

Abstract
Integrin receptors connect the extracellular matrix to the cell cytoskeleton to provide essential forces and signals. To examine the contributions of the β1 integrin cytoplasmic tail to adhesive forces, we generated cell lines expressing wild-type and tail mutant β1 integrins in β1-null fibroblasts. Deletion of β1 significantly reduced cell spreading, focal adhesion assembly, and adhesive forces, and expression of hβ1 integrin in these cells restored adhesive functions. Cells expressing a truncated tail mutant had impaired spreading, fewer and smaller focal adhesions, reduced integrin binding to fibronectin, and lower adhesion strength and traction forces compared to hβ1-expressing cells. All these metrics were equivalent to those for β1-null cells, demonstrating that the β1 tail is essential to these adhesive functions. Expression of the constitutively-active D759A hβ1 mutant restored many of these adhesive functions in β1-null cells, although with important differences when compared to wild-type β1. Even though there were no differences in integrin-fibronectin binding and adhesion strength between hβ1- and hβ1-D759A-expressing cells, hβ1-D759A-expressing cells assembled more but smaller adhesions than hβ1-expressing cells. Importantly, hβ1-D759A-expressing cells generated lower traction forces compared to hβ1-expressing cells. These differences between hβ1- and hβ1-D759A-expressing cells suggest that regulation of integrin activation is important for fine-tuning cell spreading, focal adhesion assembly, and traction force generation.
Introduction
Cell adhesion to extracellular matrices (ECMs) is central to tissue organization, maintenance, repair and pathogenesis by providing forces and signals that direct cell survival, migration, cell cycle progression, and differentiation (1–3). Heterodimeric (αβ) integrin transmembrane receptors constitute the principal mechanism of cell-ECM adhesion (1). The β1 integrin subfamily binds to fibronectin (FN), collagens, and laminins, and genetic deletion of the β1 subunit results in early embryonic lethality (4, 5). Both α and β integrin subunits form the extracellular domain that conveys ECM ligand binding and specificity, whereas binding sites in the β integrin tail mediate interactions with numerous cytoskeletal components and regulate adhesive functions (6–8). For example, two conserved NPxY motifs bind talin, kindlin, and other cytoskeletal adapters required for integrin activation and localization to focal adhesion (FA) complexes (9–14).
Early work demonstrated that binding sites in the integrin β1 tail mediate interactions with structural cytoskeletal components that regulate diverse adhesive functions. The β1 tail is required for integrin localization to FAs (15). COOH-terminal truncation of β1 eliminating the distal NPxY motif disrupted its ability to mediate cell spreading, and a more proximal truncation (5 amino acids) also disrupted talin binding (16). A truncation of only five amino acids from the COOH-terminal end of the β1 cytoplasmic domain abrogated the ability of the integrin to activate tyrosine phosphorylation (17). Using site directed mutagenesis, Horwitz et al. identified three clusters of amino acids, including the two NPxY motifs, within the β1 subunit tail that regulate integrin localization to FAs (18). These regions are well-conserved among different β subunits and across species (1). In addition, D759 in the membrane proximal β1 tail forms a salt bridge with a conserved arginine in the α subunit to stabilize a default inactive conformation of the receptor (19), and mutation of this residue (D759A) results in high affinity, ligand binding integrin (9). More recent work has established a critical role for the NPxY motifs in diverse cellular functions in development and tumorigenesis (9, 12, 20–22). Interestingly, mutations of tyrosines to alanine in NPxY resulted in developmental defects, whereas mutation of these amino acids to phenylalanine (to prevent phosphorylation) or the D759A mutation had no deleterious effects. These studies establish important roles for β1 tail residues in integrin activation, FA assembly and cellular functions. However, it is not clear the extent to which the β1 tail contributes to adhesive force generation. In this study, we analyzed the contributions of the integrin β1 tail to adhesive forces. Stable cell lines expressing wild-type and mutant human β1 integrins in β1-null fibroblasts were generated. We demonstrate that the β1 tail differentially regulates adhesion strength and traction forces.
Materials and Methods
Antibodies and reagents
PE-Cy7-conjugated anti-mouse β1 (25-0291-82) was obtained from eBioscience. FITC-labeled anti-integrin β3 (ab36437) and rat anti-mouse αv (ab64639) antibodies, as well as isotype controls (rat IgM (ab35774), rat IgG (ab18446, ab37368), goat IgG (ab37377) and hamster IgG (ab32662)) were purchased from Abcam. APC-conjugated anti-human β1 (559883), anti-mouse integrin α1 (555000), anti-mouse integrin α2 (557017), and anti-mouse integrin α4 (553β14) were purchased from BD Pharmingen, and polyclonal anti-mouse integrin α3 (FAB2787P) was purchased from R&D systems. Anti-mouse integrin α5 (sc-19668) was purchased from Santa Cruz Biotechnology. Isotype control APC-conjugated mouse IgG (554681) and PE/Cy7 Armenian hamster IgG (#25-4888-81) were purchased from BD Pharmingen and eBioscience, respectively. Blocking antibodies against mouse β1 (555002) and mouse β3 (553343) and isotype controls (553958, 553968) were from BD Pharmingen, whereas the blocking antibody against human β1 (MAB2253Z) was purchased from Millipore. For immunostaining, antibodies against β1 (MAB1952, Chemicon), vinculin (hVIN-1, Sigma), phosphoY397 FAK (ab39976, Abcam), vimentin (ab45939, Abcam), and cytokeratin (ab9026, Abcam) were used. AlexaFluor488-conjugated antibodies against mouse, rat and rabbit IgG were obtained from Invitrogen, and PE-conjugated goat anti-Armenian hamster IgG (sc-3733) was from Santa Cruz Biotechnology. AlexaFluor555-conjugated phalloidin (A34055) and the Live/Dead kit (L3224) was from Invitrogen. Magnetic anti-Cy7 microbeads (130-091-652) were from Miltenyi Biotec. All other reagents were from Sigma.
