

Abstract
Mammalian cells are able to sense environmental oxidative and genotoxic conditions such as the environmental low dose ionizing radiation (LDIR) present naturally on earth surface. The stressed cells then can induce a so-called radioadaptive response with an enhanced cellular homeostasis and repair capacity against subsequent similar genotoxic conditions such as a high dose radiation. MnSOD, a primary mitochondrial antioxidant in mammals, has long been known to play a crucial role in the radioadaptive protection through detoxifying O2·- generated by mitochondrial oxidative phosphorylation. Contrasted to the well-studied mechanisms of SOD2 gene regulation, the mechanisms underlying post-translational regulation of MnSOD for radioprotection remain to be defined. Herein, we demonstrate that Cyclin D1-cyclin-dependent kinase 4 (CDK4) serves as the messenger to deliver the stress signal to mitochondria to boost mitochondrial homeostasis in human skin keratinocytes under LDIR adaptive radioprotection. Cyclin D1/CDK4 is found to relocate to mitochondria at the same time as MnSOD enzymatic activation peaks without significant changes of total MnSOD protein level. The mitochondrial-localized CDK4 directly phosphorylates MnSOD at Serine 106 (S106), causing enhanced MnSOD enzymatic activity and mitochondrial respiration. Expression of mitochondria-targeted dominant negative CDK4 or the MnSOD-S106A mutant reverses LDIR-induced mitochondrial enhancement and adaptive protection. The CDK4-mediated MnSOD activation and mitochondrial metabolism boost are also detected in skin tissues of mice receiving in vivo whole body LDIR. These results demonstrate a unique CDK4-mediated mitochondrial communication that allows cells to sense environmental genotoxic stress and boost mitochondrial homeostasis via enhancing phosphorylation and activation of MnSOD.
Keywords: Cyclin D1/CDK4, mitochondrial homeostasis, MnSOD, phosphorylation, radioadaptive response
Introduction
Low level oxidative and genotoxic conditions such as low dose ionizing radiation (LDIR) that is naturally present in the human living environment and in medical radiation use are potential cancer risks and have been public health concerns. Mammalian cells are able to generate an adaptive and homeostatic response to LDIR [1] with an enhanced ability to maintain genomic integrity and immuno-responsibility as well as to inhibit cell transformation [2-4]. Elucidation of the mechanisms underlying the LDIR-induced homeostatic response in mammalian cells will reveal new mechanistic insights on how cells coordinate with the environmental toxic conditions which will help to invent effective approaches to reduce and prevent environmental toxin-associated diseases.
Manganese superoxide dismutase (MnSOD) is a primary mitochondrial antioxidant enzyme in mammalian cells catalyzing the conversion of two molecules of superoxide anion (O2·-) generated either as by-products of mitochondria oxidative phosphorylation or under oxidative stress into hydrogen peroxide (H2O2) which is further oxidized to water [5, 6]. In mouse epithelial skin cells, increased MnSOD activity is associated with LDIR-induced adaptive protection against subsequent lethal high dose radiation [7]. In human mammary epithelia cells, LDIR induces MnSOD phosphorylation and enzymatic activation, which is required for the development of radioadaptive protection [8]. Increased MnSOD expression and enzymatic activity are linked to radioadaptive response in either cultured cells or mice [9, 10] and the MnSOD-NF-κB-MnSOD loop is found to contribute to LDIR-induced radioprotection in brain and gut tissues from mice receiving whole body LDIR [11]. In addition, MnSOD significantly reduces drug-induced cardiac injury through protecting mitochondria from damage caused by superoxide radicals [12] and protects normal esophageal, pancreatic and bone marrow cells from radiation-induced genomic instability and apoptosis [13, 14]. These findings suggest that the regulation of MnSOD expression and activation is implicated in cellular genotoxic response. MnSOD is coded by nuclear SOD2 gene, and NF-κB and TNF signaling pathways have been identified as important components in the regulation of SOD2 expression [10, 15]. The mitochondrial targeting sequence at the N terminus allows a direct transport of the newly synthesized MnSOD protein into the mitochondrion, where it self-associates into an active homotetramer containing Mn3+ ion and functions [16]. Recently, the post-translational modification has been reported playing a pivotal role in the regulation of MnSOD enzymatic activation. For example, SIRT3-mediated deacetylation at multiple amino acid residues is found to elevate MnSOD antioxidant activity under the oxidative stress in vitro and in vivo [17, 18]. In LDIR-treated MCF10A cells, MnSOD is found to be phosphorylated at S106 site, which stabilizes MnSOD protein, leading to increased enzymatic activity [8]. While, site-specific nitration at tyrosine-34 of MnSOD results in significant inactivation of its enzyme activity [19]. However, the mechanisms underlying maintenance of mitochondrial homeostasis via post-translational modification of MnSOD in cellular adaptive response remain to be elucidated.
Cyclin D1 is involved in genotoxic stress through the regulation of mitochondrial function and cellular metabolism. In B cells, Cyclin D1 inhibits mitochondria-mediated apoptosis induced by drug cytotoxic stress through altering intracellular distribution of cell death regulators [20]. In LDIR treated human skin keratinocytes, cytoplasmic Cyclin D1 contributes to adaptive radioprotection through enhancing mitochondria-mediated anti-apoptosis function [21]. As the key regulator of G1/S transition, in addition to regulating cell cycle progression [22], Cyclin D1/CDK4 complex is involved in the regulation of mitochondria function and cellular metabolism through different subcellular localization [23-25]. Silencing CDK4 sensitizes cells to radiation-induced apoptosis through inhibiting phosphorylation of apoptosis factor, which is independent of cell cycle progression [26]. In this study, we demonstrate that LDIR enhances mitochondrial homeostasis via mitochondrial-localized Cyclin D1/CDK4 that directly phosphorylates MnSOD at Ser106 causing increased MnSOD activity and mitochondria respiration along with enhanced cell survival. These findings reveal a unique mechanism by which CDK4 communicates radiation stress to mitochondrial MnSOD, which allows cell to adapt the low level genotoxic stress via CDK4-MnSOD controlled mitochondrial function and cell survival.
Materials and methods
Ethics statement
Female BALB/c mice aged 6–8 weeks were irradiated with the indicated dose or dose combination following the protocol approved by the Institutional Animal Care and Use Committee of University of California Davis (IACUC No. 15315) and then dissected at the indicated time after radiation.
Antibodies and reagents
Rabbit polyclonal anti-Cyclin D1, rabbit polyclonal anti-MnSOD and Mouse monoclonal anti-CDK4 antibodies were from Santa Cruz. Mouse monoclonal anti-α-tubulin, anti-β-actin, anti-flag antibodies were from Sigma. Mouse monoclonal anti-phospho-serine antibody was from Millipore. Rabbit polyclonal anti-COX IV antibody was from Cell Signaling. Lipofectamine RNAiMAX and 2000 reagents, MitoSOX and JC-1 were from Invitrogen. QuikChange® site-directed mutagenesis kit was from Agilent Technologies. Phosphatase inhibitor cocktail was from Sigma. The mitochondria isolation kit and protein A/G-Sepharose beads were from Pierce. siRNA construction kit was from Ambion.
Cells and treatment
Human skin keratinocytes (HK18) were cultured in DMEM medium supplemented with 10% FBS and 1% penicillin/streptomycin (HyClone) in a humidified incubator (5% CO2), and irradiated with a cabinet x-ray System Faxitron Series at dose rate 0.028 Gy/min (Hewlett Packard).
