

Abstract
“Insulin resistance” is a widely used clinical term. It is usually defined as a state characterised by reduced glucose-lowering activity of insulin, but is also sometimes used as a shorthand label for a clinical syndrome encompassing major pathologies such as type 2 diabetes, polycystic ovary syndrome, fatty liver disease, and atherosclerosis. Nevertheless the precise cellular origins of insulin resistance (IR), the causal links among these phenomena, and the mechanisms underlying them, remain poorly understood or contentious. Prevalent IR usually results from a genetic predisposition interacting with acquired obesity, however even in some lean individuals very severe degrees of IR are seen. It is important to identify these people as they often harbour identifiable single gene defects, and they may benefit from molecular diagnosis, genetic counselling, and sometimes tailored therapies. Observation of people with known single gene defects also offers the opportunity to make inferences about the mechanistic links between IR and common pathologies. We now summarise the currently known monogenic forms of severe IR, with an emphasis on practical aspects of their recognition, diagnosis and management. In particular, we draw distinctions among the biochemical subphenotypes of IR that arise from primary adipose tissue dysfunction or from primary insulin signalling defects, and discuss the implications of this dichotomy for management.
Keywords: Severe Insulin Resistance, Hyperandrogenism, Pseudoacromegaly, Hypoglycaemia, Lipodystrophy, Genetics, Diabetes
Introduction
Obesity and the metabolic syndrome pose a substantial threat to health and longevity in contemporary society. Integral to this is the propensity of obesity to induce a state of insulin resistance (IR), or reduced responsiveness to the glucose-lowering action of insulin, in some, but not all people. Evidence from twin studies suggests that a significant part of this individual susceptibility is genetically determined[1][2][3][4], however population-based, genome-wide association studies have been largely ineffective to date in uncovering the precise genetic variants accounting for this. Improving understanding of both the mechanisms leading to IR susceptibility, and the mechanisms linking it to disease is imperative, as IR is strongly associated with, and in some cases the driver of, major morbidity and mortality in the form of diabetes mellitus[5], the spectrum of fatty liver disease[6][7], atherosclerosis[8], ovulatory dysfunction[9] and malignancy[10].
Syndromes of Severe IR (SSIR) will be used in this article to denote a group of rare disorders that feature severe IR that is not accounted for by commensurately severe obesity. In fact in the majority of conditions to be described, affected individuals are usually lean. No formal criteria for diagnosing SIR biochemically exist. In our own practice we use fasting insulin >150pmol/l and/or a peak insulin on glucose tolerance testing of >1,500pmol/l in individuals with a BMI < 30kg/m2 AND with normal glucose tolerance as a useful indicator, while in those with absolute insulin deficiency and a BMI < 30kg/m2 exogenous insulin requirement of greater than 3 units/kg/day is suggestive. However it is important to emphasise that such biochemical criteria should not be applied rigidly. This is partly because of the difficulty in interpreting insulin levels once partial beta cell decompensation has occurred, as in many patients at first evaluation, and partly because of the influence of age, pubertal status and adiposity on insulin levels in the general population. Thus, while some biochemical testing is of great value, it should be interpreted in the light of clinical features including acanthosis nigricans, oligomenorrhoea and hyperandrogenism, or features specific to individual syndromes such as abnormal adipose distribution. The prevalence of SSIR has not been formally assessed, though quaternary referral practice in our centre suggests a prevalence of the order of 0.1-0.5% amongst patients attending hospital diabetes clinics [11].
It is important to understand that, while biochemically confirmed severe IR is a convenient defining feature of SSIR, severe IR per se is a biochemical state shared by many heterogeneous disorders. These may either be acquired, with severe IR being attributable to critical illness or pharmacotherapy, or in rare cases to anti-insulin receptor antibodies[12][13], or to destruction of some or all adipose tissue[14][15]. As these disorders have been thoroughly reviewed[13][16][17][18][19], the focus of this article will be on genetic SSIR.
Over the past 25 years, the pace of genetic discovery in the field of SSIR has been intensifying as the power and reach of genetic technologies has advanced[20][21]. However the genetic landscape of SSIR has not evolved as initially anticipated, for although pathogenic mutations have been identified in several critical molecules directly involved in insulin signalling, the majority of patients have proved to have primary genetic defects affecting adipose tissue development or function, or indeed affecting other cellular processes such as DNA repair, whose link to severe IR has yet to be fully elucidated. We have incorporated these discoveries into a classification scheme for SSIR that distinguishes primary insulin signalling disorders, primary lipodystrophies, and complex, more pleiotropic disorders[11] (Table 1). This has been facilitated by a growing ability to subphenotype SSIR clinically and biochemically prior to genetic testing[22][23][24], and this approach holds further promise for the future in permitting customised therapeutic approaches to each condition.
Table 1.
Classification of Syndromes of Severe Insulin Resistance
| 1. PRIMARY INSULIN SIGNALLING DEFECTS | ||
| A. Generalised (INSR (AR or AD)) |
Discriminating Features
|
|
|
| ||
| B. Partial (AKT2 (AD), TBC1D4 (AD) others to be defined) |
|
|
|
| ||
| 2. SECONDARY TO ADIPOSE TISSUE ABNORMALITIES | ||
|
| ||
| A. Severe Obesity (acquired or genetic; e.g. MC4R (AD), LEP (AR), POMC (AR), SH2B1 (AD)) |
Discriminating Features
|
Selected Features of Genetic Forms
|
|
| ||
B. Lipodystrophy
|
|
|
|
| ||
| 2. AS A FEATURE OF COMPLEX SYNDROMES* | ||
|
| ||
|
|
|
Single cases reported only.
Summary of clinical features in Table 2
In many SSIR, abnormal glucose homeostasis is not the presenting problem, with the first clinical contact often being with paediatricians, endocrinologists, dermatologists, lipidologists, or gynaecologists among others. This review aims first to highlight classic presentations of genetic SSIR, and second to outline the principles of investigation and management that have emerged from specialised clinical practice. SSIR presenting peri- or postpubertally will be considered first, followed by the much rarer, predominantly autosomal recessive SSIRs that usually present in infancy or in prepubertal children.
Severe Insulin Resistance Syndromes Presenting Peri- or Postpubertally
SSIR most commonly present to clinical attention in the peripubertal period, although in many cases severe hyperinsulinaemia is congenital. Usually it is the symptoms arising from ovarian dysfunction which trigger medical consultation. Puberty is often accelerated in those with SSIR, most likely due to the action of hyperinsulinaemia, which exerts synergistic effects with gonadotrophins on the ovary[25][26]. Many SSIR are biochemically and clinically more severe in females, so that index cases in the large majority of affected families are female. To illustrate some of the diagnostic and therapeutic issues that arise in SSIR, several representative case vignettes will now be described:
Case 1: A lean adolescent with hirsutism, polycystic ovaries and acanthosis nigricans
A 15-year-old girl was referred to a specialist severe IR clinic with hirsutism and primary amenorrhoea. Coarse facial hair had first been noticed at 12 years old, and had subsequently become progressively more severe and cosmetically distressing. There was a history of diabetes in a paternal grandmother.