Integrin hβ1 plasmids and retrovirus production
The human β1 integrin sequence (plasmid 16042, Addgene) was cloned into a gateway pENTR/DEST plasmid (Invitrogen) using primers (forward primer 5’-CAACATGAATTTACAACCAATTTTCT-3’, reverse primer 5’-TCATTTTCCCTCATACTTCGGATT-3’). The hβ1 sequence was ligated to a retroviral pMSCV-puro gateway vector using a Gateway LR Clonase II reaction. Mutations were produced using the QuikChange II Site Directed Mutagenesis kit (Promega). All plasmids were sequenced and verified.
Retrovirus was produced by transient transfection of helper virus-free Phoenix amphotropic producer cells (23) with wild-type or mutant hβ1 DNA plasmids. Phoenix cells were transfected using a Nucleofector II (Amaxa). For each sample, 106 cells were resuspended in 100 μL of nucleofector solution MEF 2 with 2 μg of DNA plasmid and transfected using program Q-01. Retroviral supernatants were collected and stored at −80°C.
Dermal fibroblast isolation
All protocols were approved by the Institutional Animal Care and Use Committee in adherence to federal guidelines for animal care. β1-floxed (B6;129-Itgb1tm1Efu/J, itgβ1fl/fl) and control B6129SF2/J mice were obtained from Jackson Laboratories. Mice (7–10 weeks old mice) were euthanized, the dorsa were shaved and cleaned with ethanol, and full-thickness skin tissue was harvested. Skin sections were incubated in 0.25% trypsin for 20 min at 37°C in order to strip off the skin at dermal-epidermal junction. The epidermis was mechanically separated from the dermis and cut into small pieces. Strips of dermis were washed, diced and incubated for 15 min at 37°C in 0.3% collagenase (type I, Worthington). The resulting cell suspension was filtered through 100 μm cell strainers, and cells were washed with ice-cold DMEM, 20% fetal bovine serum (FBS), and 1% penicillin/streptomycin (P/S). After centrifugation at 1300 rpm for 10 min, cells were re-suspended in high glucose DMEM with 10% FBS and 1% P/S. Cells were counted and plated at a density of 4000 cells/cm2 and passaged using standard methods.
β1-null and hβ1-expressing cell lines
β1-floxed cells were transduced with Ad5CMV-Cre-GFP virus (Vector Development Lab) at a MOI 400 and after a week of culture, β1-null cells were purified by negative selection using a Miltenyi Biotec sorting kit and anti-Cy7 microbeads. To generate cells expressing human β1 integrins, β1-null cells were transduced with retrovirus encoding for hβ1 variants as described previously (23). Cells were plated on tissue culture polystyrene at 2 × 104 cells/cm2 24 h prior to retroviral transduction. Cells were transduced with 0.2 mL/cm2 of retroviral supernatant supplemented with 4 μg/mL hexadimethrine bromide and 10% fetal bovine serum, and centrifuged at 1200g for 30 min in Beckman model GS-6R centrifuge with a swinging bucket rotor. Retroviral supernatant was replaced with growth media (DMEM, 10% FBS, 1%P/S). Five days after transduction, cells were switched to puromycin (2.5 μg/cm2)-supplemented growth media and maintained under selective pressure during culture and expansion.
Cell plating on micropatterned FN substrates
Micropatterned substrates were generated by microcontact printing of self-assembled monolayers of alkanethiols on gold (24) using a PDMS stamp (Sylgard 184/186 elastomer kit). Arrays of methyl-terminated alkanethiol (HS-(CH2)11-CH3; Sigma) islands (10 μm diameter circles, 75 μm center to center spacing) were stamped onto gold-coated glass coverslips. The remaining exposed areas were functionalized with tri(ethylene glycol)-terminated alkanethiol (HS-(CH2)11-(CH2CH2O)3-OH; ProChimia Surfaces) to generate a cell adhesive-resistant background. Patterned substrates were coated with human plasma FN (25 μg/mL), blocked with 1% heat-denatured bovine serum albumin, incubated in PBS (Ca2+/Mg2+). Cells were seeded at 40,000 cells/cm2 in serum-containing media.
For antibody blocking studies, cells were resuspended in appropriate blocking antibody or isotype control for 30 min with gentle rocking. Cells were seeded on FN-coated micropatterned substrates for 15 min prior to washing three times with PBS. Adherent cells were counted using ImageJ.
Cell adhesion strength
Cell adhesion to FN-coated islands was measured using a hydrodynamic spinning disk system (24, 25). Cells were cultured overnight on coverslips with FN-coated islands in serum-containing media. Micropatterned substrates with adherent cells then were spun in PBS supplemented with 2 mM dextrose for 5 min at constant speed. The applied shear stress (τ) is given by the formula τ = 0.8r(ρμω3)1/2, where r is the radial position and ρ, μ and ω are the fluid density, viscosity and rotational speed respectively. After spinning, cells were fixed in 3.7% formaldehyde, permeabilized in 1% Triton X-100, stained with ethidium homodimer-1. Adherent cells were counted at specific radial positions using a 10X objective lens in a Nikon TE300 microscope equipped with a Ludl motorized stage, Spot-RT camera and Image-Pro analysis system. A total of 61 fields (80–100 cells per field before spinning) were analyzed and cell counts were normalized to the number of cell counts at the center of the disk. The fraction of adherent cells (f) as a function of shear stress τ (force/area) was then fitted to a sigmoid curve f = 1/(1 + exp b[τ - τ50]), where τ50 is the shear stress for 50% detachment and b is the inflection slope, using in-house macros in MATLAB. τ50 represents the mean adhesion strength for the cell population. More than 8 samples were analyzed for each condition.
Integrin binding and focal adhesion assembly
Integrin binding was quantified via a cross-linking/extraction procedure (26, 27). Cells were cultured overnight on coverslips with FN-coated islands in serum-containing media. After rinsing cultures three times with PBS, DTSSP (1.0 mM in cold PBS + 2 mM dextrose) was incubated for 30 minutes to cross-link integrins bound to FN. The cross-linking reaction was quenched by addition of Tris (50 mM in PBS) for 15 minutes. Uncross-linked cellular components were then extracted in 0.1% SDS containing 10 μg/mL leupeptin, 10 μg/mL aprotinin and 350 μg/mL PMSF. Cross-linked integrins to their bound ligands were visualized by immunostaining with β1 integrin-specific antibodies and analyzed using ImageJ. More than 20 cells were analyzed for each condition.
