

Abstract
Tumor-initiating cells (TICs) perpetuate tumor growth, enable therapeutic resistance, and drive initiation of successive tumors. Virtually nothing is known about the role of mechanotransductive signaling in controlling TIC tumorigenesis, despite the recognized importance of altered mechanics in tissue dysplasia and the common observation that extracellular matrix (ECM) stiffness strongly regulates cell behavior. To address this open question, we cultured primary human glioblastoma (GBM) TICs on laminin-functionalized ECMs spanning a range of stiffnesses. Surprisingly, we found that these cells were largely insensitive to ECM stiffness cues, evading the inhibition of spreading, migration, and proliferation typically imposed by compliant ECMs. We hypothesize that this insensitivity may result from insufficient generation of myosin-dependent contractile force. Indeed, we found that both pharmacologic and genetic activation of cell contractility through RhoA GTPase, Rho-associated kinase (ROCK), or myosin light chain kinase (MLCK) restored stiffness-dependent spreading and motility, with TICs adopting the expected rounded and non-motile phenotype on soft ECMs. Moreover, constitutive activation of RhoA restricted three-dimensional invasion in both spheroid implantation and transwell paradigms. Orthotopic xenotransplantation studies revealed that control TICs formed tumors with classical GBM histopathology including diffuse infiltration and secondary foci, whereas TICs expressing a constitutively active mutant of RhoA produced circumscribed masses and yielded a 30% enhancement in mean survival time. This is the first direct evidence that manipulation of mechanotransductive signaling can alter the tumor-initiating capacity of GBM TICs, supporting further exploration of these signals as potential therapeutic targets and predictors of tumor initiating capacity within heterogeneous tumor cell populations.
Keywords: Brain tumor, Cellular mechanotransduction, Rho GTPases, Cytoskeleton
Introduction
Glioblastoma (GBM) is the most aggressive primary brain tumor and is characterized by poor survival even in the setting of surgery, radiation, and chemotherapy (1). Diffuse invasion of single cells within the parenchyma and along vascular structures frequently renders complete surgical resection impossible and leads to recurrence and eventual mortality. This in turn has motivated much effort to understand mechanisms of GBM invasion, an important goal of which is to discover potential molecular targets that could be manipulated to slow disease progression (2,3). While soluble and cell-bound factors have long been recognized as important regulators of tumor invasion, it has only recently become clear that biochemical and biophysical cues encoded in the extracellular matrix (ECM) can also strongly regulate tumor invasion. For example, in previous work we showed that ECM rigidity can regulate GBM cell adhesion, motility, and proliferation, which in turn requires the contractile activity of myosin II (4), as well as the cell-ECM adhesion proteins α-actinin (5) and talin-1 (6). More specifically, when GBM tumor cells are cultured on comparatively stiff ECMs, they spread and migrate very readily whereas they adopt a rounded and immotile phenotype on highly compliant (soft) ECMs. This is consistent with observations in other tumor types that ECM stiffening can promote tissue dysplasia and local invasion through integrin-dependent potentiation of cell-ECM adhesion (7–9).
Much of our understanding of the importance of cell-ECM mechanotransduction in tumor progression is based on the use of continuous cell lines or, less commonly, heterogeneous primary tumor samples that are derived from the bulk tumor. While these studies have provided much valuable mechanistic insight, it has become clear over the past decade that a very specific and comparatively rare subpopulation of cells plays especially key roles in populating the tumor and driving tumor recurrence following chemo- and radiotherapy. In the case of GBM, these tumor-initiating cells (TICs) are formally defined by their ability to recapitulate the original tumor when orthotopically xenografted into immunocompromised mice and are characterized by expression of a specific complement of molecular markers (e.g., CD133, nestin) and stem cell-like properties of self-renewal and differentiation into various tissue lineages. In addition to repopulating tumors, TICs directly participate in the invasion process in vivo. For example, both primary GBM TICs (10) and H-Ras-transduced neural stem cells (11) invade brain tissue prior to forming the tumor mass. Moreover, hypoxia, which is often associated with the TIC niche (12) and the necrotic tumor core in which GBM TICs may reside, can enhance migration of GBM tumor cells through induction of the family of hypoxia-inducible factors (13,14).
Invasive motility through tissue is a physically integrated process that requires tumor cells to sense ECM-based mechanical inputs, dramatically change their shape, and exert propulsive forces against the microenvironment (2,15). Interestingly, many TIC markers such as Oct 3/4 (16) and SOX2 (17,18) have been shown to regulate cell behaviors that require cell-ECM mechanotransduction, such as motility and invasion. Conversely, several proteins long known for their adhesive or cytoskeletal function have more recently been discovered to be highly enriched and functionally important in TICs, such as the actin-binding protein girdin (19), the α6 integrin subunit (20,21), and the hyaluronan receptor CD44 (22) and its effector moesin (23).
These and other studies led us to speculate that cell-ECM mechanotransductive signaling systems play key roles in the ability of GBM TICs to interact with brain ECM and infiltrate tissue, and that targeting these systems may limit tumor growth and progression in vivo. Here, we explore this important open question by combining materials fabrication, single-cell biophysical tools, in vitro characterization of primary GBM TICs, and mouse xenograft models. We show that GBM TICs are capable of evading restrictions on spreading, motility, and self-renewal normally imposed by ECMs with compliance comparable to brain tissue, and that this mechanosensitivity may be restored by activation of myosin-dependent contractility. This contractile activation has the additional effect of limiting tumor invasion in a mouse orthotopic xenograft model and dramatically enhancing survival. Our work establishes the importance of cell-ECM mechanotransductive signaling in the initiation of GBM tumors and suggests a new set of molecular targets that may be manipulated to limit tumor infiltration.
