

ABSTRACT
Proper kinetochore recruitment and regulation of dynein and the Mad1–Mad2 complex requires the Rod–Zw10–Zwilch (RZZ) complex. Here, we describe rodZ3, a maternal-effect Drosophila mutation changing a single residue in the Rough Deal (Rod) subunit of RZZ. Although the RZZ complex containing this altered subunit (denoted RZ3ZZ) is present in early syncytial stage embryos laid by homozygous rodZ3 mothers, it is not recruited to kinetochores. Consequently, the embryos have no spindle assembly checkpoint (SAC), and syncytial mitoses are profoundly perturbed. The polar body (residual meiotic products) cannot remain in its SAC-dependent metaphase-like state, and decondenses into chromatin. In neuroblasts of homozygous rodZ3 larvae, RZ3ZZ recruitment is only partially reduced, the SAC is functional and mitosis is relatively normal. RZ3ZZ nevertheless behaves abnormally: it does not further accumulate on kinetochores when microtubules are depolymerized; it reduces the rate of Mad1 recruitment; and it dominantly interferes with the dynein-mediated streaming of RZZ from attached kinetochores. These results suggest that the mutated residue of rodZ3 is required for normal RZZ kinetochore recruitment and function and, moreover, that the RZZ recruitment pathway might differ in syncytial stage embryos and post-embryonic somatic cells.
KEY WORDS: RZZ, Polar body, Spindle assembly checkpoint, Mad1, Syncytial embryo, Kinetochore
INTRODUCTION
Cell division is driven by oscillations in the activity of cyclin-dependent kinases (Cdks) and their associations with regulatory cyclin subunits. Mitotic entry is driven by high Cdk1–Cyclin-B activity, and mitotic exit is dependent on mechanisms that inactivate this complex and dephosphorylate its targets (reviewed in O'Farrell, 2001; Lindqvist et al., 2009; Mochida and Hunt, 2012). The initiation of mitotic exit at the metaphase-anaphase transition is under the control of the spindle assembly checkpoint (SAC), which indirectly regulates the proteolytic degradation of Cyclin B by controlling its ubquitinylation by the E3 ubiquitin ligase known as the anaphase promoting complex (APC/C) (reviewed in Musacchio, 2011; Lara-Gonzalez et al., 2012; Jia et al., 2013). Among the key SAC proteins in this process are Mad1 and Mad2, which form a complex at unattached kinetochores and help generate an inhibitor of the APC/C. Upon proper kinetochore attachment to the spindle, several SAC components, including Mad1, Mad2 and the Rough Deal (Rod)–Zw10–Zwilch (RZZ) complex are removed from the kinetochore by a dynein-dependent process (Howell et al., 2000; Hoffman et al., 2001; Howell et al., 2001; Wojcik et al., 2001; Basto et al., 2004), thus dismantling the SAC apparatus and inactivating the source of the APC/C inhibitor.
RZZ is an important component of the outer kinetochore (Karess, 2005). It is a determinant of Mad1–Mad2 kinetochore levels, as it promotes both Mad1–Mad2 recruitment (Buffin et al., 2005; Kops et al., 2005) and recruitment of dynein and its associated regulatory proteins (Starr et al., 1998; Griffis et al., 2007; Chan et al., 2009). RZZ is also involved in regulating kinetochore–microtubule attachments (Gassmann et al., 2008; Gassmann et al., 2010; Cheerambathur et al., 2013), although how it performs these functions, and how it is itself recruited to kinetochores is poorly understood.
In the early division cycles of Drosophila embryogenesis, nuclei divide synchronously in a syncytium, using a streamlined cell cycle in which S- and M-phase oscillate, driven by stockpiles of maternal mRNAs and proteins (reviewed in Foe et al., 1993; Lee and Orr-Weaver, 2003). During these syncytial cycles, the unfertilized nuclear products of female meiosis remain in M-phase at the periphery of the embryo. These nuclear products, referred to as polar bodies, coalesce to form a ‘starburst’ of condensed chromosomes and maintain this state until they are culled from the embryo during cellularization.
Defective polar body M-phase maintenance has been reported in mutants with either reduced Cdk1 activity or misregulated levels of Cyclin B. For example, the Pan Gu (Png) kinase complex promotes the translation of Cyclin B during female meiosis and syncytial embryogenesis. Females mutant for the png kinase or its regulatory subunits lay eggs with abnormally low Cyclin B levels. As a result, polar bodies do not remain in M-phase but instead re-enter interphase and undergo unregulated rounds of DNA replication, resulting in polyploid interphase-like polar bodies (Fenger et al., 2000; Lee et al., 2001). The SAC, which also regulates levels of Cyclin B, is another proposed mechanism by which polar body M-phase is maintained. Indeed, eggs laid by females homozygous for certain viable mutations of the SAC genes Mps1 (also known as ald in Drosophila) and BubR1 present similar polar body defects (Fischer et al., 2004; Pérez-Mongiovi et al., 2005).
In a screen for new cell-cycle regulators specifically required during the syncytial stages of embryogenesis, we identified a novel, maternal-effect allele of the rough deal (rod) gene, called here rodZ3. This mutation only slightly perturbs RZZ activity in homozygotes, which are viable and have relatively normal mitoses. However, in eggs laid by rodZ3 mothers, RZZ is incapable of kinetochore recruitment. As a result, such embryos present decondensed polar bodies, profoundly perturbed syncytial mitoses, and a non-functional SAC. The phenotype of this mutation suggests that aspects of kinetochore assembly may differ in maternal and zygotic Drosophila mitosis.
RESULTS
rodZ3 is a new maternal-effect lethal allele of rod with abnormal syncytial mitoses and defective polar body M-phase maintenance
In a screen for new genes in Drosophila affecting polar body maintenance, we identified a novel allele of rod in a large maternal-effect lethal collection (Koundakjian et al., 2004). Flies homozygous for this allele, called rodZ3, are viable and appear normal. However, they are completely sterile; the females lay eggs that fail to hatch (Table 1). Examination of 3–5-hour-old eggs revealed a number of early developmental defects. None of the embryos had reached gastrulation and most presented highly abnormal distribution of blastoderm nuclei and unusually large internal nuclei, suggesting problems with the syncytial mitoses (Table 1; Fig. 1A; supplementary material Fig. S1). In contrast, nearly all the wild-type (WT) embryos at this stage had either begun gastrulating (73%) or were cellularizing (22%) (Table 1).
Table 1. Maternal-effect embryo phenotype of rodZ3.

Fig. 1.

Embryos laid by rodZ3 mothers are defective in syncytial mitosis, polar body maintenance and the SAC. (A) Representative images of DNA-stained early (left) and late (right) syncytial embryos from wild-type (WT) and rodZ3 females. rodZ3-derived embryos contain large decondensed polar bodies (left). Also note the asynchrony of mitotic stages and the irregular distribution of nuclei in the rodZ3-derived embryos (right). Scale bars: 50 µm. Insets, larger images of polar bodies (marked by arrows). (B) Representative images of prophase and metaphase figures from WT and rodZ3-derived syncytial embryos stained for tubulin (green), centrosomin (red) and DNA (blue). rodZ3 mitotic figures have abnormal centrosome attachments. Broad acentrosomal and multipolar spindles are common. Scale bar: 10 µm. (C) Details of polar bodies from WT and rodZ3-derived syncytial embryos stained for PH3 and DNA. rodZ3 polar bodies are either PH3-positive (partially decondensed) or PH3-negative (fully decondensed). Scale bar: 10 µm. (D) The SAC is not functional in rodZ3-derived embryos. Relative change in the fraction of mitotic syncytial embryos after 30 min incubation in colchicine (compared with untreated, set at 1) for the indicated genotypes. rodZ3-derived embryos, like those derived from Mad2 null mothers (Mad2P, negative control), show no significant increase after treatment. *P<0.005; **P<0.0001 (Fisher's exact test; n = 80 embryos, except rescue-derived embryos where n = 140).
In younger (0–2 h) syncytial stage rodZ3-derived embryos, the nuclei were frequently unevenly spaced and asynchronously cycling (Fig. 1A,B; Table 1). The majority of the mitotic figures in these embryos were abnormal, including multipolar spindles with multiple centrosomes and poorly focused spindles lacking one or both centrosomes (Fig. 1B). In fact, the overall phenotype of these embryos is similar to that reported for germline clones of zw10-null mutants (Williams and Goldberg, 1994).
In nearly all the eggs derived from rodZ3 females, the polar body formed a large indistinct mass of interphase-like decondensed chromatin instead of the starburst configuration of condensed chromosomes found in WT embryos (Table 2; Fig. 1A,C). The increased size of these polar bodies and the intensity of their DNA stain compared to WT suggest that they have an increase in DNA content, presumably owing to additional rounds of DNA replication. Expression of the rod+tC3L9 transgene, which contains the WT rod genomic region, in the rodZ3 background rescued this polar body defect, the embryonic lethality (Table 1) and the mitotic phenotypes described below, confirming that the mutation in rod was responsible for the phenotypes.