MnSOD activity assay
MnSOD activity assay was performed as described previously [27] with modifications. Briefly, cells were lysed in DETAPAC buffer (50 mM potassium phosphate buffer containing 1.34 mmol/L diethylenetriaminepentaacetic acid), sonicated with six 5-sec bursts on ice, and centrifuged at 8000g for 5 min at 4°C. The assay reaction was initiated in DETAPAC buffer supplemented with 0.13 mg/ml BSA, 1.25 U/ml of Catalase, 50 μM xanthine, 74.6 μM Nitroblue tetrazolium (NBT), 50 μM Bathocuproinedisulfonic acid (BCS) with or without 5 mM NaCN by adding xanthine oxidase and incubated at RT for 2 min. The absorbance was monitored at 560 nm every 15 sec for 2.5 min with the Spectra Max M2e plate reader (Molecular Devices Co.). One unit of SOD activity was calculated as the amount of protein that inhibits NBT reduction by 50%.
Preparation of mitochondria fractions from mouse tissues and cultured cells
Mitochondria fractions were prepared using the published protocol [28] with modifications. Briefly, fresh mice liver tissues were minced into small pieces and homogenized using a Teflon-pestle in ice-cold IBc buffer (containing 0.2 M sucrose, 10 mM Tris/MOPS and 1 mM EGTA/Tris, pH 7.4) for 20 min, and the homogenates were centrifuged at 600g for 10 min at 4°C. The supernatant was collected and centrifuged at 7,000g for 10 min at 4°C. The pellets were washed with ice-cold IBc buffer and re-suspended in cell lysis buffer for either immediate use or storage at -80°C for future use. Mitochondrial and cytosolic fractions of HK18 cells were obtained using a mitochondria isolation kit (Pierce) following the manufacturer's instructions.
Alkaline extraction
The alkaline extraction was performed as previously reported [29]. Briefly, the mitochondrial fraction was incubated in 0.1 M Na2CO3 (pH 11) for 20 min at 4°C. The membrane was then pelleted by centrifugation at 100,000g. Proteins in the supernatants were precipitated using a final volume of 10% trichloroacetic acid (TCA). The pellets were re-suspended in dissolving buffer containing 7 M urea, 3 M thiourea, 2% CHAPS, 30 mM Tris, pH 8.5.
Cyclin D1/CDK4 kinase assay
For in vitro CDK4 kinase assay, wt- and mut-MnSOD were isolated from stable transfected cells by immunoprecipitation with anti-flag antibody and incubated with 2 units of Cyclin D1/CDK4 complex (New England Biolabs) in 30 μl of 1 × kinase assay buffer supplemented with 10 mM ATP and 100 μCi/umol of [γ-32P] at 30°C for 30 min. Samples were separated on SDS-PAGE gel followed by autoradiography.
Δψm assay
The Δψm was monitored using JC-1 staining method (Invitrogen) following manufacturer's instructions. Briefly, cells seeded in 96-well plates were incubated for 30 min at 37°C in medium containing 2 μg/ml of JC-1. Following washing with PBS (pH 7.4), the fluorescence intensity of JC-1 red and JC-1 green was detected using a Spectra Max M2e plate reader at excitation/emission wavelengths of 560 nm/595 nm and 485 nm/525 nm respectively. The ratio of fluorescent intensity of JC-1 red to JC-1 green was used as an indicator of Δψm.
Mitochondrial superoxide generation
Mitochondrial superoxide level was measured following the protocol published previously [27, 30] with modifications. In brief, cells seeded in a 96-well plate were treated with desired dose of radiation and washed with pre-warmed 1× PBS, pH 7.4 at indicated time points after radiation. 100 μl of 5 μM MitoSOX Red working solution (diluted from a 5 mM stock solution in DMSO with 1 × PBS, pH 7.4) were added to cells and incubated for 10 min at 37°C. Wash cells with PBS twice and then the fluorescence intensity was detected directly in PBS using microplate reader of SpectraMax M2e at excitation/emission wavelength of 510 nm/580 nm. The mitochondrial superoxide was shown as percent of fluorescence generated in control cells.
Oxygen consumption
Oxygen consumption was measured in HK18 cells or mitochondria fractions isolated from fresh mice tissues using Clarke-type oxygen electrode (Rank Brothers Ltd) as described previously [28]. Exponentially growing cells were collected and 2×106 cells were resuspended in 300 μl Oxygen consumption buffer containing 25 mM sucrose, 15 mM KCl, 1 mM EGTA, 0.5 mM MgCl2 and 30 mM K2HPO4. Oxygen consumption was monitored at 30 °C with succinate as complex II substrate. For oxygen consumption of mice liver, mitochondria fractions in oxygen consumption buffer at a final concentration of 1 mg/ml were used.
Mitochondria ATP production
Mitochondria ATP production was tested using published method [31] with modifications. In brief, cells seeded in a 96-well plate were permeablized by adding digitonin (25μg/ml) and shaken on ice for 1 min. 30 μl of buffer containing P1,P5-di(adenosine pentaphosphate) (0.15 mM), ADP (0.1 mM), malate (1 mM) and pyruvate (1 mM) with and without oligomycin (1 μg/ml) was added to the cells and incubated at RT for 5 min. 5% trichloroacetic acid was then added to the cells and ATP extracts were neutralized by adding Tris-Acetate (pH 7.75) buffer and monitored using an ATP determination kit (Invitrogen).
Apoptosis assay
HK18 cells with desired dose of irradiation or no-radiation sham were collected and resuspended in 500 μl of Annexin-binding buffer (Biosource, Invitrogen) at 2 × 106 cells/ml. Cells were stained by adding 1 μl of FITC conjugated Annexin-V (Biosource, Invitrogen) and propidium iodide (Sigma) to 100 μl of cell suspensions followed by incubation at room temperature for 15 min in the dark. Refilled 400 μl of Annexin-binding buffer to each samples and percentage of apoptotic cells was then determined using flow cytometry immediately by counting 10,000 cells per sample.
Clonogenic survival assay
Cells treated with desired dose or dose combinations were plated in 60 mm plates with varied cell numbers and cultured for 10 days. The colonies formed were then stained with crystal violet and the numbers of colonies (with more than 50 cells) were counted and the clonogenic survival ability of irradiated cells was represented as survival fraction after normalized with controls.
Plasmid constructs and cell transfection
For mitochondria targeted gene expression, mitochondria targeting sequence (MTS) derived from the precursor of human cytochrome c oxidase subunit 8A (COX8) was cloned into pEGFP-N1 and pERFP-N1 vectors (Clontech) at NheI and BamHI sites. Cyclin D1 or CDK4 cDNA was then cloned into pEGFP-N1-COX8 or pERFP-N1-COX8 plasmid at BamHI site in frame with COX8 sequence. MnSOD cDNA was cloned into pcDNA3.1-flag vector (Invitrogen) at EcoRI and XhoI sites. CDK4 D158N dominant negative and MnSOD S106A inactive mutant were generated using QuikChange® site-directed mutagenesis kit. The primer sequences for all plasmid constructs were listed in Table S1. Exponentially growing HK18 cells were transfected using Lipofectamine 2000 and the stable transfectants were selected by G418 (500 μg/ml).
