

Abstract
Purpose
While the overall cure rate for pediatric acute lymphoblastic leukemia (ALL) approaches 90%, infants with ALL harboring translocations in the mixed-lineage leukemia (MLL) oncogene (infant MLL-ALL) experience shorter remission duration and lower survival rates (∼50%). Mutations in the p53 tumor suppressor gene are uncommon in infant MLL-ALL, and drugs that release p53 from inhibitory mechanisms may be beneficial. The purpose of this study was to assess the efficacy of the orally available nutlin, RG7112, against patient-derived MLL-ALL xenografts.
Experimental Design
Eight MLL-ALL patient-derived xenografts were established in immune-deficient mice, and their molecular features compared with B-lineage ALL and T-ALL xenografts. The sensitivity of MLL-ALL xenografts to RG7112 was assessed in vitro and in vivo, and the ability of RG7112 to induce p53, cell cycle arrest and apoptosis in vivo was evaluated.
Results
Gene expression analysis revealed that MLL-ALL, B-lineage ALL and T-ALL xenografts clustered according to subtype. Moreover, genes previously reported to be over-expressed in MLL-ALL, including MEIS1, CCNA1, and members of the HOXA family, were significantly up-regulated in MLL-ALL xenografts, confirming their ability to recapitulate the clinical disease. Exposure of MLL-ALL xenografts to RG7112 in vivo caused p53 up-regulation, cell cycle arrest and apoptosis. RG7112 as a single agent induced significant regressions in infant MLL-ALL xenografts. Therapeutic enhancement was observed when RG7112 was assessed using combination treatment with an induction-type regimen (vincristine/dexamethasone/L-asparaginase) against an MLL-ALL xenograft.
Conclusion
The utility of targeting the p53-MDM2 axis in combination with established drugs for the management of infant MLL-ALL warrants further investigation.
Keywords: acute lymphoblastic leukemia, xenografts, MLL translocations, p53/mdm2 axis
Introduction
Overall cure rates for pediatric acute lymphoblastic leukemia (ALL) have improved considerably in the last 50 years due to improvements in the use of multi-agent chemotherapy and advances in supportive care, such that almost 90% of patients now experience long-term survival (1, 2). Despite this success, subsets of patients are associated with a poor prognosis. Infants (<12 months of age) diagnosed with ALL frequently present with a range of high-risk features, including high leukocyte count at diagnosis, an immature CD10-negative phenotype, and co-expression of myeloid antigens. However, the most distinctive genetic feature of infant ALL is the presence of rearrangements involving the MLL (mixed lineage leukemia) oncogene at the 11q23 chromosomal region (3-5). MLL translocations are found in nearly 80% of infants diagnosed with ALL compared to 2-4% of older children, and confer a poorer prognosis than for infants with germline MLL (6-8).
Between 90-95% of infants with ALL achieve remission following intensive induction therapy using established drugs including glucocorticoids, vincristine, L-asparaginase, cytarabine, daunorubicin and methotrexate (6). However, despite this intensive treatment there is a high rate of relapse and the overall survival rate for infants with ALL harboring an MLL-rearrangement is approximately 50% (9, 10). This is in part due to the fact that patients with confirmed MLL translocations are often particularly resistant to glucocorticoids such as prednisone and dexamethasone, which are key components in current ALL chemotherapy treatments (6, 11, 12). Studies in vitro have also shown that MLL-ALL has a distinct drug resistance profile in comparison to childhood ALL, with high levels of resistance to glucocorticoids and L-asparaginase observed (13). These results highlight the need for treatment protocols that are more specifically tailored for MLL-ALL and the need for targeted therapies that could be incorporated to strengthen current combination chemotherapy regimens.
The p53 tumor suppressor has long been an attractive therapeutic target for anti-cancer strategies. Once p53 is activated in response to cellular stress it initiates the transcription of p53-related genes that are involved in cell cycle arrest, senescence and apoptosis, thereby preventing the proliferation of genetically unstable cells in its function as a key suppressor of tumorigenesis (14). Since errant activation of p53 could have disastrous consequences for multicellular organisms, it is tightly regulated primarily through its interaction with the ubiquitin E3 ligase MDM2 (mouse double minute 2), which suppresses p53 transcriptional activity and promotes its proteasomal degradation (15-17). It is estimated that p53 mutations are present in approximately 50% of all human cancers (14). However, they are relatively infrequent in pediatric ALL, being detected in approximately 2% and 6-19% of diagnosis and relapse cases, respectively (18-20). Although p53 mutations may be less prevalent in pediatric cancer, loss of p53 function is characteristic of virtually all cancers as even those that retain wild type p53 utilize alternative mechanisms to impede its function (21). One such mechanism is the over expression of MDM2 (22), present in 20-30% of ALL patients and is often associated with chemoresistance and a poor prognosis (23-25).
Within the past decade several strategies have been developed to reactivate p53 function in hematological malignancies, including targeting the MDM2-p53 interaction (26-30). RG7112 is an orally available cis-imidazole that binds to the hydrophobic pocket of MDM2, blocking its interaction with p53 resulting in p53-mediated induction of apoptosis (31, 32). RG7112 was previously reported by the Pediatric Preclinical Testing Program (PPTP) to exhibit broad anti-leukemic efficacy against a heterogeneous panel of patient-derived xenografts, with enhanced activity against a single infant MLL-ALL xenograft tested (31). Gene expression analysis revealed that the ALL panel had relatively high basal levels of p53 compared with other pediatric cancer histotypes, indicating that pediatric ALL may be primed for unleashing p53 from MDM2 inhibition to initiate apoptosis. Due to the relatively poor outcome for infant MLL-ALL, our preliminary report of significant in vivo RG7112 efficacy against a single infant MLL-ALL xenograft (31) clearly warranted additional evaluation against a larger panel of infant MLL-ALL patient-derived xenografts. We now report the molecular characterization of a panel of patient-derived infant MLL-ALL xenografts, their in vivo responses to single agent RG7112, and the ability of RG7112 to exert therapeutic synergy with an induction-type regimen of vincristine, dexamethasone and L-asparaginase (VXL).
Materials and Methods
Development of an infant MLL-ALL xenograft panel
All studies had prior approval from the Human Research Ethics Committees of the University of New South Wales and the Dana-Farber Cancer Institute, and the Animal Care and Ethics Committee of the University of New South Wales. Human leukemia cells used in this study were from peripheral blood or bone marrow biopsy specimens of infants with MLL-ALL and were kindly provided by Dr. Scott Armstrong (Dana-Farber Cancer Institute, Boston, MA, USA). Detailed methods used to establish patient-derived xenografts from pediatric ALL biopsies in NOD/SCID (NOD.CB17-Prkdcscid/J) mice have been previously described (33). Briefly, leukemia cells from patients were inoculated intravenously into 6-8 week old NOD/SCID mice (Australian BioResources, Moss Vale, NSW, Australia). Leukemia engraftment was monitored by flow cytometric quantification of the proportion of human CD45-positive (huCD45+) cells versus total CD45+ leukocytes (human + murine) in the peripheral blood and tissues as described previously (33, 34). The rate of engraftment was measured as the time in days following transplantation for leukemia cells to disseminate and reach 5% huCD45+ in peripheral blood. To establish continuous xenografts spleen-derived cells (usually at >95% huCD45+ purity) from engrafted mice were inoculated into secondary and tertiary recipient mice. MLL translocations were confirmed by long distance inverse-PCR as previously described (35) and serial passage xenografts were validated using a single-nucleotide polymorphism array assay.