Materials and Methods
Tumor sample and primary cell culture
The two patient-specific human brain tumor samples used in this study, L0 and L2, were collected in a previous study (10) after informed consent from male patients who underwent surgical treatment and Institutional Review Board approval. Briefly, the extracted tissue was placed in an enzymatic cocktail containing trypsin/ethylenediaminetetraacetic acid (0.05%) for 10 minutes at 37°C and filtered through a 40 μm filter. Cells were then propagated in neurosphere assay growth conditions (24) with serum-free media (Neurocult NS-A Proliferation kit, Stem Cell Technologies) that contained epidermal growth factor (EGF, 20 ng/ml, R&D), basic fibroblast growth factor (bFGF, 10 ng/ml, R&D), and heparin (0.2% diluted in phosphate buffered saline, Sigma). The tumor cells form gliomaspheres in suspension under these culture conditions and were serially passaged every 5–7 days when spheres reached a diameter of about 150 μm according to a previously established protocol (10). Gliomaspheres were dissociated with trypsin/ethylenediaminetetraacetic acid (0.05%) for 2 minutes and then re-plated in fresh media with addition of EGF, bFGF, and heparin. Both lines used were only passaged less than 20 times. These cells have been transcriptionally characterized and classified as the Classical subtype of GBM (25). Short tandem repeat (STR) analysis (University of Arizona Genetics Core) confirmed that these cells had not been contaminated by any known cell lines.
Continuous cell line culture
U373-MG human glioblastoma cells were obtained from the University of California, Berkeley Tissue Culture Facility in 2007, which obtained its cultures directly from the American Type Culture Collection (ATCC) in 1995. Frozen stocks were made immediately upon receipt, and cultured for less than six months for experiments. We note that STR analysis has recently revealed ATCC U373-MG cells share a common origin with the U251-MG glioma cell line (26), although these lines may have subsequently diverged to exhibit differential drug sensitivities (27). The tumor cells were cultured adherently in DMEM (Life Technologies) supplemented with 10% calf serum (J.R. Scientific) and 1% penicillin/streptomycin, MEM nonessential amino acids, and sodium pyruvate (Life Technologies).
Differentiation of brain tumor-initiating cells
A previously established protocol for differentiating tumor-initiating cells was followed (10). Briefly, 5% calf serum was added to basal culture media that lacked growth factors. After 7–10 days, expression of lineage markers was assayed using immunofluorescence. For BMP-4 induced differentiation (Fig. S2), TICs were incubated with BMP-4 (100 ng/ml, R&D Systems) in the absence of growth factors (bFGF, EGF, heparin) in adherent culture for 48h (28).
Xenotransplantation of human primary brain tumor-initiating cells into mice
Female eight-week old non-obese diabetic/severe combined immunodeficient gamma (NSG) mice (NOD.Cg-Prkdc(scid)Il2rg(tm1Wjl)/SzJ) were implanted intracranially with 200,000 L0 TICs (CA RhoA or Control) following institutional and national regulations, and according to a previously establish protocol (10). The mice were treated daily with doxycyline (625 mg/kg) 4 days post implant until endpoint. Five mice were used for each cohort and followed until death.
Statistical methods
Data are reported as mean ± standard error. All pairwise statistical comparisons were performed with Student’s unpaired t test, except as noted. Details of comparisons and replicates are provided in the appropriate figure legend.
Results
Tumor-initiating population is maintained on soft and stiff ECMs
Before asking whether ECM-encoded stiffness cues play instructive roles in regulating GBM TIC behavior, we first assessed the degree to which ECM stiffness might exert selective pressures that could compromise GBM TIC tumor-initiating or differentiation properties. To do this, we isolated two GBM TIC lines, L0 and L2, from human primary GBM tumors as previously described; unlike continuous culture models of GBM, these TICs generate tumors with pathologic hallmarks and invasion patterns of GBM when orthotopically xenografted in immunocompromised mice, even after multiple passages in culture (10). We then cultured these GBM TICs in neurobasal growth medium on laminin-coated polyacrylamide gels of rigidity varying from 0.08 kPa to 119 kPa (4). In a previous study, it was shown that the top 5% most slowly cycling GBM TIC fraction contained the most tumorigenic cells (10). To rule out the possibility that extracellular matrix (ECM) rigidity might be selecting for or against this key subpopulation, we first applied a serial dye dilution assay to measure the distribution of cell cycling time (Fig. 1A). Flow cytometry of carboxyfluorescein-treated cells revealed fluorescence distributions similar to previously reported results and highly overlapping across all ECM rigidities. This result indicates that a slow cycling, tumor-initiating subpopulation is preserved across all ECM rigidities (Fig. 1A, arrow), prompting us to use the overall unsorted population for all subsequent studies. As additional evidence for an absence of selection, 100% of GBM TICs were found to express the self-renewal markers nestin and SOX2 (Fig. 1B) and could be differentiated by serum into glial and neuronal marker-positive cells across all ECM rigidities (Fig. 1C). Both cell lines demonstrated qualitatively similar behavior. Thus, alterations in ECM rigidity do not preferentially select for or against slow-cycling highly tumorigenic cells, alter stem cell marker expression, or compromise differentiation.
Figure 1. Human primary glioblastoma tumor-initiating cells (GBM TICs) retain stem-like properties when cultured on soft and stiff polyacrylamide gels coated with laminin.

(A) ECM stiffness does not select against slow-cycling GBM TICs. GBM TICs were treated with carboxyfluorescein diacetate succinimidyl ester (CFSE) and analyzed by flow cytometry to determine distribution of cycling rate. Based on the persistence of the high-CFSE shoulder (arrow), the slow cycling population is retained on all gel stiffnesses. L2 curves are shown; L0 displayed similar results. (B) GBM TICs also express the stem cell markers,nestin (top row; red; 100% positive) and SOX2 (bottom row; red; 100% positive) on all ECM stiffnesses. (C) All ECM stiffnesses permit neuronal and glial differentiation. Addition of 5% serum to GBM TIC cultures results in differentiation into glial (GFAP, green) and neuronal lineages (βIII tubulin, red).