Investigation at initial presentation revealed enlarged ovaries with multiple peripheral cysts (Figure 1A), an elevated LH:FSH ratio and a strikingly high testosterone of 8.2 nmol/l. DHEAS levels were normal, and subsequent ovarian vein sampling confirmed the serum testosterone to be of ovarian origin. Fasting insulin was extremely elevated at 1088 pmol/l (reference < 60 pmol/l) with a concomitant glucose of 3.4 mmol/l. Pharmacological therapy including metformin, rosiglitazone, acarbose and antiandrogenic therapy was used over the next 2 years, with gradual improvement of hyperandrogenaemia and hirsutism (Figure 1B).
Figure 1.

Case 1 A. Ultrasound appearance of the ovary at 12 years old showing multiple circumferential cysts B. Serial testosterone measurements and concurrent therapeutic regimen. Spiro = spironolactone; grey shaded area represents normal female range. C: Axillary acanthosis nigricans and acrochordons
On re-evaluation at 16 years old her body mass index was 21 kg/m2 and height 1.52 m. She had moderately severe acne and severe hirsutism affecting her face, chest and lower abdomen. Puberty was well advanced (Tanner stage 4). Acanthosis nigricans was prominent in both axillae (Figure 1C), with numerous skin tags, and the patient volunteered that her mother had always scrubbed her neck because it was “dirty” as a child. General examination was otherwise normal.
Severe hyperinsulinaemia persisted (fasting plasma insulin 745 pmol/l), however a lipid profile was normal and there was neither biochemical nor radiological evidence of fatty liver. Adiponectin was elevated at 17.7ug/l (2.4-14.9). A genetic defect in the INSR gene, encoding the insulin receptor was diagnosed, and sequencing identified a well known dominant negative heterozygous missense mutation, Pro1178Leu.
Comment
Hyperandrogenism with primary amenorrhoea or oligomenorrhoea is the commonest presentation of severe IR, with acanthosis nigricans and hyperglycaemia often only noticed on subsequent evaluation. Hyperandrogenism is usually particularly severe in the second decade, when pubertal IR interacts with the underlying genetic defect. Testosterone levels may be extremely high (up to 15 nmol/l), and may induce some virilisation. Such hyperandrogenaemia commonly triggers a search for a virilising tumour, however clinical experience suggests that testosterone may usually be lowered to the normal female range by use of GnRH analogues to suppress gonadotrophins, as has been reported in postmenopausal hyperthecosis with severe hyperandrogenism and IR[27][28][29]. Nevertheless bona fide virilising tumours have been described in the context of congenital severe insulin resistance [30] and Semple R.K. (unpublished), raising the possibility that sustained hyperinsulinaemia is a risk factor for development of autonomous androgen-secreting tumours. More formal evaluation of the relative risks and benefits of a “block and replace” strategy in young women with SIR-related hyperandrogenism, using GnRH agonists in conjunction with hormone replacement, has yet to be reported. Nevertheless in our own practice several examples of autonomous virilising tumours arising in the context of many years of severe IR suggest that IR-related hyperandrogenaemia may not be an entirely benign entity, at least in the long term.
Although these ovarian features of IR are generic to nearly all forms of SSIR, other aspects of insulin receptoropathy are distinct: Dyslipidaemia and fatty liver are almost never a feature of proximal insulin signalling disorders[24], while inappropriately normal or high plasma adiponectin levels are also characteristic[23][31][32], and can be used to triage patents with SSIR for INSR gene screening[33]. Whether this apparently benign lipid profile and lack of fatty liver translates into long term protection of patients with INSR mutations from atherosclerosis and fibrotic liver disease despite their severe IR has yet to be determined.
Case 2: The “asymptomatic” male relative
The 46-year-old father of a 15 year old girl recently diagnosed with an INSR mutation was referred for genetic counselling. He insisted he was in excellent health but on detailed enquiry a long-standing history of post-prandial light-headedness and sweating was established. On examination he was lean, with marked acanthosis nigricans in the axillae and nuchal region. Fasting glucose was 7.8 mmol/l and HBA1c was suggestive of occult diabetes at 52 mmol/mol. On prolonged glucose tolerance testing, his blood glucose fell to 1.9 mmol/l at 150 minutes, with concomitant sweating and confusion, and plasma insulin was subsequently found to be severely elevated at 1872 pmol/l (Figure 2). A lipid profile and indices of liver function were normal, and the presence of the INSR mutation was confirmed. A low glycaemic index diet, acarbose, and advice on snacking between meals helped to alleviate post-prandial hypoglycaemia.
Figure 2.
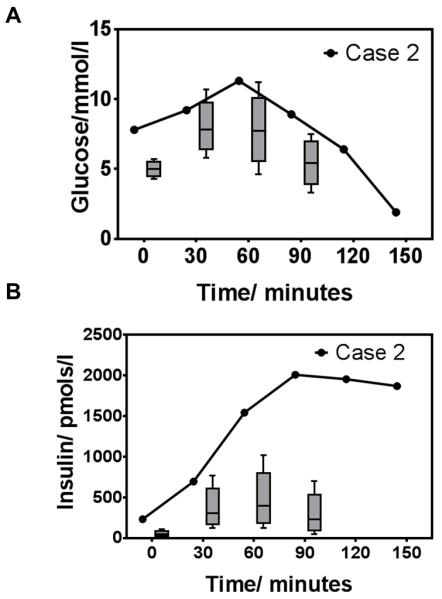
75g oral glucose tolerance test in Case 2. A. Glucose and B. Insulin levels in comparison to age and sex-matched non diabetic controls. Box and whiskers represent 5th and 95th centiles respectively (control data from Professor Nicholas J. Wareham, MRC Epidemiology Unit, Cambridge[67])
Comment
IR is commonly less severe in men than in women, even in the face of the same underlying genetic defect. Compounding this, the lack of the “early warning” signs of oligomenorrhoea and clinical hyperandrogenism means that men are commonly either undiagnosed, or simply diagnosed with “type 2 diabetes” in mid life. SSIRs are thus identified far more frequently in females than males, and after diagnosis of a girl with severe IR, it is very common in autosomal dominant cases later to diagnose occult severe IR and diabetes in her father. As in this case, hypoglycaemia may be a major feature of severe IR, whether due to insulin receptor dysfunction or lipodystrophy, and may be sufficiently severe to cause neuroglycopaenia and loss of consciousness. This may be the presenting feature of a SSIR, although as beta cells decompensate hypoglycaemia lessens and hyperglycaemia comes to dominate. It is not uncommon for detailed work ups for primary hyperinsulinism (tumoral or otherwise) to be undertaken, though the presence of acanthosis nigricans and severe hyperinsulinaemia should point towards a SSIR.