Microarray analysis of gene expression
Gene expression profiling on RNA extracted from spleen-derived cells was performed using the Illumina Human Ref-12 Expression BeadChip (Illumina Inc., San Diego, CA). The sample gene profiles obtained were normalized using quantile normalization and log2 transformed using GenomeStudio (Version 1.6.0, Illumina Inc.). Differential gene expression was established using limma, based on a moderate t-statistic. After analysis in limma, the false discovery rate (FDR) was used to adjust the P-value for multiple testing. In this study, differences in gene expression were deemed significant with an FDR ≤ 0.05. All statistical analysis and heatmaps were generated using GenePattern (36). Gene expression data sets can be accessed at www.ncbi.nlm.nih.gov/geo (accession no. GSE52991).
In vitro cell culture and cytotoxicity assays
RS4;11, Jurkat, CEM, NALM6 cell lines were all obtained from commercial suppliers and used within 3 months of culture following validation by short tandem repeat analysis. Cell lines were maintained in RPMI supplemented with 10% heat inactivated fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine (Life Technologies, Carlsbad, CA). ALL cell lines were validated by Short Tandem Repeat analysis, verified mycoplasma-free and cultured for <3 months. Xenograft cells were retrieved from cryostorage and resuspended in QBSF-60 medium (Quality Biological, Gaithersburg, MD) supplemented with 20 ng/ml Flt-3 ligand, 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine. Prior to treatment, cells were plated in 96-well plates (100 μl/well) at a density previously optimized and were equilibrated overnight at 37°C with 5% CO2. Cells were then treated with 10-fold serial dilutions of RG7112 (10 μM – 1 pM) for 48 h, at which point AlamarBlue reagent [0.6 mM Resazurin, 0.07 mM Methylene Blue, 1 mM potassium hexacyanoferrate (III), 1 mM potassium hexacyanoferrate (II) trihydrate] was added. Fluorescence was measured at 0 and 6 h following addition of AlamarBlue using a fluorescent plate reader (VICTOR3™, PerkinElmer, MA) with excitation at 560 nm and emission at 590 nm. Data are expressed as a percentage of untreated controls.
Combination cytotoxicity assays
For fixed ratio combination assays, cells were treated with RG7112 and vincristine, dexamethasone or L-asparaginase at 0.25, 0.5, 1, 2 and 4 times the relevant half maximal inhibitory concentration (IC50) values for each cell line/xenograft. Drug effects were assessed by AlamarBlue as described in the Materials and Methods, and combination indices (CIs) were calculated using CalcuSyn software (Version 2.0, Biosoft, Cambridge, UK) to determine possible drug synergies. CI values <0.9 and >1.1 were considered synergistic and antagonistic, respectively.
Cell cycle and apoptosis assays
RS4;11 cells were seeded in 96-well plates (105 cells/ml) and incubated with 5 μM RG7112 for up to 72 h, and 10 μM of EdU (5-ethynyl-2´-deoxyuridine) (Life Technologies, Carlsbad, CA) was added to each well 2 h prior to analysis. MLL-5 and MLL-14 xenograft cells were seeded in 96-well plates at 1.5-2.5 × 106 cells/ml and exposed to 0.5 μM RG7112 for up to 24 h. The pan-caspase inhibitor, Q-VD-Oph (QVD) (Sigma-Aldrich, St Louis, MO), was included in some experiments. Apoptosis was determined using Annexin V/7-AAD staining and cell cycle analysis was assessed using the Click-iT EdU cell proliferation assay according to the manufacturer's instructions (Life Technologies, Carlsbad, CA). Samples were run on a FACSCanto and analyzed using FlowJo (version X.0.6) software (Tree Star Inc.).
Protein expression analysis
Methods for preparation of whole-cell extracts, determination of protein concentrations, and analysis of cellular proteins by immunoblotting have been described in detail previously (37). Membranes were probed with specific antibodies for the following proteins: p53 (sc-126), MDM2 (SMP14), p21WAF1 (sc-817) (Santa Cruz Biotechnology, Dallas, TX), PUMA (D30C10; Cell Signaling, Beverly, MA) and Actin (Sigma-Aldrich, St Louis, MO). The secondary antibody used was horseradish peroxidase (HRP) conjugates of anti-rabbit IgG (GE Healthcare, Little Chalfont, UK). Signals were detected by Immobilon Western Chemiluminescent HRP Substrate (Merck Millipore, Billerica, MA) and visualized using VersaDoc 5000 Imaging System (Bio-Rad, Berkeley, CA).
In vivo drug treatments
MLL-ALL xenograft cells were transplanted into 6-8 week old female NOD/SCID mice by intravenous injection. When the median %huCD45+ cells in the peripheral blood was above 1% for the entire cohort, mice were randomized and allocated to treatment groups (6-9 mice per group). RG7112 was supplied pre-formulated by Roche Pharmaceuticals Inc. through the Cancer Therapy Evaluation Program (NCI). For single agent efficacy studies RG7112 was administered orally (P.O.) at a dose of 100 mg/kg daily for 14 days. For combination studies RG7112 was given daily for 5 days at 100 mg/kg (P.O.). VXL (Prince of Wales Hospital Pharmacy, NSW, Australia) was administered via intraperitoneal injection over the same 5 day period, consisting of vincristine (0.15 mg/kg) on Day 0 followed by daily dexamethasone (5 mg/kg) and L-asparaginase (1000 U/kg) (38). Due to side effects observed during a prior tolerability experiment, standard mouse chow was replaced with a nutrient fortified water gel [DietGel recovery, (Techniplast, Rydalmere, NSW, AU)] for the duration of the combination treatment.
Determination of in vivo treatment response
Individual mouse event-free survival (EFS) was calculated as the time in days from treatment initiation until the %huCD45+ cells in the peripheral blood reached 25%, or until mice reached a humane end-point with evidence of leukemia-related morbidity. EFS time was represented graphically by Kaplan-Meier analysis and survival curves were compared by log-rank test. The response to drug treatment was evaluated by 2 methods: (1) leukemia growth delay (LGD), calculated as the difference between the median EFS of the drug-treated cohort and the median EFS of the vehicle-treated cohort; and (2) using an objective response measure (ORM) modeled after stringent clinical criteria, which was assessed at Day 42 post treatment initiation as previously described (39) and detailed in the Supplementary Methods.