Tumor-initiating cells can spread, migrate, and proliferate on soft and stiff ECMs
In previous studies with continuous human GBM cell lines, we demonstrated that soft ECMs comparable to brain tissue (0.08–0.8 kPa) can strongly limit cell spreading, motility, and proliferation (4). We therefore asked whether similar regulatory effects would be observed for GBM TICs. To our surprise, L2 TICs adopted the same elongated morphology on soft ECMs as on stiff ECMs (Fig. 2A), with no significant differences in projected spread area (Fig. 2B). Moreover, GBM TICs did not exhibit prominent stress fibers or mature focal adhesions (Fig. 2C), structures strongly promoted by stiff ECMs in many other cell types (4,8,29). Qualitatively similar results were obtained for the L0 TIC line (data not shown). For both L0 and L2 TICs, random cell migration speed was only weakly sensitive to ECM rigidity (Fig. 2D), whereas proliferation was completely insensitive to ECM rigidity, with GBM TICs robustly migrating and proliferating even on the most compliant matrices (Fig. 2E). It occurred to us that the reduced stiffness sensitivity of GBM TICs relative to continuous GBM cell lines might simply be a consequence of the fact that GBM TICs are cultured in a neurobasal, serum-free medium whereas continuous lines are typically cultured in a DMEM-based medium supplemented with 10% serum, which contains both ECM components and activators of adhesion and contractility (4,10). To rule out this possibility, we cultured the continuous human GBM cell line U373-MG in both neurobasal and DMEM-based medium with or without 10% serum supplementation and measured average cell speed across a range of ECM stiffnesses (Supplementary Fig. S1). To assess correlations between stiffness-sensitivity and tumor-initiating capacity, we repeated these experiments with TICs after a brief (48 h) treatment with bone morphogenetic protein 4 (BMP4), which has previously been shown to strongly inhibit tumorigenic potential (28) (Supplementary Fig. S2). Notably, BMP4 treatment restored ECM mechanosensitivity with respect to cell spread area but not motility, with the exception of L2 on the stiffest ECM.
Figure 2. Tumor-initiating cells can spread, migrate, and proliferate on soft and stiff 2-D ECMs.

(A) Effect of ECM stiffness on cell spreading. GBM TICs were cultured on laminin-coated polyacrylamide matrices with stiffnesses ranging from 0.08kPa to 119 kPa, and on laminin-coated glass. Phase contrast imaging reveals that GBM TICs can spread on ECMs of all stiffnesses. (B) Quantification of projected cell area shows that ECM rigidity does not regulate L2 TIC spreading area. n = 20 cells (pooled from at least 3 technical and 3 biological experiments) for all conditions. L0 TICs (not shown) exhibited qualitatively similar data. (C) GBM TICs do not form prominent stress fibers or focal adhesions on stiff ECMs. Cells were fixed and stained for the focal adhesion marker vinculin (red), F-actin (green), and nuclear DNA (blue). (D) Effect of ECM stiffness on cell motility. Time-lapse phase contrast imaging of random cell motility over 8–12 hours indicate that the migration speed depends very weakly on ECM stiffness, with a modest optimum at 0.8 kPa. *p<0.05 and **p<0.001 relative to glass; n > 80 cells for all conditions. (E) Effect of ECM stiffness on cell proliferation. Cells were incubated with BrdU for 45 minutes and stained with 7AAD before analysis by flow cytometry. Plots show sample means with standard error. n > 3 independent experiments for all conditions.
Myosin II-dependent contractility restricts rigidity dependent spreading, motility, and invasion
Previously, we had shown that myosin II-dependent contractility is required for GBM tumor cells to sense ECM rigidity, in as much as pharmacologic inhibition of myosin rescues spreading and motility on compliant ECMs (4). This observation is consistent with a model in which cells must exert traction forces through myosin motors to deform the ECM and sample local stiffness (30,31). We therefore hypothesized that the ability of GBM TICs to evade motility limitations imposed by soft ECMs derives from insufficient generation of myosin-based contractile forces. Myosin-dependent cell contractility is activated by RhoA GTPase, which promotes myosin phosphorylation by activating Rho-associated kinase (ROCK) (32). We thus performed gain-of-function studies in which we overexpressed a constitutively activated (CA) mutant of RhoA (Fig. 3A) to determine if increased contractility could restore suppression of motility on soft ECMs. Western blots confirmed elevation of phosphorylated myosin light chain expression in the constitutively active GBM TIC lines (Fig. 3B). To investigate the effect of CA RhoA on cellular mechanical properties, we used atomic force microscopy (AFM) to measure cell elasticity (Fig. 3C). We found that transduction with CA RhoA conserved cell stiffness for L0 while pushing the observed stiffness range to higher values and significantly increased cell stiffness for L2 relative to control GBM TICs transduced with the control empty vector.
Figure 3. Increased Rho GTPase activation leads to stiffness dependent spreading and motility.

(A) Genetic strategy for inducible activation of contractility. A constitutively active (CA) mutant of RhoA (or CA ROCK/CA MLCK as noted) was placed under the control of a doxycycline-inducible promoter, placed into a lentiviral vector, and used to transduce GBM TICs to yield stable cells lines. (B) RhoA induction increases myosin light chain phosphorylation. Western blot analysis confirms that induction of CA RhoA increases myosin light chain phosphorylation relative to control cells transduced with an empty vector. Data shown are for the L0 TIC line. n = 3 independent experiments. (C) CA RhoA preserves or enhances cell stiffness. AFM measurements reveal that overexpression of CA RhoA preserves mean cell stiffness in L0 cells (while pushing the range of observed stiffness values to higher values) and increases mean cell stiffness in L2 cells, consistent with an increase in cell contractility. n = 10–20 cells pooled from at least 3 biological and technical replicates. (D) CA RhoA restores ECM stiffness-dependent spreading. Phase contrast imaging reveals that overexpression of CA RhoA produces cell rounding on soft ECMs, in contrast to control-transduced cells, which spread on ECMs spanning the entire range of stiffness. (E) Quantification of projected cell area shows that CA RhoA rescues stiffness dependent cell spreading, with significantly decreased spread area for CA RhoA TIC on soft ECMs. *p<0.05 and +p<0.001 relative to glass. n = 30 cells (pooled from at least three technical and three biological experiments) for all conditions. (F) CA RhoA rescues stiffness dependent cell motility. Time-lapse phase contrast imaging of random cell motility over 8–12 hours show significantly decreased motility of these cells on soft ECMs relative to stiff ECMs. *p<0.05 and **p<0.001 relative to glass. n=30 cells for all conditions. (G) CA RhoA does not alter cell proliferation. Cells were incubated with BrdU for 45 minutes and stained with 7AAD before analysis by flow cytometry. The data reveal that overexpression of CA RhoA does not produce statistically significant changes in proliferation for any given ECM stiffness nor does it render proliferation sensitive to ECM stiffness. n = 3 independent experiments. (H) CA RhoA TICs express the stem cell markers nestin (top row; red; 100% positive) and SOX2 (bottom row; red; 100% positive) on all ECM stiffnesses. (I) CA RhoA does not compromise the ability of TICs to form neurospheres *p<0.001 relative to control, n = 384 TICs. (J) Quantification of neurosphere size for L0 and L2 cells as a function of RhoA expression. *p<0.001 and +p<0.05 relative to control, n = ~ 1500 spheres for all conditions.