Table 2. Polar body phenotype of rodZ3.
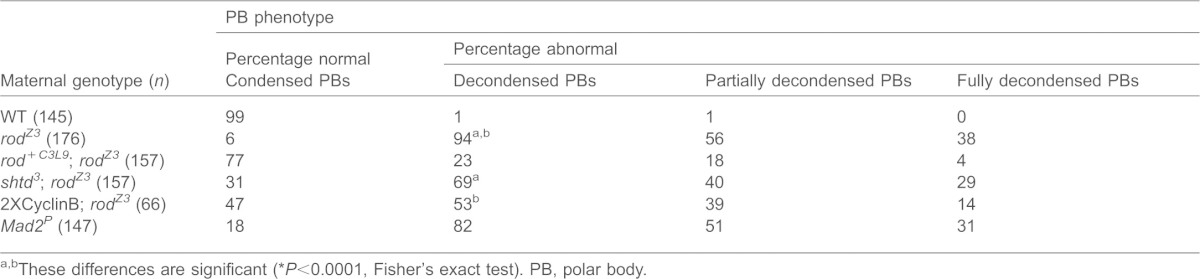
These differences are significant (*P<0.0001, Fisher's exact test). PB, polar body.
Immunostaining rodZ3-derived embryos for phosphorylated Histone H3 (PH3), a marker of M-phase chromosome condensation, distinguished two classes of decondensed polar bodies. Roughly 40% of the polar bodies were PH3-negative with rounded, interphase-like chromatin (Fig. 1C; Table 2); the rest were PH3-positive with irregularly-shaped and partially decondensed chromatin. WT polar bodies were PH3-positive as has been reported by others (Fischer et al., 2004; Pérez-Mongiovi et al., 2005). The irregularly-shaped, semi-decondensed and PH3-positive polar bodies of rodZ3-derived embryos also labeled with Spc25–RFP (a marker of the outer kinetochore, present only during M-phase), indicating that kinetochores were intact. Interestingly, these polar bodies contained more than the expected 12 kinetochore signals (Fig. 3A), suggesting that they had at least partially replicated their DNA and returned to an M-phase-like state. The more rounded interphase-like polar bodies lacked Spc25–RFP (Fig. 3A), consistent with the interpretation that they were in interphase. These results suggest that the polar bodies of rodZ3-derived embryos might be cycling in and out of M-phase, as has been proposed for other mutants (Fischer et al., 2004; Pérez-Mongiovi et al., 2005).
Fig. 3.

RZZ kinetochore recruitment is absent in rodZ3-derived embryos but present, although reduced, in homozygous rodZ3 neuroblasts. (A) RZZ does not localize to polar bodies in rodZ3-derived embryos. Polar bodies from fixed WT and rodZ3-derived syncytial embryos expressing Spc25–RFP (a kinetochore marker, red), GFP–Zw10 (an RZZ component, green) and stained for DNA (blue). Scale bar: 10 µm. (n = 12 polar bodies examined, all negative.) (B) RZZ does not localize to metaphase kinetochores or spindle fibers in rodZ3-derived embryos. Scale bar: 10 µm. (n = 30 metaphases, all negative, from 12 embryos). (C) RZZ is recruited to kinetochores in rodZ3 larval neuroblasts, but at reduced levels. In WT or rodZ3 heterozygous neuroblasts GFP–Zw10 localizes to kinetochores (marked by Spc25–RFP) and spindle fibers. In homozygous rodZ3 cells (bottom), GFP–Zw10 is detectable at kinetochores, but at lower intensity. Particles of RZZ are visible between the kinetochores (white arrow) and the poles (yellow arrows) in WT and heterozygous rodZ3, but not in homozygous rodZ3, suggesting a problem with dynein-dependent streaming (see also Fig. 5). Scale bars: 2 µm. (D) Quantification of RZZ levels on kinetochores. The GFP–Zw10 signal, normalized to that of Spc25–RFP, was determined for WT (blue, n = 7, ncolch = 26), rodZ3 homozygous (red, n = 7, ncolch = 8) or rodZ3/+ heterozygous (green) cells. Closed circles and open circles indicate cells treated or untreated, respectively, with colchicine for 15 min. Results are mean±s.e.m. *P<0.05, **P<0.01 (Student's t-test). (E) The SAC is functional in rodZ3 larval brains. Relative mitotic density in larval brains as a function of time in colchicine. WT (black), rodZ3 (light gray), rod null (dark gray). Results are mean±s.e.m. *P<0.05 (Student's t-test, using at least five larval brains per experiment). In rodZ3, the mitotic density increases as a function of time in colchicine similar to WT, whereas in SAC-defective rod null brains, mitotic cells do not accumulate, even after 1 h in colchicine. (F) Lagging chromatids are common in rodZ3 neuroblast mitosis. Representative images of single live neuroblasts of the indicated genotypes in metaphase and anaphase. Kinetochores are visualized with Spc25–RFP (red) and the spindle is marked with GFP–Jupiter (green). Indicated times (min:seconds) are relative to anaphase onset (see also Table 3). Scale bars: 2 µm.
The SAC is required for polar body maintenance
Decondensed polar bodies have been reported previously in mutants of mps1 and BubR1 (Fischer et al., 2004; Pérez-Mongiovi et al., 2005), two other genes encoding SAC components. rod is the third SAC gene to be associated with this phenotype. To determine whether rodZ3-derived syncytial embryos could undergo SAC-dependent mitotic arrest in response to spindle damage, syncytial-stage embryos were incubated with or without colchicine for 30 min, and the fraction of mitotic embryos was determined (see Materials and Methods for details). Whereas nearly all WT embryos had become mitotic (Fig. 1D), neither rodZ3 nor Mad2-null (Mad2P)-derived embryos (a negative control) displayed any increase in the fraction of mitotic embryos in response to colchicine treatment. These results indicate that the SAC is defective in rodZ3-derived embryos and suggests that the failure of polar bodies to remain in M-phase is due to the defective SAC.
However, Mps1, BubR1 and the RZZ complex all participate in other mitotic functions that do not involve the SAC (Starr et al., 1998; Ditchfield et al., 2003; Jones et al., 2005; Lampson and Kapoor, 2005; Maure et al., 2007; Jelluma et al., 2008; Hewitt et al., 2010; Santaguida et al., 2010; Sliedrecht et al., 2010; Althoff et al., 2012; Wainman et al., 2012). It is therefore conceivable that the polar body decondensation is not specifically connected to a defect in SAC activity. To confirm the role of the SAC in the maintenance of polar body condensation, we examined embryos laid by Mad2P females. Drosophila Mad2-null mutants have a defective SAC, but do not exhibit the mitotic defects associated with mutations in other SAC genes (Buffin et al., 2007). We found that most of the polar bodies in Mad2P-derived embryos were large with semi- or fully decondensed chromatin (Table 2), similar to those of rodZ3. This result supports the hypothesis that SAC activity is required to maintain polar body condensation, presumably by stabilizing Cyclin B (Fischer et al., 2004; Pérez-Mongiovi et al., 2005). To more rigorously test this latter point, we generated rodZ3 mutants carrying one copy of a lethal allele of the shattered (shtd) gene encoding APC/C subunit APC1 in Drosophila. We also directly increased Cyclin B levels in rodZ3 flies by introducing two additional copies of the WT Cyclin B (CycB) gene. Both modifications partially suppressed the polar body phenotype of rodZ3-derived embryos (Table 2). The frequency of abnormal polar bodies fell to 69% in shtd3/+; rodZ3-derived embryos, and to 53% in 2×Cyclin B/+; rodZ3-derived embryos. These results argue that WT rod is required to maintain polar body M-phase through its role in the SAC, which in turn inhibits APC/C-regulated degradation of Cyclin B.
rodZ3 does not affect RZZ assembly
Sequencing the rodZ3 genomic DNA revealed a point mutation in rod where glycine 1973 was replaced by a glutamic acid residue, predicted to disrupt a short α-helix in the relatively conserved C-terminal region of the 2089-residue Rod protein. Drosophila Rod has an overall predicted structure similar to that described for human Rod (also known a KNTC1) (Çivril et al., 2010) with N-terminal β-folds and a long α-solenoid region extending to the C-terminus. Although not conserved in vertebrate Rod (where it is a tyrosine or a serine residue), the glycine at position 1973 has been conserved since the divergence of mosquitos and Drosophila, over 250 million years (Wiegmann et al., 2011) (Fig. 2A).
Fig. 2.