For staining of FAs, cells cultured overnight on FN-coated surfaces were rinsed and permeabilized in cytoskeleton-stabilizing buffer (0.5% Triton X-100, 10 mM PIPES buffer, 50 mM NaCl, 150 mM sucrose, 3mM MgCl2, 1 mM PMSF, 1 μg/mL leupeptin, 1 μg/mL aprotinin, 1 μg/mL pepstatin) for 10 min, fixed in 3.7% formaldehyde for 5 min, blocked in 5% goat serum, and incubated with primary antibodies against FA components followed by AlexaFluor-labeled secondary antibodies. Images were captured using a Nikon Eclipse E400 equipped with a 60X APO (1.4 NA) TIRF objective and Spot RT Camera. FAs were quantified using ImageJ and custom image analysis macros.
Traction force
Microfabricated postarray deflection device (mPAD) silicon masters were prepared as previously described (28). Elastomeric micropost arrays were then fabricated using PDMS replica molding. Briefly, PDMS prepolymer was cast on top of mPAD silicon masters, cured at 110°C for 1 h, peeled off and oxidized with oxygen plasma (Plasma-Preen II-862, Terra Universal). Immediately following oxidization, molds were then silanized with (tridecafluoro-1,1,2,2-tetrahydroocytl)-1-trichlorosilane vapor overnight under vacuum. Following silanization, PDMS prepolymer was cast over the template and stamped onto 1.5 thickness 25-mm diameter coverslips, degassed under vacuum and cured at 110°C for 24 h. Collapsed posts were recovered by sonicating in ethanol for 5 min followed by supercritical drying in liquid CO2 using a critical point dryer (Samdri-PVT-3D, Tousimis).
Flat PDMS stamps were generated by casting PDMS prepolymer on silanized silicon wafers and curing for 1 h. Stamps were coated in 50 μg/mL FN for 1 h. These stamps were then washed in distilled water and dried with N2 gas. FN-coated stamps were placed in contact with plasma-treated mPAD substrates for 5 minutes to allow FN transfer to the mPAD device posts. mPAD substrates were labeled with 2.5 μg/mL of Δ9-DiI (Invitrogen) in distilled water for 1 h. Substrates were then transferred to a solution of 0.1% Pluronics F127 for 30 min to prevent non-specific protein absorption. Cells were seeded in growth medium and allowed to attach and spread overnight.
Following overnight incubation, mPAD substrates were transferred to a coverslip holder and placed in a stage top incubator maintained at 37°C, 90% humidity and 5% CO2 (Live Cell, Pathology Devices). Confocal images were taken with a Nikon C2 module connected to a Nikon Eclipse Ti inverted microscope with a high magnification objective (60X Apochromat TIRF, NA 1.49, Nikon). Bottoms and tops of posts were imaged using a 561-nm laser and deflections between the two regions of the posts were measured. Resulting forces for each post, F, were calculated using Euler-Bernoulli beam theory, , where E, D, L and δ are the Young’s modulus, post diameter, post height and post deflection respectively. More than 25 cells were analyzed for each condition.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 6.0. For normally distributed data with equal variances, one-way ANOVA with Tukey’s multiple comparison test was used and results are displayed as mean ± standard error of the mean. For data that did not satisfy the requirements for ANOVA, non-parametric Kruskal-Wallis tests with Dunn’s multiple comparison tests were used and results are displayed using box-whisker plots. A p-value < 0.05 was considered significant. Non-linear regression analyses were also performed in GraphPad Prism 6.0.
Results
Engineered cells provide a suitable system to analyze the function of target integrins in the absence of endogenous integrins
We generated stable cell lines expressing wild-type and mutant human β1 integrins in β1-null fibroblasts. A major advantage of this system is the lack of endogenous, wild-type murine β1 (mβ1) integrins that could confound interpretation of the data. Dermal fibroblasts from β1-floxed mice (carrying the murine ItgB1 gene flanked by loxP sites (29)) were isolated by enzymatic digestion. Immunostaining for vimentin and cytokeratin confirmed a high purity population of fibroblasts without appreciable keratinocyte contamination (Fig. S1). These cells (flox) were transduced with adenovirus encoding for Cre-GFP to delete the ItgB1 gene. After a week in culture, β1-null cells were purified by negative selection using magnetic beads coated with antibodies against mβ1. Flow cytometry analyses showed that >98% of the cells lacked mβ1 integrin expression (Fig. 1A). Additionally, expression of murine α1, α2, and α5 subunits was also reduced whereas expression of αv integrin remained unchanged (Fig. S2).
Figure 1. Deletion of mβ1 and expression of hβ1 in cells.

(A) Flow cytometry histograms (black: integrin antibody, red: isotype control) for mβ1 (top) and hβ1 (bottom) in wild-type (WT), β1-floxed (flox), β1-null (null), and hβ1-expressing (hβ1) cells. (B) Cell adhesion to FN in the presence of blocking antibodies. Cell counts are normalized to counts for cells incubated in isotype control. * p<0.01 vs. isotype control. (C) Cell spreading area on FN. * p<0.01 vs. all other cell types. (D) Immunostained images for FA components (top) vinculin (green) and actin (red) and (bottom) phosphoY397 FAK (green). Scale bar, 5 μm.
β1-null cells were then transduced with retrovirus encoding for human β1A (hβ1) integrin and puromycin resistance. A stable polyclonal population of hβ1-expressing cells was established following culture in the presence of puromycin. Flow cytometry analyses showed high levels of hβ1 integrin expression in the absence of mβ1 (Fig. 1A). Expression levels for hβ1 integrin were comparable to expression levels of mβ1 in floxed cells and control cells (WT) isolated from wild-type mice. Furthermore, expression of hβ1 resulted in expression of α1 and α5 integrin subunits (Fig. S2), suggesting expression of integrin heterodimers. The expressed hβ1 is functional as adhesion to FN was completely blocked by antibodies against hβ1 (p<0.01), whereas antibodies against mβ1 had no effect (Fig. 1B). Integrin β3 provided residual adhesion to FN in these cells as anti-β3 antibodies reduced adhesion by 20% but this was not statistically significant. Importantly, the strong dependence of adhesion on β1 integrin for hβ1-expressing cells is consistent with results for control WT and floxed cells. As expected, adhesion of β1-null cells to FN was completely blocked by antibodies against integrin β3 (p<0.01), whereas antibodies against mβ1 had no effect (Fig. 1B).
hβ1-expressing cells spread on FN to the same extent as control floxed cells, whereas the average spread area of β1-null cells was approximately 25% lower (p<0.01) (Fig. 1C). hβ1-expressing cells assembled robust vinculin-containing FAs and actin stress fibers and phosphorylated FAK localized to FAs (Fig. 1D), similar to results for floxed cells. These results demonstrate that the expressed hβ1 integrin is functional and these engineered cells recapitulate many of the adhesive characteristics of control floxed and WT cells that express endogenous mβ1.