In vivo pharmacodynamic analysis
Mice were inoculated with MLL-ALL xenograft cells and monitored until peripheral blood engraftment reached >70% huCD45+, at which stage a single dose of RG7112 (100 mg/kg) was administered. Groups of 3 mice were culled and spleens harvested at each time point of 6, 12 and 24 h post-treatment. Protein expression was analyzed by immunoblotting and cell cycle status by flow cytometry. For EdU cell cycle analysis, 1.25 mg was administered via intraperitoneal injection to mice 2 h prior to sample collection.
Statistical analysis
The exact log-rank test as implemented using Proc Stat-Xact for SAS was used to compare EFS distributions between treatment and control groups (2-tailed), with P ≤ 0.05 considered significant. To evaluate interactions between drugs in vivo, therapeutic enhancement was considered if the EFS of mice treated with the combination of RG7112 and VXL was significantly greater (P < 0.01) than those induced by both single arms of the study (40, 41).
Results
Development and characterization of a panel of MLL-rearranged xenografts
The engraftment and characterization of a panel of ALL primary patient biopsies in immune-deficient NOD/SCID mice has been previously described (33, 34). To expand the ALL xenograft panel to include other high-risk subtypes, 8 patient biopsies were obtained from infants diagnosed with MLL-ALL and inoculated into NOD/SCID mice. Seven of these showed robust evidence of engraftment and dissemination into the peripheral blood, while another caused hind-limb paralysis and was not used in subsequent experiments. The engraftment rates of these MLL-ALL xenografts over 3 passages are detailed in Supplementary Figure S1A and Supplementary Table S1. Three xenografts showed no significant changes in engraftment rates at 2° and 3° passage compared with 1° passage (MLL-3, -6 and -8); 2 showed acceleration at 2° and 3° passage (MLL-5 and -7); one accelerated only at 3° passage (MLL-2); and one showed variable rates of engraftment at 2° and 3° passage (MLL-1). At first passage all xenografts showed high level infiltration of bone marrow and spleen (Supplementary Figure 1B), with variable infiltration of peripheral organs consistent with the clinical experience in this disease (42). In addition, one MLL-ALL xenograft (MLL-14) with a t(11;19) (MLL-ENL translocation) that had previously been established in our laboratory was also used in this study (43). MLL translocations were confirmed by long distance inverse-PCR and are detailed in Supplementary Table S1.
Previous studies have clearly demonstrated that MLL translocations specify a gene expression profile that is distinct from other ALL subtypes, including B-cell precursor ALL (BCP-ALL) and acute myeloid leukemia (4, 44). Unsupervised hierarchical clustering revealed that a large xenograft panel clustered according to ALL subtype (Figure 1A), indicating the genes that differentiated each leukemia subtype were conserved through serial xenografting. The BCP-ALL xenograft ALL-3 clustered with the MLL-ALL xenografts, in accordance with it harboring a t(11;19) and MLL-ENL translocation (33, 34). Also, MLL-ALL xenografts loosely clustered according to their specific MLL translocation (Figure 1B; Supplementary Table S1). Of the approximate 34,600 genes tested, 1419 were up-regulated and 1235 down-regulated in the 9 MLL-ALL xenografts compared with 17 BCP-ALL xenografts (defined by FDR < 0.05). The top 100 genes that were most highly correlated with each class distinction are shown in Figure 1C; additional information on these genes, including FDR values, is detailed in Supplementary Table S2. Genes previously reported (4, 11, 44) to be under-expressed in MLL-translocated versus non-translocated leukemia [including MME (CD10), LARGE, ID3 and ALOX5] are represented in the list (Figure 1C, Supplementary Figure 2A, Supplementary Table S2). Genes reported to be over-expressed in MLL-translocated leukemia, including MEIS1 (a co-factor of HOXA9 and HOXA7), RNASE3 and IGF2BP2 were also significantly up-regulated in the MLL-ALL xenografts (Figure 1C, Supplementary Figure 2B, Supplementary Table S2). Moreover the HOXA genes, which are reported to be over-expressed in the majority of infant MLL-ALL cases (4, 45, 46), were overexpressed in 6/9 MLL-ALL xenografts (Supplementary Figure S3). The differences in expression of all of these HOXA genes were significant between the MLL-ALL and BCP-ALL xenografts (Supplementary Figure S3B).
Figure 1. Characterization of an MLL-ALL xenograft panel by gene expression profiling.

A, unsupervised hierarchical clustering of MLL-ALL (n=9, red), BCP-ALL (n=17, yellow) and T-ALL (blue, n=9) xenografts. B, dendrogram representing hierarchical clustering of the MLL-ALL xenograft panel: t(11;19), green; t(4;11), yellow; t(1;11), purple; t(10;11), blue; t(11;17), red. C, the top 100 genes most highly correlated with either the MLL-ALL or BCP-ALL subtype. Each column represents an individual leukemia xenograft sample and each row represents a unique gene. Expression levels are normalized for each gene, where the mean is 0. Expression levels greater than the mean are shown in red and less than the mean are in blue. (D) Heat map separating the MLL-rearranged from BCP-ALL xenografts based on the MLL-rearranged gene expression signature published by Armstrong et al (4). Red arrows indicate genes that were differentially expressed (FDR ≤ 0.05) between MLL-ALL and BCP-ALL xenografts (see Supplementary Table S3 for statistical analysis). Color scaling is as indicated in (C).
We then asked whether the unique gene expression signature for MLL-translocated ALL reported by Armstrong et al (4) was represented in the MLL-ALL xenografts that had undergone serial passaging in vivo. The Armstrong signature clearly separated the MLL-ALL and BCP-ALL xenografts (Figure 1D) with 70% of the genes identified by Armstrong et al being significantly differentially expressed between the two xenograft panels (Figure 1D with statistical analysis shown in Supplementary Table S3). Collectively, these results confirm that the unique gene expression signature distinguishing MLL-translocated leukemia is maintained during serial passage through NOD/SCID mice.
RG7112 activates p53 and induces cell cycle arrest and apoptosis in leukemia cells in vitro
In preparation for assessing the efficacy of RG7112 against the panel of MLL-ALL xenografts described above, we first tested its in vitro cytotoxicity against leukemia cell lines of known p53 status. Consistent with the literature (32), the cytotoxic effects of RG7112 appeared to be dependent on p53 status; NALM6 and RS4;11 (p53 wild-type) exhibited IC50s of 3.2 μM and 1.4 μM, respectively, while those of JURKAT and CEM (p53 mutant) were >10 μM (Figure 2A). When assessed using the same in vitro assay MLL-ALL xenograft cells exhibited IC50s of 0.1-0.35 μM, consistent with wild-type p53 status (Figure 2A). The wild-type p53 status of the infant MLL-ALL xenografts was also confirmed by exome sequencing (data not shown).