The rodZ3 mutation changes a conserved glycine in the Rod_C domain, but does not affect RZZ assembly. (A) The mutation in rodZ3 results in a G to E change at residue 1973 in a conserved C-terminal region of the protein. A Clustal alignment around the mutated region of Rod from mammals and dipterans shows that G1973 (red box) has been conserved since the divergence of mosquitoes and flies. The corresponding position in mammalian Rod is always a polar residue. (B) Levels of Rod, Zw10 and Zwilch are similar in extracts of WT and rodZ3-derived syncytial embryos, and immunoprecipitation (IP) of Rod brings down Zw10 and Zwilch equally well in both WT and rodZ3 extracts. Immunoblots are for Rod, Zw10 and Zwilch in input lysates of WT or rodZ3 derived embryos (left) (Tubulin is a loading control). Immunoprecipitates using anti-Rod, (middle), or control non-immune rabbit serum are shown on the right). The lower molecular mass in the Zwilch blots is an unrelated contaminant. (C) RZZ from WT and rodZ3-derived syncytial embryos both form high-molecular-mass complexes. Fractions of a sucrose density gradient centrifugation of embryo extracts immunoblotted for Rod. Arrows indicate migration of size markers.
The RZZ complex, with an apparent molecular mass of 600–700 kDa, is believed to contain two copies of each of the three subunits Rod, Zw10 and Zwilch, with Rod serving as the scaffold (Scaërou et al., 2001; Williams et al., 2003; Çivril et al., 2010). Immunoblotting revealed that levels of Rod, Zw10 and Zwilch in total extracts of rodZ3-derived embryos were similar to those in WT embryos (Fig. 2B). More importantly, Zw10 and Zwilch co-immunoprecipitated with Rod equally well in extracts of WT or rodZ3-derived embryos (Fig. 2B). Fractionating lysates from WT and rodZ3-derived syncytial embryos by sucrose density gradient centrifugation revealed that the rodZ3 mutation does not affect the capacity of Rod to form high molecular mass complexes (Fig. 2C). These data argue that the rodZ3 mutation does not affect RZZ complex formation or steady-state levels.
RZZ is not recruited to kinetochores in rodZ3-derived embryos but is recruited at reduced levels in rodZ3 neuroblasts
With the knowledge that the RZZ complex was intact, we next examined its behavior in rodZ3-derived syncytial embryos by following GFP-tagged Zw10. WT embryos expressing GFP–Zw10 and Spc25–RFP contained RZZ foci at the kinetochores of the polar body and mitotic chromosomes (Fig. 3A,B) as well as along metaphase spindle fibers (Fig. 3B), reflecting the streaming of RZZ from attached kinetochores (Wojcik et al., 2001; Basto et al., 2004). In contrast, no GFP signal was detected on any mitotic structure in rodZ3-derived embryos (Fig. 3A,B). These data suggest that the rodZ3 mutation interferes with kinetochore recruitment of the RZZ complex.
Although the absence of RZZ recruitment provided an explanation for the severity of the phenotype in embryos laid by homozygous rodZ3 mothers, it was also intriguing because the homozygous rodZ3 offspring of heterozygous parents were apparently normal. A mutation blocking RZZ kinetochore recruitment would normally be a zygotic lethal. Indeed, null mutations in any component of RZZ are homozygous lethal, dying as a consequence of profound defects in chromosome segregation during postembryonic development (Karess and Glover, 1989; Williams et al., 1992; Williams and Goldberg, 1994; Williams et al., 2003; Karess, 2005). We therefore examined RZZ behavior in dividing tissues of homozygous rodZ3 third-instar larvae (Fig. 3C). Unlike the rodZ3-derived embryos, rodZ3 larval neuroblasts displayed substantial but limited recruitment of RZZ to kinetochores during unperturbed mitosis. Average RZZ kinetochore levels (normalized to the Spc25–RFP signal) were approximately one-third that of WT (Fig. 3D). In addition to the reduction of RZZ recruitment to kinetochores, there was little or no RZZ on the spindle fibers and poles of rodZ3 neuroblasts, suggesting a problem with streaming (see below and Fig. 5).
Fig. 5.
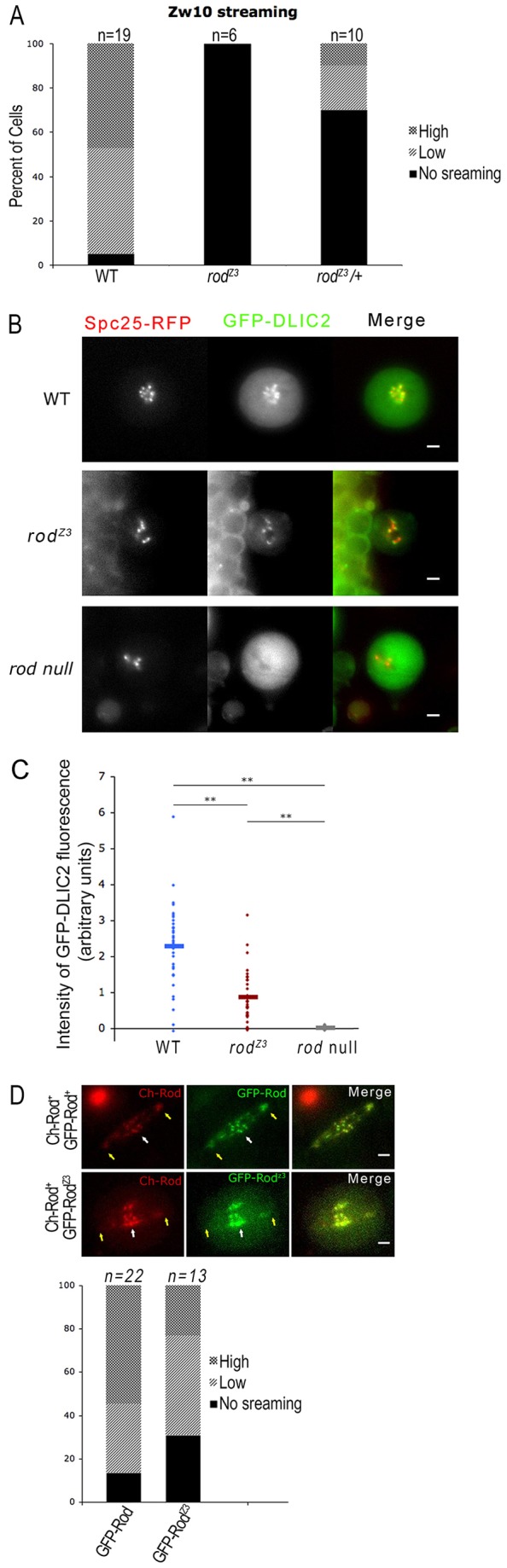
The mutation in rodZ3 affects RZZ streaming in neuroblasts. (A) Quantification of RZZ streaming in WT, rodZ3, and rodZ3/+ neuroblasts. Cells were classed by the intensity of GFP–Zw10 streaming: high, low, and no streaming. RZZ streaming is not observed in homozygous rodZ3 cells and is substantially reduced in rodZ3/+ cells. (B) Dynein recruitment is reduced but not eliminated in rodZ3 neuroblasts. Images of GFP–DLIC2 at kinetochores in live WT, rodZ3 and rod-null cells treated with colchicine. Dynein is recruited to unattached kinetochores in WT and rodZ3 cells, but not in rod-null cells. Scale bars: 2 µm. (C) Quantification of GFP–DLIC2 at kinetochores, normalized to Spc25–RFP, in colchicine-treated WT (blue, n = 36), rodZ3 (red, n = 34) or rod-null (gray, n = 18) cells. In 90% of rodZ3 cells, GFP–DLIC2 is detectable at kinetochores but its signal is reduced compared to that in WT cells. Horizontal bars represent the mean. **P<0.01 (Student's t-test). (D) RodZ3 dominantly disrupts RZZ streaming. Top, metaphase spindles of neuroblasts expressing two different colors of WT Rod [Cherry (Ch)–Rod+ and GFP–Rod+, first row, or one WT copy and one mutant copy (Ch–Rod+ and GFP–RodZ3, second row] in a rod-null background. In cells expressing only WT Rod, streaming is normal, and particles containing both colors are found on the spindle fibers. When GFP–RodZ3 is co-expressed with Ch–Rod+, the streaming of both is reduced. Scale bars: 2 µm. Bottom, quantification of Rod streaming, using criteria described in A. The fraction of cells with robust streaming is substantially reduced when GFP–RodZ3 is present.
In WT cells, a brief colchicine or nocodazole treatment to depolymerize kinetochore microtubules greatly increases the levels of several outer kinetochore proteins, including RZZ, Mad1 and Mad2 (Hoffman et al., 2001; Williams and Goldberg, 1994; Basto et al., 2004; Buffin et al., 2005). Strikingly, colchicine treatment did not significantly increase the kinetochore signal of GFP–Zw10 in rodZ3 neuroblasts; whereas in WT cells, the signal more than doubled (Fig. 3D). This reduced recruitment of RZZ in rodZ3 neuroblasts was confirmed by quantification of Rod levels by immunostaining with anti-Rod antibody (data not shown).