As a final validation, we measured the adhesion strength to FN of these stable lines using our spinning disk device that applies hydrodynamic detachment forces. The adhesion strength is the force required to detach cells from the FN-coated surface and provides a sensitive measure of adhesive function (24). Because cell spreading alters adhesion strength, cells were cultured on micropatterned FN islands to control cell shape and adhesive area. For the spinning disk assay, a coverslip containing adherent cells is exposed to a gradient of hydrodynamic forces with detachment forces increasing linearly with distance from the center of the coverslip. After spinning, cells are counted at radial positions of known applied force. Fig. 2A presents typical detachment profiles showing sigmoidal decreases in the number of adherent cells with increasing shear stress. The rightward shift in the adhesion profile for the floxed cells compared to the β1-null cells indicates higher adhesive forces. The shear stress for 50% detachment, which represents the mean adhesion strength, was measured for the cell lines (Fig. 2B). Deletion of β1 integrin resulted in a 50% decrease in adhesion strength to FN compared with floxed and WT cells (p<0.001). The residual adhesion in β1-null cells is attributed to β3-dependent adhesion (Fig. 1). Importantly, expression of hβ1 in null cells rescued the deficits in adhesion strength and restored adhesion strength to the levels of floxed and WT cells. Taken together, these results demonstrate that deletion of β1 integrin significantly reduces cell spreading and adhesion strength to FN and expression of hβ1 integrin in these cells restores adhesive functions, including adhesion strength, to the levels of control cells.
Figure 2. hβ1 reconstitutes adhesion strength to FN in β1-null cells.

(A) Detachment profiles showing the fraction of adherent cells (f) vs. surface shear stress (τ) for β1-null and β1-floxed cells. (B) Cell adhesion strength, box-whisker plot (mean, 10th, 25th, 75th, and 90th percentile). * p<0.001 vs. all other cell types.
The β1 tail regulates cell spreading, morphology and FA assembly
We developed stable lines for β1-null cells expressing integrin mutants following the strategy outlined above (Fig. 3A). A cell line expressing a truncation mutant at position 760 (hβ1-tr) was generated. This mutant severely reduces integrin recruitment and FA assembly (30). We note that a stable cell line expressing the truncation mutant at position 754 previously described (β1) could not be established as these cells did not grow. We also generated a stable cell line expressing the D759A mutant (hβ1-D759A); this mutant results in constitutively active β1 integrin (9, 19). Flow cytometry analysis showed that expression levels for these mutants were comparable to levels in the cell line expressing wild-type hβ1 and approximately 15-fold higher than levels in the β1-null cell (p<0.001) (Fig. 3B). Antibodies against hβ1 integrin completely blocked adhesion to FN for hβ1- and hβ1-D759A-expressing cells (p<0.01), whereas antibodies against mβ1 or mβ3 had no effect (Fig. 3C). Adhesion of β1-null cells to FN was completely mediated by mβ3 integrin (p<0.01), whereas mβ3 integrin partially contributed to the adhesion of hβ1-tr-expressing cells to FN (p<0.05).
Figure 3. Integrin expression and cell adhesion for β1 tail mutants.

(A) Schematic of integrin constructs engineered. (B) Flow cytometry analysis for expression levels of hβ1 normalized to expression levels of β1-null cells. * p<0.001 vs. all other cell types. (C) Cell adhesion to FN in the presence of blocking antibodies. Cell counts are normalized to counts for cells incubated in isotype control. § p<0.05 vs. isotype control, * p<0.01 vs. isotype control.
Significant differences in cell shape, spread area, and FA assembly were evident among the cell lines cultured on FN-coated glass (Fig. 4). β1-null cells spread to compact shapes with high circularity and exhibited well-defined actin stress fibers terminating at vinculin-containing FAs. hβ1-expressing cells had larger spread areas (p<0.001) and were elongated with long projections that contained large, well-defined FAs. Cells expressing the truncated β1 mutant spread more than β1-null cells (p<0.001) but to similar levels as hβ1-expressing cells. hβ1-tr-expressing cells had fewer and smaller FAs and less defined stress fibers than hβ1-expressing cells (p<0.01). Cells expressing the constitutively active D759A mutant exhibited the highest spread area and were more circular than hβ1-expressing cells (p<0.01). Interestingly, hβ1-D759A-expressing cells had a higher number of vinculin-containing FAs compared with hβ1-expressing cells (p<0.001), but the FAs were significantly smaller although the total focal adhesion areas were equivalent. These results demonstrate that the β1 integrin tail regulates cell spreading and shape as well as the number and size of FAs. In addition, the differences between hβ1- and hβ1-D759A-expressing cells indicate that regulation of integrin activation contributes to cell spreading, morphology and FA assembly.
Figure 4. β1 tail regulates cell spreading, morphology and FA assembly.

(A) Immunostained images for vinculin (green) and actin (red). Scale bar, 10 μm. (B) Cell spread area, box-whisker plot (mean, 10th, 25th, 75th, and 90th percentile). * p<0.001 vs. null, § p<0.001 vs. hβ1-tr, † p<0.01 vs. hβ1. (C) Cell circularity. * p<0.001 vs. null, § p<0.001 vs. hβ1-tr. (D) Number of FA, box-whisker plot (mean, 10th, 25th, 75th, and 90th percentile). * p<0.001 vs. null, § p<0.01 vs. hβ1-tr, † p<0.01 vs. hβ1. (E) Total FA area, box-whisker plot (mean, 10th, 25th, 75th, and 90th percentile). * p<0.001 vs. null, § p<0.001 vs. hβ1-tr.