Figure 2. RG7112 activates p53 signaling and induces cell cycle arrest and apoptosis in leukemia cells in vitro.
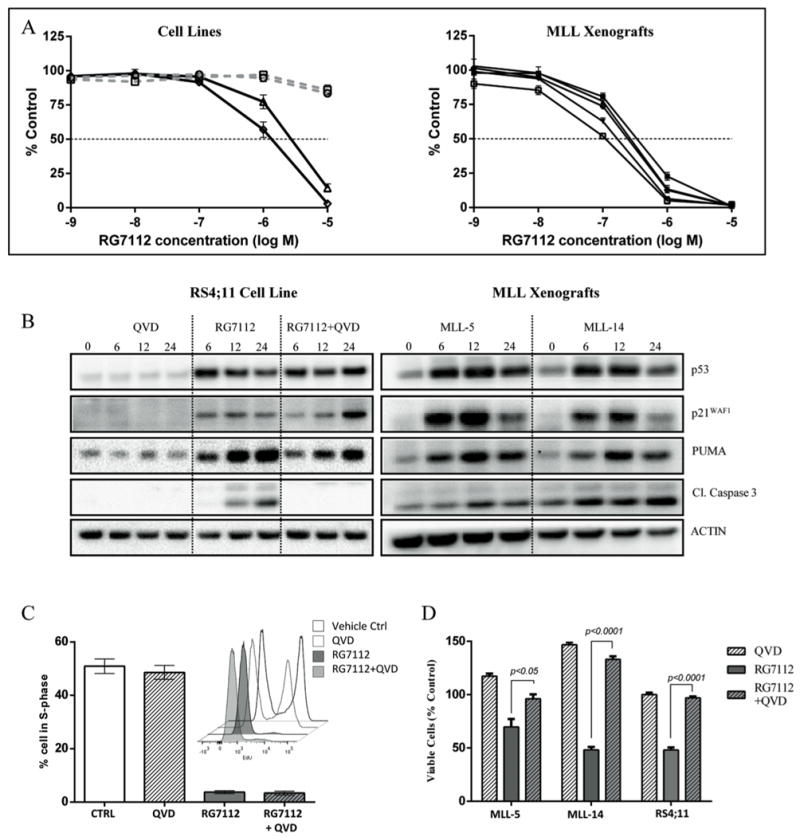
A, RG7112 cytotoxicity depends on the p53 status of leukemia cells. Two p53 wild-type (NALM6, triangles; RS4;11, diamonds) or mutant (JURKAT, circles; CEM, squares) cell lines and 5 MLL-ALL xenografts were incubated with RG7112 for 48 h and cell viability measured by AlamarBlue assay. B, protein levels of p53, p21WAF1,PUMA, and cleaved caspase 3 (Cl.caspase 3) were analyzed by immunoblotting in RS4;11 cells after treatment with 5 μM RG7112, or MLL xenograft cells exposed to 0.5 μM RG7712 for 6, 12 and 24 h. C, RS4;11 cells were incubated with RG7112 (5 μM), QVD (10 μM) or RG7112+QVD for 24 h and the % cells in S-phase was determined by EdU labeling and cell cycle analysis. Insert is a representative histogram showing EdU uptake in RS4;11 cells at 24 h post treatment. D, RG7112 induces caspase-dependent apoptosis in leukemia cells. MLL-ALL xenograft cells and the RS4;11 cell line were treated with RG7112 (0.5 and 5 μM, respectively), 10 μM QVD or RG7112 + QVD for 24 hours. Data represent the mean ± SEM from ≥ 3 independent experiments.
In order to define the mechanism of RG7112-induced cell death of MLL-ALL cells we next showed that exposure of RS4;11 cells to RG7112 resulted in profound up-regulation of the p53 protein within 6 h, as well as its downstream targets p21WAF1 and PUMA (Figure 2B). This activation of p53 was associated with a profound decrease in cells in S-phase, and accumulation of cells in G1 (Figure 2C). This cell cycle arrest was followed by induction of apoptosis, as assessed by annexin V/7AAD staining (Figure 2D and Supplementary Figure S4), and cleavage of caspase 3 (Figure 2B). Since the IC50 values of the MLL-ALL xenograft cells were approximately 10-fold lower than RS4;11 cells, we next exposed MLL-5 and MLL-14 xenograft cells to a 10-fold lower concentration of RG7112 than RS4;11 cells in vitro, and observed similar findings. Immunoblot analysis revealed marked upregulation of p53 within 6 h, as well as the downstream proteins p21WAF1, PUMA and cleaved caspase 3 in both xenografts (Figure 2B).
Since RG7112 has been previously reported to induce apoptosis in a caspase-independent manner (32), we treated RS4;11 cells with RG7112 in the presence of the pan-caspase inhibitor QVD. QVD alone had no effect on the viability of RS4;11 cells for up to 72 h (Figure 2D and Supplementary Figure S4). However, it substantially delayed and inhibited RG7112-induced cell death via apoptosis, as measured by its effects on RG7112-induced loss of cell viability and its ability to prevent cleavage of caspase 3 (Figure 2B). In contrast, QVD did not inhibit the ability of RG7112 to induce p53, p21WAF1 or PUMA (Figure 2B), and cell cycle arrest (Figure 2C). QVD also partially inhibited RG7112-induced apoptosis in both MLL-5 and MLL-14 xenograft cells after 24 h treatment (Figure 2D). These results show that RG7112 induces caspase-dependent apoptosis in both MLL-ALL cell lines and xenograft cells, in contrast to studies with cancer cells of different histotypes (32).
RG7112 activates the p53 pathway in vivo and induces significant regressions in infant MLL-ALL xenografts
The in vivo efficacy of RG7112 was next tested against the infant MLL-ALL xenograft panel described above. Once daily administration of RG7112 for 14 days induced significant and prolonged regressions of 7/7 xenografts (Figure 3A, Table 1 and Supplementary Figure S5), including the previously reported results for MLL-7 (31). Using stringent objective response criteria modeled after the clinical setting, 6 xenografts achieved complete responses (CRs), and one (MLL-7) had previously sustained a maintained complete response (MCR) (31). RG7112 also significantly prolonged mouse EFS compared to vehicle controls in all evaluable xenografts, with LGDs ranging from 17.1-44.7 days (Table 1). The 14-day RG7112 treatment was well tolerated and caused no marked effects on mouse weight or hematological parameters (Supplementary Figure S6). A complete summary of results is provided in Supplementary Table S4, including total numbers of mice, numbers of mice that died (or were otherwise excluded), numbers of mice with events and median times to events, as well as numbers in each of the ORM categories and “treated over control” (T/C) values. Of note, only 2/51 (<4%) of RG7112 mice experienced toxicity-related events, with 45/51 (88%) of mice analyzed for leukemia-related events.