Cyclin D1 silencing by siRNA
siRNA targeting Cyclin D1 mRNA was synthesized with the Silencer siRNA Construction Kit (Ambion, Austin, TX, USA). Cells were seeded in 35mm plates to achieve 30–50% confluence and cultured for 24h before transfection performed using Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA, USA). Scramble RNA Duplex (Ambion) was included as control. Inhibition of Cyclin D1 expression was determined by western blotting at 24h post-transfection. The primers used to synthesize the siRNA were listed in Table S2.
Immunoblot and immunoprecipitation
For immunoblot, 20 μg of total cell lysates, cytosolic and mitochondrial fractions were separated on SDS-PAGE gel and transferred to the polyvinylidene difluoride membrane followed by immunoblot with antibodies specific to the proteins of interest. The purity of the fractions was verified by immunoblot with COXIV and α-tubulin as markers of mitochondria and cytosol protein respectively.
For co-IP/IB analysis, total and mitochondrial proteins (200 μg) were pre-cleaned with normal mouse or rabbit IgG and 20 μl of 1:1 slurry of protein A/G-Sepharose beads for 1 h at 4°C. Pre-cleaned samples were immunoprecipitated with antibodies against proteins of interest overnight at 4°C, followed by 3 h incubation with protein A/G beads. Beads were then collected by brief spin down, washed 3 times with pre-ice cold 1× PBS, boiled in SDS-PAGE gel loading buffer. Samples were then fractionated on SDS-PAGE gel followed by immunoblot with desired antibodies. Immunoprecipitation with normal rabbit or mouse IgG was used as a negative control.
Immunohistochemistry
The dorsal skin tissues from female BALB/c mice either untreated or irradiated were embedded, frozen and sliced into tissue sections. For H&E staining, the tissue sections were deparaffinized in xylene 3×5 min and re-hydrated in 100%, 95%, 85% and 75% ethanol 5 min each, followed by rinsing with tap water for 5 min. Sections were stained with hematoxylin for 5 min at room temperature and rinsed with deionized H2O. Sections were rinsed with running tap water for 5 min and dunked in acid alcohol (1% HCl in 70% ETOH) until the sections turned pink. Sections were washed with running tap water for 5 min and stained with eosin for 5 min followed by dehydration with 95% ethanol for 3×5 min and 100% ethanol for 3×5 min. Sections were soaked in xylene for 3×15 min to clean the water. The sections were then ready for imaging. Radiation induced skin tissue injury was scored by people blinded to the treatment of each sample.
Statistical analysis
SPSS13.0 software was used to analyze the data. The variances are presented as the mean ± S.E.M. Statistical significance among groups was determined by using the two-tailed Students t-test or ANOVA along with a post hoc Student's Newman-Keul test. The data were considered significant at P value<0.05.
Results
Low dose ionizing radiation induces adaptive protection in immortalized human skin keratinocytes
To investigate the mechanisms by which mammalian cells respond to low level of genotoxic stress, we first tested the response of cells to 5 Gy irradiation with/without 10 cGy low dose irradiation pretreatment using HK18 cells as an in vitro model system. We found that apoptosis rate of the cells caused by 5 Gy lethal high dose irradiation was significantly reduced by 10 cGy low dose irradiation pretreatment (Fig. 1A), which was paralleled with the lowered mitochondrial O2.- (Fig. 1B). This result is consistent with our previous findings showing that 10 cGy pre-treatment increases the clonogenic survival ability in HK18 cells exposing to the following challenging dose of 5 Gy radiation compared to cells exposed to 5 Gy radiation alone [21], confirming that LDIR is indeed a reliable stress source to induce the adaptive response in mammalian cells. Furthermore, MnSOD activity was increased in cells exposed to 10 cGy + 5 Gy IR compared to that of cells treated with 5 Gy alone (Fig. 1C), indicating that LDIR-induced adaptive protection might be mediated by MnSOD enzymatic activation.
Fig. 1.

Enhanced MnSOD enzymatic activity in LDIR-induced adaptive radioprotection in human skin keratinocytes. Human skin keratinocyte HK18 cells exposed to different dose or dose combinations of radiation (sham, 10 cGy, 10 cGy + 5 Gy and 5 Gy) were collected at desired time points for measuring (A) apoptosis, (B) mitochondrial superoxide level and (C) MnSOD enzymatic activity. Mean ± S.E.M.; *P < 0.05; n ≥ 3.
Enhanced MnSOD activity and mitochondrial metabolism in LDIR-induced adaptive response
Next, we tested the response of HK18 cells to 10 cGy LDIR. The mitochondria membrane potential was dramatically enhanced in the HK18 cells in response to LDIR with a peak at 6 h after radiation (Fig. 2A). MnSOD activity was induced, also peaking at 6 h after radiation with a 36% increase compare to the sham no-radiation control (Fig. 2B); however, the total cellular MnSOD and mitochondrial MnSOD protein levels did not show significant increase (Fig. 2C).
Fig. 2.

MnSOD post-translational modification is involved in LDIR-induced adaptive radioprotection. (A-E) HK18 cells were exposed to 10 cGy irradiation and collected at indicated times for measuring (A) mitochondria membrane potential, (B) MnSOD activity, (C) MnSOD protein levels in total cell lysate and mitochondrial fractions by western blot, (D) oxygen consumption and (E) mitochondria ATP generation. Sham, non-radiation control. Mean ± S.E.M.; *P < 0.05; **P < 0.01; n ≥ 3. (F) Enhanced MnSOD serine phosphorylation in LDIR-treated HK18 cells detected by Co-IP with anti-p-serine following by IB with anti-MnSOD and vice versa.
Since the MnSOD protein level did not correlate with the increased MnSOD enzymatic activity in these cells, we were wondering if the enhanced MnSOD activity could be linked to the potential post-translational modification. Indeed, co-IP analysis detected a substantial amount of serine-phosphorylated MnSOD in LDIR-treated HK18 cells (Fig. 2F), which, together with our previous observation of CDK1-mediated MnSOD activation [8], suggests the serine-phosphorylated MnSOD is required for enhancing MnSOD enzymatic activity and mitochondrial homeostasis in LDIR-induced adaptive protection. The cellular oxygen consumption (Fig. 2D) and mitochondrial ATP generation (Fig. 2E) were also peaked at 6 h, indicating that post-translational modification of MnSOD is linked with LDIR-induced mitochondria homeostasis.
Cyclin D1/CDK4 translocates to mitochondria and phosphorylates MnSOD
We then sought for the kinase mediating LDIR-induced MnSOD serine phosphorylation. Although Cyclin D1 is shown to regulate mitochondrial function [24] and MnSOD activity [32], the exact mechanism in Cyclin D1-controlled MnSOD activity is unknown. Here, we found that a substantial amount of Cyclin D1 and CDK4 was localized in mitochondria in HK18 cells after LDIR with a peak accumulation similar to MnSOD enzymatic activation without evident increase in cytosolic fractions (Fig. 3A and B). The total cellular Cyclin D1 and CDK4 levels were significantly enhanced (Fig. S1), indicating that most of LDIR-induced Cyclin D1/CDK4 proteins are localized in the mitochondria. Alkaline extraction analysis revealed that Cyclin D1 and CDK4 were localized in mitochondrial matrix and/or inter-membrane space in mitochondria, but not on the outer- or inner-membrane (Fig. 3C). Furthermore, co-immunoprecipitation assays showed an increased Cyclin D1/CDK4/MnSOD interaction in cells collected at 6 h after 10 cGy irradiation (Fig. 3D), which together with results of the in vitro CDK4 kinase assay using either wild type or S106A mutant MnSOD (Fig. 3E), suggesting that Cyclin D1/CDK4 directly phosphorylates MnSOD at S106, a minimal motif of serine/threonine kinase substrate.
