05), Institut National de la Santé et de la Recherche Médicale (INSERM) and Centre National de la Recherche Scientifique (CNRS).
REFERENCES
- Altschuler SM, Bao XM, Bieger D, Hopkins DA, Miselis RR. Viscerotopic representation of the upper alimentary tract in the rat: sensory ganglia and nuclei of the solitary and spinal trigeminal tracts. J Comp Neurol. 1989;283:248–268. doi: 10.1002/cne.902830207. [DOI] [PubMed] [Google Scholar]
- Alvarez de la Rosa D, Zhang P, Shao D, White F, Canessa CM. Functional implications of the localization and activity of acid-sensitive channels in rat peripheral nervous system. Proc Natl Acad Sci USA. 2002;99:2326–2331. doi: 10.1073/pnas.042688199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson CJ, Xie J, Wemmie JA, Price MP, Henss JM, Welsh MJ, Snyder PM. Heteromultimers of DEG/ENaC subunits form H+-gated channels in mouse sensory neurons. Proc Natl Acad Sci USA. 2002;99:2338–2343. doi: 10.1073/pnas.032678399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boscan P, Pickering AE, Paton JF. The nucleus of the solitary tract: an integrating station for nociceptive and cardiorespiratory afferents. Exp Physiol. 2002;87:259–266. doi: 10.1113/eph8702353. [DOI] [PubMed] [Google Scholar]
- Danzer M, Jocic M, Samberger C, Painsipp E, Bock E, Pabst MA, Crailsheim K, Schicho R, Lippe IT, Holzer P. Stomach-brain communication by vagal afferents in response to luminal acid backdiffusion, gastrin, and gastric acid secretion. Am J Physiol. 2004;286:G403–G411. doi: 10.1152/ajpgi.00308.2003. [DOI] [PubMed] [Google Scholar]
- Diochot S, Baron A, Rash LD, Deval E, Escoubas P, Scarzello S, Salinas M, Lazdunski M. A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. EMBO J. 2004;23:1516–1525. doi: 10.1038/sj.emboj.7600177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube GR, Lehto SG, Breese NM, Baker SJ, Wang X, Matulenko MA, Honore P, Stewart AO, Moreland RB, Brioni JD. Electrophysiological and in vivo characterization of A-317567, a novel blocker of acid sensing ion channels. Pain. 2005;117:88–96. doi: 10.1016/j.pain.2005.05.021. [DOI] [PubMed] [Google Scholar]
- Ettaiche M, Guy N, Hofman P, Lazdunski M, Waldmann R. Acid-sensing ion channel 2 is important for retinal function and protects against light-induced retinal degeneration. J Neurosci. 2004;24:1005–1012. doi: 10.1523/JNEUROSCI.4698-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamboa-Esteves FO, Kaye JC, McWilliam PN, Lima D, Batten TF. Immunohistochemical profiles of spinal lamina I neurones retrogradely labelled from the nucleus tractus solitarii in rat suggest excitatory projections. Neuroscience. 2001;104:523–538. doi: 10.1016/s0306-4522(01)00071-9. [DOI] [PubMed] [Google Scholar]
- Graff G, Gamache DA, Brady MT, Spellman JM, Yanni JM. Improved myeloperoxidase assay for quantitation of neutrophil influx in a rat model of endotoxin-induced uveitis. J Pharmacol Toxicol Methods. 1998;39:169–178. doi: 10.1016/s1056-8719(98)00023-9. [DOI] [PubMed] [Google Scholar]
- Holzer P. Acid-sensitive ion channels in gastrointestinal function. Curr Opin Pharmacol. 2003;3:618–625. doi: 10.1016/j.coph.2003.06.008. [DOI] [PubMed] [Google Scholar]
- Holzer P, Danzer M, Schicho R, Samberger C, Painsipp E, Lippe IT. Vagal afferent input from the acid-challenged rat stomach to the brainstem: enhancement by interleukin-1β. Neuroscience. 2004;129:439–445. doi: 10.1016/j.neuroscience.2004.07.040. [DOI] [PubMed] [Google Scholar]