Figure 3. RG7112 exhibits significant single-agent efficacy against MLL-ALL xenografts in vivo.

A, individual (left panels) and median (middle panels) %huCD45+ cells in the peripheral blood and Kaplan–Meier curves for EFS (right panels) for MLL-5 and MLL-6. Vehicle controls, dashed lines; RG7112 treated, solid lines. B, representative immunoblots of protein expression following in vivo RG7112 treatment of groups of 3 mice engrafted with MLL-5 or MLL-14. Lysates were prepared from spleen-derived cells (>95% huCD45+) after treatment with 100 mg/kg RG7112 for various time points and immunoblotted for p53, MDM2, p21WAF1, PUMA and survivin. Blots are representative of spleens from n=3 mice, see Supplementary Figure S7 for immunoblots of all MLL-14 engrafted mice. C, flow cytometry analysis of in vivo EdU incorporation into huCD45+ spleen-derived cells following RG7112 treatment. N ≥ 3 mice/timepoint, data represent the mean ± SEM. D, representative histogram of EdU staining of huCD45+ spleen-derived cells 24 h post RG7112 treatment. In (C) and (D) mice were administered a single dose of vehicle or RG7112 (100 mg/kg) and pulsed with EdU 2 h before sample collection.
Table 1. In vivo activity of RG7112 against MLL-ALL xenografts.
| Xenograft | Median EFS (Days) | LGD (Days) | Log-rank P-value (treated versus control) | Median ORM | |
|---|---|---|---|---|---|
|
| |||||
| Control | RG7112 | ||||
| MLL-2 | 9.7 | 41.4 | 31.7 | 0.0002 | CR |
| MLL-3 | 10.5 | 32.2 | 21.7 | <0.0001 | CR |
| MLL-5 | 6.4 | 23.5 | 17.1 | <0.0001 | CR |
| MLL-6 | 4.7 | 34.8 | 30.1 | <0.0001 | CR |
| MLL-7a | 6.8 | 51.5 | 44.7 | <0.0001 | MCR |
| MLL-8 | 6.8 | 26.6 | 26.6 | 0.0011 | CR |
| MLL-14 | 6.3 | 27.3 | 27.3 | <0.0001 | CR |
Values in bold signify statistically significant drug effects (P<0.05).
Data for this xenograft previously published (31).
To investigate mechanisms associated with the pronounced in vivo efficacy of RG7112 against infant MLL-ALL xenografts, mice highly engrafted with MLL-5 or MLL-14 received a single dose of RG7112 or vehicle control and spleen-derived cells (>95% huCD45+) were harvested 6-48 h later. Immunoblot analysis showed a marked increase in p53 and its transcriptional targets MDM2 and p21WAF1 proteins within 6 h of treatment (see Figure 3B for immunoblots representative of a single mouse from each group of 3 mice, and Supplementary Figure S7 for replicate immunoblots of all MLL-14 engrafted mice). The highest levels of p53 protein were observed at 6 h post treatment, but p53 remained elevated until at least 24 h post treatment. This activation of p53 was followed by marked cell cycle arrest; flow cytometry detection of EdU incorporation demonstrated a significant reduction in the proportion of MLL-5 cells (gated on huCD45+ cells) in S-phase in RG7112 treated mice compared to controls (Figure 3C and D). After 24 h only 10.9 ± 1.3% of RG7112 treated MLL-5 cells incorporated EdU compared to 28.1 ± 1.0% of control cells (Figure 3C). Up-regulation of p53 was followed by induction of PUMA, and down-regulation of survivin (Figure 3B). These results indicate that RG7112 effectively induces cell cycle arrest and pro-apoptotic protein expression patterns in vivo in infant MLL-ALL xenografts.
In order to further understand the underlying basis for the impressive in vivo efficacy of RG7112 against infant MLL-ALL xenografts, we compared the relative expression of the TP53 and MDM2 genes with respect to in vivo RG7112 efficacy between infant MLL-ALL xenografts in this study, and BCP-ALL xenografts in our previous report (31). Infant MLL-ALL xenografts expressed significantly higher levels of TP53, and significantly lower MDM2 levels, than BCP-ALL xenografts (Supplementary Figure S8A-C). Basal expression of MDM2 (R2=0.52, P<0.005, Supplementary Figure S8D) but not TP53 (R2=0.026, P=0.58, Supplementary Figure S8E) significantly correlated with in vivo sensitivity of ALL xenografts to RG7112, with the paradoxical finding that xenografts expressing lower levels of MDM2 exhibited a longer progression delay. The infant MLL-ALL xenografts had a significantly better response to RG7112 in vivo compared to BCP-ALL xenografts, as assessed by both treated/control EFS and treated-control EFS values (Supplementary Figure S8F-G).
Synergistic in vitro and in vivo effects of RG7112 with established drugs against MLL-translocated leukemia cells
Approximately 90% of infants with MLL-ALL initially enter complete remission when treated with combination chemotherapy, although 5-year EFS is, at best, 50% for these patients (6, 9, 10). Therefore, we next assessed whether RG7112 exhibited the potential to augment the anti-leukemic effects of the established drugs vincristine, dexamethasone and L-asparaginase. Fixed-ratio combination cytotoxicity assays were carried out using ex vivo cultured MLL-ALL xenograft cells and the RS4;11 cell line (Figure 4). RG7112 exhibited synergistic cytotoxicity with vincristine in 4/4 models, with dexamethasone in 2/4 and L-asparaginase in 3/4 (Figure 4 and Supplementary Table S5).
Figure 4. Combination cytotoxicity of RG7112 with vincristine, dexamethasone or L-asparaginase.

Dose response curves of the RS4;11 cell line and infant MLL-ALL xenografts to RG7112 (closed circles), vincristine, dexamethasone, or L-asparaginase (open triangles) and the relevant combinations (gray triangles). The concentrations used were 0.25, 0.5 1, 2, 4 × the IC50 values for each drug/cell type. Results shown are the mean ± SEM of 3 biological repeats.
Since RG7112 exhibited broad in vitro synergistic effects with vincristine, dexamethasone and L-asparaginase across 4 MLL-translocated leukemia models, we next assessed whether these results could be replicated in the in vivo setting. A 4-week VXL induction-type regimen that was recently optimized in our lab (38) induced significant progression delays in all MLL-ALL xenografts (Supplementary Table S6). Since RG7112 administered daily for 14 days also exerted impressive single-agent efficacy against all MLL-ALL xenografts tested (Figure 3, Table 1, Supplementary Figure S5, Supplementary Table S4), we devised an abbreviated 5-day treatment schedule in order to achieve measurable responses and indications of therapeutic synergy (see Materials and Methods).