To assess whether this limited RZZ recruitment is compatible with a functional SAC, we assayed the ability of colchicine-treated rodZ3 larval brains to accumulate mitotic cells (Fig. 3E). Both rodZ3 and WT larval brains more than doubled their density of mitotic cells after 1 h incubation in colchicine, whereas, rod-null brains did not. These results indicate that the SAC is properly functioning in rodZ3 larval neuroblasts.
Examination of karyotypes of mitotic cells in third-instar rodZ3 larval brains revealed relatively little aneuploidy (Table 3). Live imaging of dividing rodZ3 neuroblasts (Fig. 3F), however, showed that there was an elevated frequency of anaphases in which some kinetochores migrated poleward more slowly than the others. Indeed, the fraction of anaphases with at least one lagging kinetochore was nearly as high in rodZ3 as in rod or Mad1-null mutants (Emre et al., 2011), although only rarely was more than one kinetochore involved (Table 3). As is the case for Mad1-null mutants (Emre et al., 2011), these lagging chromatids were in most cases correctly segregated, despite their delay in migration (given that overall aneuploidy levels are low). In several cases, the lagging chromatids appeared to be attached to spindle fibers emanating from both poles (merotelic attachment), which is also a feature associated with mitosis in Mad1-null mutants (Emre et al., 2011).
Table 3. Mitotic phenotype of rodZ3 larval neuroblasts.

The percentage of aneuploidy (>2n only) was determined on aceto-orcein-stained fixed tissue.
Anaphases were determined on live cells.
From Buffin et al. (Buffin et al., 2007).
From Emre et al. (Emre et al., 2011).
In summary, RZZ retains a limited capacity to associate with the mitotic apparatus in rodZ3 larval neuroblasts, but this is apparently adequate for mounting a SAC-mediated mitotic arrest and providing sufficient functionality for maintaining reasonably accurate mitosis. In contrast, RZZ appears to be incapable of kinetochore recruitment in early embryos derived from rodZ3 mothers. These results largely explain the different mitotic phenotypes of these two tissues.
Mad1 kinetochore recruitment rate and accumulation is reduced in rodZ3 neuroblasts
We next asked whether rodZ3 affected the recruitment of Mad1 and Mad2 given that kinetochore levels of the Mad1–Mad2 complex depend in part on the presence of functional RZZ (Buffin et al., 2005; Kops et al., 2005). In untreated rodZ3 neuroblasts, levels of Mad1 (Fig. 4A) and Mad2 (data not shown) were often low, and sometimes undetectable on prometaphase mitotic figures. On average, the Mad1–GFP kinetochore signal in rodZ3 was not significantly different from that in rod-null cells (Fig. 4B). The low levels of Mad1 might therefore help explain why rodZ3 displays the elevated rate of lagging chromatids also seen in Mad1 mutants. However, when treated with colchicine, rodZ3 neuroblasts did accumulate Mad1 and Mad2 at kinetochores (Fig. 4C). Both the rate of this accumulation and the final level attained were reduced; reaching approximately one-third of the levels found in WT after 8–10 min. This degree of Mad1 and Mad2 recruitment must be sufficient to elicit the robust SAC response of colchicine-treated rodZ3 larval brains (Fig. 3E).
Fig. 4.

Mad1 kinetochore recruitment is slow, but its steady state dynamics are normal in rodZ3 neuroblasts. (A) Images of live WT (top) or rodZ3 (bottom) prometaphase neuroblasts expressing Mad1–GFP and Spc25–RFP. Scale bars: 2 µm. (B) Quantification of Mad1–GFP kinetochore recruitment, normalized to Spc25–RFP in untreated WT (blue, n = 43), rodZ3 (red, n = 44), or rod-null (gray, n = 29) cells. Horizontal bars represent the mean. In most rodZ3 cells, little or no Mad1 is detectable at kinetochores, similar to rod null. **P<0.01; Student's t-test. (C) Mad1–GFP (top) and GFP–Mad2 (bottom) accumulate slowly on kinetochores, but reach substantial levels in colchicine-treated rodZ3 neuroblasts. GFP signals of kinetochore Mad1 or Mad2 were monitored in individual, colchicine-treated WT (blue) or rodZ3 (red) neuroblasts, normalized to Spc25–RFP, and plotted as a function of time after nuclear envelope breakdown (NEB). The maximum accumulation is about 35% that of WT. P<0.05 (Student's t-test). (D) The relationship between Mad1 and RZZ levels is unchanged in rodZ3 cells. Larval brains expressing Cherry (Ch)–Mad1 and GFP–Zw10 were imaged after a 30-min colchicine treatment. The intensities of kinetochore Cherry and GFP were quantified in individual WT (blue) or rodZ3 (red) cells. Levels of GFP–-Zw10, representing RZZ, were plotted on the x-axis and those of Ch–Mad1 were plotted on the y-axis. The two data sets show a similar correlation between Mad1 and RZZ. The regression curves shown for WT and rodZ3 fit the data points within the black box, which correspond to kinetochores displaying the range of GFP–Zw10 signals common to the two genotypes. The difference in slope is not significant (ANOVA test). (E) rodZ3 does not alter turnover dynamics of Mad1–GFP on unattached kinetochores. Left, FRAP analysis of WT (blue) or rodZ3 (red) neuroblasts expressing Mad1–GFP (top) or GFP–Spc25 (bottom) treated with colchicine for 15 min. Fluorescence recovery was monitored over 200 s and normalized to the pre-bleach signal. Neither the size of the fast-turnover pool of Mad1 (∼40%) nor its half-life (∼10 s) are altered in rodZ3 cells. The stable outer kinetochore component Spc25 was used as a control. Right, representative images of Mad1–GFP in (a) WT and (b) rodZ3 cells, and (c) GFP–Spc25 in WT cells, before and after photobleaching kinetochores. The bleached area is circled in yellow. Scale bars: 5 µm.
The relatively low levels of Mad1 recruitment after treatment with colchicine might be a quantitative consequence of the reduced RZZ at rodZ3 kinetochores or a qualitative consequence of the structural change caused by the rodZ3 mutation (or both). To gain insight into this question, we simultaneously measured the fluorescent signals from GFP–Zw10 and Cherry–Mad1 on the kinetochores of individual colchicine-treated neuroblasts and displayed the values in a scatter plot (Fig. 4D). The plot shows that the highest RZZ and Mad1 signals are (not surprisingly) only found in the WT cells. However, considering only the range of RZZ values overlapping between WT and rodZ3 (included within the box of Fig. 4D), the distribution of Mad1 values was not significantly different and the linear regression curves that fitted the two data sets had similar slopes. This result indicates that the recruitment levels of Mad1 are ‘normal’ in rodZ3 cells, given the reduced amount of RZZ on unattached kinetochores.
To determine whether the rodZ3 mutation is affecting steady-state turnover of Mad1 at unattached kinetochores, we subjected Mad1–GFP to fluorescence recovery after photobleaching (FRAP) analysis (Fig. 4E). The Mad1–GFP that had accumulated on rodZ3 kinetochores after 15 min in colchicine displayed the same turnover dynamics as that in WT. A fast turnover pool, ∼40% of the total, recovered with a half-life of 10–12 seconds, whereas the remaining 60% was essentially stable, showing little or no recovery. Thus, the steady-state turnover properties of Mad1 are not disrupted in rodZ3 neuroblasts.
In summary, the primary cause of the reduced kinetochore Mad1 signal in colchicine-treated rodZ3 neuroblasts is the low levels of RZZ recruitment itself, and not a specific effect of the rodZ3 mutation on the ability of RZZ to recruit Mad1. Nevertheless, the systematic reduction in the kinetochore Mad1 signal seen in unperturbed mitosis (Fig. 4A) to levels found in rod-null cells, and the fact that Mad1 is slower to accumulate on unattached kinetochores (Fig. 4C), suggests the rodZ3 mutation might specifically alter the rate of Mad1 accumulation.
RZZ streaming is disrupted in rodZ3 neuroblasts
Several transient outer kinetochore components, including RZZ and Mad1–Mad2, display a characteristic movement from kinetochores to poles along kinetochore fibers called shedding, stripping or streaming (Howell et al., 2000; Howell et al., 2001; Wojcik et al., 2001; Karess, 2005; Famulski et al., 2011). This dynein-dependent process is believed to contribute to the extinction of the SAC signal following proper kinetochore–microtubule attachment. In live images of GFP–Zw10 expressed in rodZ3 neuroblasts (Fig. 3C), this streaming appeared to be substantially reduced. Because streaming is highly variable, even in WT, we quantified this apparent difference by scoring mitotic cells into three classes according to their degree of streaming: none (all GFP–Zw10 signal restricted to kinetochores), low (GFP–Zw10 signal mostly on kinetochores) and high (signal on kinetochores, spindle and poles). By these criteria, rodZ3 neuroblasts consistently failed to show any streaming (Fig. 5A).