The β1 integrin tail is essential for β1 integrin-FN binding and adhesion strength
We next quantified binding of these integrin tail mutants to FN using a cross-linking and detergent extraction method to selectively retain integrin-FN complexes in cells adhering to micropatterned FN islands (24, 32). Fig. 5A presents images of immunostained β1 integrin for single cells adhering to 10 μm-diameter FN islands. Fig. 5B presents frequency maps for integrin binding generated by stacking and color coding multiple images; this analysis exploits the spatial arrangement of the FN islands to extract the dominant spatial localization of FN-bound integrins across multiple cells (33). Fig. 5C and D plot the fraction of the adhesive area occupied by integrin-FN complexes and the mean intensity of integrin staining over the micropatterned area, respectively. β1-null cells had very low levels of bound β1 integrins, consistent with the antibody-blocking results (Fig. 1B). hβ1-expressing cells exhibited punctate integrin-FN clusters localized to the periphery of the FN island that occupied 8-fold higher area (p<0.001) and had 2-fold higher mean intensity (p<0.01) compared with β1-null cells. Cells expressing the truncated tail mutant had low levels of integrin-FN complexes equivalent to β1-null cells. This result shows that the β1 tail is essential for the formation of stable integrin-FN complexes. Cells expressing the D759A mutant formed robust integrin-FN complexes localized to the periphery of the FN island, and there were no differences in area occupied or mean intensity between hβ1 and hβ1-D759A.
Figure 5. β1 tail is required for integrin β1-FN binding on micropatterned islands.
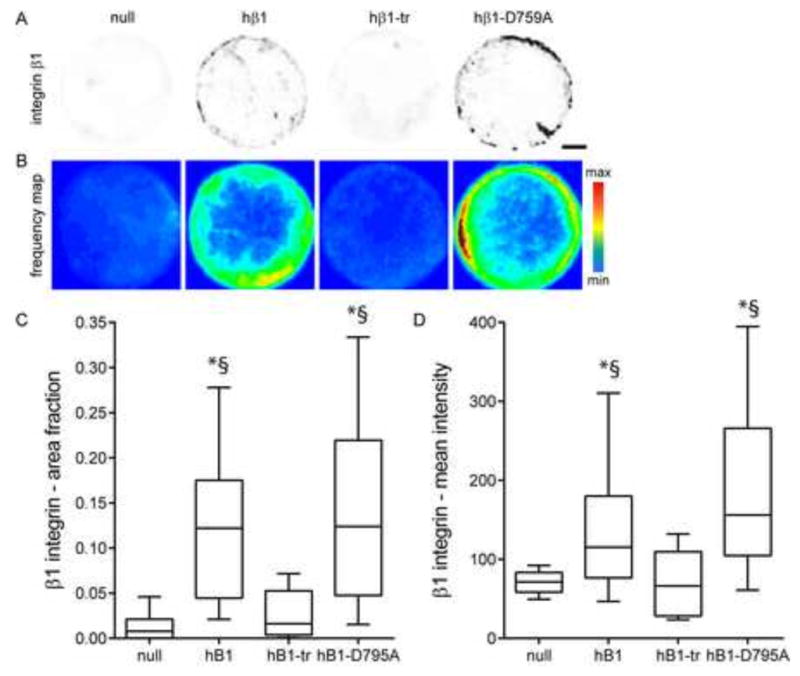
(A) Immunostained images for hβ1 integrin in cell adhering FN islands. Staining is shown as grayscale on white background to facilitate visualization. Scale bar, 2 μm. (B) Frequency maps for stacked images of immunostained hβ1. (C) Fraction of FN island stained for hβ1, box-whisker plot (mean, 10th, 25th, 75th, and 90th percentile). * p<0.001 vs. null, § p<0.001 vs. hβ1-tr. (D) Mean intensity of hβ1 staining, box-whisker plot (mean, 10th, 25th, 75th, and 90th percentile). * p<0.01 vs. null, § p<0.01 vs. hβ1-tr.
We next analyzed the adhesion strength of these cell lines to FN micropatterned islands (Fig. 6). The adhesion strength of hβ1 was approximately 2.3-fold higher than β1-null cells (p<0.001). hβ1-tr-expressing cells had low levels of adhesion strength to FN, equivalent levels as β1-null cells. Adhesion strength for the D759A mutant was not different from wild-type hβ1 but was significantly higher from the null control and truncated tail mutant (p<0.001). The differences in adhesion strength among the integrin mutants are consistent with the differences in integrin-FN complex formation. We expect a significant contribution of endogenous β3 integrin to the adhesion strength of β1-null and hβ1-tr-expressing cells to FN as antibodies against this integrin subunit significantly reduced adhesion to FN (Fig. 3B). In contrast, adhesion of hβ1- and hβ1-D759A-expressing cells to FN was completely mediated by integrin β1 (p<0.01) (Fig. 3B). Taken together, these results demonstrate that the β1 tail is essential for adhesion strength to FN and the D759A mutation has no effect on integrin-FN complexes or adhesion strength for cells on micropatterned FN islands.
Figure 6. β1 tail is essential for adhesion strength to FN.
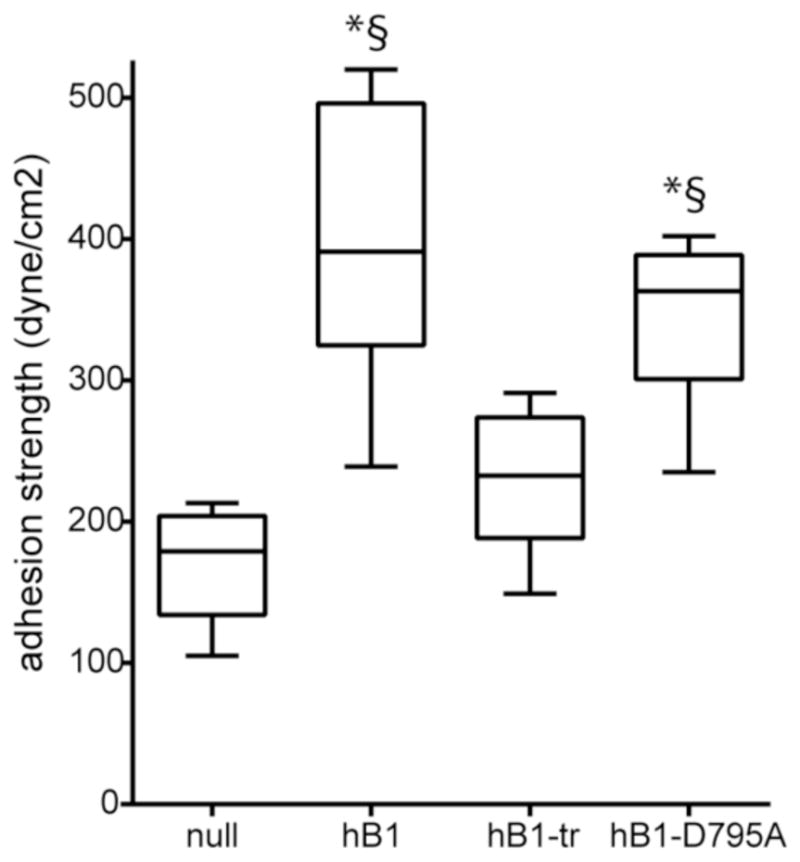
Cell adhesion strength, box-whisker plot (mean, 10th, 25th, 75th, and 90th percentile). * p<0.01 vs. null, § p<0.01 vs. hβ1-tr.