VXL and RG7112 significantly delayed the progression of MLL-5 and MLL-14 compared to vehicle controls (Figure 5, Supplementary Table S7). RG7112 achieved progressive disease 2 (PD2) in MLL-5 (P < 0.0001) and a CR in MLL-14 (P = 0.001), with LGDs of 11.1 and 17.3 days, respectively. VXL also achieved a PD2 in MLL-5 and CR in MLL-14, with LGDs of 8.0 and 23.5 days, respectively. The combination of RG7112 and VXL significantly extended disease remission to 19.0 days for MLL-5 and increased the ORM from PD2 for each of RG7112 or VXL to a CR for the combination, while the LGD for MLL-14 increased to 60.1 days with an ORM of MCR (compared to CRs for RG7112 and VXL alone). Using strict criteria defined in the Materials and Methods RG7112 plus VXL caused therapeutic enhancement in MLL-14 (P < 0.01 for the combination versus both the RG7112 and VXL treatment arms) and a trend for therapeutic enhancement for MLL-5 (P = 0.04 for the combination versus VXL). These data suggest that combining RG7112 with current remission-induction chemotherapy could prove useful in the management of infant MLL-ALL.
Figure 5. In vivo efficacy of RG7112 in combination with VXL against infant MLL-ALL xenografts.

A and B, individual (left panels) and median (middle panels) %huCD45+ cells in the peripheral blood and Kaplan–Meier curves for EFS (right panels), for MLL-5 (A) and MLL-14 (B). Vehicle controls, black dotted lines; VXL, green lines; RG7112, blue lines; RG7112/VXL combination, red lines. C, distribution of ORMs of individual mice engrafted with MLL-5 or MLL-14 and treated with RG7112, VXL or RG7112 plus VXL.
Discussion
The NOD/SCID mouse model has previously been shown to be a useful tool for the engraftment and serial passage of primary childhood ALL patient biopsies (33, 34). In addition, it has been demonstrated that in vivo responses of childhood ALL xenografts to commonly used chemotherapeutic agents, dexamethasone and vincristine, significantly correlated with the clinical outcome of the patients from whom the xenografts were originally derived (34). Here we report on the establishment and characterization of a new panel of xenografts derived from infant ALL harboring MLL translocations. Seven of nine samples successfully engrafted and were further expanded to quaternary passage, demonstrating the robustness of this xenograft model. Gene expression profiling revealed that MLL-ALL xenografts cluster separately from BCP-ALL and T-ALL xenografts and that there was a subset of 2,654 genes that were differentially expressed between the MLL-ALL and BCP-ALL xenografts. These results demonstrate that the unique signature distinguishing MLL-translocated leukemias from BCP-ALL and T-ALL was maintained during serial passage through NOD/SCID mice; an important requirement for useful pre-clinical models to study novel therapies specifically targeted against this disease.
Not only are p53 mutations uncommon in infant MLL-ALL, but some of the most frequently detected MLL fusion proteins are reported to inhibit p53 transcriptional activity, which may contribute to MLL-induced leukemogenesis and drug resistance (47). Therefore, drugs that release p53 from inhibitory mechanisms could be of therapeutic benefit against this high-risk ALL subtype. We previously examined the in vivo efficacy of RG7112 as a single agent against 8 ALL xenografts (5 BCP-ALL, 2 T-ALL and 1 infant MLL-ALL) (31). RG7112 demonstrated strong anti-leukemic activity against all ALL subtypes, however the most impressive response was observed in the single infant MLL-ALL xenograft (MLL-7) which achieved an LGD of >44 days and an MCR. Here we report that RG7112 induces significant regressions against a large panel of MLL-ALL xenografts with diverse MLL translocation partners in vivo. RG7112 efficiently stabilized and activated p53 in MLL-ALL xenografts leading to induction of p53-dependent cell cycle arrest and apoptosis in vivo. In addition to the induction of p53-dependent pro-apoptotic targets such as PUMA, we also observed down regulation of the anti-apoptotic protein survivin after RG7112 treatment. This is consistent with a previous study reporting that the survivin gene contains a p53 response element, which is activated upon cellular stress resulting in reduced survivin expression (48). The mechanism of death induced by RG7112 in an MLL-ALL cell line and xenografts appeared highly dependent on apoptosis, in contrast to studies with cancer cells of different histotypes (32), which may reflect the greater propensity of lymphoid cells to undergo apoptosis compared with other cell lineages. Taken together our results suggest that RG7112-mediated cell death in infant MLL-ALL is due to activation of the p53 protein and subsequent p53-mediated transcriptional activity, both through induction of pro-apoptotic factors and repression of anti-apoptotic proteins such as survivin.
Infants diagnosed with MLL-rearranged ALL usually respond to initial induction therapy, but the majority achieves only a shallow remission with detectable molecular disease and these patients frequently relapse on therapy (49). In this study we showed that combining RG7112 with an induction-type regimen (VXL) significantly enhanced objective responses and prolonged leukemia regressions in vivo, which reflected the in vitro synergistic interactions between paired combinations of RG7112 with the individual established drugs. While the precise mechanism(s) associated with these in vitro and in vivo synergistic interactions were not defined, this study provides an important proof of concept for the use of nutlin-based p53 activators in combination with established induction schedules to improve the treatment of p53 wild-type infant MLL-ALL. The pharmacodynamic studies in immune-deficient mice engrafted with infant MLL-ALL patient-derived xenografts reported herein also agree with a recently completed clinical study that confirmed the ability of RG7112 to activate p53 and induce cell cycle arrest and apoptosis in human tumors in vivo (50). Taken together, these findings support further investigations targeting the p53/MDM2 axis to improve the treatment of infant MLL-ALL.
Supplementary Material
Translational Relevance.
Infants with acute lymphoblastic leukemia (ALL) harboring translocations in the mixed-lineage leukemia (MLL) oncogene (infant MLL-ALL) experienced lower survival rates than children with ALL, and more effective treatments are urgently required. Due to the low incidence of p53 mutations in infant MLL-ALL, the p53/MDM2 axis represents a potential target to improve its treatment. This study describes the development and molecular characterization of a panel of patient-derived infant MLL-ALL xenografts, and their in vivo sensitivity to the nutlin derivative and MDM2 inhibitor RG7112. RG7112 induced significant regressions and prolonged progression delay in a panel of infant MLL-ALL xenografts, concomitant with p53 up-regulation, cell cycle arrest, and induction of apoptosis. Moreover, RG7112 significantly enhanced the in vivo efficacy of an induction-type regimen of vincristine, dexamethasone and L-asparaginase against infant MLL-ALL xenografts. These results support the rationale for targeting the p53/MDM2 axis in combination with established drugs in the treatment of infant MLL-ALL.
Acknowledgments
The authors thank Drs Scott Armstrong and Lewis Silverman for providing infant MLL-ALL biopsy samples (Dana-Farber Cancer Institute, Boston, MA, USA), and Roche Pharmaceuticals Inc. for providing RG7112. Children's Cancer Institute is affiliated with the University of New South Wales and The Sydney Children's Hospitals Network.