Given that streaming is a dynein-dependent process and RZZ is required for the recruitment of the dynein–dynactin complex to kinetochores (Starr et al., 1998; Bader and Vaughan, 2010; Raaijmakers et al., 2013), we asked whether rodZ3 specifically reduced kinetochore dynein levels. We measured the accumulation of GFP–DLIC2, a dynein subunit, relative to Spc25–RFP, on mutant or WT cells incubated 15 min in colchicine (Fig. 5B,C). In 90% of rodZ3 cells, GFP–DLIC2 was detectable at kinetochores, averaging about half the signal found in WT. As was the case for kinetochore Mad1–GFP, the range of GFP–DLIC2 levels was very broad and probably reflects the variation in RZZ recruitment levels.
Given that dynein recruitment is only partially reduced, whereas dynein-dependent streaming is entirely absent, we considered the possibility that the RodZ3 protein was directly interfering with the ability of RZZ to stream. Consistent with this idea, rodZ3/+ heterozygous neuroblasts also displayed reduced streaming relative to WT (Fig. 5A), suggesting that rodZ3 has a dominant effect in disrupting RZZ function. To test this idea, we co-expressed two Rod proteins with different fluorescent tags in a rod-null background: Cherry–Rod+ and either GFP–Rod+ or GFP–RodZ3, expressed from a transgene containing the identical mutation as the rodZ3 allele (Fig. 5D). When two WT transgenes were expressed, both green and red particles of RZZ streamed on kinetochore-attached microtubules, whereas when GFP–RodZ3 was co-expressed with Cherry–Rod+ the streaming of both the red (WT) and green (mutant) RZZ was greatly reduced (Fig. 5D). These results are consistent with a partially functional GFP–RodZ3 subunit assembling into the multi-subunit RZZ complex and inhibiting its ability to be transported from kinetochores.
DISCUSSION
The SAC is required for polar body M-phase maintenance in Drosophila
In a search for maternal effect mutants affecting the regulation of early embryonic divisions, we found a unique allele of rod, called rodZ3, which profoundly affects the ability of RZZ to be recruited to kinetochores in the early embryo.
Although previous studies have shown that Cdk1 and Cyclin B activity are required for polar body condensation, we have established that polar bodies maintain Cdk1–Cyclin-B activity through activation of the SAC. We show here that the RZZ complex, like Mps1 and BubR1, localizes to kinetochores in polar bodies and is required to maintain polar body condensation in Drosophila. rodZ3-derived embryos, like those of Mad2, Mps1 and BubR1 mutants, contain large interphase-like polar bodies.
Pérez-Mongiovi et al. (Pérez-Mongiovi et al., 2005) found evidence that in BubR1Rev1-derived embryos, polar bodies cycle between periods of condensation and decondensation with accompanying DNA replication. They suggested that these polar body cycles are under the control of the embryonic mitotic oscillator. Because polar bodies in rodZ3 embryos are not always fully decondensed, it is possible that they undergo similar cycles of DNA replication and condensation. The fact that the semi-decondensed polar bodies have more than the expected 12 kinetochores (labeled with the outer kinetochore protein Spc25; Fig. 3A), is consistent with this idea.
Directly or indirectly increasing Cyclin B levels in rodZ3-derived embryos partially suppresses the polar body condensation defects. This result argues that Rod and the other SAC components maintain polar body condensation by inhibiting the APC/C-mediated degradation of Cyclin B. The Png kinase complex is also required to maintain Cyclin B and thus polar body condensation (Lee et al., 2001). However, rod and png regulate Cyclin B levels through different mechanisms. During embryogenesis, Png kinase promotes the translation of Cyclin B by regulating the polyadenylation of its mRNA and antagonizing the translational repressor Pumilio (Vardy and Orr-Weaver, 2007). The SAC components, by contrast, recognize the polar body kinetochores as improperly attached to the spindle, and generate the inhibitor of APC/C-mediated proteolytic degradation of Cyclin B.
rodZ3 sterility is due to defects in RZZ function beyond its role in the SAC
rodZ3-derived embryos undergo aberrant syncytial mitosis and a developmental arrest prior to gastrulation. These embryos accumulate asynchronously dividing nuclei with centrosome attachment defects throughout the syncytial divisions. Defects in mitotic spindle function during syncytial embryogenesis can uncouple the nuclear and centrosomes cycles (Archambault and Pinson, 2010). The resulting replication of detached centrosomes and fusion of free centrosomes to neighboring mitoses leads to additional aberrant mitoses. Furthermore, centrosome detachment during syncytial embryogenesis inhibits the nuclear positioning and migration required for morphogenesis, all of which might contribute to the developmental arrest of rodZ3-derived embryos.
In Drosophila, inactivation of the SAC has only minor consequences on mitosis and on embryonic viability (Buffin et al., 2007). Comparing the embryonic phenotype of rodZ3 with that of Mad2 nulls is instructive. Both lack a functional SAC in embryos and consequently the polar body fails to arrest in M-phase. However, although 60–70% of Mad2-null embryos hatch and reach adulthood (Buffin et al., 2007), no rodZ3-derived embryos reach the gastrulation stage. The different phenotypes of Mad2-null and rodZ3-derived embryos argue strongly that it is not the absence of the SAC per se that is responsible for the lethality of rodZ3-derived embryos, but rather the combination of defects caused by simultaneously removing the SAC and non-SAC functions of RZZ, the latter regulating dynein and kinetochore–microtubule attachments (Starr et al., 1998; Yang et al., 2007; Gassmann et al., 2008; Gassmann et al., 2010).
rodZ3 affects the recruitment and behavior of RZZ and Mad1
The primary defect we have identified in RZ3ZZ is its failure to associate with syncytial and polar body kinetochores in the early embryo. Most of the other phenotypes can be explained as secondary consequences of this recruitment failure. The striking difference between the maternal and zygotic phenotypes of rodZ3 can be ascribed to the fact that RZ3ZZ partially retains its ability to be recruited to kinetochores in dividing larval tissues.
Why is RZ3ZZ capable of kinetochore recruitment in larval neuroblasts and not in maternally-derived early embryos? One possibility is that the unusual rapidity of syncytial mitosis might render it more sensitive to the structural change in RZ3ZZ. However, the fact that RZ3ZZ is also absent from polar bodies argues that this is unlikely to be a complete explanation because the polar body does not rapidly cycle but remains in M-phase. It would, in this scenario, still have enough time to recruit RZ3ZZ.
We suggest that there are factors involved in recruiting RZZ to kinetochores that subtly differ between the syncytial stage and postembryonic mitoses. For example, there might be two, partially redundant, recruitment pathways for RZZ. One would be employed in postembryonic mitoses, such as in larval neuroblasts, but not in the early embryo. The other recruitment pathway would normally be used in both tissues, but is damaged in rodZ3. These two ‘pathways’ could be as simple as two different kinetochore-binding domains of RZZ and two corresponding docking sites on kinetochores. This idea might also provide an explanation for the second striking feature of RZ3ZZ behavior: in larval neuroblasts, it shows no further kinetochore accumulation beyond the basal level when microtubules are depolymerized with colchicine. This supplemental RZZ binding might also depend on the second recruitment pathway, explaining why it is defective in rodZ3. The mutation in rodZ3 might thus affect a residue important to RZZ kinetochore binding, about which very little is known, particularly in Drosophila. Kinetochore proteins implicated in RZZ recruitment in vertebrates such as ZWINT (Starr et al., 2000; Famulski et al., 2008) and CENP-I (Matson and Stukenberg, 2014), have not been found in the fly genome (Przewloka et al., 2007).
During unperturbed mitosis, kinetochore Mad1 levels are as low in rodZ3 as they are in rod-null larval neuroblasts (Fig. 4A,B). We have previously reported that Mad1-null neuroblasts display a relatively mild mitotic phenotype. Despite having a defective SAC, they display little aneuploidy, but have a high incidence of lagging chromatids and merotely (Emre et al., 2011). It is therefore not surprising that we find a similar phenotype in rodZ3 cells, which have greatly reduced kinetochore Mad1 levels.
The mutation in rodZ3 does not seem to alter the ratio of Mad1 recruited per unit of RZZ. On unattached kinetochores, given enough time, Mad1 accumulates to levels approximately proportional to the amount of RZZ present (Fig. 4C,D). The recruited Mad1 also displays the same steady-state turnover dynamics as in WT cells (Fig. 4E). However, the time required to reach that maximal recruitment is longer than in WT (Fig. 4C). We speculate that the slower recruitment rate of Mad1 might explain its low levels on kinetochores during unperturbed mitosis. There is little known about the molecular mechanism by which RZZ helps to recruit Mad1. It is not even known if they interact directly, although a fraction of RZZ and Mad1 can be co-immunoprecipitated from mitotic cells (our unpublished work).