Hypoglycaemia is most commonly symptomatic in the postprandial state, though may commonly also be seen in the fasting state, and may be a feature of any form of severe IR[34][35][36]. Because of this, fasting glucose may be normal or frankly low in the early stages of evaluation of a patient with a SSIR, contrasting with a blood glucose level in the diabetic range after an oral glucose challenge. The mechanism of hypoglycaemia in SSIR is not clear, but a low glycaemic index diet and acarbose, which blunts post-prandial glucose and thus insulin excursion, may be helpful. Comparative studies of these strategies have however, yet to be performed.
Case 3: A muscular young adult with severe dyslipidaemia and abdominal pain
A 21-year-old woman presented with a 12 hour history of severe central abdominal pain radiating to her back. She reported previous similar but milder episodes overt the preceding two to three years. A family history of non-insulin dependent diabetes in her father was noted. She drank minimal quantities of alcohol and exercised infrequently. On inspection she was lean with a BMI 23.5 kg/m2. There was generalised paucity of subcutaneous adipose tissue on the limbs and torso, with evidence of previous breast augmentation surgery. Adipose tissue was increased in the head and neck, giving a somewhat Cushingoid appearance, but there were no other clinical features of Cushing’s syndrome. Indeed far from showing muscle wasting peripherally, she was conspicuously muscular, most strikingly in her calves. There was moderate facial hirsutism and both nuchal and axillary acanthosis nigricans. Examination of the abdomen revealed pronounced epigastric tenderness, some distension, and a palpable liver edge, with very quiet bowel sounds. There were no stigmata of chronic liver disease. On insertion of a urinary catheter, pronounced accumulation of labial adipose tissue, but no cliteromegaly, were noted.
Acute pancreatitis was suspected, but serum amylase levels were normal. Markedly lipaemic serum was reported, with evidence of a systemic inflammatory response, and abdominal CT imaging showed a swollen pancreas consistent with pancreatitis. Fasting plasma triglycerides were later found to be 38mmol/l, with a fasting blood glucose of 8.6 mmol/l and plasma insulin of 2,300pmol/l.
After recovery from this acute episode, further evaluation showed adiponectin and leptin levels to be low at 2.3 mg/l (normal range 2.6-14.9) and 12 ug/l (normal range 14.9-60.2) respectively, while a liver ultrasound study was suggestive of hepatic steatosis. A T1-weighted MRI abdomen depicted marked expansion of visceral adipose stores in stark contrast to lack of abdominal subcutaneous adipose tissue. A clinical diagnosis of familial partial lipodystrophy type 2 was made, and a heterozygous mis-sense mutation, p.Arg482Trp, in the LMNA gene was identified.
Longer term management centred on dietary “off-loading” of adipose tissue through a low fat, energy balanced diet, with adjunctive use of metformin. Fenofibrate was initiated to help control persistent hypertriglyceridaemia, but leptin therapy was not considered initially in view of the base-line serum leptin level of greater than 12 ug/l.
Comment
This case illustrates several classical features of lipodystrophy. There may be a delay in recognising partial lipodystrophy, especially the commonest type described here, associated with LMNA mutations, because adipose tissue in the head and neck, and often in visceral depots is increased (Figure 3). Adiposity of the labia majora may also be markedly expanded and distressing in this condition. In other forms of partial lipodystrophy some patients can have BMIs in the obese range. The adipose topography may have a Cushingoid appearance, and it is common for biochemical screening for Cushing’s syndrome to have taken place prior to definitive diagnosis, although thin skin, easy bruising, striae and muscle atrophy are not features of lipodystrophy.
Figure 3.

Familial Partial Lipodystrophy Type 2 A) Representative patient with FPLD2 at 21 years demonstrating lipoatrophy of limbs and calf hypertrophy with preservation of head and neck adiposity giving rise to a “Cushingoid” adipose topography (reproduced from[41]) B) Sagittal and transverse T1-weighted MRI images of abdomen depicting reduced subcutaneous fat (highlighted in red) and abundant visceral fat (highlighted in yellow) (reproduced from[68])
Although imaging modalities such as MRI, or body composition analysis using, for example, DXA or skin fold thickness, are sometimes used to substantiate a diagnosis of lipodystrophy, it remains predominantly a clinical diagnosis, made by inspection of a patient in their underwear. This is especially true in partial lipodystrophies, where not uncommonly it is disproportion between different adipose depots that is more striking than absolute deficiency in any one depot, manifest as relatively lean limbs with preserved or slightly increased truncal adiposity.
In view of the difficulty that may be encountered in confidently clinically discerning lipodystrophy, being alert to collateral clinical features that may aid diagnosis is of great importance, especially when evaluating the descriptions of other clinicians. The first such feature is unusually severe dyslipidaemia (low HDL cholesterol, high triglyceride), sometimes complicated by eruptive xanthomata and episodes of acute pancreatitis. Notably, serum amylase levels may be normal in the context of hypertriglyceridaemia-induced pancreatitis[37], and so should not be used as the basis for ruling it out. A linked feature of lipodystrophy is unusually severe and early onset fatty liver disease, which may even progress to bridging fibrosis within the first decade of life in severe cases. A “muscular appearance” is often commented on in women with lipodystrophy, which is due to the composite effects of the lack of limb adipose tissue and true muscular hypertrophy[38], the pathogenesis of which has yet to be elucidated.
Dietary management is the most critical element of the management of lipodystrophy, with the aim being to “offload” adipose tissue. This is achieved with low fat, energy balanced or sometimes hypocaloric diets, which may have a dramatic effect on metabolic derangement. Indeed a particular risk to a lipodystrophic patient is the misinterpretation of their lack of adipose tissue as a sign of malnourishment, as well meaning efforts to “build them up” with calorie supplements may have severely adverse metabolic consequences. Thus an experienced dietician is a critical part of the multidisciplinary team approach to lipodystrophy. Measures targeted wholly or in part at weight loss, including orlistat, glucagon-like-peptide 1 analogues and bariatric surgery[39] should be considered in poorly controlled lipodystrophic patients even when, as is likely, they are not obese, however despite the strong rationale for this approach, larger scale evidence for its efficacy is awaited.