Fig. 3.

Cyclin D1/CDK4 relocates into mitochondria in response to LDIR. (A) LDIR induces mitochondria translocation of Cyclin D1 and CDK4 in HK18 cells. (B) Mitochondrial but not cytosolic Cyclin D1/CDK4 complex was increased in response to LDIR. COX IV and α-tubulin were used to confirm the purity of mitochondria and cytosol fractions respectively. (C) Identification of sub-mitochondria localization of Cyclin D1/CDK4. T, total mitochondrial fraction; S, soluble matrix and intermembrane proteins; P, membrane pellets. IMS, inter-membrane space; OM, outer membrane. Hsp60, Timm13 and Tom40 serve as markers of mitochondria matrix, inter-membrane space and outer membrane respectively. (D) LDIR enhances protein interaction between MnSOD and Cyclin D1/CDK4 complex in mitochondria. MnSOD without IP serves as the equal input control. (E) Cyclin D1/CDK4 complex phosphorylates MnSOD on S106. Immunoblot of immunoprecipitation reactions with MnSOD antibody were used to confirm the equal amount of MnSOD used for each kinase assay reaction.
Serine-106 phosphorylation is required for enhanced MnSOD activity and mitochondria respiration in adaptive radioprotection in HK18 cells
To confirm if S106 phosphorylation is required for MnSOD activation and enhanced mitochondria metabolism, we constructed wild type and S106A mutant MnSOD expressing plasmids and established stable transfectants in HK18 cells. Plasmid constructs and confirmation of stable transfectants are shown in Fig. S2A and B. We found that MnSOD activity was enhanced in wt- but not S106A mutant MnSOD transfectants (Fig. 4A) and a substantial amount of LDIR-enhanced oxygen consumption and mitochondrial ATP generation was lost in the S106A mutant MnSOD transfectants (Fig. 4B and C), indicating that MnSOD S106 phosphorylation is required for its enzymatic activation and mitochondrial oxidative respiration. In addition, 10 cGy pretreatment of S106A mutant MnSOD transfected HK18 cells didn't show significant protection from following 5 Gy radiation compared to empty vector control cells tested by clonogenic survival assay (Figure 4D), suggesting that S106 phosphorylation contributes to MnSOD-mediated cell adaptive response and might be the only phosphorylation event functions in this radioadaptive response, supporting that S106 phosphorylation of MnSOD is required by LDIR-induced adaptive response.
Fig. 4.

Serine106 phosphorylation is required for MnSOD activation and LDIR-induced mitochondria respiration. HK18 cells were transfected with either wild type or S106A-mutant MnSOD and stable transfected cells were collected at 6 h after LDIR for measuring (A) MnSOD activity, (B) oxygen consumption, (C) mitochondrial ATP generation and (D) clonogenic survival assay. Mean ± S.E.M.; *P < 0.05; n ≥ 3.
Cyclin D1 is required for MnSOD activation in LDIR-induced radioprotection
To determine whether or not Cyclin D1 is required for MnSOD activation and LDIR-induced adaptive protection, we performed siRNA-mediated knockdown in HK18 cells. We found that Cyclin D1 siRNA transfected cells had a lower level of LDIR-induced p-serine-MnSOD (Fig. 5A) and MnSOD activity (Fig. 5B), along with increased mitochondrial superoxide (Fig. 5C), reduced mitochondrial membrane potential (Δψm) (Fig. 5D) and increased apoptosis (Fig. 5E) compared to scrambled siRNA and transfection reagent only control cells. Combined with our published data showing that HK18 cells treated with Cyclin D1 siRNA are deficient in LDIR-induced clonogenic radioprotection ability compared to scrambled siRNA treated control cells [21]. These results suggest that Cyclin D1 is required for MnSOD activation and adaptive response in HK18 cells.
Fig. 5.
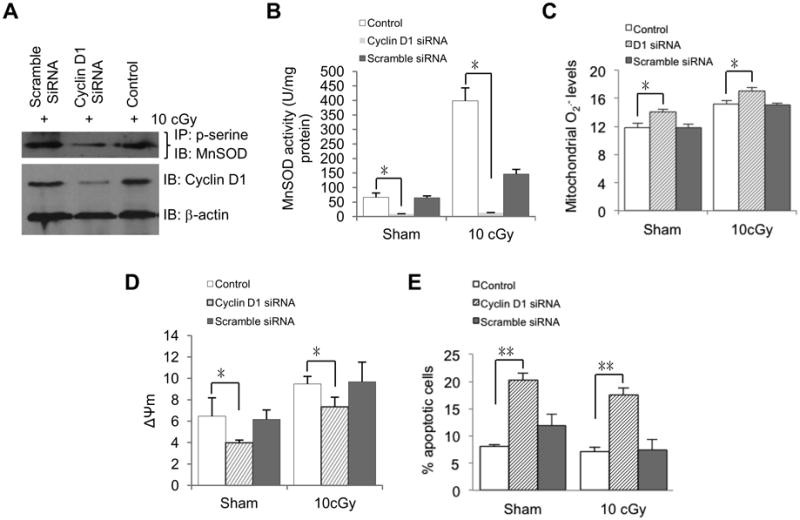
Cyclin D1 is required for MnSOD activation and enhanced mitochondria respiration in LDIR-induced radioprotection. HK18 cells were transfected with Cyclin D1-specific siRNA, scrambled siRNA or reagents only, respectively, and irradiated with 10 cGy 48 h after transfection. At 6 h after radiation, cells were collected to measure (A) p-serine-MnSOD, (B) MnSOD activity, (C) mitochondria superoxide, (D) mitochondria membrane potential and (E) apoptosis. Control, no siRNA transfection. Mean ± S.E.M.; *P < 0.05; n ≥ 3.
CDK4 kinase domain is required in LDIR-induced adaptive protection
To further confirm the role of Cyclin D1/CDK4-mediated MnSOD S106 phosphorylation in MnSOD activation and adaptive radioprotection in HK18 cells, we constructed mitochondria-targeted Cyclin D1 and CDK4 (either wild type or D158N kinase domain dominant negative mutant) expressing plasmids (Fig. S3A). Mitochondrial localization of MTS-Cyclin D1 and MTS-CDK4-wt in the stable transfectants was verified by fluorescent microscopy (Fig. S3B). The stable transfected cells were treated with 10 cGy low dose radiation and collected at 6h after radiation for each experiment. Consistent with the higher level of p-serine MnSOD (Fig. 6A), MnSOD activity was remarkably enhanced (∼7 fold) (Fig. 6B) along with decreased mitochondrial O2.- (Fig. 6C) in MTS-CDK4-wt cells but not MTS-CDK4-dn cells compared to empty vector control cells. Contrasted with MTS-CDK4-dn cells, MTS-CDK4-wt cells showed reduced apoptosis (Fig. 6D) and increased mitochondria membrane potential (Fig. 6E). To further confirm CDK4-mediated adaptive resistance, we did the clonogenic survival assay and found that colony formation ability of MTS-CDK4-dn cells receiving 5 Gy radiation were not significantly increased by 10 cGy pretreatment (Fig. 6F). Compared with wild type CDK4, the oxygen consumption (Fig. 6G) and mitochondrial ATP generation (Fig. 6H) were inhibited in the MTS-CDK4-dn cells.