Expression of CA RhoA remarkably restored sensitivity to ECM stiffness, with CA RhoA GBM TICs significantly rounding up on soft ECMs and spreading on stiff ECMs (Fig. 3D, E; L2 CA RhoA GBM TICs shown, with L0 CA RhoA GBM TICs yielding similar results).Additionally, CA RhoA strongly retarded GBM TIC motility on soft ECMs compared to stiff ECMs (Fig. 3F). Notably, GBM TIC proliferation (Fig. 3G) and the universally positive expression of nestin and SOX2 (Fig. 3H) remained relatively insensitive to ECM rigidity. In addition, both control and CA RhoA TICs could still form neurospheres (Fig. 3I, J), which is a predictor of glioma tumor progression and is associated with poor clinical outcome (33). Altogether, these data confirm that constitutive activation of RhoA does not compromise GBM TIC self-renewal properties.
To verify that the observed increase in contractility is due to myosin activation per se as opposed to other downstream effects of RhoA activation, we treated CA RhoA GBM TICs with the myosin II ATPase inhibitor blebbistatin, which rescued spreading of CA RhoA TICs on the softest ECM (Fig. 4A). Consistent with this finding, treatment with blebbistatin did not yield systematic dependences of spreading area (Fig. 4B) or migration speed (Fig. 4C) as a function of matrix stiffness for either control or RhoA TICs. To investigate potential effects of blebbistatin on growth, we measured TIC proliferation (Fig. 4D) and viability (Fig. 4E) as a function of ECM stiffness and CA RhoA expression, which did not reveal systematic effects of the drug. Given the demonstrated importance of the myosin II isoforms A, B, and C to glioma invasion (2, 34), we also measured isoform expression as a function of ECM stiffness and CA RhoA status. Western blots (Fig. 4F,G) revealed that TICs express all three isoforms under all stiffnesses and in the presence or absence of CA RhoA induction. To confirm and quantify these results, we obtained mass spectrometry data (Fig. 4H), which revealed myosin IIA to be the dominant isoform and the relative ratio of isoforms A, B, and C to be largely unchanged across all conditions. Thus, stiffness- and RhoA-mediated effects in this system do not appear to be a consequence of changes in the myosin II isoform ratio.
Figure 4. Non-muscle myosin II regulates tumor-initiating cell mechanosensitivity.

(A) CA RhoA-mediated suppression of cell spreading can be rescued with pharmacologic inhibition of myosin-II. Addition of 25 μM of blebbistatin results in spreading of CA RhoA GBM TICs on soft ECMs, similar to the control GBM TICs. (B) Quantification of projected cell area shows that addition of blebbistatin reverses CA RhoA effects, and caused CA RhoA TICs to spread on soft ECMs. *p<0.05 relative to glass, n=30 cells pooled from at least three technical and three biological experiments for all conditions. (C) Quantification of blebbistatin effects on cell migration speed. Time-lapse phase contrast imaging of random cell motility was conducted over 8–12 hours. *p<0.05 relative to glass, n=30 cells for all conditions. (D) CA RhoA TIC proliferation is not systemically altered by addition of blebbistatin. Cells were incubated with BrdU for 45 minutes and stained with 7AAD before analysis by flow cytometry. Plots show sample means with standard error. n= 3 independent experiments for all conditions. (E) Live/dead assay with propidium iodide staining shows that TICs remain viable in the presence of blebbistatin. (F) Western blot analysis of myosin II heavy chain isoforms A, B, and C shows that control and CA RhoA TICs express all three isoforms on all ECM stiffnesses (L2 shown; L0 had similar results). (G) Quantificatin of Western blot data, n>2 independent experiments. (H) Relative levels of myosin II heavy chain isoforms measured by liquid chromatography tandem mass spectroscopy. n=2 experiments per condition.
To test the generality of the effect of increased myosin activity on cell migration, we developed additional GBM TIC lines overexpressing CA mutants of the contractile activators ROCK1 (Fig. 5A) and MLCK (Fig. 5B), the latter of which activates myosin through an orthogonal, RhoA-independent mechanism. Indeed, overexpression of either molecule produced highly rigidity-dependent motility, with soft ECMs strongly suppressing motility. Thus, suppression of motility on these highly compliant ECMs is consistently associated with generation of high contractile forces against the ECM.
Figure 5. Tumor-initiating cells overexpressing other contractile activators also exhibit stiffness dependent motility and spreading.
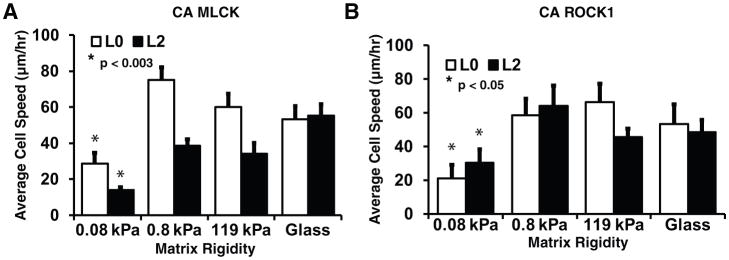
Lentiviral transduction of L0 TICs with (A) CA myosin light chain kinase (MLCK) and (B) CA Rho-associated kinase 1 (ROCK1) resulted in increased average cell speed on stiff ECMs compared to soft ECMs and restore mechanosensitive migration speed. *p<0.003 relative to glass, n = 10–20 cells pooled from at least 3 biological and technical replicates per condition.