Pharmacological therapies in lipodystrophic SSIR should be aimed first at insulin sensitisation. Metformin is best established and should be titrated to the maximum tolerated dose. Thiazolidinediones may sometimes be effective[40][41][42], but the best available evidence suggests that they require residual adipose depots to achieve metabolic benefit[43], and this may occur at the expense of expansion of cosmetically distressing depots such as those in the head and neck. Thus they should be used only with caution. Drugs aimed at reduction of hypertriglyceridaemia (e.g. fibrates) also have an important place in management.
In patients with lipodystrophy and low levels of serum leptin subcutaneous administration of recombinant human leptin may be dramatically beneficial in improving glycaemic control, dyslipidaemia and hepatic lipid accumulation[44][45][46][47][43]. The patients to benefit most from this are those with generalised lipodystrophy and consequently extremely low or undetectable serum leptin levels. Some patients with partial lipodystrophy also benefit, however what the upper limit of pretreatment serum leptin is which predicts a clinically meaningful response to therapy remains to be precisely defined. To date it has been demonstrated that patients with partial lipodystrophy and features of severe metabolic derangement and leptin levels up to around 7 μg/l do benefit from leptin replacement, but further work is required to clarify the role of leptin in patients with higher leptin levels[48]. It is important to appreciate that leptin is not a panacea for the metabolic treatment of lipodystrophy, however, and dietary non compliance will severely attenuate the response to its use.
Leptin has generally been found to be well tolerated. The most serious adverse events associated with its use have been progressive renal disease, and T cell lymphoma, which has been reported in 2 patients[47]. It remains uncertain whether these adverse events are causally related to leptin therapy, as affected patients all had acquired lipodystrophy and pre-existing active autoimmune disease. Nevertheless as replacing leptin in hypoleptinaemic patients has been shown to exert stimulatory effects on T lymphocyte function[49], due caution should be exercised when it is used in the context of active evidence of autoimmune disease.
Case 4: A diabetic patient with acromegalic features
A 50-year-old man with longstanding type 2 diabetes was evaluated for deteriorating glycaemic control. Initial presentation had been at 25 years old with eruptive xanthomata, polyuria and polydipsia. Subsequently he had developed treatment-resistant hypertension, renal dysfunction and hepatic steatosis. Jaw prognathism and large hands were repeatedly noted (Figure 4), but no supporting biochemical evidence for acromegaly was found on multiple evaluations. Medications included 380 units of insulin per day, metformin, fenofibrate, fish oil and six different antihypertensive agents.
Figure 4.

Case 4 showing pseudoacromegalic appearance of A. Face B & C. Hands D. Mandible and E. Tongue. F. Axillary acanthosis nigricans
On inspection acral enlargement, widely spaced teeth, prognathism, and large hands were noted. His Body Mass Index was 33.6kg/m2 with predominantly central adipose distribution and relatively lean, muscular limbs. Mild acanthosis nigricans was present in both axillae. HbA1c was 78 mmol/mol and lipids were well controlled. Serum IGF1 was again within the normal range. A clinical diagnosis of severe IR with pseudoacromegaly was made, and sequencing of the PPARG gene identified a frameshift mutation, confirming a diagnosis of familial partial lipodystrophy type 3 (FPLD3).
Comment
Among the most striking differences between SSIR and insulin deficiency are the range of overgrowth phenomena that are seen in the presence of hyperinsulinaemia. Acanthosis nigricans is the most common of these, giving the appearance of thickened, brown, “velvety” skin in flexures, often associated with skin tags or acrochordons. In the most severe cases it may be seen in perioral, periocular and buccal regions, or even on planar surfaces. Unlike dyslipidaemia and fatty liver, acanthosis nigricans is seen in all known monogenic forms of severe IR. Pseudoacromegalic soft tissue overgrowth is also well described, although seen in only some cases. It is most obvious in settings where there is concomitant lack of adipose tissue, but it is also seen in the context of obesity-related severe IR. It is most likely that severe IR/hyperinsulinaemia drives these growth phenomena by augmenting mitogenic signalling through the IGF1 or other tyrosine kinase growth factor receptors, as there is cross talk between both endocrine and paracrine IGF1 signalling at multiple levels. For example, very high levels of insulin can act directly on the IGF1 receptor, while insulin action may also modulate expression of the IGF1 receptor and bioavailability of IGF1 itself through alterations in binding protein levels. Nevertheless formal proof of this notion, and understanding of the precise mechanism involved, is currently lacking.
Severe Insulin Resistance Syndromes Presenting in Prepubertal Children
Deranged blood glucose levels are often not the first feature of prepubertal SSIR that are recognised, unless hypoglycaemia is severe, as is often seen in infantile insulin receptor defects. Rather, problems with growth, or other syndromic features, tend to be the earliest problems to trigger investigation.
Donohue Syndrome (formerly “leprechaunism”)[50] and Rabson Mendenhall Syndrome[51], whose eponyms reflect their early clinical descriptions many decades before identification of the insulin receptor, represent different parts of the spectrum of abnormality caused by autosomal recessive insulin receptor defects. In Donohue syndrome little or no residual insulin receptor function is found, while in Rabson Mendenhall syndrome receptor function is slightly less severely impaired. Clinical features of both syndromes have been thoroughly reviewed elsewhere[52][51][11][53][54]. In brief, it is often failure to thrive, with a combination of impaired linear growth, underdevelopment of fat and muscle, and overgrowth of soft tissues including skin, hair, teeth and viscera that is first noticed. Although postprandial hyperglycaemia, which may be severe, is the norm, fasting hypoglycaemia is also very common, and may dominate the metabolic picture. Nephrocalcinosis, rectal prolapse, and ventricular hypertrophy are common, and in girls ovarian enlargement may be massive, in some cases complicated by development of tumours. Surprisingly infants with Donohue syndrome are protected from ketoacidosis initially[55], although this eventually supervenes after the first 2 or 3 years. In Donohue syndrome death most commonly occurs during intercurrent infection, while in Rabson Mendenhall syndrome microvascular complications of diabetes in the second decade are the major threat.
Congenital generalised lipodystrophy is also usually apparent in infancy, sometimes being picked up in the early weeks of life by clinicians, but commonly first brought to their attention by mothers concerned about the “wrinkly” and thin appearance of their babies, which may be coupled to muscular enlargement, accelerated growth, and abdominal distension due to hepatic engorgement with triglyceride. Where adequate control is not achieved, congenital generalised lipodystrophy may give rise to some degree of irreversible liver fibrosis within the first one to two decades, and soft tissue overgrowth may also require surgical intervention. In general familial partial lipodystrophy does not become overtly clinically obvious until puberty, when body adipose tissue accretion gathers pace, especially in girls. However few genetically affected individuals have been studied prepubertally, and, at least in the case of FPLD3 due to PPARG mutations, diabetes and dyslipidaemia may present well before ten years old[35][56].