Fig. 6.

CDK4 kinase activity is required for enhanced mitochondrial respiration and mitochondria-mediated anti-apoptosis. (A) HK18 cells were co-transfected with MTS-CDK4-wt or MTS-CDK4-dn along with Cyclin D1, and p-serine-MnSOD levels were measured at 6h after radiation in stable transfectants further transfected with MnSOD-flag. (B-G) HK18 cells co-transfected with MTS-CDK4-wt or MTS-CDK4-dn along with Cyclin D1 were treated with 10 cGy radiation and collected at 6h after radiation to measure (B) MnSOD activity, (C) mitochondria superoxide, (D) apoptosis, (E) mitochondrial membrane potential, (F) clonogenic survival assay, (G) oxygen consumption and (H) mitochondrial ATP production. MTS, mitochondria targeting sequence; wt, wild type CDK4; dn, D158N dominant negative mutant. GFP and RFP were used to confirm equal amount of inputs. Mean ± S.E.M.; *P < 0.05; n ≥ 3.
Cyclin D1/CDK4/MnSOD pathway in LDIR-induced adaptive protection in vivo
Wondering if Cyclin D1/CDK4/MnSOD pathway causes LDIR-induced adaptive response in vivo, we treated BALB/c mice by whole body radiation with the established regimen of radiation dose or doses combination (sham, 10 cGy, 10 cGy + 5 Gy, 5 Gy), and dissected mice at desired time points after radiation. Samples of whole skin tissues were extracted, fixed, embedded and sliced for H&E staining. Radiation induced skin injury was analyzed with the following criteria: 1) the layers of the skin especially dermis turn thinner; 2) the regular histological structure of skin is destroyed; 3) cells undergo apoptosis or necrosis; and 4) in some cases, inflammatory cells infiltrated in the dermis layer were accounted. As is shown in Figure 7A, we found that the mice receiving LDIR pre-treatment showed an improved radiation injury in skin with reduction of inflammatory cells and fibroblast proliferation after exposure to a challenging single high dose of radiation. In the skin epidermal tissues isolated from mice exposed to whole body LDIR, MnSOD activity was increased ∼40% (Fig. 7C), paralleled with an enhanced CDK4/MnSOD interaction and increased level of p-serine-MnSOD (Fig. 7B). Consistent with the features of mitochondrial respiration in irradiated cultured cells, mitochondrial oxygen consumption and ATP production were increased in the mitochondrial fractions isolated from liver tissues (Fig. 7D and E) of LDIR-treated mice.
Fig. 7.

Cyclin D1/CDK4/MnSOD pathway in LDIR-induced adaptive protection in vivo. Female BALB/c mice were exposed to whole body irradiation with different dose combination (sham, 10 cGy, 5 Gy or 10cGy+5Gy) or single low dose (10 cGy). Mice were then dissected at the desired time point after irradiation and tissues were used for immunohistochemistry assay or preparation of mitochondria fractions. Mean ± S.E.M.; *P < 0.05; n ≥ 3. (A) Representative histological changes and quantitative results of inflammatory cells in skin epidermal tissues from mice received indicated dose combination of whole body irradiation. Mice were exposed to 10 cGy, 10 cGy + 5 Gy or 5 Gy irradiation and dissected at the desired time points after radiation. Mouse skin tissues were embedded and sliced. The tissue sections were subjected to HE staining and the images were presented to show the skin injury by irradiation. Black arrow, lymphocyte infiltration. Blue arrow (5 Gy), abundant fibroblast proliferation. Sham, no radiation control. Scale bar, 50 μm. (B) Enhanced CDK4 interaction with MnSOD and increased MnSOD phosphorylation in mitochondria of skin epidermal layers from 10 cGy-irradiated mice. (C) MnSOD activity measured in skin epidermal tissues from mice treated with 10 cGy irradiation. (D) Enhanced mitochondria oxygen consumption in livers from 10 cGy irradiated mice. (E) Enhanced mitochondria ATP production in livers from 10 cGy irradiated mice. (F) Schematic show of CDK4/MnSOD signaling pathway in LDIR-induced cellular adaptive protection. LDIR induces Cyclin D1/CDK4 mitochondria relocation and phosphorylation of MnSOD at serine-106, causing an enhanced mitochondrial homeostasis via MnSOD activation and oxidative respiration.
Discussion
Mitochondrial homeostasis is believed to play a key role in stress-induced adaptive response. In this study, we demonstrated for the first time that Cyclin D1/CDK4 complex can relocate to mitochondria in radiation-induced adaptive response, contributing to mitochondrial homeostasis via MnSOD phosphorylation and enzymatic activation. The CDK4-mediated post-translational modification of MnSOD may serve as a critical pathway for a quick enhancement of mitochondrial metabolism for ATP generation and cell survival. CDK4 is also a critical cell cycle regulator controlling G1/S transition [33] and in glucose metabolism and insulin secretion, through regulating the associated gene expression [34]. Nuclear Cyclin D1 accompanied with its kinase partner CDK4 induces cell cycle progression but in the postmitotic cells Cyclin D1 is mainly localized in cytoplasm [35]. Although the cytoplasmic sequestration of Cyclin D1 is required for the survival of postmitotic neurons and nuclear relocalization of Cyclin D1 induces apoptosis [25], there is no direct evidence for CDK4-mediated mitochondrial targets. However, cytosolic accumulation of Cyclin D1 is required to enhance anti-apoptosis with improved mitochondrial membrane potential [21]. The current study reveals that a fraction of CDK4 is physically localized in the mitochondria. The mitochondrial Cyclin D1/CDK4 complex is able to reduce mitochondria superoxide levels via phosphorylation (S106) and activation of MnSOD and boosting the overall mitochondrial bioenergetics. A cluster of mitochondrial respiration chain complex I subunits has just been identified to be subtracts of CyclinB1/CDK1 for enhancing mitochondrial ATP generation required for cell cycle G2/M progression [36]. The exact mechanism underlying CDK4-MnSOD activation-mediated mitochondrial homeostasis and ATP generation is to be further investigated.