To determine whether activation of contractile signaling could also limit motility in three-dimensional ECMs that better capture steric and architectural features of tissue ECM, we performed spheroid invasion assays in soft collagen I gels (Fig. 6A). Consistent with our 2-D migration studies, RhoA activation almost completely abrogated invasion relative to control cells transduced with an empty vector. RhoA activation also produced a 39% reduction in migration through laminin coated 3μm-pore size transwell inserts (Fig. 6B). Notably, treatment of TICs with BMP-4 also significantly reduced invasion in both cell lines (Supplementary Fig. S2C).
Figure 6. Increased Rho GTPase activation limits invasion and motility in 3-D.

(A) CA RhoA prevents GBM TICs from invading 3D collagen ECMs. Images depict spheroids formed from CA RhoA or control GBM TICs, implanted in 1.0 mg/ml collagen gels, and captured by phase contrast imaging 3 days later. n > 8 spheroids per condition for all conditions. (B) CA RhoA reduces transwell migration of GBM TICs. Transwell inserts were coated with laminin, and CA RhoA and control cells were allowed to migrate through pores for 24 hours before fixation. A 2x gradient of soluble EGF was used to drive chemotaxis through the pores, and the total number of cells migrated per transwell was counted. Data shown are for the L2 TIC line; L0 TICs exhibited qualitatively similar data.
Increased cellular contractility extends survival time in an orthotopic mouse model
Finally, to test whether contractility-mediated restriction of GBM TIC motility could reduce tumor invasion and initiation in vivo, we compared the tumorigenicity of CA RhoA and control GBM TICs in a mouse orthotopic xenograft model. We intracranially injected GBM TICs transduced with either CA RhoA or an empty vector into immunocompromised (NSG) mice and followed each cohort until death (Fig. 7A). Remarkably, RhoA activation prolonged survival time by 30% (Fig. 7B).
Figure 7. Constitutive activation of RhoA in GBM TICs extends survival time and reduces tumor invasion in an orthotopic xenograft model.

(A) Mice orthotopically implanted with L0 CA RhoA GBM TICs live significantly longer than mice implanted with L0 control TICs. Eight-week-old NSG mice were implanted intracranially with 200,000 cells. Mice were treated daily with doxycycline (625 mg/kg) 4 days post implant until endpoint. **p=0.0018, log rank test. n = 5 mice per condition. (B) CA RhoA results in a 30% increase in overall mouse survival. **p=0.0042, t-test. (C) Tumors formed from L0 CA RhoA GBM TICs do not infiltrate brain tissue as extensively as L0 control TICs. H&E stain shows (i) a large area of infiltration and secondary tumor formation (*) in mice implanted with L0 control TICs; and (ii) a defined area of infiltration with clear borders and no secondary tumor formation in mice implanted with L0 CA RhoA GBM TICs. (D) CA RhoA reduces diffuse invasion in GBM. H&E (i,v) and human nestin stains (iv,viii) reveal a clear border between CA RhoA tumors and the parenchyma whereas control GBM TICs infiltrate readily. Ipsilateral (ii) and contralateral (vi) hemisphere H&E stains show secondary tumor formation (*) for control cells and not for CA RhoA cells. (E) Human GBM cells were identified using anti-human Nestin antibody and visulaized using the ABC-Elite peroxidase method (Vector Laboratories). Counterstaining of the nuclei was performed using hematoxylin. Analysis of cros-sectional area shows significantly decreased tumor occupancy for mice brains implanted with L0 CA RhoA TICs compared to L0 control TICs. Graph shows average +/− SE over 5–8 sections per group. ***p<0.0001. (F) Ki67 staining and quantification shows no difference in cell proliferation across multiple regions in control and CA RhoA GBM TICs.
Immunohistochemical analysis further revealed that tumors seeded by CA RhoA GBM TICs had reduced invasion capacity and highly circumscribed growth patterns (Fig. 7C), whereas control GBM TICs diffusely spread throughout both hemispheres and generated a secondary tumor in the contralateral hemisphere, consistent with human GBMs (Fig. 7D(i–iii, v–vii)). Moreover, although both CA RhoA and control GBM TICs were able to form tumors in vivo, only tumors derived from the control GBM TICs exhibited all of the defining morphological characteristics of GBM: pseudo-palisading necrosis, nest-like formations, angiogenesis, and mitotic figures. Notably, both control and CA RhoA GBM TICs also continued to express nestin (Figure 7D(iv,viii)), illustrating retention of an important TIC marker throughout the growth and invasion process. Quantification of tumor cross-sectional area confirmed that the area occupied by CA RhoA TIC-induced tumors is >3.5-fold lower than control TIC-induced tumors (Fig. 7E). To verify that the difference in tumor occupancy was not a result of cellular proliferation, tissue sections were stained with the antibody Ki67 (Fig. 7F). We found no difference in proliferation between control and CA RhoA GBM TICs, lending further support to the notion that the reduction in tumor invasion more likely stemmed from limited migration than altered cell division.
Discussion
Mechanical signals encoded in the ECM are increasingly recognized as important regulators of tumor progression (8,35,36). It has also been shown that continuous culture models of GBM require myosin II and its upstream regulators to sense and respond to ECM rigidity (4,5,37,38). However, it has remained unclear if these principles apply to primary TICs, or if manipulation of the mechanosensing machinery can influence tumor initiation and progression in vivo. Here we have addressed these questions by elucidating connections between mechanosensitivity in vitro, activation of contractile signals, and tumor progression and survival in vivo. We discovered that primary GBM TICs can evade limits on spreading and motility normally imposed by compliant matrices by generating low contractile forces. Constitutive activation of myosin II increased GBM TIC contractility, severely restricted GBM TIC motility on compliant ECMs and limited invasion through three-dimensional matrices. Moreover, mice orthotopically implanted with GBM TICs in which contractile signaling had been activated developed smaller and less invasive tumors in vivo relative to control, and exhibited 30% increased overall survival. These findings provide strong support for the notion that GBM can be regarded as a disease of cell migration, and that targeting signals that influence this process in vitro can also influence disease progression in vivo.