The final group of disorders which are commonly identified in the prepubertal years are those where severe IR is only part of a larger constellation of problems. These include Alström syndrome[57], some forms of primordial dwarfism including osteodysplastic primordial dwarfism of Majewski type 2[58], due to defects in the pericentrosomal protein pericentrin[59], and several forms of progeria or DNA damage repair disorders including Werner[60][61] and Bloom[62] syndromes and mandibuloacral dysplasia[63] (Table 2). The early natural history of the metabolic derangement in these conditions has not been studied in great detail, but cross sectional assessments suggest that severe IR is not congenital, but rather appears in the first few years of life. The metabolic phenotype in each of these conditions resembles that of lipodystrophy rather than that associated with insulin receptor defects. That is, extremely elevated levels of insulin are accompanied often by severe metabolic dyslipidaemia and fatty liver. However while suggestive that the key defect may lie in the adipose tissue in these conditions, this evidence is circumstantial only, and the role of other key insulin responsive tissues remains to be investigated.
Table 2.
Complex Syndromes Associated with Severe Insulin Resistance
| Syndrome | Gene(s) | Inheritance | Core Clinical Features | IR Subphenotype |
|---|---|---|---|---|
| Alström (OMIM #203800) | ALMS1 | AR | Rod-cone dystrophy, deafness, cardiomyopathy, pulmonary/ hepatic/ renal dysfunction | Severe fatty liver and dyslipidaemia |
| MOPDII* (OMIM #210720) | PCNT | AR | Short stature, microcephaly, osteodysplasia, Moya moya vascular anomalies | Severe fatty liver and dyslipidaemia |
| Bloom (OMIM #210900) | RECQ2 | AR | Telangiectases, photosensitivity, short stature, immunodeficiency, increased susceptibility to cancer | Severe fatty liver and dyslipidaemia |
| Werner (OMIM #277700) | RECQL2 LMNA | AR | Premature ageing, osteoporosis, cataracts, atherosclerosis, increased susceptibility to cancer Limb contractures | Severe fatty liver and dyslipidaemia |
| Mandibuloacral dysplasia (OMIM #248370) | LMNA ZMPSTE 24 | AR | Postnatal growth retardation, craniofacial and skeletal abnormalities, cutaneous pigmentation | Severe fatty liver and dyslipidaemia |
MOPDII, Osteodysplastic primordial dwarfism of Majewski type II.
Management of Prepubertal SSIR
The principles guiding management of prepubertal SSIR are broadly similar to those guiding management of disease in older patients. Thus, in generalised lipodystrophy, minimising the dietary load of fat is essential, and careful nutritional follow up is required to balance this with the demands of growth. As in older patients with absent adipose tissue leptin replacement may have a major beneficial effect, and indeed there is a strong argument that early treatment from infancy may protect children with generalised lipodystrophy from accruing complications of poor metabolic control. However case literature is only now beginning to emerge in this group[64].
In infants and children with SSIR due to insulin signalling defects, recombinant human IGF1 has been widely used and appears to improve glycaemia and perhaps survival in some cases[65][66]. It has variously been proposed to exert benefit through insulin mimetic activity, through action as a trophic factor for pancreatic beta cells, or by enhancing insulin sensitivity through postreceptor cross talk between insulin and IGF-I signalling pathways. Nevertheless its dominant mode of action, optimal dosing, and precise clinical indications for its use remain unclear.
Acknowledgements
We are grateful to Professor Steve O’Rahilly, Dr David Savage and colleagues in the UK National Severe Insulin Resistance Service for discussion and review of this manuscript.
Funding
VERP and RKS are funded by research fellowships from the Wellcome Trust (Clinical Research Training Fellowship 097721/Z/11/Z and Senior Research Fellowship in Clinical Science 098498/Z/12/Z respectively)
Footnotes
Declaration of interest
No interests to declare
References
- 1.Souren NY, Paulussen ADC, Loos RJF, Gielen M, Beunen G, Fagard R, Derom C, Vlietinck R, Zeegers MP. Anthropometry, carbohydrate and lipid metabolism in the East Flanders Prospective Twin Survey: heritabilities. Diabetologia. 2007;50(10):2107–16. doi: 10.1007/s00125-007-0784-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freeman MS, Mansfield MW, Barrett JH, Grant PJ. Heritability of features of the insulin resistance syndrome in a community-based study of healthy families. Diabetic Medicine: a Journal of the British Diabetic Association. 2002;19(12):994–9. doi: 10.1046/j.1464-5491.2002.00843.x. [DOI] [PubMed] [Google Scholar]
- 3.Mills GW, Avery PJ, McCarthy MI, Hattersley AT, Levy JC, Hitman GA, Sampson M, Walker M. Heritability estimates for beta cell function and features of the insulin resistance syndrome in UK families with an increased susceptibility to type 2 diabetes. Diabetologia. 2004;47(4):732–8. doi: 10.1007/s00125-004-1338-2. [DOI] [PubMed] [Google Scholar]
- 4.Falchi M, Wilson SG, Paximadas D, Swaminathan R, Spector TD. Quantitative linkage analysis for pancreatic B-cell function and insulin resistance in a large twin cohort. Diabetes. 2008;57(4):1120–4. doi: 10.2337/db07-0708. [DOI] [PubMed] [Google Scholar]
- 5.Hanson RL, Imperatore G, Bennett PH, Knowler WC. Components of the “metabolic syndrome” and incidence of type 2 diabetes. Diabetes. 2002;51(10):3120–7. doi: 10.2337/diabetes.51.10.3120. [DOI] [PubMed] [Google Scholar]
- 6.Utzschneider KM, Kahn SE. Review: The role of insulin resistance in nonalcoholic fatty liver disease. The Journal of Clinical Endocrinology and Metabolism. 2006;91(12):4753–61. doi: 10.1210/jc.2006-0587. [DOI] [PubMed] [Google Scholar]