Contributions of the β1 integrin tail to traction forces
We used microfabricated post array deflection devices (mPADs) to measure traction forces generated by cells expressing these integrins. In this system, traction forces generated by adhering cells deflect the tips of the posts in the array; these deflections can then be used to determine the traction forces because the elastic modulus and geometry of the posts are defined (28, 32). During overnight culture on these devices, all cell lines attached to, spread on the posts, and generated traction forces. Fig. 7A presents images for FN-coated posts (red) with the cell outlined in yellow. Post deflections were measured and converted into traction forces, and Fig. 7B presents the traction force vectors (cyan). On these deformable substrates, hβ1-expressing cells spread considerably more than the other cell lines (p<0.01) (Fig. 7C). hβ1-D759A-expressing cells spread to the same extent as β1-null and hβ1-truncated-expressing cells. This result contrasts with our observations for cell spreading on glass (Fig. 4) and may be due to the difference in stiffness between glass and the mPADs. The magnitude of traction forces varied significantly across a single cell, with the highest forces at the cell periphery. Fig. 7D presents box-whisker plots for the total traction force per cell, which represents the sum of the magnitudes of the force vectors for each cell. hβ1-expressing cells exerted 3.7-fold higher traction forces compared to β1-null cells (p<0.001). Cells expressing the integrin mutant with a truncated tail generated equivalent traction forces as β1-null cells. This result shows that the β1 tail is essential for traction forces on FN. Surprisingly, cells expressing the constitutively activated D759A mutant exerted higher forces than the β1-null cells (p<0.01), but the levels were only 33% of the total traction force generated by the hβ1-expressing cells.
Figure 7. β1 tail and traction force.

(A) Cells (yellow outline) spread on mPADs (posts labeled red). Scale bar, 50 μm. (B) Traction force vectors (cyan) with cell outline (yellow). Scale bar, 10 nN. (C) Cell spread area, box-whisker plot (mean, 10th, 25th, 75th, and 90th percentile). * p<0.001 vs. null, § p<0.001 vs. hβ1-tr, † p<0.05 vs. hβ1-D759A. (D) Total traction force per cell, box-whisker plot (mean, 10th, 25th, 75th, and 90th percentile). * p<0.001 vs. null, § p<0.001 vs. hβ1-tr, † p<0.01 vs. hβ1-D759A. (E) Traction force per post, box-whisker plot (mean, 10th, 25th, 75th, and 90th percentile). * p<0.001 vs. null, § p<0.001 vs. hβ1-tr, † p<0.01 vs. hβ1-D759A.
Because there are significant differences in cell spreading and total traction force among the cell lines, we also evaluated the traction force per post (Fig. 7E). Similar to the measurements for total traction force, hβ1-expressing cells generated significantly higher traction force per post compared with β1-null and hβ1-tr-expressing cells (p<0.001). There was no difference in traction force per post between β1-null and hβ1-tr-expressing cells, confirming our conclusion from the total traction force analysis. Cells expressing the hβ1-D759A mutant generated higher traction forces per post than β1-null and hβ1-tr-expressing cells (p<0.001), but these forces were approximately 40% lower than forces exerted by hβ1-expressing cells (p<0.01).
Finally, we examined the relationship between cell area and traction force because we previously showed that cell area-traction force coupling represents a robust metric to analyze the role of FA components on force transfer (32). Fig. S3 plots cell area and the corresponding traction force for individual cells. Linear regression analyses revealed a strong correlation between cell area and traction force for each cell line (p<0.0001). There was no difference in regression slope among cell lines, indicating that the integrin β1 tail does not alter cell area-traction force coupling. Taken together, these results demonstrate that the β1 tail is required for the generation of traction forces and D759 is important for high traction force generation.
Discussion
In this study, we generated stable cell lines expressing wild-type and tail mutant β1 integrins in β1-null fibroblasts to analyze the contributions of the β1 tail to adhesive forces. A major advantage of this system is the lack of endogenous, wild-type murine β1 integrins that could confound interpretation of the data. Deletion of β1 integrin significantly reduced cell spreading, focal adhesion assembly, and adhesive forces, and expression of hβ1 integrin in these cells restored adhesive functions to the levels of control wild-type and floxed cells. Cells expressing the truncated tail mutant had impaired spreading, fewer and smaller FAs, reduced integrin binding to FN, and significantly lower adhesion strength and traction forces compared to hβ1-expressing cells. All these metrics were equivalent to those for β1-null cells, demonstrating that the β1 tail is essential to these adhesive functions. Expression of the constitutively-active D759A mutant restored many of these adhesive functions in β1-null cells, although with important differences when compared to wild-type β1. Even though there were no differences in integrin-FN binding and adhesion strength to FN between hβ1- and hβ1-D759A-expressing cells, hβ1-D759A-expressing cells assembled more but smaller FAs and the spread area and shape were different from hβ1-expressing cells. Importantly, hβ1-D759A-expressing cells generated considerably lower traction forces compared to hβ1-expressing cells.