Grant Support: This research was funded by grants from the National Cancer Institute (NOI-CM-42216 and NOI-CM-91001-03), and by grant DKS 2011.09 from the German Children Cancer Aid to RM. RBL is supported by a Fellowship from the National Health and Medical Research Council.
Footnotes
Disclosure of Potential Conflicts of Interest: The authors declare no conflicts of interest.
Authors’ Contributions: Concept and design: J. Richmond, H. Carol, L. High, P.J. Houghton, M.A. Smith, R.B. Lock
Acquisition of data: J. Richmond, K. Evans, H. Carol, L. High, A. Mendomo, A. Robbins, C. Meyer, N. Venn, R. Marschalek, M. Henderson and R. Sutton
Analysis and interpretation of data; J. Richmond, H. Carol, L. High, C. Meyer, R. Marschalek, M. Henderson, R. Sutton and R.T. Kurmasheva
Writing, review, and/or revision of the manuscript; J. Richmond, U.R. Kees, P.J. Houghton, M.A. Smith and R.B. Lock
References
- 1.Pui CH, Pei D, Campana D, Bowman WP, Sandlund JT, Kaste SC, et al. Improved prognosis for older adolescents with acute lymphoblastic leukemia. J Clin Oncol. 2011;29:386–91. doi: 10.1200/JCO.2010.32.0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hunger SP, Lu X, Devidas M, Camitta BM, Gaynon PS, Winick NJ, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. J Clin Oncol. 2012;30:1663–9. doi: 10.1200/JCO.2011.37.8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Popovic R, Zeleznik-Le NJ. MLL: how complex does it get? J Cell Biochem. 2005;95:234–42. doi: 10.1002/jcb.20430. [DOI] [PubMed] [Google Scholar]
- 4.Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30:41–7. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- 5.Li ZY, Liu DP, Liang CC. New insight into the molecular mechanisms of MLL-associated leukemia. Leukemia. 2005;19:183–90. doi: 10.1038/sj.leu.2403602. [DOI] [PubMed] [Google Scholar]
- 6.Pieters R, Schrappe M, De Lorenzo P, Hann I, De Rossi G, Felice M, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet. 2007;370:240–50. doi: 10.1016/S0140-6736(07)61126-X. [DOI] [PubMed] [Google Scholar]
- 7.Hilden JM, Dinndorf PA, Meerbaum SO, Sather H, Villaluna D, Heerema NA, et al. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the Children's Oncology Group. Blood. 2006;108:441–51. doi: 10.1182/blood-2005-07-3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jansen MW, Corral L, van der Velden VH, Panzer-Grumayer R, Schrappe M, Schrauder A, et al. Immunobiological diversity in infant acute lymphoblastic leukemia is related to the occurrence and type of MLL gene rearrangement. Leukemia. 2007;21:633–41. doi: 10.1038/sj.leu.2404578. [DOI] [PubMed] [Google Scholar]
- 9.Kotecha RS, Gottardo NG, Kees UR, Cole CH. The evolution of clinical trials for infant acute lymphoblastic leukemia. Blood Cancer J. 2014;4:e200. doi: 10.1038/bcj.2014.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silverman LB. Acute lymphoblastic leukemia in infancy. Pediatr Blood Cancer. 2007;49:1070–3. doi: 10.1002/pbc.21352. [DOI] [PubMed] [Google Scholar]
- 11.Yeoh EJ, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1:133–43. doi: 10.1016/s1535-6108(02)00032-6. [DOI] [PubMed] [Google Scholar]
- 12.Stam RW, den Boer ML, Pieters R. Towards targeted therapy for infant acute lymphoblastic leukaemia. Br J Haematol. 2006;132:539–51. doi: 10.1111/j.1365-2141.2005.05909.x. [DOI] [PubMed] [Google Scholar]
- 13.Ramakers-van Woerden NL, Beverloo HB, Veerman AJ, Camitta BM, Loonen AH, van Wering ER, et al. In vitro drug-resistance profile in infant acute lymphoblastic leukemia in relation to age, MLL rearrangements and immunophenotype. Leukemia. 2004;18:521–9. doi: 10.1038/sj.leu.2403253. [DOI] [PubMed] [Google Scholar]
- 14.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–31. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 15.Yang Y, Li C-CH, Weissman AM. Regulating the p53 system through ubiquitination. Oncogene. 2004;23:2096–106. doi: 10.1038/sj.onc.1207411. [DOI] [PubMed] [Google Scholar]
- 16.Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–3. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 17.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–45. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 18.Blau O, Avigad S, Stark B, Kodman Y, Luria D, Cohen IJ, et al. Exon 5 mutations in the p53 gene in relapsed childhood acute lymphoblastic leukemia. Leuk Res. 1997;21:721–9. doi: 10.1016/s0145-2126(97)80032-x. [DOI] [PubMed] [Google Scholar]
- 19.Hof J, Krentz S, van Schewick C, Korner G, Shalapour S, Rhein P, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29:3185–93. doi: 10.1200/JCO.2011.34.8144. [DOI] [PubMed] [Google Scholar]
- 20.Wada M, Bartram C, Nakamura H, Hachiya M, Chen D, Borenstein J, et al. Analysis of p53 mutations in a large series of lymphoid hematologic malignancies of childhood. Blood. 1993;82:3163–9. [PubMed] [Google Scholar]
- 21.Lane DP. p53, guardian of the genome. Nature. 1992;358:15–6. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 22.Bueso-Ramos C, Yang Y, deLeon E, McCown P, Stass S, Albitar M. The human MDM-2 oncogene is overexpressed in leukemias. Blood. 1993;82:2617–23. [PubMed] [Google Scholar]
- 23.Marks D, Kurz B, Link M, Ng E, Shuster J, Lauer S, et al. Altered expression of p53 and mdm-2 proteins at diagnosis is associated with early treatment failure in childhood acute lymphoblastic leukemia. J Clin Oncol. 1997;15:1158–62. doi: 10.1200/JCO.1997.15.3.1158. [DOI] [PubMed] [Google Scholar]
- 24.Gustafsson B, Axelsson B, Christensson B, Winiarski J. MDM2 and p53 in childhood acute lymphoblastic leukemia: higher expression in childhood leukemias with poor prognosis compared to long-term survivors. Pediatr Hematol Oncol. 2001;18:497–508. doi: 10.1080/088800101753328466. [DOI] [PubMed] [Google Scholar]
- 25.Zhou M, Gu L, Abshire TC, Homans A, Billett AL, Yeager AM, et al. Incidence and prognostic significance of MDM2 oncoprotein overexpression in relapsed childhood acute lymphoblastic leukemia. Leukemia. 2000;14:61–7. doi: 10.1038/sj.leu.2401619. [DOI] [PubMed] [Google Scholar]