- Holzer P, Painsipp E, Jocic M, Heinemann A. Acid challenge delays gastric pressure adaptation, blocks gastric emptying and stimulates gastric fluid secretion in the rat. Neurogastroenterol Motil. 2003;15:45–55. doi: 10.1046/j.1365-2982.2003.00382.x. [DOI] [PubMed] [Google Scholar]
- Hughes P, Dragunow M. Induction of immediate-early genes and the control of neurotransmitter-regulated gene expression within the nervous system. Pharmacol Rev. 1995;47:133–178. [PubMed] [Google Scholar]
- Jones RC, Xu L, Gebhart GF. The mechanosensitivity of mouse colon afferent fibers and their sensitization by inflammatory mediators require transient receptor potential vanilloid 1 and acid-sensing ion channel 3. J Neurosci. 2005;25:10981–10989. doi: 10.1523/JNEUROSCI.0703-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev. 2002;82:735–767. doi: 10.1152/physrev.00007.2002. [DOI] [PubMed] [Google Scholar]
- Krawisz JE, Sharon P, Stenson WF. Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models. Gastroenterology. 1984;87:1344–1350. [PubMed] [Google Scholar]
- Kress M, Waldmann R. Acid sensing ionic channels. Curr Top Membranes. 2006;57:241–276. [Google Scholar]
- Lamb K, Kang YM, Gebhart GF, Bielefeldt K. Gastric inflammation triggers hypersensitivity to acid in awake rats. Gastroenterology. 2003;125:1410–1418. doi: 10.1016/j.gastro.2003.07.010. [DOI] [PubMed] [Google Scholar]
- Lingueglia E, de Weille JR, Bassilana F, Heurteaux C, Sakai H, Waldmann R, Lazdunski M. A modulatory subunit of acid sensing ion channels in brain and dorsal root ganglion cells. J Biol Chem. 1997;272:29778–29783. doi: 10.1074/jbc.272.47.29778. [DOI] [PubMed] [Google Scholar]
- Medda BK, Sengupta JN, Lang IM, Shaker R. Response properties of the brainstem neurons of the cat following intra-esophageal acid-pepsin infusion. Neuroscience. 2005;135:1285–1294. doi: 10.1016/j.neuroscience.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Menetrey D, Basbaum AI. Spinal and trigeminal projections to the nucleus of the solitary tract: a possible substrate for somatovisceral and viscerovisceral reflex activation. J Comp Neurol. 1987;255:439–450. doi: 10.1002/cne.902550310. [DOI] [PubMed] [Google Scholar]
- Michl T, Jocic M, Heinemann A, Schuligoi R, Holzer P. Vagal afferent signaling of a gastric mucosal acid insult to medullary, pontine, thalamic, hypothalamic and limbic, but not cortical, nuclei of the rat brain. Pain. 2001;92:19–27. doi: 10.1016/s0304-3959(00)00467-x. [DOI] [PubMed] [Google Scholar]
- Munglani R, Hunt SP. Proto-oncogenes: basic concepts and stimulation induced changes in the spinal cord. Prog Brain Res. 1995;104:283–298. doi: 10.1016/s0079-6123(08)61796-3. [DOI] [PubMed] [Google Scholar]
- Norgren R, Smith GP. Central distribution of subdiaphragmatic vagal branches in the rat. J Comp Neurol. 1988;273:207–223. doi: 10.1002/cne.902730206. [DOI] [PubMed] [Google Scholar]
- Ozaki N, Bielefeldt K, Sengupta JN, Gebhart GF. Models of gastric hyperalgesia in the rat. Am J Physiol. 2002;283:G666–G676. doi: 10.1152/ajpgi.00001.2002. [DOI] [PubMed] [Google Scholar]
- Page AJ, Brierley SM, Martin CM, Martinez-Salgado C, Wemmie JA, Brennan TJ, Symonds E, Omari T, Lewin GR, Welsh MJ, Blackshaw LA. The ion channel ASIC1 contributes to visceral but not cutaneous mechanoreceptor function. Gastroenterology. 2004;127:1739–1747. doi: 10.1053/j.gastro.2004.08.061. [DOI] [PubMed] [Google Scholar]
- Page AJ, Brierley SM, Martin CM, Price MP, Symonds E, Butler R, Wemmie JA, Blackshaw LA. Different contributions of ASIC channels 1a, 2, and 3 in gastrointestinal mechanosensory function. Gut. 2005;54:1408–1415. doi: 10.1136/gut.2005.071084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. Academic Press; San Diego: 2001. [Google Scholar]