Previous studies have established that truncations at different locations along the β1 tail impair cell spreading, integrin recruitment to FAs, FA assembly, and signaling (16, 18, 30, 34). Our results confirm the critical importance of the β1 tail domain to these adhesive functions. Furthermore, we demonstrate that the β1 tail is essential for integrin-FN binding and the transmission of adhesive forces related to adhesion strength and traction force. Our previous analyses of adhesion strength to FN indicated that binding of β1 integrin to FN in the absence of FA assembly provided the major contribution to adhesion strength (24). Based on this result, we expected that the truncated β1 mutant would provide partial adhesion strength. However, the lack of any adhesion strength for this mutant can be explained by the abrogation of β1 integrin binding to FN in this mutant.
The effects of the D759A mutation on adhesive functions are not well understood and appear to be cell context-dependent. Expression of this mutant in β1-deficient GD25 fibroblastic cells results in a constitutively active β1 integrin with high binding affinity for FN, increased cell adhesion and motility, and more numerous and larger FAs (9, 35). In contrast, mice carrying the D759A mutation had no overt phenotype, and keratinocytes isolated from these mice exhibit normal integrin activation levels, adhesion and migration (12). In the present study, we observed significant reductions in spread area and traction forces on deformable substrates for hβ1-D759A-expressing cells compared to hβ1-expressing cells, indicating a defect in traction force generation. This defect in traction force generation accounts for the reduced cell spreading on the deformable substrates as there are no differences in traction force-area coupling between hβ1- and hβ1-D759A-expressing cells. Indeed, the traction force per post was reduced in hβ1-D759A-expressing cells compared to hβ1-expressing cells. However, it is not clear whether the reduced traction force arises from a defect in force transmission at the single receptor level. Alternatively, the reduced traction force generation could be a consequence of altered receptor clustering/FA assembly or interactions with cytoskeletal partners due to the inability of this receptor to regulate its affinity. The smaller FAs assembled by hβ1-D759A-expressing cells could account for the reduced adhesive force (28, 33, 36). These differences between hβ1- and hβ1-D759A-expressing cells suggest that regulation of integrin activation is important for fine-tuning cell spreading, FA assembly, and traction force generation.
Supplementary Material
Highlights.
Stable lines expressing mutant human β1 integrins in β1-null fibroblasts generated.
Complementary force-sensing platforms used to analyze cell adhesive forces.
β1 tail is essential for integrin-fibronectin binding and focal adhesion assembly.
β1 integrin tail differentially regulates adhesion strength and traction forces.
Acknowledgments
Funding was provided by the National Institutes of Health (R01-GM065918). J.R.G. was partially supported by the National Science Foundation IGERT program on Stem Cell Biomanufacturing (DGE 0965945). The pMSCV-puro gateway vector was kindly provided by Jing Chen (Emory University School of Medicine). No conflicts of interest are declared.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 2.Wickstrom SA, Radovanac K, Fassler R. Genetic analyses of integrin signaling. Cold Spring Harb Perspect Biol. 2011;3:a005116. doi: 10.1101/cshperspect.a005116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fassler R, Meyer M. Consequences of lack of beta 1 integrin gene expression in mice. Genes Dev. 1995;9:1896–1908. doi: 10.1101/gad.9.15.1896. [DOI] [PubMed] [Google Scholar]
- 5.Stephens LE, Sutherland AE, Klimanskaya IV, Andrieux A, Meneses J, Pedersen RA, Damsky CH. Deletion of beta 1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes Dev. 1995;9:1883–1895. doi: 10.1101/gad.9.15.1883. [DOI] [PubMed] [Google Scholar]
- 6.Calderwood DA, Campbell ID, Critchley DR. Talins and kindlins: partners in integrin-mediated adhesion. Nat Rev Mol Cell Biol. 2013;14:503–517. doi: 10.1038/nrm3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu S, Calderwood DA, Ginsberg MH. Integrin cytoplasmic domain-binding proteins. J Cell Sci. 2000;113:3563–3571. doi: 10.1242/jcs.113.20.3563. [DOI] [PubMed] [Google Scholar]
- 8.Harburger DS, Calderwood DA. Integrin signalling at a glance. J Cell Sci. 2009;122:159–163. doi: 10.1242/jcs.018093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakai T, Zhang Q, Fassler R, Mosher DF. Modulation of beta1A integrin functions by tyrosine residues in the beta1 cytoplasmic domain. J Cell Biol. 1998;141:527–538. doi: 10.1083/jcb.141.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirsch E, Barberis L, Brancaccio M, Azzolino O, Xu D, Kyriakis JM, Silengo L, Giancotti FG, Tarone G, Fassler R, Altruda F. Defective Rac-mediated proliferation and survival after targeted mutation of the beta1 integrin cytodomain. J Cell Biol. 2002;157:481–492. doi: 10.1083/jcb.200111065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wennerberg K, Armulik A, Sakai T, Karlsson M, Fassler R, Schaefer EM, Mosher DF, Johansson S. The cytoplasmic tyrosines of integrin subunit beta1 are involved in focal adhesion kinase activation. Mol Cell Biol. 2000;20:5758–5765. doi: 10.1128/mcb.20.15.5758-5765.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Czuchra A, Meyer H, Legate KR, Brakebusch C, Fassler R. Genetic analysis of {beta}1 integrin “activation motifs” in mice. J Cell Biol. 2006;174:889–899. doi: 10.1083/jcb.200604060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nieves B, Jones CW, Ward R, Ohta Y, Reverte CG, LaFlamme SE. The NPIY motif in the integrin beta1 tail dictates the requirement for talin-1 in outside-in signaling. J Cell Sci. 2010;123:1216–1226. doi: 10.1242/jcs.056549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Margadant C, Kreft M, de Groot DJ, Norman JC, Sonnenberg A. Distinct roles of talin and kindlin in regulating integrin alpha5beta1 function and trafficking. Curr Biol. 2012;22:1554–1563. doi: 10.1016/j.cub.2012.06.060. [DOI] [PubMed] [Google Scholar]