- 7.Siddique A, Kowdley KV. Insulin resistance and other metabolic risk factors in the pathogenesis of hepatocellular carcinoma. Clinics in Liver Disease. 2011;15(2):281–96. vii–x. doi: 10.1016/j.cld.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Resnick HE, Jones K, Ruotolo G, Jain AK, Henderson J, Lu W, Howard BV. Insulin resistance, the metabolic syndrome, and risk of incident cardiovascular disease in nondiabetic american indians: the Strong Heart Study. Diabetes Care. 2003;26(3):861–7. doi: 10.2337/diacare.26.3.861. [DOI] [PubMed] [Google Scholar]
- 9.Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes. 1989;38(9):1165–74. doi: 10.2337/diab.38.9.1165. [DOI] [PubMed] [Google Scholar]
- 10.Pollack MN. Insulin, insulin-like growth factors, insulin resistance, and neoplasia. The American Journal of Clinical Nutrition. 2007;86(3):s820–2. doi: 10.1093/ajcn/86.3.820S. [DOI] [PubMed] [Google Scholar]
- 11.Semple RK, Savage DB, Cochran EK, Gorden P, O’Rahilly S. Genetic syndromes of severe insulin resistance. Endocrine Reviews. 2011;32(4):498–514. doi: 10.1210/er.2010-0020. [DOI] [PubMed] [Google Scholar]
- 12.Flier JS, Kahn CR, Jarrett DB, Roth J. Characterization of antibodies to the insulin receptor: a cause of insulin-resistant diabetes in man. The Journal of Clinical Investigation. 1976;58(6):1442–9. doi: 10.1172/JCI108600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arioglu E, Andewelt A, Diabo C, Bell M, Taylor SI, Gorden P. Clinical course of the syndrome of autoantibodies to the insulin receptor (type B insulin resistance): a 28-year perspective. Medicine. 2002;81(2):87–100. doi: 10.1097/00005792-200203000-00001. [DOI] [PubMed] [Google Scholar]
- 14.Sissons JG, West RJ, Fallows J, Williams DG, Boucher BJ, Amos N, Peters DK. The complement abnormalities of lipodystrophy. The New England Journal of Medicine. 1976;294(9):461–5. doi: 10.1056/NEJM197602262940902. [DOI] [PubMed] [Google Scholar]
- 15.Savage DB, Semple RK, Clatworthy MR, Lyons PA, Morgan BP, Cochran EK, Gorden P, Raymond-Barker P, Murgatroyd PR, Adams C, Scobie I, Mufti GJ, Alexander GJM, Thiru S, Murano I, Cinti S, Chaudhry AN, Smith KGC, O’Rahilly S. Complement abnormalities in acquired lipodystrophy revisited. The Journal of Clinical Endocrinology and Metabolism. 2009;94(1):10–6. doi: 10.1210/jc.2008-1703. [DOI] [PubMed] [Google Scholar]
- 16.Misra A, Peethambaram A, Garg A. Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: report of 35 cases and review of the literature. Medicine. 2004;83(1):18–34. doi: 10.1097/01.md.0000111061.69212.59. [DOI] [PubMed] [Google Scholar]
- 17.Misra A, Garg A. Clinical features and metabolic derangements in acquired generalized lipodystrophy: case reports and review of the literature. Medicine. 2003;82(2):129–46. doi: 10.1097/00005792-200303000-00007. [DOI] [PubMed] [Google Scholar]
- 18.Garg A. Acquired and inherited lipodystrophies. The New England Journal of Medicine. 2004;350(12):1220–34. doi: 10.1056/NEJMra025261. [DOI] [PubMed] [Google Scholar]
- 19.Larsen J, Goldner W. Approach to the hospitalized patient with severe insulin resistance. The Journal of Clinical Endocrinology and Metabolism. 2011;96(9):2652–62. doi: 10.1210/jc.2011-0255. [DOI] [PubMed] [Google Scholar]
- 20.Raffan E, Semple RK. Next generation sequencing--implications for clinical practice. British Medical Bulletin. 2011;99:53–71. doi: 10.1093/bmb/ldr029. [DOI] [PubMed] [Google Scholar]
- 21.Parker VER, Savage DB, O’Rahilly S, Semple RK. Mechanistic insights into insulin resistance in the genetic era. Diabetic Medicine : a Journal of the British Diabetic Association. 2011;28(12):1476–86. doi: 10.1111/j.1464-5491.2011.03463.x. [DOI] [PubMed] [Google Scholar]
- 22.Savage DB, Semple RK, Chatterjee VKK, Wales JKH, Ross RJM, O’Rahilly S. A clinical approach to severe insulin resistance. Endocrine Development. 2007;11:122–32. doi: 10.1159/000111067. [DOI] [PubMed] [Google Scholar]
- 23.Semple RK, Soos MA, Luan J, Mitchell CS, Wilson JC, Gurnell M, Cochran EK, Gorden P, Chatterjee VKK, Wareham NJ, O’Rahilly S. Elevated plasma adiponectin in humans with genetically defective insulin receptors. The Journal of Clinical Endocrinology and Metabolism. 2006;91(8):3219–23. doi: 10.1210/jc.2006-0166. [DOI] [PubMed] [Google Scholar]
- 24.Semple RK, Sleigh A, Murgatroyd PR, Adams CA, Bluck L, Jackson S, Vottero A, Kanabar D, Charlton-Menys V, Durrington P, Soos MA, Carpenter TA, Lomas DJ, Cochran EK, Gorden P, O’Rahilly S, Savage DB. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. The Journal of Clinical Investigation. 2009;119(2):315–22. doi: 10.1172/JCI37432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poretsky L, Cataldo NA, Rosenwaks Z, Giudice LC. The insulin-related ovarian regulatory system in health and disease. Endocrine Reviews. 1999;20(4):535–82. doi: 10.1210/edrv.20.4.0374. [DOI] [PubMed] [Google Scholar]
- 26.Nestler JE, Jakubowicz DJ. Decreases in ovarian cytochrome P450c17 alpha activity and serum free testosterone after reduction of insulin secretion in polycystic ovary syndrome. The New England Journal of Medicine. 1996;335(9):617–23. doi: 10.1056/NEJM199608293350902. [DOI] [PubMed] [Google Scholar]
- 27.Koroscil TM, Harter SB, Ouweleen J, Blauer KL. Use of a gonadotropin-releasing hormone agonist in the evaluation of postmenopausal virilization due to ovarian hyperthecosis. A case report. The Journal of Reproductive Medicine. 1996;41(4):259–62. [PubMed] [Google Scholar]
- 28.Pascale MM, Pugeat M, Roberts M, Rousset H, Déchaud H, Dutrieux-Berger N, Tourniaire J. Androgen suppressive effect of GnRH agonist in ovarian hyperthecosis and virilizing tumours. Clinical Endocrinology. 1994;41(5):571–6. doi: 10.1111/j.1365-2265.1994.tb01820.x. [DOI] [PubMed] [Google Scholar]
- 29.Chico A, García JL, Matías-Guiu X, Webb SM, Rodríguez J, Prat J, Calaf J. A gonadotrophin dependent stromal luteoma: a rare cause of post-menopausal virilization. Clinical Endocrinology. 1995;43(5):645–9. doi: 10.1111/j.1365-2265.1995.tb02931.x. [DOI] [PubMed] [Google Scholar]