Contrasted with well-studied aspects of MnSOD including gene transcription and protein folding, potential MnSOD post-translational modifications and their physiologic functions are not well understood. O2·- is generated as the by-products of mitochondrial oxidative respiration as well as by different forms of oxidative stress including ionizing radiation, both can cause the primary damages in mtDNA and other mitochondrial macromolecules [37]. Mitochondrial O2·- is the major source of cellular ROS which is considered as redox signaling molecules [38] and may serve as a sensor for genotoxic stress and aging [39, 40]. MnSOD is encoded by genomic DNA but its antioxidant ability is fully activated after mitochondria translocation through a set of post-translational modifications, including acetylation/deacetylation, phosphorylation/dephosphorylation and nitration [41]. The current study using in vitro irradiated human skin keratinocytes and in vivo irradiated mouse tissues demonstrates that Cyclin D1/CDK4-mediated phosphorylation of mitochondria MnSOD is required for the elevated antioxidant ability of MnSOD, which compensates the increased cytotoxic ROS due to enhanced mitochondrial respiration. How phosphorylation of MnSOD affects its enzyme activity is still not clear. Our recent study shown that Cyclin B1/CDK1 links cell cycle G2/M transition via CDK1-mediated phosphorylation and activation of mitochondrial respiration chain [36] and CDK1 also phosphorylates MnSOD at the same site S106, which stabilizes MnSOD protein and enzymatically activates MnSOD possibly in LDIR induced adaptive response [8]. Although the exact mechanism of phosphorylation-mediated MnSOD enzymatic activation remains to be solved, the negative charges introduced by phosphorylation might enhance the affinity of the SOD2 protein to Mn2+ ion whose incorporation into the protein is necessary for SOD2 enzymatic activation during its mitochondrial translocation. In addition, S106 phosphorylation may enhance and stabilize MnSOD homotetramer conformation, which has proven contribute to the enzyme stability and activity of human mitochondrial MnSOD [16]. Our current data also suggest that the post-translational modifications of MnSOD proteins may be able to induce a quick adaptive reaction without the need of substantially regulation of SOD2 gene transcription in response to genotoxic stresses. We have previously observed that in LDIR-induced adaptive response of mouse skin epithelial cells, MnSOD protein level is significantly induced by either single LDIR or fractionated LDIR compared to no-radiation control paralleled with enhanced enzyme activity [7]. In human skin keratinocytes, however in the present studies, MnSOD enzymatic activity was found significantly enhanced without detectable increase in the protein level, indicating that different mechanisms might be involved in mouse and human cells for mitochondrial antioxidant regulation. Interestingly, MnSOD phosphorylation on the serine/threonine site is reported to decrease its enzyme activity in plant and bacteria [41] leaving specific phosphorylation site unidentified, indicating that phosphorylation of MnSOD at different amino acid might lead to different effects on its enzyme activity. Thus, it is highly possible that different mechanisms are adopted by varied species to enhance mitochondrial antioxidants and homeostasis under genotoxic stress conditions. The mechanisms underlying cell cycle regulators (CDK1 and CDK4)-mediated MnSOD S106 phosphorylation and enzymatic activation in LDIR-induced adaptive response and mitochondrial homeostasis need to be further investigated.
As is shown in our model in Figure 7F, MnSOD activity is induced by pre-exposure of 10 cGy radiation which is in agreement with our previously reported results [7, 8], leading to the increased mitochondrial homeostasis. Increased mitochondrial superoxide is reported contribute to the release of redox active metal ions, such as Fe(II) and Cu(I), which can be deleterious to cells due to the generation of highly reactive hydroxyl radical •OH and OH− by Fenton Reaction [42, 43] and multiple human diseases including cancer and neuronal diseases [44-46]. We didn't test these redox active metal ions in this study, but it is highly possible that LDIR induced MnSOD activation protects cells the excessive release of the redox active metal ions through scavenging increased mitochondrial superoxide. Neither our current data could not exclude the possibility of CDK4-mediated mitochondrial homeostasis is linked with DNA repair capacity. However, the increased DNA repair capacity has been reported in LDIR-induced radioadaptive resistance [47, 48]. Also the MnSOD activation detoxifies the superoxide that otherwise could generate additional DNA damages. On the other hand, the enhanced mitochondrial respiration may provide additional ATP required for repair of DNA and other large molecules in cells. These results support a cooperative mechanism between DNA repair system and mitochondrial bioenergetics, which is activated to meet the increased cellular energy demand for quickly DNA repair. Identification of the CDK4-mediated mitochondrial homeostasis will help to invent effective targets for radioprotection of normal cells and radiosensitize tumor cells.
Increasing evidence shows that cell cycle regulators are implicated in the regulation of mitochondria function and cellular energy metabolism either dependent or independent of cell cycle progression under the normal and stress conditions. CDK4 is the first member of cell cycle regulators proven implicated in the control of energy and glucose homeostasis through the regulation of genes participating in glucose metabolism via CDK4/pRB/E2F pathway [34]. Cyclin D1 represses mitochondrial metabolism through regulation of mitochondria gene expression, leading to reduced mitochondria activity and metabolism shift to glycolysis in vivo [23]. While CDK4-mediated pRB phosphorylation releases E2F to bind to the promoter region of target genes implicated in metabolism, leading to switch of energy metabolism from glycolysis to mitochondria oxidative phosphorylation under the stress of cold and fasting [49]. Nuclear-localized Cyclin D1/CDK4 has been found be able to control glucose homeostasis in postmitotic cell through regulation of gluconeogenic genes independently of cell cycle progression [50]. In normal physiological conditions, Cyclin D1 negatively regulates aerobic glycolysis and mitochondrial size and activity through inducing expression of a subset of genes governing mitochondrial activity [24], while under low dose radiation stress, cytoplasmic retained Cyclin D1 is required for enhanced mitochondria-mediated anti-apoptosis function, leading to adaptive radioprotection [21]. Besides Cyclin D1/CDK4, our previous studies have shown that Cyclin B1/CDK1 also participates in the regulation of mitochondria metabolism either in normal conditions or under low dose irradiation-induced cellular adaptive response. Under normal conditions, Cyclin B1/CDK1 kinase activity is involved in the regulation of mitochondrial metabolism through phosphorylation of mitochondria electron transport chain complex I subunits in mitochondria, providing energy for cells to G2/M transition [36], while in LDIR-induced cellular adaptive response, mitochondria localized Cyclin B1/CDK1 can phosphorylate multiple mitochondrial targets, such as p53 and MnSOD [8, 51]. Also our unpublished data indicate that mitochondria localized Cyclin B1/CDK1 contributes to regulation of SIRT3 enzyme activity through phosphorylation. Our current data extend these findings and demonstrate, for the first time, a new Cyclin D1/CDK4/MnSOD network, which communicates cell cycle kinases and mitochondrial homeostasis via phosphorylation of mitochondria key factors in LDIR induced cellular adaptive protection. These results support the concept that an overall status of cell cycle kinase-mediated phosphorylation of mitochondrial targets, such as MnSOD, p53, SIRT3 and complex I subunits, plays a pivotal role in regulation of mitochondrial homeostasis in cell adaptive response and survival to low levels of genotoxic stress. In addition to Cyclin B1/CDK1 and Cyclin D1/CDK4 complexes, there might be other currently unknown cell cycle dependent kinases that are also able to localize to mitochondria and phosphorylate mitochondrial targets to enhance mitochondrial homeostasis under various genotoxic stress conditions, especially sublethal levels of radiation, to allow cells to quickly respond to oxidative stress to adapt and survive. Further elucidation of such a cluster of kinases that control the mitochondrial respiration and antioxidant capacity under different genotoxic conditions may reveal additional new information on the mechanism underlying mitochondrial homeostasis during cell survival. Since CDK4 is activated in G1/S transition whereas CDK1 is required for G2/M transition, it is highly possible that Cyclin D1/CDK4-mediated MnSOD phosphorylation and mitochondrial activation is related to the mitochondrial regulation during different cellular energy demands during cell cycle progression using the same phosphorylation site of MnSOD.
In summary, this study demonstrates a unique CDK4-MnSOD activation pathway in mitochondrial homeostasis (Figure 7F), in which mitochondrial-localized Cyclin D1/CDK4 phosphorylates MnSOD at Serine 106 (S106) to quickly increase MnSOD enzymatic activity required for the redox balancing and boosted mitochondria respiration in cellular adaptive response to environmental genotoxic stress conditions such as low dose ionizing radiation.
Supplementary Material
Highlights.
Radiation enhances CDK4 mitochondrial relocation and MnSOD activity.
Mitochondrial CDK4 phosphorylates MnSOD Ser106.
CDK4-mediated MnSOD activation increases mitochondrial respiration.