Several studies have explored regulatory roles of the Rho GTPases and myosin II in GBM tumor progression, although different experimental systems have produced a somewhat equivocal view of whether activation of these pathways promotes or suppresses tumorigenesis. For example, pharmacologic and/or genetic suppression of myosin II has been shown to reduce glioma invasion in xenograft, genetic, and slice culture models, which has been attributed to the importance of myosin II in squeezing the nucleus through tight tissue spaces during invasion (2,34). Consistent with this finding, simultaneous activation of Rac and RhoA has been found to increase the invasive capacity of GBM TICs (39). In contrast, another study revealed that hyperactivation of Rac1 and Cdc42 through silencing of β8 integrin diminishes GBM invasion with concomitant increases in tumor volume in a mouse xenograft model (40). While these studies may appear to be mutually contradictory, they can be reconciled by considering the notion that activation of these pathways is likely to be highly dynamic and regulated both within and across tumor cells, as well as significantly variable from one physical microenvironment to another. This may give rise to highly nonlinear phenotypic effects that may defy easy prediction. For example, while perinuclear myosin II activation may be critical to nuclear deformation during migration, myosin activation can limit the cytoskeletal plasticity needed for process extension (32). Indeed, recent in vivo florescence resonance energy transfer studies demonstrate that Rho, Rac, and Cdc42 activation vary dramatically in glioma tumor cells as they invade the brain, with different anatomical modes of invasion recruiting different molecular machinery (41). Constitutive activation of myosin and its activators, as we do in this study, would override these regulatory effects and prevent tumor cells from attenuating and enhancing contractility as needed for productive migration. Moreover, Shin and colleagues (42) recently showed that inhibition of myosin II in hematopoietic stem and progenitor cells preferentially enriched the stem cell population, which is broadly consistent with our observations that TICs evade restrictions on adhesion and proliferation normally imposed by highly compliant ECMs. Nonetheless, it is important to note that RhoA has diverse functions and that our in vivo results may not derive exclusively from altered mechanotransductive signaling. Additional in vivo genetic and pharmacological gain- and loss-of-function studies targeting other proteins that enhance and suppress contractility should help to clarify this issue.
While the patient-derived L0 and L2 TIC lines used here behave in a broadly similar fashion across the vast majority of our assays, they do differ in some respects, most notably with respect to neurosphere formation frequency and size (Fig. 3I–J). Although both lines are derived from classical-subtype GBM tumors, these cell lines are derived from different patients and thus would be expected to exhibit some phenomenological differences in specific assays. In addition, neurosphere-forming properties are expected to be a complex function of cell-cell adhesion, proliferation rate, cell contractility, and other factors, small perturbations in any one of which could unpredictably alter the endpoint readout.
Interestingly, while we find that activation of contractile signaling reduces motility, invasion, and tissue infiltration, neither RhoA activation nor ECM stiffness significantly influences proliferation, expression of TIC markers, or differentiation potential. This would suggest that these manipulations do not exert a significant selective effect, which is consistent with our finding that the slowest-cycling (and most highly tumorigenic) subpopulation is preserved across all matrix rigidities.
Finally, our study points to the potential importance of the cell-matrix mechanosensing machinery in GBM TIC physiology. A number of matrix adhesion-related molecules have been identified as GBM TIC markers, including α6 integrin (20) and CD44/moesin (23), and laminin α2 was recently shown to support GBM TIC growth and self-renewal (44). In addition to being correlative predictors of tumor-initiating potential, these molecules may play important functional roles that directly promote tumor growth and invasion. For example, pharmacologic disruption of the CD44/moesin interface strongly reduces GBM TIC proliferation (23), and inhibition of integrin α6 promotes tumor latency and survival (20). Complementary to these studies, our work demonstrates that GBM TICs can evade ECM stiffness-induced suppression of cell motility, analogous to their widely observed resistance to radiation and chemotherapeutic inputs. We hypothesize that tumor cells making up the bulk of the tumor may have relatively high contractility, and are thus prevented by the soft microenvironment from invading into the parenchyma. Previous cortical stiffness measurements of the continuous cell line U373-MG average at 2 kPa (32), which is more than two times higher than the value of our GBM TIC stiffness measurement of 0.8 kPa (Figure 3C). GBM TICs seem to have adapted to the soft environment by generating lower contractility and can thus aggressively invade through brain tissue to initiate new tumors. One method to override this GBM TIC adaptation is to hyperactivate the underlying mechanotransductive actuation mechanisms, which renders GBM TICs susceptible to soft matrix suppression both in vitro and in vivo. In the future, it would be valuable to systematically compare sensitivity to ECM stiffness and RhoA activation across matched, explanted tumor subpopulations at well-defined stages of differentiation, and across cultured TICs at various times after morphogen-induced differentiation. It would also be highly informative to repeat these studies with a panel of TICs derived from all transcriptional subtypes of GBM to determine if these phenotypes are specific to classical GBMs or a more generic feature of TICs. Collectively, previous studies and our findings raise the exciting possibility that mechanisms through which GBM TICs physically engage the ECM can serve as a novel set of druggable targets.
Supplementary Material
Acknowledgments
We would like to thank the following individuals: Dr. David Schaffer and Dr. Song Li for sharing equipment and reagents for Western blot and immunohistochemistry analysis; Dr. Robert Adelstein, Dr. Xuefei Ma, and Dr. Antoine Smith at NIH for mass spectrometric analysis of myosin II isoforms; Hector Nolla for technical assistance with flow cytometry; Dr. Mary West and Dr. Pingping He for assistance with instrumentation in the CIRM/QB3 Stem Cell Shared Facility; and Kevin Yamauchi and Amy Herr’s Laboratory at UC Berkeley for use of the Typhoon FLA 9500 biomolecular imager for Western blot visualization. This work was supported by grants to S.K. from the National Institutes of Health (1DP2OD004213, Director’s New Innovator Award; 1U54CA143836, Physical Sciences Oncology Center Grant, 1R21EB016359), the W. M. Keck Foundation, and a Samsung Global Research Outreach Program. S.Y.W. was supported by a California Institute for Regenerative Medicine pre-doctoral fellowship and a Cancer Research Coordinating Committee fellowship.