Finally, rodZ3 has a perceptible dominant effect on the dynein-dependent streaming of RZZ. This can best be seen in Fig. 5D, where RFP–RodZ3 co-expressed with GFP-Rod+ interferes with the streaming of both tagged proteins from kinetochores. The RZZ complex is believed to contain two copies of each of its three subunits, with Zw10 and Zwilch binding to the N-terminal half of the Rod protein (Starr et al., 1998; Scaërou et al., 2001; Williams et al., 2003; Çivril et al., 2010; Cheerambathur et al., 2013). This stoichiometry means that a given RZZ particle could contain one WT and one mutant copy of RZZ, if both versions are present. Given that the effect of rodZ3 on streaming is more severe than its effect on dynein recruitment, we suggest that the mutation in rodZ3 might also perturb an interaction with the dynein motor complex.
In summary, the mutation in rodZ3 affects three activities of RZZ: its own kinetochore recruitment, its capacity to rapidly recruit Mad1 and its capacity to be removed from attached kinetochores by dynein-dependent streaming. The G to E mutation of rodZ3 at residue 1973 resides in the most highly conserved portion of the Rod protein, known as Rod_C domain (pfam10493), which is predicted to fold into several short α-helices. This domain is only found in known or suspected homologs of Rod, although no specific function or interacting proteins have been are associated with it. Thus, characterization of rodZ3 provides a unique opportunity to examine how the Rod C-terminus contributes to RZZ function.
MATERIALS AND METHODS
Drosophila stocks
Flies were maintained at 25°C using standard techniques. A y w stock was used as wild type for embryo experiments. The stock bw; rodZ3 st/TM6B was a gift from Charles Zuker (Columbia University, New York, NY). The rodZ3 allele had designation Z3-0733 in the Zuker stock collection. We shortened and superscripted it to indicate that it is an allele of rod. The following mutations and transgenes have been previously described: rod-null alleles rodAG1 and rodX2 and the transgene rod+tC3L9 (Scaërou et al., 1999), GFP-Rod (Basto et al., 2004), RFP-Rod (Buffin et al., 2005), the null allele Mad11, the transgene Mad1-GFP and Cherry-Mad1 (Emre et al., 2011), the null allele Mad2P (Buffin et al., 2007), the 2XCyclin B stocks, and transgenes Spc25-GFP and Spc25-RFP (Schittenhelm et al., 2007) (gifts from Christian Lehner, University of Zurich, Zurich, Switzerland), the shtd3 stock (from the Bloomington stock center), transgene GFP-DLIC2 (gift from Jordan Raff, Oxford, UK).The GFP-Zw10 construct will be described elsewhere. Transgenic lines Cherry-Rod, and GFP-RodZ3 were obtained by P-element transposition. The transgenes correspond to genomic sequence (including the promoter region) as originally described previously (Basto et al., 2004). Fly transformation was performed by BestGene (Chino Hills, CA).
Embryo immunostaining and colchicine treatment
0–2-h embryos (unless otherwise noted) were collected as described previously (Rothwell and Sullivan, 2000). For Tubulin, Centrosomin and DNA staining, embryos were dechorionated in 50% bleach, and fixed and devitellinized by shaking in a 1∶1 mixture of methanol and heptane. For visualizing RFP or GFP fluorescence during cortical divisions, 0–30-min embryos were collected, aged for 1 h 15 min, dechorionated and fixed and devitellinized as above. For PH3 immunostaining, embryos were dechorionated, fixed in a 1∶1 mixture of 4% formaldehyde (in PBS) and heptane, and devitellinized as described above. For colchicine treatment, 0–1-h embryos were collected, dechorionated and incubated in 1∶1 mixture of 250 µM colchicine (in PBS) and heptane for 30 min. Treated embryos were then fixed with formaldehyde and devitellinized as above. The SAC response in embryos was determined by comparing the percentage of mitotic embryos following a 30 min incubation with or without colchicine. ‘Mitotic’ embryos were defined as those having at least 50% PH3-positive nuclei. For immunostaining, fixed embryos were rehydrated in PBS and incubated in 1 mg/ml RNaseA for 1 h, then incubated in primary antibody overnight at 4°C, washed, then incubated in Cy2- and/or Cy5-conjugated secondary antibodies (Jackson ImmunoResearch) for 3 h at room temperature. Embryos were stained with propidium iodide and mounted in clearing solution (Fenger et al., 2000) or in Prolong-Gold with DAPI (Life Technologies). Embryos were visualized with a Nikon Eclipse 80i microscope equipped with a CoolSNAP ES camera (Photometrics). Statistical analysis of imaging quantifications was performed using the Fisher Exact test. Primary antibodies used were anti-α-Tubulin YL1/2 (AbD Serotec) or anti-Tubulin DM1α (Sigma-Aldrich), anti-Centrosomin (gift from William Theurkauf, University of Massachusetts Medical School, Worcester, MA), anti-PH3 (06-570, Millipore), anti-Rod (Scaërou et al., 1999), anti-Zw10 and anti-Zwilch (gifts from Michael Goldberg, Cornell University, Ithaca, NY).
Embryo immunoblotting
0–1-h embryos were homogenized in non-denaturing lysis buffer (NDLB) (50 mM Tris-HCl pH 7.4, 300 mM NaCl, 5 mM EDTA, 1% Triton X-100). For immunoprecipitations, 500 µg of embryo lysate was incubated with 5 µl anti-Rod serum (Scaërou et al., 1999) or normal rabbit serum overnight at 4°C. Lysates were then incubated with 25 µl of Protein-G–Sepharose beads (GE Healthcare) for 3 h at 4°C. Beads were pelleted, washed three times in NDLB, and boiled in 30 µl 6× sample buffer. The resulting supernatant and 20 µg of input lysate were analyzed by SDS-PAGE and by immunoblotting using standard techniques. Horseradish peroxidase (HRP)-conjugated secondary antibodies and chemiluminescence were used to detect primary antibodies.
For sucrose gradient analysis, 2 mg of embryo lysate was layered on top of a sucrose gradient column of 5%, 10%, 20% and 30% sucrose, then centrifuged in a L8-70M ultracentrifuge (Beckman) with a SW55Ti rotor for 4 h at 250,000 g and 4°C. A total of 19 fractions were collected and 8% of each fraction was analyzed by SDS-PAGE and immunoblotting.
Neuroblast cytology
Immunostaining used the method described previously (Williams et al., 2003). Drosophila third-instar larval brains expressing Spc25–RFP were dissected in 0.7% NaCl and treated for 15 min in 10−4 M colchicine. After formaldehyde fixation and methanol dehydration, brains were immunostained with a mouse monoclonal anti-RFP (Abcam, ab658556; 1∶500) and rabbit anti-Rod (1∶1000) antibodies. Secondary antibodies were Alexa-Fluor-594-conjugated anti-mouse-IgG and Alexa-Fluor-488-conjugated anti-rabbit-IgG (1∶500; Life Technologies). Images were acquired with a Zeiss Axio Imager.Z1 equipped with a Plan-Apo 100× NA 1.4 oil objective and an apotome module. Acquisition times were 400 ms for Alexa Fluor 594 and 300 ms for Alexa Fluor 488 and images were stacks of five planes at 1-µm intervals. Fluorescence was quantified using ImageJ.
For determination of aneuploidy rates and to assay the SAC response, third-instar larval brains were fixed and stained in aceto-orcein following incubation in colchicine (10−4 M) for 5, 30 or 60 min, and the mitotic density (the average number of mitotic figures per microscope field with at least one mitosis) was determined as described previously (Buffin et al., 2007).
In vivo observation of larval neuroblasts
Larval brains were prepared as described previously (Emre et al., 2011). Fluorescence time-lapse videos were acquired with an Olympus IX-70 inverted microscope, a focused xenon lamp and an OrcaER camera (Hammamatsu), piloted by the Cell-R hardware and software system (Olympus). Acquisition times per frame were 100 ms for GFP–Zw10 and 150 ms for RFP–Rod, and images were obtained as stacks of five planes at 1-µm intervals taken every 15 s with a 60× NA 1.4 oil objective.
FRAP analysis
Neuroblasts from third instar larval brains were treated 15 min with colchicine at 10−4 M. FRAP experiments were performed using a confocal microscopy system (Zeiss Axiovert with LSM780) equipped with a Plan-Apo 63× NA 1.4 oil objective (during experiments with Mad1–GFP, Spc25–GFP and GFP–Rod) or a Plan-Apo 40× NA 1.3 oil objective (during experiments with GFP–Zw10). Imaging was controlled by Zeiss confocal software (Zen 2012). A circular area of 0.9 µm (GFP–Zw10) or 4 µm (Mad1–GFP, Spc25–GFP) diameter was bleached with the 488 nm and 514 nm lines of an argon laser at 25 mW. Images were acquired with a temporal resolution of 1 s (for Mad1–GFP) or 5 seconds (for Spc25–GFP and GFP–Zw10) with a GaAsP detector (490–650 nm). Fluorescence intensity in a region of interest was quantified using ImageJ. The exponential kinetics of FRAP was analyzed by nonlinear regression fitting using Origin software.