CDK4/MnSOD pathway is activated in in vivo radiation induced adaptive protection.
Acknowledgments
We appreciate the active discussion from graduate students at the Jian Jian Li's lab and the Comparative Pathology Program of the University of California at Davis. This work was supported by National Institutes of Health (CA152313 to J.J.L.) and the Department of Energy Office of Science (DE-SC0001271 to G.W., J.J.L., D.J.G.).
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
Author Contributions: C. J., L.Q. performed most of the experiments; Y.S., D.C., C.L., M.F. conducted the experiments of mitochondrial functions; C.J., L.Q., R.L., M.F. performed the in vivo mouse experiments; R.S. studied pathological sections of mouse tissues; A.T.V., L.S.W., R.F.L., J.S.M., G.W., D.J.G., J.J.L. discussed the data and edited the paper; C.J., L.Q., and J.J.L. designed the study and wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Feinendegen LE, Bond VP, Sondhaus CA, Muehlensiepen H. Radiation effects induced by low doses in complex tissue and their relation to cellular adaptive responses. Mutat Res. 1996;358:199–205. doi: 10.1016/s0027-5107(96)00121-2. [DOI] [PubMed] [Google Scholar]
- 2.Wolff S. Are radiation-induced effects hormetic? Science. 1989;245:575. doi: 10.1126/science.2762808. [DOI] [PubMed] [Google Scholar]
- 3.Feinendegen LE, Bond VP, Sondhaus CA, Altman KI. Cellular signal adaptation with damage control at low doses versus the predominance of DNA damage at high doses. C R Acad Sci III. 1999;322:245–251. doi: 10.1016/s0764-4469(99)80051-1. [DOI] [PubMed] [Google Scholar]
- 4.Liu SZ. Biological effects of low level exposures to ionizing radiation: theory and practice. Hum Exp Toxicol. 2010;29:275–281. doi: 10.1177/0960327109363967. [DOI] [PubMed] [Google Scholar]
- 5.Oberley LW, Buettner GR. Role of superoxide dismutase in cancer: a review. Cancer Res. 1979;39:1141–1149. [PubMed] [Google Scholar]
- 6.McCord JM, Keele BB, Jr, Fridovich I. An enzyme-based theory of obligate anaerobiosis: the physiological function of superoxide dismutase. Proc Natl Acad Sci USA. 1971;68:1024–1027. doi: 10.1073/pnas.68.5.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fan M, Ahmed KM, Coleman MC, Spitz DR, Li JJ. Nuclear factor-kappaB and manganese superoxide dismutase mediate adaptive radioresistance in low-dose irradiated mouse skin epithelial cells. Cancer Res. 2007;67:3220–3228. doi: 10.1158/0008-5472.CAN-06-2728. [DOI] [PubMed] [Google Scholar]
- 8.Candas D, Fan M, Nantajit D, Vaughan AT, Murley JS, Woloschak GE, Grdina DJ, Li JJ. CyclinB1/Cdk1 phosphorylates mitochondrial antioxidant MnSOD in cell adaptive response to radiation stress. J Mol Cell Biol. 2013;5:166–175. doi: 10.1093/jmcb/mjs062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grdina DJ, Murley JS, Miller RC, Mauceri HJ, Sutton HG, Thirman MJ, Li JJ, Woloschak GE, Weichselbaum RR. A Manganese Superoxide Dismutase (SOD2)-Mediated Adaptive Response. Radiat Res. 2012 doi: 10.1667/RR3126.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murley JS, Baker KL, Miller RC, Darga TE, Weichselbaum RR, Grdina DJ. SOD2-mediated adaptive responses induced by low-dose ionizing radiation via TNF signaling and amifostine. Free Radic Biol Med. 2011;51:1918–1925. doi: 10.1016/j.freeradbiomed.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veeraraghavan J, Natarajan M, Herman TS, Aravindan N. Low-dose gamma-radiation-induced oxidative stress response in mouse brain and gut: regulation by NFkappaB-MnSOD cross-signaling. Mutat Res. 2011;718:44–55. doi: 10.1016/j.mrgentox.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 12.Yen HC, Oberley TD, Vichitbandha S, Ho YS, St Clair DK. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J Clin Invest. 1996;98:1253–1260. doi: 10.1172/JCI118909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Epperly MW, Sikora C, Defilippi S, Bray J, Koe G, Liggitt D, Luketich JD, Greenberger JS. Plasmid/liposome transfer of the human manganese superoxide dismutase transgene prevents ionizing irradiation-induced apoptosis in human esophagus organ explant culture. Int J Cancer. 2000;90:128–137. doi: 10.1002/1097-0215(20000620)90:3<128::aid-ijc2>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 14.Niu Y, Wang H, Wiktor-Brown D, Rugo R, Shen H, Huq MS, Engelward B, Epperly M, Greenberger JS. Irradiated esophageal cells are protected from radiation-induced recombination by MnSOD gene therapy. Radiat Res. 2010;173:453–461. doi: 10.1667/RR1763.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murley JS, Kataoka Y, Weydert CJ, Oberley LW, Grdina DJ. Delayed radioprotection by nuclear transcription factor kappaB -mediated induction of manganese superoxide dismutase in human microvascular endothelial cells after exposure to the free radical scavenger WR1065. Free Radic Biol Med. 2006;40:1004–1016. doi: 10.1016/j.freeradbiomed.2005.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borgstahl GE, Parge HE, Hickey MJ, Beyer WF, Jr, Hallewell RA, Tainer JA. The structure of human mitochondrial manganese superoxide dismutase reveals a novel tetrameric interface of two 4-helix bundles. Cell. 1992;71:107–118. doi: 10.1016/0092-8674(92)90270-m. [DOI] [PubMed] [Google Scholar]
- 17.Chen Y, Zhang J, Lin Y, Lei Q, Guan KL, Zhao S, Xiong Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011;12:534–541. doi: 10.1038/embor.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, Olivier AK, Spitz DR, Gius D. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell. 2010;40:893–904. doi: 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quijano C, Hernandez-Saavedra D, Castro L, McCord JM, Freeman BA, Radi R. Reaction of peroxynitrite with Mn-superoxide dismutase. Role of the metal center in decomposition kinetics and nitration. J Biol Chem. 2001;276:11631–11638. doi: 10.1074/jbc.M009429200. [DOI] [PubMed] [Google Scholar]
- 20.Roue G, Pichereau V, Lincet H, Colomer D, Sola B. Cyclin D1 mediates resistance to apoptosis through upregulation of molecular chaperones and consequent redistribution of cell death regulators. Oncogene. 2008;27:4909–4920. doi: 10.1038/onc.2008.126. [DOI] [PubMed] [Google Scholar]
- 21.Ahmed KM, Fan M, Nantajit D, Cao N, Li JJ. Cyclin D1 in low-dose radiation-induced adaptive resistance. Oncogene. 2008;27:6738–6748. doi: 10.1038/onc.2008.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agami R, Bernards R. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell. 2000;102:55–66. doi: 10.1016/s0092-8674(00)00010-6. [DOI] [PubMed] [Google Scholar]