Footnotes
Conflict of Interest Disclosure: The authors do not have any potential conflicts of interest.
References
- 1.Nakada M, Nakada S, Demuth T, Tran NL, Hoelzinger DB, Berens ME. Molecular targets of glioma invasion. Cell Mol Life Sci. 2007;64:458–78. doi: 10.1007/s00018-007-6342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beadle C, Assanah MC, Monzo P, Vallee R, Rosenfeld SS, Canoll P. The Role of Myosin II in Glioma Invasion of the Brain. Mol Biol Cell. 2008;19:3357–68. doi: 10.1091/mbc.E08-03-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Demuth T, Berens ME. Molecular mechanisms of glioma cell migration and invasion. J Neurooncol. 2004;70:217–28. doi: 10.1007/s11060-004-2751-6. [DOI] [PubMed] [Google Scholar]
- 4.Ulrich TA, de Juan Pardo EM, Kumar S. The Mechanical Rigidity of the Extracellular Matrix Regulates the Structure, Motility, and Proliferation of Glioma Cells. Cancer Res. 2009;69:4167–4174. doi: 10.1158/0008-5472.CAN-08-4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sen S, Dong M, Kumar S. Isoform-Specific Contributions of α-Actinin to Glioma Cell Mechanobiology. PLoS ONE. 2009;4:e8427. doi: 10.1371/journal.pone.0008427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sen S, Ng WP, Kumar S. Contributions of talin-1 to glioma cell-matrix tensional homeostasis. J R Soc Interface. 2012;9:1311–7. doi: 10.1098/rsif.2011.0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–54. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 9.Samuel MS, Lopez JI, McGhee EJ, Croft DR, Strachan D, Timpson P, et al. Actomyosin-Mediated Cellular Tension Drives Increased Tissue Stiffness and β-Catenin Activation to Induce Epidermal Hyperplasia and Tumor Growth. Cancer Cell. 2011;19:776–91. doi: 10.1016/j.ccr.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deleyrolle LP, Harding A, Cato K, Siebzehnrubl FA, Rahman M, Azari H, et al. Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain. 2011;134:1331–43. doi: 10.1093/brain/awr081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sampetrean O, Saga I, Nakanishi M, Sugihara E, Fukaya R, Onishi N, et al. Invasion Precedes Tumor Mass Formation in a Malignant Brain Tumor Model of Genetically Modified Neural Stem Cells. Neoplasia N Y N. 2011;13:784–91. doi: 10.1593/neo.11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Filatova A, Acker T, Garvalov BK. The cancer stem cell niche(s): The crosstalk between glioma stem cells and their microenvironment. Biochim Biophys Acta BBA - Gen Subj. 2013;1830:2496–508. doi: 10.1016/j.bbagen.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 13.Coma S, Shimizu A, Klagsbrun M. Hypoxia induces tumor and endothelial cell migration in a semaphorin 3F- and VEGF-dependent manner via transcriptional repression of their common receptor neuropilin 2. Cell Adhes Migr. 2011;5:266–75. doi: 10.4161/cam.5.3.16294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Persano L, Rampazzo E, Basso G, Viola G. Glioblastoma cancer stem cells: Role of the microenvironment and therapeutic targeting. Biochem Pharmacol. 2013;85:612–22. doi: 10.1016/j.bcp.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 15.Pathak A, Kumar S. Biophysical regulation of tumor cell invasion: moving beyond matrix stiffness. Integr Biol. 2011;3:267–78. doi: 10.1039/c0ib00095g. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi K, Takahashi H, Inoue A, Harada H, Toshimori S, Kobayashi Y, et al. Oct-3/4 promotes migration and invasion of glioblastoma cells. J Cell Biochem. 2012;113:508–17. doi: 10.1002/jcb.23374. [DOI] [PubMed] [Google Scholar]
- 17.Alonso MM, Diez-Valle R, Manterola L, Rubio A, Liu D, Cortes-Santiago N, et al. Genetic and Epigenetic Modifications of Sox2 Contribute to the Invasive Phenotype of Malignant Gliomas. PLoS ONE. 2011;6:e26740. doi: 10.1371/journal.pone.0026740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oppel F, Müller N, Schackert G, Hendruschk S, Martin D, Geiger KD, et al. SOX2-RNAi attenuates S-phase entry and induces RhoA-dependent switch to protease-independent amoeboid migration in human glioma cells. Mol Cancer. 2011;10:137. doi: 10.1186/1476-4598-10-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Natsume A, Kato T, Kinjo S, Enomoto A, Toda H, Shimato S, et al. Girdin maintains the stemness of glioblastoma stem cells. Oncogene. 2012;31:2715–24. doi: 10.1038/onc.2011.466. [DOI] [PubMed] [Google Scholar]
- 20.Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, MacSwords J, et al. Integrin Alpha 6 Regulates Glioblastoma Stem Cells. Cell Stem Cell. 2010;6:421–32. doi: 10.1016/j.stem.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Velpula KK, Rehman AA, Chelluboina B, Dasari VR, Gondi CS, Rao JS, et al. Glioma stem cell invasion through regulation of the interconnected ERK, integrin α6 and N-cadherin signaling pathway. Cell Signal. 2012;24:2076–84. doi: 10.1016/j.cellsig.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Xu Y, Stamenkovic I, Yu Q. CD44 Attenuates Activation of the Hippo Signaling Pathway and Is a Prime Therapeutic Target for Glioblastoma. Cancer Res. 2010;70:2455–64. doi: 10.1158/0008-5472.CAN-09-2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu X, Morales FC, Agarwal NK, Dogruluk T, Gagea M, Georgescu M-M. Moesin Is a Glioma Progression Marker That Induces Proliferation and Wnt/β-Catenin Pathway Activation via Interaction with CD44. Cancer Res. 2013;73:1142–55. doi: 10.1158/0008-5472.CAN-12-1040. [DOI] [PubMed] [Google Scholar]