Assay for perdurance of maternally supplied Rod
To test the hypothesis that perdurance of a small amount of WT Rod (protein or mRNA), initially loaded into the cytoplasm of the egg laid by a rodZ3/+ heterozygous mother, might be responsible for the relatively mild phenotype of homozygous rodZ3 larvae, we assayed for GFP–Rod+ of strictly maternal origin in third-instar larval brains. From the cross of y w; GFP-Rod/y+Cy; +/+ females to y w/Y males, phenotypically y+ larvae should not inherit the GFP–Rod transgene. Third-instar larval brains from these y+ larvae were examined both by immunoblotting, probing with anti-GFP antibody, and by fluorescence microscopy following colchicine treatment, for evidence of GFP signal on kinetochores. Neither approach detected a positive signal of GFP–Rod.
Supplementary Material
Acknowledgments
We thank the ImagoSeine facility, a member of France BioImaging infrastructure, supported by the French National Research Agency [grant number ANR-10-INSB-04, ‘Investments of the Future’].
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
All authors contributed to the conception and design of experiments, analysis of data, and manuscript preparation. Experiments were performed principally by L.D. and S.G.H. A.M. contributed to the initial analysis of the rodZ3 zygotic phenotype.
Funding
This work was supported by the National Institutes of Health [grant numbers GM074044 to L.A.L., GM008554 to S.G.H.]. L.D. was supported by fellowships from the French ‘Ministère Français de l'Enseignement Supérieur et de la Recherche’, and the Association pour la Recherche sur le Cancer. The R.K. laboratory was supported by the Centre National de la Recherche Scientifique, and La Ligue National Contre le Cancer ‘Equipe Labellisée’ program 2012, and is a member of the Laboratory of Excellence program No. ANR-11-LABX-0071/Investments for the Future program No. ANR-11-IDEX-0005-01. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.165712/-/DC1
References
- Althoff F., Karess R. E., Lehner C. F. (2012). Spindle checkpoint-independent inhibition of mitotic chromosome segregation by Drosophila Mps1. Mol. Biol. Cell 23, 2275–2291 10.1091/mbc.E12-02-0117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archambault V., Pinson X. (2010). Free centrosomes: where do they all come from? Fly (Austin) 4, 172–177 10.4161/fly.4.2.11674 [DOI] [PubMed] [Google Scholar]
- Bader J. R., Vaughan K. T. (2010). Dynein at the kinetochore: timing, interactions and functions. Semin. Cell Dev. Biol. 21, 269–275 10.1016/j.semcdb.2009.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto R., Scaërou F., Mische S., Wojcik E., Lefebvre C., Gomes R., Hays T., Karess R. (2004). In vivo dynamics of the rough deal checkpoint protein during Drosophila mitosis. Curr. Biol. 14, 56–61 10.1016/j.cub.2003.12.025 [DOI] [PubMed] [Google Scholar]
- Buffin E., Lefebvre C., Huang J., Gagou M. E., Karess R. E. (2005). Recruitment of Mad2 to the kinetochore requires the Rod/Zw10 complex. Curr. Biol. 15, 856–861 10.1016/j.cub.2005.03.052 [DOI] [PubMed] [Google Scholar]
- Buffin E., Emre D., Karess R. E. (2007). Flies without a spindle checkpoint. Nat. Cell Biol. 9, 565–572 10.1038/ncb1570 [DOI] [PubMed] [Google Scholar]
- Chan Y. W., Fava L. L., Uldschmid A., Schmitz M. H., Gerlich D. W., Nigg E. A., Santamaria A. (2009). Mitotic control of kinetochore-associated dynein and spindle orientation by human Spindly. J. Cell Biol. 185, 859–874 10.1083/jcb.200812167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheerambathur D. K., Gassmann R., Cook B., Oegema K., Desai A. (2013). Crosstalk between microtubule attachment complexes ensures accurate chromosome segregation. Science 342, 1239–1242 10.1126/science.1246232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Çivril F., Wehenkel A., Giorgi F. M., Santaguida S., Di Fonzo A., Grigorean G., Ciccarelli F. D., Musacchio A. (2010). Structural analysis of the RZZ complex reveals common ancestry with multisubunit vesicle tethering machinery. Structure 18, 616–626 10.1016/j.str.2010.02.014 [DOI] [PubMed] [Google Scholar]
- Ditchfield C., Johnson V. L., Tighe A., Ellston R., Haworth C., Johnson T., Mortlock A., Keen N., Taylor S. S. (2003). Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J. Cell Biol. 161, 267–280 10.1083/jcb.200208091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emre D., Terracol R., Poncet A., Rahmani Z., Karess R. E. (2011). A mitotic role for Mad1 beyond the spindle checkpoint. J. Cell Sci. 124, 1664–1671 10.1242/jcs.081216 [DOI] [PubMed] [Google Scholar]
- Famulski J. K., Vos L., Sun X., Chan G. (2008). Stable hZW10 kinetochore residency, mediated by hZwint-1 interaction, is essential for the mitotic checkpoint. J. Cell Biol. 180, 507–520 10.1083/jcb.200708021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famulski J. K., Vos L. J., Rattner J. B., Chan G. K. (2011). Dynein/Dynactin-mediated transport of kinetochore components off kinetochores and onto spindle poles induced by nordihydroguaiaretic acid. PLoS ONE 6, e16494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenger D. D., Carminati J. L., Burney-Sigman D. L., Kashevsky H., Dines J. L., Elfring L. K., Orr-Weaver T. L. (2000). PAN GU: a protein kinase that inhibits S phase and promotes mitosis in early Drosophila development. Development 127, 4763–4774. [DOI] [PubMed] [Google Scholar]
- Fischer M. G., Heeger S., Häcker U., Lehner C. F. (2004). The mitotic arrest in response to hypoxia and of polar bodies during early embryogenesis requires Drosophila Mps1. Curr. Biol. 14, 2019–2024 10.1016/j.cub.2004.11.008 [DOI] [PubMed] [Google Scholar]
- Foe V. E., Odell G. M., Edgar B. A. (1993). Mitosis and morphogenesis in the Drosophila embryo: point and counterpoint. The Development of Drosophila Melanogaster Bate M, Arias A M, ed149–300Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Gassmann R., Essex A., Hu J. S., Maddox P. S., Motegi F., Sugimoto A., O'Rourke S. M., Bowerman B., McLeod I., Yates J. R., III et al. (2008). A new mechanism controlling kinetochore-microtubule interactions revealed by comparison of two dynein-targeting components: SPDL-1 and the Rod/Zwilch/Zw10 complex. Genes Dev. 22, 2385–2399 10.1101/gad.1687508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassmann R., Holland A. J., Varma D., Wan X., Çivril F., Cleveland D. W., Oegema K., Salmon E. D., Desai A. (2010). Removal of Spindly from microtubule-attached kinetochores controls spindle checkpoint silencing in human cells. Genes Dev. 24, 957–971 10.1101/gad.1886810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffis E. R., Stuurman N., Vale R. D. (2007). Spindly, a novel protein essential for silencing the spindle assembly checkpoint, recruits dynein to the kinetochore. J. Cell Biol. 177, 1005–1015 10.1083/jcb.200702062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt L., Tighe A., Santaguida S., White A. M., Jones C. D., Musacchio A., Green S., Taylor S. S. (2010). Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J. Cell Biol. 190, 25–34 10.1083/jcb.201002133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman D. B., Pearson C. G., Yen T. J., Howell B. J., Salmon E. D. (2001). Microtubule-dependent changes in assembly of microtubule motor proteins and mitotic spindle checkpoint proteins at PtK1 kinetochores. Mol. Biol. Cell 12, 1995–2009 10.1091/mbc.12.7.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell B. J., Hoffman D. B., Fang G., Murray A. W., Salmon E. D. (2000). Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J. Cell Biol. 150, 1233–1250 10.1083/jcb.150.6.1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell B. J., McEwen B. F., Canman J. C., Hoffman D. B., Farrar E. M., Rieder C. L., Salmon E. D. (2001). Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J. Cell Biol. 155, 1159–1172 10.1083/jcb.200105093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelluma N., Brenkman A. B., van den Broek N. J., Cruijsen C. W., van Osch M. H., Lens S. M., Medema R. H., Kops G. J. (2008). Mps1 phosphorylates Borealin to control Aurora B activity and chromosome alignment. Cell 132, 233–246 10.1016/j.cell.2007.11.046 [DOI] [PubMed] [Google Scholar]