- Piqueras L, Corpa JM, Martinez J, Martinez V. Gastric hypersecretion associated to iodoacetamide-induced mild gastritis in mice. Naunyn-Schmiedeberg’s Arch Pharmacol. 2003;367:140–150. doi: 10.1007/s00210-002-0670-7. [DOI] [PubMed] [Google Scholar]
- Price MP, McIlwrath SL, Xie J, Cheng C, Qiao J, Tarr DE, Sluka KA, Brennan TJ, Lewin GR, Welsh MJ. The DRASIC cation channel contributes to the detection of cutaneous touch and acid stimuli in mice. Neuron. 2001;32:1071–1083. doi: 10.1016/s0896-6273(01)00547-5. [DOI] [PubMed] [Google Scholar]
- Roza C, Puel JL, Kress M, Baron A, Diochot S, Lazdunski M, Waldmann R. Knockout of the ASIC2 channel in mice does not impair cutaneous mechanosensation, visceral mechanonociception and hearing. J Physiol. 2004;558:659–669. doi: 10.1113/jphysiol.2004.066001. (London) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schicho R, Florian W, Liebmann I, Holzer P, Lippe IT. Increased expression of TRPV1 receptor in dorsal root ganglia by acid insult of the rat gastric mucosa. Eur J Neurosci. 2004;19:1811–1818. doi: 10.1111/j.1460-9568.2004.03290.x. [DOI] [PubMed] [Google Scholar]
- Schuligoi R, Jocic M, Heinemann A, Schöninkle E, Pabst MA, Holzer P. Gastric acid-evoked cfos messenger RNA expression in rat brainstem is signaled by capsaicin-resistant vagal afferents. Gastroenterology. 1998;115:649–660. doi: 10.1016/s0016-5085(98)70144-1. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Dang K, Lamb K, Bielefeldt K, Gebhart GF. Acid-sensing properties in rat gastric sensory neurons from normal and ulcerated stomach. J Neurosci. 2005;25:2617–2627. doi: 10.1523/JNEUROSCI.2894-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Ota H, Sasagawa S, Sakatani T, Fujikura T. Assay method for myeloperoxidase in human polymorphonuclear leukocytes. Anal Biochem. 1983;132:345–352. doi: 10.1016/0003-2697(83)90019-2. [DOI] [PubMed] [Google Scholar]
- Voilley N, de Weille J, Mamet J, Lazdunski M. Nonsteroid anti-inflammatory drugs inhibit both the activity and the inflammation-induced expression of acid-sensing ion channels in nociceptors. J Neurosci. 2001;21:8026–8033. doi: 10.1523/JNEUROSCI.21-20-08026.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann R, Lazdunski M. H+-gated cation channels: neuronal acid sensors in the NaC/DEG family of ion channels. Curr Opin Neurobiol. 1998;8:418–424. doi: 10.1016/s0959-4388(98)80070-6. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Price MP, Welsh MJ. Acid-sensing ion channels: advances, questions and therapeutic opportunities. Trends Neurosci. 2006;29:578–586. doi: 10.1016/j.tins.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Wultsch T, Painsipp E, Thoeringer CK, Herzog H, Sperk G, Holzer P. Endogenous neuropeptide Y depresses the afferent signaling of gastric acid challenge to the mouse brainstem via neuropeptide Y type Y2 and Y4 receptors. Neuroscience. 2005;136:1097–1107. doi: 10.1016/j.neuroscience.2005.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiangou Y, Facer P, Smith JA, Sangameswaran L, Eglen R, Birch R, Knowles C, Williams N, Anand P. Increased acid-sensing ion channel ASIC-3 in inflamed human intestine. Eur J Gastroenterol Hepatol. 2001;13:891–896. doi: 10.1097/00042737-200108000-00003. [DOI] [PubMed] [Google Scholar]
- Xie J, Price MP, Berger AL, Welsh MJ. DRASIC contributes to pH-gated currents in large dorsal root ganglion sensory neurons by forming heteromultimeric channels. J Neurophysiol. 2002;87:2835–2843. doi: 10.1152/jn.2002.87.6.2835. [DOI] [PubMed] [Google Scholar]