- 30.Brisigotti M, Fabbretti G, Pesce F, Gatti R, Cohen A, Parenti G, Callea F. Congenital bilateral juvenile granulosa cell tumor of the ovary in leprechaunism: a case report. Pediatric Pathology / Affiliated with the International Paediatric Pathology Association. 13(5):549–58. doi: 10.3109/15513819309048242. [DOI] [PubMed] [Google Scholar]
- 31.Antuna-Puente B, Boutet E, Vigouroux C, Lascols O, Slama L, Caron-Debarle M, Khallouf E, Lévy-Marchal C, Capeau J, Bastard JP, Magré J. Higher adiponectin levels in patients with Berardinelli-Seip congenital lipodystrophy due to seipin as compared with 1-acylglycerol-3-phosphate-o-acyltransferase-2 deficiency. The Journal of Clinical Endocrinology and Metabolism. 2010;95(3):1463–8. doi: 10.1210/jc.2009-1824. [DOI] [PubMed] [Google Scholar]
- 32.Hattori Y, Hirama N, Suzuki K, Hattori S, Kasai K. Elevated plasma adiponectin and leptin levels in sisters with genetically defective insulin receptors. Diabetes Care. 2007;30(11):e109. doi: 10.2337/dc07-1342. [DOI] [PubMed] [Google Scholar]
- 33.Semple RK, Cochran EK, Soos MA, Burling KA, Savage DB, Gorden P, O’Rahilly S. Plasma adiponectin as a marker of insulin receptor dysfunction: clinical utility in severe insulin resistance. Diabetes Care. 2008;31(5):977–9. doi: 10.2337/dc07-2194. [DOI] [PubMed] [Google Scholar]
- 34.Højlund K, Hansen T, Lajer M, Henriksen JE, Levin K, Lindholm J, Pedersen O, Beck-Nielsen H. A novel syndrome of autosomal-dominant hyperinsulinemic hypoglycemia linked to a mutation in the human insulin receptor gene. Diabetes. 2004;53(6):1592–8. doi: 10.2337/diabetes.53.6.1592. [DOI] [PubMed] [Google Scholar]
- 35.Agostini M, Schoenmakers E, Mitchell C, Szatmari I, Savage D, Smith A, Rajanayagam O, Semple R, Luan J, Bath L, Zalin A, Labib M, Kumar S, Simpson H, Blom D, Marais D, Schwabe J, Barroso I, Trembath R, et al. Non-DNA binding, dominant-negative, human PPARgamma mutations cause lipodystrophic insulin resistance. Cell Metabolism. 2006;4(4):303–11. doi: 10.1016/j.cmet.2006.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, Soos MA, Murgatroyd PR, Williams RM, Acerini CL, Dunger DB, Barford D, Umpleby AM, Wareham NJ, Davies HA, Schafer AJ, Stoffel M, O’Rahilly S, Barroso I. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304(5675):1325–8. doi: 10.1126/science.1096706. (New York, NY) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma P, Lim S, James D, Orchard RT, Horne M, Seymour CA. Pancreatitis may occur with a normal amylase concentration in hypertriglyceridaemia. BMJ. 1996;313(7067):1265–1265. doi: 10.1136/bmj.313.7067.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ji H, Weatherall P, Adams-Huet B, Garg A. Increased Skeletal Muscle Volume in Women with Familial Partial Lipodystrophy, Dunnigan Variety. The Journal of Clinical Endocrinology and Metabolism. 2013 doi: 10.1210/jc.2013-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ciudin A, Baena-Fustegueras JA, Fort JM, Encabo G, Mesa J, Lecube A. Successful treatment for the Dunnigan-type familial partial lipodystrophy with Roux-en-Y gastric bypass. Clinical Endocrinology. 2011;75(3):403–4. doi: 10.1111/j.1365-2265.2011.04057.x. [DOI] [PubMed] [Google Scholar]
- 40.Owen KR, Donohoe M, Ellard S, Hattersley AT. Response to treatment with rosiglitazone in familial partial lipodystrophy due to a mutation in the LMNA gene. Diabetic Medicine. 2003;20(10):823–827. doi: 10.1046/j.1464-5491.2003.01034.x. [DOI] [PubMed] [Google Scholar]
- 41.Gambineri A, Semple RK, Forlani G, Genghini S, Grassi I, Hyden CSS, Pagotto U, O’Rahilly S, Pasquali R. Monogenic polycystic ovary syndrome due to a mutation in the lamin A/C gene is sensitive to thiazolidinediones but not to metformin. European Journal of Endocrinology / European Federation of Endocrine Societies. 2008;159(3):347–53. doi: 10.1530/EJE-08-0272. [DOI] [PubMed] [Google Scholar]
- 42.Sleilati GG, Leff T, Bonnett JW, Hegele RA. Efficacy and Safety of Pioglitazone in Treatment of a Patient with an Atypical Partial Lipodystrophy Syndrome. Endocrine Practice. 2007;13(6) doi: 10.4158/EP.13.6.656. [DOI] [PubMed] [Google Scholar]
- 43.Simha V, Rao S, Garg A. Prolonged thiazolidinedione therapy does not reverse fat loss in patients with familial partial lipodystrophy, Dunnigan variety. Diabetes, Obesity & Metabolism. 2008;10(12):1275–6. doi: 10.1111/j.1463-1326.2008.00978.x. [DOI] [PubMed] [Google Scholar]
- 44.Javor ED, Cochran EK, Musso C, Young JR, Depaoli AM, Gorden P. Long-term efficacy of leptin replacement in patients with generalized lipodystrophy. Diabetes. 2005;54(7):1994–2002. doi: 10.2337/diabetes.54.7.1994. [DOI] [PubMed] [Google Scholar]
- 45.Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, Cline GW, DePaoli AM, Taylor SI, Gorden P, Shulman GI. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. The Journal of Clinical Investigation. 2002;109(10):1345–50. doi: 10.1172/JCI15001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chong AY, Lupsa BC, Cochran EK, Gorden P. Efficacy of leptin therapy in the different forms of human lipodystrophy. Diabetologia. 2010;53(1):27–35. doi: 10.1007/s00125-009-1502-9. [DOI] [PubMed] [Google Scholar]