- Jia L., Kim S., Yu H. (2013). Tracking spindle checkpoint signals from kinetochores to APC/C. Trends Biochem. Sci. 38, 302–311 10.1016/j.tibs.2013.03.004 [DOI] [PubMed] [Google Scholar]
- Jones M. H., Huneycutt B. J., Pearson C. G., Zhang C., Morgan G., Shokat K., Bloom K., Winey M. (2005). Chemical genetics reveals a role for Mps1 kinase in kinetochore attachment during mitosis. Curr. Biol. 15, 160–165 10.1016/j.cub.2005.01.010 [DOI] [PubMed] [Google Scholar]
- Karess R. (2005). Rod-Zw10-Zwilch: a key player in the spindle checkpoint. Trends Cell Biol. 15, 386–392 10.1016/j.tcb.2005.05.003 [DOI] [PubMed] [Google Scholar]
- Karess R. E., Glover D. M. (1989). rough deal: a gene required for proper mitotic segregation in Drosophila. J. Cell Bio. 109, 2951–2961 10.1083/jcb.109.6.2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops G. J., Kim Y., Weaver B. A., Mao Y., McLeod I., Yates J. R., III, Tagaya M., Cleveland D. W. (2005). ZW10 links mitotic checkpoint signaling to the structural kinetochore. J. Cell Biol. 169, 49–60 10.1083/jcb.200411118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koundakjian E. J., Cowan D. M., Hardy R. W., Becker A. H. (2004). The Zuker collection: a resource for the analysis of autosomal gene function in Drosophila melanogaster. Genetics 167, 203–206 10.1534/genetics.167.1.203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson M. A., Kapoor T. M. (2005). The human mitotic checkpoint protein BubR1 regulates chromosome-spindle attachments. Nat. Cell Biol. 7, 93–98 10.1038/ncb1208 [DOI] [PubMed] [Google Scholar]
- Lara-Gonzalez P., Westhorpe F. G., Taylor S. S. (2012). The spindle assembly checkpoint. Curr. Biol. 22, R966–R980 10.1016/j.cub.2012.10.006 [DOI] [PubMed] [Google Scholar]
- Lee L. A., Orr-Weaver T. L. (2003). Regulation of cell cycles in Drosophila development: intrinsic and extrinsic cues. Annu. Rev. Genet. 37, 545–578 10.1146/annurev.genet.37.110801.143149 [DOI] [PubMed] [Google Scholar]
- Lee L. A., Elfring L. K., Bosco G., Orr-Weaver T. L. (2001). A genetic screen for suppressors and enhancers of the Drosophila PAN GU cell cycle kinase identifies cyclin B as a target. Genetics 158, 1545–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist A., Rodríguez-Bravo V., Medema R. H. (2009). The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J. Cell Biol. 185, 193–202 10.1083/jcb.200812045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson D. R., Stukenberg P. T. (2014). CENP-I and Aurora B act as a molecular switch that ties RZZ/Mad1 recruitment to kinetochore attachment status. J. Cell Biol. 205, 541–554 10.1083/jcb.201307137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maure J. F., Kitamura E., Tanaka T. U. (2007). Mps1 kinase promotes sister-kinetochore bi-orientation by a tension-dependent mechanism. Curr. Biol. 17, 2175–2182 10.1016/j.cub.2007.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida S., Hunt T. (2012). Protein phosphatases and their regulation in the control of mitosis. EMBO Rep. 13, 197–203 10.1038/embor.2011.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A. (2011). Spindle assembly checkpoint: the third decade. Philos. Trans. R. Soc. B 366, 3595–3604 10.1098/rstb.2011.0072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Farrell P. H. (2001). Triggering the all-or-nothing switch into mitosis. Trends Cell Biol. 11, 512–519 10.1016/S0962-8924(01)02142-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Mongiovi D., Malmanche N., Bousbaa H., Sunkel C. (2005). Maternal expression of the checkpoint protein BubR1 is required for synchrony of syncytial nuclear divisions and polar body arrest in Drosophila melanogaster. Development 132, 4509–4520 10.1242/dev.02028 [DOI] [PubMed] [Google Scholar]
- Przewloka M. R., Zhang W., Costa P., Archambault V., D'Avino P. P., Lilley K. S., Laue E. D., McAinsh A. D., Glover D. M. (2007). Molecular analysis of core kinetochore composition and assembly in Drosophila melanogaster. PLoS ONE 2, e478 10.1371/journal.pone.0000478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaijmakers J. A., Tanenbaum M. E., Medema R. H. (2013). Systematic dissection of dynein regulators in mitosis. J. Cell Biol. 201, 201–215 10.1083/jcb.201208098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell W. F., Sullivan W. (2000). Fluorescent analysis of Drosophila embryos. Drosophila Protocols Sullivan W, Ashburner M, Hawley R S, ed141–157Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Santaguida S., Tighe A., D'Alise A. M., Taylor S. S., Musacchio A. (2010). Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J. Cell Biol. 190, 73–87 10.1083/jcb.201001036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaërou F., Aguilera I., Saunders R., Kane N., Blottière L., Karess R. (1999). The rough deal protein is a new kinetochore component required for accurate chromosome segregation in Drosophila. J. Cell Sci. 112, 3757–3768. [DOI] [PubMed] [Google Scholar]
- Scaërou F., Starr D. A., Piano F., Papoulas O., Karess R. E., Goldberg M. L. (2001). The ZW10 and Rough Deal checkpoint proteins function together in a large, evolutionarily conserved complex targeted to the kinetochore. J. Cell Sci. 114, 3103–3114. [DOI] [PubMed] [Google Scholar]
- Schittenhelm R. B., Heeger S., Althoff F., Walter A., Heidmann S., Mechtler K., Lehner C. F. (2007). Spatial organization of a ubiquitous eukaryotic kinetochore protein network in Drosophila chromosomes. Chromosoma 116, 385–402 10.1007/s00412-007-0103-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliedrecht T., Zhang C., Shokat K. M., Kops G. J. (2010). Chemical genetic inhibition of Mps1 in stable human cell lines reveals novel aspects of Mps1 function in mitosis. PLoS ONE 5, e10251 10.1371/journal.pone.0010251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr D. A., Williams B. C., Hays T. S., Goldberg M. L. (1998). ZW10 helps recruit dynactin and dynein to the kinetochore. J. Cell Biol. 142, 763–774 10.1083/jcb.142.3.763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr D. A., Saffery R., Li Z., Simpson A. E., Choo K. H., Yen T. J., Goldberg M. L. (2000). HZwint-1, a novel human kinetochore component that interacts with HZW10. J. Cell Sci. 113, 1939–1950. [DOI] [PubMed] [Google Scholar]
- Vardy L., Orr-Weaver T. L. (2007). The Drosophila PNG kinase complex regulates the translation of cyclin B. Dev. Cell 12, 157–166 10.1016/j.devcel.2006.10.017 [DOI] [PubMed] [Google Scholar]
- Wainman A., Giansanti M. G., Goldberg M. L., Gatti M. (2012). The Drosophila RZZ complex – roles in membrane trafficking and cytokinesis. J. Cell Sci. 125, 4014–4025 10.1242/jcs.099820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegmann B. M., Trautwein M. D., Winkler I. S., Barr N. B., Kim J. W., Lambkin C., Bertone M. A., Cassel B. K., Bayless K. M., Heimberg A. M. et al. (2011). Episodic radiations in the fly tree of life. Proc. Natl. Acad. Sci. USA 108, 5690–5695 10.1073/pnas.1012675108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams B. C., Goldberg M. L. (1994). Determinants of Drosophila zw10 protein localization and function. J. Cell Sci. 107, 785–798. [DOI] [PubMed] [Google Scholar]
- Williams B. C., Karr T. L., Montgomery J. M., Goldberg M. L. (1992). The Drosophila l(1)zw10 gene product, required for accurate mitotic chromosome segregation, is redistributed at anaphase onset. J. Cell Biol. 118, 759–773 10.1083/jcb.118.4.759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams B. C., Li Z., Liu S., Williams E. V., Leung G., Yen T. J., Goldberg M. L. (2003). Zwilch, a new component of the ZW10/ROD complex required for kinetochore functions. Mol. Biol. Cell 14, 1379–1391 10.1091/mbc.E02-09-0624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik E., Basto R., Serr M., Scaërou F., Karess R., Hays T. (2001). Kinetochore dynein: its dynamics and role in the transport of the Rough deal checkpoint protein. Nat. Cell Biol. 3, 1001–1007 10.1038/ncb1101-1001 [DOI] [PubMed] [Google Scholar]
- Yang Z., Tulu U. S., Wadsworth P., Rieder C. L. (2007). Kinetochore dynein is required for chromosome motion and congression independent of the spindle checkpoint. Curr. Biol. 17, 973–980 10.1016/j.cub.2007.04.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.