- 24.Deleyrolle LP, Reynolds BA. Identifying and enumerating neural stem cells: application to aging and cancer. In: Joost Verhaagen EMH, Huitenga Inge, Wijnholds Jan, Bergen Arthur B, Boer Gerald J, Swaab Dick F, editors. Prog Brain Res [Internet] Elsevier; 2009. pp. 43–51. [cited 2014 Jun 7] Available from: http://www.sciencedirect.com/science/article/pii/S0079612309175040. [DOI] [PubMed] [Google Scholar]
- 25.Siebzehnrubl FA, Silver DJ, Tugertimur B, Deleyrolle LP, Siebzehnrubl D, Sarkisian MR, et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med. 2013;5:1196–212. doi: 10.1002/emmm.201302827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Torsvik A, Røsland GV, Svendsen A, Molven A, Immervoll H, McCormack E, et al. Spontaneous Malignant Transformation of Human Mesenchymal Stem Cells Reflects Cross-Contamination: Putting the Research Field on Track – Letter. Cancer Res. 2010;70:6393–6. doi: 10.1158/0008-5472.CAN-10-1305. [DOI] [PubMed] [Google Scholar]
- 27.Stepanenko AA, Kavsan VM. Karyotypically distinct U251, U373, and SNB19 glioma cell lines are of the same origin but have different drug treatment sensitivities. Gene. 2014;540:263–5. doi: 10.1016/j.gene.2014.02.053. [DOI] [PubMed] [Google Scholar]
- 28.Piccirillo SGM, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–5. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- 29.Frey MT, Wang Y. A photo-modulatable material for probing cellular responses to substrate rigidity. Soft Matter. 2009;5:1918–24. doi: 10.1039/b818104g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell. 2006;126:677–89. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 31.Chan CE, Odde DJ. Traction Dynamics of Filopodia on Compliant Substrates. Science. 2008;322:1687–91. doi: 10.1126/science.1163595. [DOI] [PubMed] [Google Scholar]
- 32.MacKay JL, Keung AJ, Kumar S. A Genetic Strategy for the Dynamic and Graded Control of Cell Mechanics, Motility, and Matrix Remodeling. Biophys J. 2012;102:434–42. doi: 10.1016/j.bpj.2011.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laks DR, Masterman-Smith M, Visnyei K, Angenieux B, Orozco NM, Foran I, et al. Neurosphere Formation Is an Independent Predictor of Clinical Outcome in Malignant Glioma. STEM CELLS. 2009;27:980–7. doi: 10.1002/stem.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ivkovic S, Beadle C, Noticewala S, Massey SC, Swanson KR, Toro LN, et al. Direct inhibition of myosin II effectively blocks glioma invasion in the presence of multiple motogens. Mol Biol Cell. 2012;23:533–42. doi: 10.1091/mbc.E11-01-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kümper S, Marshall CJ. ROCK-Driven Actomyosin Contractility Induces Tissue Stiffness and Tumor Growth. Cancer Cell. 2011;19:695–7. doi: 10.1016/j.ccr.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 36.Yu H, Mouw JK, Weaver VM. Forcing form and function: biomechanical regulation of tumor evolution. Trends Cell Biol. 2011;21:47–56. doi: 10.1016/j.tcb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tilghman RW, Cowan CR, Mih JD, Koryakina Y, Gioeli D, Slack-Davis JK, et al. Matrix Rigidity Regulates Cancer Cell Growth and Cellular Phenotype. PLoS ONE. 2010;5:e12905. doi: 10.1371/journal.pone.0012905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sen S, Ng WP, Kumar S. Contractility Dominates Adhesive Ligand Density in Regulating Cellular De-adhesion and Retraction Kinetics. Ann Biomed Eng. 2011;39:1163–73. doi: 10.1007/s10439-010-0195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruiz-Ontañon P, Orgaz JL, Aldaz B, Elosegui-Artola A, Martino J, Berciano MT, et al. Cellular Plasticity Confers Migratory and Invasive Advantages to a Population of Glioblastoma-initiating Cells that Infiltrate Peritumoral Tissue. STEM CELLS. 2013:N/A–N/A. doi: 10.1002/stem.1349. [DOI] [PubMed] [Google Scholar]
- 40.Reyes SB, Narayanan AS, Lee HS, Tchaicha JH, Aldape KD, Lang FF, et al. αvβ8 integrin interacts with RhoGDI1 to regulate Rac1 and Cdc42 activation and drive glioblastoma cell invasion. Mol Biol Cell. 2013;24:474–82. doi: 10.1091/mbc.E12-07-0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirata E, Yukinaga H, Kamioka Y, Arakawa Y, Miyamoto S, Okada T, et al. In vivo fluorescence resonance energy transfer imaging reveals differential activation of Rho-family GTPases in glioblastoma cell invasion. J Cell Sci. 2012;125:858–68. doi: 10.1242/jcs.089995. [DOI] [PubMed] [Google Scholar]
- 42.Shin J-W, Buxboim A, Spinler KR, Swift J, Christian DA, Hunter CA, et al. Contractile Forces Sustain and Polarize Hematopoiesis from Stem and Progenitor Cells. Cell Stem Cell. 2014;14:81–93. doi: 10.1016/j.stem.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Corcoran A, Del Maestro RF. Testing the “Go or Grow” hypothesis in human medulloblastoma cell lines in two and three dimensions. Neurosurgery. 2003;53:174–184. doi: 10.1227/01.neu.0000072442.26349.14. discussion 184–185. [DOI] [PubMed] [Google Scholar]
- 44.Lathia JD, Li M, Hall PE, Gallagher J, Hale JS, Wu Q, et al. Laminin alpha 2 enables glioblastoma stem cell growth. Ann Neurol. 2012;72:766–78. doi: 10.1002/ana.23674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.