- 47.Chan JL, Lutz K, Cochran E, Huang W, Peters Y, Weyer C, Gorden P. Clinical effects of long-term metreleptin treatment in patients with lipodystrophy. Endocrine Practice : Official Journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 17(6):922–32. doi: 10.4158/EP11229.OR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simha V, Subramanyam L, Szczepaniak L, Quittner C, Adams-Huet B, Snell P, Garg A. Comparison of efficacy and safety of leptin replacement therapy in moderately and severely hypoleptinemic patients with familial partial lipodystrophy of the Dunnigan variety. The Journal of Clinical Endocrinology and Metabolism. 2012;97(3):785–92. doi: 10.1210/jc.2011-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matarese G, La Rocca C, Moon HS, Huh JY, Brinkoetter MT, Chou S, Perna F, Greco D, Kilim HP, Gao C, Arampatzi K, Wang Z, Mantzoros CS. Selective capacity of metreleptin administration to reconstitute CD4+ T-cell number in females with acquired hypoleptinemia. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(9):E818–27. doi: 10.1073/pnas.1214554110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Donohue WL, Uchida I. Leprechaunism: a euphemism for a rare familial disorder. The Journal of Pediatrics. 1954;45(5):505–19. doi: 10.1016/s0022-3476(54)80113-2. [DOI] [PubMed] [Google Scholar]
- 51.Rabson SM, Mendenhall EN. Familial hypertrophy of pineal body, hyperplasia of adrenal cortex and diabetes mellitus; report of 3 cases. American Journal of Clinical Pathology. 1956;26(3):283–90. doi: 10.1093/ajcp/26.3.283. [DOI] [PubMed] [Google Scholar]
- 52.Musso C, Cochran E, Moran SA, Skarulis MC, Oral EA, Taylor S, Gorden P. Clinical course of genetic diseases of the insulin receptor (type A and Rabson-Mendenhall syndromes): a 30-year prospective. Medicine. 2004;83(4):209–22. doi: 10.1097/01.md.0000133625.73570.54. [DOI] [PubMed] [Google Scholar]
- 53.Accili D, Cama A, Barbetti F, Kadowaki H, Kadowaki T, Taylor SI. Insulin resistance due to mutations of the insulin receptor gene: an overview. Journal of Endocrinological Investigation. 1992;15(11):857–64. doi: 10.1007/BF03348820. [DOI] [PubMed] [Google Scholar]
- 54.Semple RK, Williams RM, Dunger DB. What is the best management strategy for patients with severe insulin resistance? Clinical Endocrinology. 2010;73(3):286–90. doi: 10.1111/j.1365-2265.2010.03810.x. [DOI] [PubMed] [Google Scholar]
- 55.Ogilvy-Stuart AL, Soos MA, Hands SJ, Anthony MY, Dunger DB, O’Rahilly S. Hypoglycemia and resistance to ketoacidosis in a subject without functional insulin receptors. The Journal of Clinical Endocrinology and Metabolism. 2001;86(7):3319–26. doi: 10.1210/jcem.86.7.7631. [DOI] [PubMed] [Google Scholar]
- 56.Madhra M, Noh RM, Zammitt NN, Patrick AW, Love CDB. A complicated pregnancy in a patient with lipodystrophic diabetes attributable to a peroxisome proliferator-activated receptor gamma (PPARG) mutation. Diabetic Medicine: a Journal of the British Diabetic Association. 2012;29(10):e398–401. doi: 10.1111/j.1464-5491.2012.03742.x. [DOI] [PubMed] [Google Scholar]
- 57.Marshall JD, Paisey RB, Carey C, Macdermott S. Alström Syndrome. 2003 [Google Scholar]
- 58.Huang-Doran I, Bicknell LS, Finucane FM, Rocha N, Porter KM, Tung YCL, Szekeres F, Krook A, Nolan JJ, O’Driscoll M, Bober M, O’Rahilly S, Jackson AP, Semple RK. Genetic defects in human pericentrin are associated with severe insulin resistance and diabetes. Diabetes. 2011;60(3):925–35. doi: 10.2337/db10-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rauch A, Thiel CT, Schindler D, Wick U, Crow YJ, Ekici AB, Van Essen AJ, Goecke TO, Al-Gazali L, Chrzanowska KH, Zweier C, Brunner HG, Becker K, Curry CJ, Dallapiccola B, Devriendt K, Dörfler A, Kinning E, Megarbane A, et al. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. 2008;319(5864):816–9. doi: 10.1126/science.1151174. (New York, NY) [DOI] [PubMed] [Google Scholar]
- 60.Yamada K, Ikegami H, Yoneda H, Miki T, Ogihara T. All patients with Werner’s syndrome are insulin resistant, but only those who also have impaired insulin secretion develop overt diabetes. Diabetes Care. 1999;22(12):2094–5. doi: 10.2337/diacare.22.12.2094. [DOI] [PubMed] [Google Scholar]
- 61.Imura H, Nakao Y, Kuzuya H, Okamoto M, Yamada K. Clinical, endocrine and metabolic aspects of the Werner syndrome compared with those of normal aging. Advances in Experimental Medicine and Biology. 1985;190:171–85. doi: 10.1007/978-1-4684-7853-2_6. [DOI] [PubMed] [Google Scholar]
- 62.Diaz A, Vogiatzi MG, Sanz MM, German J. Evaluation of short stature, carbohydrate metabolism and other endocrinopathies in Bloom’s syndrome. Hormone Research. 2006;66(3):111–7. doi: 10.1159/000093826. [DOI] [PubMed] [Google Scholar]
- 63.Jacob KN, Garg A. Laminopathies: multisystem dystrophy syndromes. Molecular Genetics and Metabolism. 2006;87(4):289–302. doi: 10.1016/j.ymgme.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 64.Beltrand J, Beregszaszi M, Chevenne D, Sebag G, De Kerdanet M, Huet F, Polak M, Tubiana-Rufi N, Lacombe D, De Paoli AM, Levy-Marchal C. Metabolic correction induced by leptin replacement treatment in young children with Berardinelli-Seip congenital lipoatrophy. Pediatrics. 2007;120(2):e291–6. doi: 10.1542/peds.2006-3165. [DOI] [PubMed] [Google Scholar]
- 65.Regan FM, Williams RM, McDonald A, Umpleby AM, Acerini CL, O’Rahilly S, Hovorka R, Semple RK, Dunger DB. Treatment with recombinant human insulin-like growth factor (rhIGF)-I/rhIGF binding protein-3 complex improves metabolic control in subjects with severe insulin resistance. The Journal of Clinical Endocrinology and Metabolism. 2010;95(5):2113–22. doi: 10.1210/jc.2009-2088. [DOI] [PubMed] [Google Scholar]
- 66.McDonald A, Williams RM, Regan FM, Semple RK, Dunger DB. IGF-I treatment of insulin resistance. European Journal of Endocrinology / European Federation of Endocrine Societies. 2007;157(Suppl):S51–6. doi: 10.1530/EJE-07-0271. [DOI] [PubMed] [Google Scholar]
- 67.Wareham NJ, Byrne CD, Williams R, Day NE, Hales CN. Fasting proinsulin concentrations predict the development of type 2 diabetes. Diabetes Care. 1999;22(2):262–70. doi: 10.2337/diacare.22.2.262. [DOI] [PubMed] [Google Scholar]
- 68.Huang-Doran I, Sleigh A, Rochford JJ, O’Rahilly S, Savage DB. Lipodystrophy: metabolic insights from a rare disorder. The Journal of Endocrinology. 2010;207(3):245–55. doi: 10.1677/JOE-10-0272. [DOI] [PubMed] [Google Scholar]