- 23.Sakamaki T, Casimiro MC, Ju X, Quong AA, Katiyar S, Liu M, Jiao X, Li A, Zhang X, Lu Y, Wang C, Byers S, Nicholson R, Link T, Shemluck M, Yang J, Fricke ST, Novikoff PM, Papanikolaou A, Arnold A, Albanese C, Pestell R. Cyclin D1 determines mitochondrial function in vivo. Mol Cell Biol. 2006;26:5449–5469. doi: 10.1128/MCB.02074-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang C, Li Z, Lu Y, Du R, Katiyar S, Yang J, Fu M, Leader JE, Quong A, Novikoff PM, Pestell RG. Cyclin D1 repression of nuclear respiratory factor 1 integrates nuclear DNA synthesis and mitochondrial function. Proc Natl Acad Sci USA. 2006;103:11567–11572. doi: 10.1073/pnas.0603363103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sumrejkanchanakij P, Tamamori-Adachi M, Matsunaga Y, Eto K, Ikeda MA. Role of cyclin D1 cytoplasmic sequestration in the survival of postmitotic neurons. Oncogene. 2003;22:8723–8730. doi: 10.1038/sj.onc.1206870. [DOI] [PubMed] [Google Scholar]
- 26.Hagen KR, Zeng X, Lee MY, Tucker Kahn S, Harrison Pitner MK, Zaky SS, Liu Y, O'Regan RM, Deng X, Saavedra HI. Silencing CDK4 radiosensitizes breast cancer cells by promoting apoptosis. Cell Div. 810:2013. doi: 10.1186/1747-1028-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spitz DR, Oberley LW. An assay for superoxide dismutase activity in mammalian tissue homogenates. Anal Biochem. 1989;179:8–18. doi: 10.1016/0003-2697(89)90192-9. [DOI] [PubMed] [Google Scholar]
- 28.Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 29.Lu G, Ren S, Korge P, Choi J, Dong Y, Weiss J, Koehler C, Chen JN, Wang Y. A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes Dev. 2007;21:784–796. doi: 10.1101/gad.1499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mukhopadhyay P, Rajesh M, Hasko G, Hawkins BJ, Madesh M, Pacher P. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat Protoc. 2007;2:2295–2301. doi: 10.1038/nprot.2007.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vives-Bauza C, Yang L, Manfredi G. Assay of mitochondrial ATP synthesis in animal cells and tissues. Methods Cell Biol. 2007;80:155–171. doi: 10.1016/S0091-679X(06)80007-5. [DOI] [PubMed] [Google Scholar]
- 32.Marchal JA, Nunez MC, Suarez I, Diaz-Gavilan M, Gomez-Vidal JA, Boulaiz H, Rodriguez-Serrano F, Gallo MA, Espinosa A, Aranega A, Campos JM. A synthetic uracil derivative with antitumor activity through decreasing cyclin D1 and Cdk1, and increasing p21 and p27 in MCF-7 cells. Breast Cancer Res Treat. 2007;105:237–246. doi: 10.1007/s10549-006-9450-2. [DOI] [PubMed] [Google Scholar]
- 33.Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer. 2001;1:222–231. doi: 10.1038/35106065. [DOI] [PubMed] [Google Scholar]
- 34.Blanchet E, Annicotte JS, Fajas L. Cell cycle regulators in the control of metabolism. Cell Cycle. 2009;8:4029–4031. doi: 10.4161/cc.8.24.10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamamori-Adachi M, Ito H, Sumrejkanchanakij P, Adachi S, Hiroe M, Shimizu M, Kawauchi J, Sunamori M, Marumo F, Kitajima S, Ikeda MA. Critical role of cyclin D1 nuclear import in cardiomyocyte proliferation. Circ Res. 2003;92:e12–19. doi: 10.1161/01.res.0000049105.15329.1c. [DOI] [PubMed] [Google Scholar]
- 36.Wang Z, Fan M, Candas D, Zhang TQ, Qin L, Eldridge A, Wachsmann-Hogiu S, Ahmed KM, Chromy BA, Nantajit D, Duru N, He F, Chen M, Finkel T, Weinstein LS, Li JJ. Cyclin B1/Cdk1 coordinates mitochondrial respiration for cell-cycle G2/M progression. Dev Cell. 2014;29:217–232. doi: 10.1016/j.devcel.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henderson JR, Swalwell H, Boulton S, Manning P, McNeil CJ, Birch-Machin MA. Direct, real-time monitoring of superoxide generation in isolated mitochondria. Free Radic Res. 2009;43:796–802. doi: 10.1080/10715760903062895. [DOI] [PubMed] [Google Scholar]
- 38.Rigoulet M, Yoboue ED, Devin A. Mitochondrial ROS generation and its regulation: mechanisms involved in H(2)O(2) signaling. Antioxid Redox Signal. 2011;14:459–468. doi: 10.1089/ars.2010.3363. [DOI] [PubMed] [Google Scholar]
- 39.Figueira TR, Barros MH, Camargo AA, Castilho RF, Ferreira JC, Kowaltowski AJ, Sluse FE, Souza-Pinto NC, Vercesi AE. Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxid Redox Signal. 2013;18:2029–2074. doi: 10.1089/ars.2012.4729. [DOI] [PubMed] [Google Scholar]
- 40.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 41.Dhar SK, St Clair DK. Manganese superoxide dismutase regulation and cancer. Free Radic Biol Med. 2012;52:2209–2222. doi: 10.1016/j.freeradbiomed.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 42.Stohs SJ, Bagchi D. Oxidative mechanisms in the toxicity of metal ions. Free Radic Biol Med. 1995;18:321–336. doi: 10.1016/0891-5849(94)00159-h. [DOI] [PubMed] [Google Scholar]
- 43.Leonard SS, Harris GK, Shi X. Metal-induced oxidative stress and signal transduction. Free Radic Biol Med. 2004;37:1921–1942. doi: 10.1016/j.freeradbiomed.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 44.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 45.Lee JC, Son YO, Pratheeshkumar P, Shi X. Oxidative stress and metal carcinogenesis. Free Radic Biol Med. 2012;53:742–757. doi: 10.1016/j.freeradbiomed.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 46.Hureaua C. Coordination of redox active metal ions to the amyloid precursor protein and to amyloid-β peptides involved in Alzheimer disease. Coordination Chemistry Reviews. 2012;256:2164–2174. [Google Scholar]
- 47.Pollycove M. Radiobiological basis of low-dose irradiation in prevention and therapy of cancer. Dose Response. 2007;5:26–38. doi: 10.2203/dose-response.06-112.Pollycove. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Phan N, De Lisio M, Parise G, Boreham DR. Biological effects and adaptive response from single and repeated computed tomography scans in reticulocytes and bone marrow of C57BL/6 mice. Radiat Res. 2012;177:164–175. doi: 10.1667/rr2532.1. [DOI] [PubMed] [Google Scholar]
- 49.Blanchet E, Annicotte JS, Lagarrigue S, Aguilar V, Clape C, Chavey C, Fritz V, Casas F, Apparailly F, Auwerx J, Fajas L. E2F transcription factor-1 regulates oxidative metabolism. Nat Cell Biol. 2011;13:1146–1152. doi: 10.1038/ncb2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee Y, Dominy JE, Choi YJ, Jurczak M, Tolliday N, Camporez JP, Chim H, Lim JH, Ruan HB, Yang X, Vazquez F, Sicinski P, Shulman GI, Puigserver P. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature. 2014 doi: 10.1038/nature13267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nantajit D, Fan M, Duru N, Wen Y, Reed JC, Li JJ. Cyclin B1/Cdk1 phosphorylation of mitochondrial p53 induces anti-apoptotic response. PLoS ONE. 2010;5:e12341. doi: 10.1371/journal.pone.0012341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.