- 15.LaFlamme SE, Akiyama SK, Yamada KM. Regulation of fibronectin receptor distribution. J Cell Biol. 1992;117:437–447. doi: 10.1083/jcb.117.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaapa A, Peter K, Ylanne J. Effects of mutations in the cytoplasmic domain of integrin beta(1) to talin binding and cell spreading. Exp Cell Res. 1999;250:524–534. doi: 10.1006/excr.1999.4533. [DOI] [PubMed] [Google Scholar]
- 17.Finkelstein LD, Reynolds PJ, Hunt SW, 3rd, Shimizu Y. Structural requirements for beta1 integrin-mediated tyrosine phosphorylation in human T cells. J Immunol. 1997;159:5355–5363. [PubMed] [Google Scholar]
- 18.Reszka AA, Hayashi Y, Horwitz AF. Identification of amino acid sequences in the integrin beta 1 cytoplasmic domain implicated in cytoskeletal associations. J Cell Biol. 1992;117:1321–1330. doi: 10.1083/jcb.117.6.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes PE, Diaz-Gonzalez F, Leong L, Wu C, McDonald JA, Shattil SJ, Ginsberg MH. Breaking the integrin hinge. A defined structural constraint regulates integrin signaling. J Biol Chem. 1996;271:6571–6574. doi: 10.1074/jbc.271.12.6571. [DOI] [PubMed] [Google Scholar]
- 20.Maginnis MS, Mainou BA, Derdowski A, Johnson EM, Zent R, Dermody TS. NPXY motifs in the beta1 integrin cytoplasmic tail are required for functional reovirus entry. J Virol. 2008;82:3181–3191. doi: 10.1128/JVI.01612-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Green JA, Berrier AL, Pankov R, Yamada KM. beta1 integrin cytoplasmic domain residues selectively modulate fibronectin matrix assembly and cell spreading through talin and Akt-1. J Biol Chem. 2009;284:8148–8159. doi: 10.1074/jbc.M805934200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meves A, Geiger T, Zanivan S, DiGiovanni J, Mann M, Fassler R. Beta1 integrin cytoplasmic tyrosines promote skin tumorigenesis independent of their phosphorylation. Proc Natl Acad Sci U S A. 2011;108:15213–15218. doi: 10.1073/pnas.1105689108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Byers BA, Pavlath GK, Murphy TJ, Karsenty G, Garcia AJ. Cell-type-dependent up-regulation of in vitro mineralization after overexpression of the osteoblast-specific transcription factor Runx2/Cbfal. J Bone Miner Res. 2002;17:1931–1944. doi: 10.1359/jbmr.2002.17.11.1931. [DOI] [PubMed] [Google Scholar]
- 24.Gallant ND, Michael KE, García AJ. Cell adhesion strengthening: contributions of adhesive area, integrin binding, and focal adhesion assembly. Mol Biol Cell. 2005;16:4329–4340. doi: 10.1091/mbc.E05-02-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.García AJ, Huber F, Boettiger D. Force required to break alpha5beta1 integrin-fibronectin bonds in intact adherent cells is sensitive to integrin activation state. J Biol Chem. 1998;273:10988–10993. doi: 10.1074/jbc.273.18.10988. [DOI] [PubMed] [Google Scholar]
- 26.García AJ, Vega MD, Boettiger D. Modulation of cell proliferation and differentiation through substrate-dependent changes in fibronectin conformation. Mol Biol Cell. 1999;10:785–798. doi: 10.1091/mbc.10.3.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keselowsky BG, García AJ. Quantitative methods for analysis of integrin binding and focal adhesion formation on biomaterial surfaces. Biomaterials. 2005;26:413–418. doi: 10.1016/j.biomaterials.2004.02.050. [DOI] [PubMed] [Google Scholar]
- 28.Fu J, Wang YK, Yang MT, Desai RA, Yu X, Liu Z, Chen CS. Mechanical regulation of cell function with geometrically modulated elastomeric substrates. Nat Methods. 2010;7:733–736. doi: 10.1038/nmeth.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raghavan S, Bauer C, Mundschau G, Li Q, Fuchs E. Conditional ablation of beta1 integrin in skin. Severe defects in epidermal proliferation, basement membrane formation, and hair follicle invagination. J Cell Biol. 2000;150:1149–1160. doi: 10.1083/jcb.150.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hayashi Y, Haimovich B, Reszka A, Boettiger D, Horwitz A. Expression and function of chicken integrin beta 1 subunit and its cytoplasmic domain mutants in mouse NIH 3T3 cells. J Cell Biol. 1990;110:175–184. doi: 10.1083/jcb.110.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vignoud L, Albiges-Rizo C, Frachet P, Block MR. NPXY motifs control the recruitment of the alpha5beta1 integrin in focal adhesions independently of the association of talin with the beta1 chain. J Cell Sci. 1997;110:1421–1430. doi: 10.1242/jcs.110.12.1421. [DOI] [PubMed] [Google Scholar]
- 32.Dumbauld DW, Lee TT, Singh A, Scrimgeour J, Gersbach CA, Zamir EA, Fu J, Chen CS, Curtis JE, Craig SW, Garcia AJ. How vinculin regulates force transmission. Proc Natl Acad Sci U S A. 2013;110:9788–9793. doi: 10.1073/pnas.1216209110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coyer SR, Singh A, Dumbauld DW, Calderwood DA, Craig SW, Delamarche E, Garcia AJ. Nanopatterning reveals an ECM area threshold for focal adhesion assembly and force transmission that is regulated by integrin activation and cytoskeleton tension. J Cell Sci. 2012;125:5110–5123. doi: 10.1242/jcs.108035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcantonio EE, Guan JL, Trevithick JE, Hynes RO. Mapping of the functional determinants of the integrin beta 1 cytoplasmic domain by site-directed mutagenesis. Cell Regul. 1990;1:597–604. doi: 10.1091/mbc.1.8.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Millon-Fremillon A, Bouvard D, Grichine A, Manet-Dupe S, Block MR, Albiges-Rizo C. Cell adaptive response to extracellular matrix density is controlled by ICAP-1-dependent beta1-integrin affinity. J Cell Biol. 2008;180:427–441. doi: 10.1083/jcb.200707142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balaban NQ, Schwarz US, Riveline D, Goichberg P, Tzur G, Sabanay I, Mahalu D, Safran S, Bershadsky A, Addadi L, Geiger B. Force and focal adhesion assembly: a close relationship studied using elastic micropatterned substrates. Nat Cell Biol. 2001;3:466–472. doi: 10.1038/35074532. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.