- 26.Gu L, Zhu N, Findley HW, Zhou M. MDM2 antagonist nutlin-3 is a potent inducer of apoptosis in pediatric acute lymphoblastic leukemia cells with wild-type p53 and overexpression of MDM2. Leukemia. 2008;22:730–9. doi: 10.1038/leu.2008.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kojima K, McQueen T, Chen Y, Jacamo R, Konopleva M, Shinojima N, et al. p53 activation of mesenchymal stromal cells partially abrogates microenvironment-mediated resistance to FLT3 inhibition in AML through HIF-1alpha-mediated down-regulation of CXCL12. Blood. 2011;118:4431–9. doi: 10.1182/blood-2011-02-334136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 29.Zhu N, Gu L, Li F, Zhou M. Inhibition of the Akt/survivin pathway synergizes the antileukemia effect of nutlin-3 in acute lymphoblastic leukemia cells. Mol Cancer Ther. 2008;7:1101–9. doi: 10.1158/1535-7163.MCT-08-0179. [DOI] [PubMed] [Google Scholar]
- 30.Drakos E, Thomaides A, Medeiros LJ, Li J, Leventaki V, Konopleva M, et al. Inhibition of p53-murine double minute 2 interaction by nutlin-3A stabilizes p53 and induces cell cycle arrest and apoptosis in Hodgkin lymphoma. Clin Cancer Res. 2007;13:3380–7. doi: 10.1158/1078-0432.CCR-06-2581. [DOI] [PubMed] [Google Scholar]
- 31.Carol H, Reynolds CP, Kang MH, Keir ST, Maris JM, Gorlick R, et al. Initial testing of the MDM2 inhibitor RG7112 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2013;60:633–41. doi: 10.1002/pbc.24235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tovar C, Graves B, Packman K, Filipovic Z, Xia BHM, Tardell C, et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013;73:2587–97. doi: 10.1158/0008-5472.CAN-12-2807. [DOI] [PubMed] [Google Scholar]
- 33.Lock RB, Liem N, Farnsworth ML, Milross CG, Xue C, Tajbakhsh M, et al. The nonobese diabetic/severe combined immunodeficient (NOD/SCID) mouse model of childhood acute lymphoblastic leukemia reveals intrinsic differences in biologic characteristics at diagnosis and relapse. Blood. 2002;99:4100–8. doi: 10.1182/blood.v99.11.4100. [DOI] [PubMed] [Google Scholar]
- 34.Liem NL, Papa RA, Milross CG, Schmid MA, Tajbakhsh M, Choi S, et al. Characterization of childhood acute lymphoblastic leukemia xenograft models for the preclinical evaluation of new therapies. Blood. 2004;103:3905–14. doi: 10.1182/blood-2003-08-2911. [DOI] [PubMed] [Google Scholar]
- 35.Meyer C, Hofmann J, Burmeister T, Groger D, Park TS, Emerenciano M, et al. The MLL recombinome of acute leukemias in 2013. Leukemia. 2013;27:2165–76. doi: 10.1038/leu.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38:500–1. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 37.Bachmann PS, Gorman R, Papa RA, Bardell JE, Ford J, Kees UR, et al. Divergent mechanisms of glucocorticoid resistance in experimental models of pediatric acute lymphoblastic leukemia. Cancer Res. 2007;67:4482–90. doi: 10.1158/0008-5472.CAN-06-4244. [DOI] [PubMed] [Google Scholar]
- 38.Szymanska B, Wilczynska-Kalak U, Kang MH, Liem NL, Carol H, Boehm I, et al. Pharmacokinetic modeling of an induction regimen for in vivo combined testing of novel drugs against pediatric acute lymphoblastic leukemia xenografts. PLoS ONE. 2012;7:e33894. doi: 10.1371/journal.pone.0033894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, et al. The pediatric preclinical testing program: Description of models and early testing results. Pediatr Blood Cancer. 2007;49:928–40. doi: 10.1002/pbc.21078. [DOI] [PubMed] [Google Scholar]
- 40.Houghton PJ, Morton CL, Gorlick R, Lock RB, Carol H, Reynolds CP, et al. Stage 2 combination testing of rapamycin with cytotoxic agents by the Pediatric Preclinical Testing Program. Mol Cancer Ther. 2010;9:101–12. doi: 10.1158/1535-7163.MCT-09-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rose WC, Wild R. Therapeutic synergy of oral taxane BMS-275183 and cetuximab versus human tumor xenografts. Clin Cancer Res. 2004;10:7413–7. doi: 10.1158/1078-0432.CCR-04-1045. [DOI] [PubMed] [Google Scholar]
- 42.Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004;350:1535–48. doi: 10.1056/NEJMra023001. [DOI] [PubMed] [Google Scholar]
- 43.Henderson MJ, Choi S, Beesley AH, Baker DL, Wright D, Papa RA, et al. A xenograft model of infant leukaemia reveals a complex MLL translocation. Br J Haematol. 2008;140:716–9. doi: 10.1111/j.1365-2141.2007.06966.x. [DOI] [PubMed] [Google Scholar]
- 44.Stam RW, Schneider P, Hagelstein JA, van der Linden MH, Stumpel DJ, de Menezes RX, et al. Gene expression profiling-based dissection of MLL translocated and MLL germline acute lymphoblastic leukemia in infants. Blood. 2010;115:2835–44. doi: 10.1182/blood-2009-07-233049. [DOI] [PubMed] [Google Scholar]
- 45.Dou Y, Hess JL. Mechanisms of transcriptional regulation by MLL and its disruption in acute leukemia. Int J Hematol. 2008;87:10–8. doi: 10.1007/s12185-007-0009-8. [DOI] [PubMed] [Google Scholar]
- 46.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–33. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 47.Wiederschain D, Kawai H, Shilatifard A, Yuan ZM. Multiple mixed lineage leukemia (MLL) fusion proteins suppress p53-mediated response to DNA damage. J Biol Chem. 2005;280:24315–21. doi: 10.1074/jbc.M412237200. [DOI] [PubMed] [Google Scholar]
- 48.Zhou M, Gu L, Li F, Zhu Y, Woods WG, Findley HW. DNA damage induces a novel p53-survivin signaling pathway regulating cell cycle and apoptosis in acute lymphoblastic leukemia cells. J Pharmacol Exp Ther. 2002;303:124–31. doi: 10.1124/jpet.102.037192. [DOI] [PubMed] [Google Scholar]
- 49.Van der Velden VH, Corral L, Valsecchi MG, Jansen MW, De Lorenzo P, Cazzaniga G, et al. Prognostic significance of minimal residual disease in infants with acute lymphoblastic leukemia treated within the Interfant-99 protocol. Leukemia. 2009;23:1073–9. doi: 10.1038/leu.2009.17. [DOI] [PubMed] [Google Scholar]
- 50.Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. The Lancet Oncology. 2012;13:1133–40. doi: 10.1016/S1470-2045(12)70474-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.