

Abstract
Although the association between cancer and venous thromboembolism (VTE) has long been known, the mechanisms are poorly understood. Circulating tissue factor–bearing extracellular vesicles have been proposed as a possible explanation for the increased risk of VTE observed in some types of cancer. The International Society for Extracellular Vesicles (ISEV) and International Society on Thrombosis and Haemostasis (ISTH) held a joint Educational Day in April 2014 to discuss the latest developments in this field. This review discusses the themes of that event and the ISEV 2014 meeting that followed.
Keywords: extracellular vesicles, thrombosis, cancer, tissue factor
The association between cancer and venous thrombosis (venous thromboembolism, VTE) was first recognized by Jean-Baptiste Bouillaud in 1823 (1). VTE is a frequent complication in cancer patients and is a leading cause of death. For example, pulmonary embolism is responsible for 14% of deaths in cancer patients who die in hospital (2). Different cancer types have different rates of thrombosis, but the underlying mechanisms for the increased risk of VTE are still incompletely understood (3). In the early 1980s, tumour-derived and tissue factor (TF)-exposing extracellular vesicles (EVs) were proposed as a possible mechanism to explain the increased risk of VTE in cancer patients (4,5). Cancer cells can express TF, which can be released on EVs into the circulation. Although such procoagulant TF-exposing EVs (TF-EVs) are associated with the increased rates of VTE reported in cancer patients (6), this mechanism remains controversial. Very few of the studies reported in literature were performed in vivo. Furthermore, isolation and measurement methods are not standardized and comparable. Nevertheless, despite these limitations we believe that there is already strong evidence that EVs play an important role in tumour growth and development.
It was against this backdrop that the International Society for Extracellular Vesicles (ISEV) and the International Society on Thrombosis and Haemostasis (ISTH) held their first joint symposium on 29th April 2014 in Rotterdam, The Netherlands, to discuss the latest developments in EVs, TF, cancer and thrombosis. As with previous ISEV meetings, cancer EV was a major topic during ISEV 2015 with 61 oral and 28 poster cancer presentations. These covered EVs in the tumour microenvironment, EVs in cancer metastasis and EVs as tumour biomarkers. Excitingly, the role of EVs as therapeutic agents or targets in cancer therapy is starting to emerge as a strong theme.
Vesiculation in cancer
The emerging molecular complexity and cellular diversity of human cancers challenge the traditional preoccupation with only intracellular signalling pathways (7). It is increasingly clear that exchange of biochemical information with the environment may contribute also to malignant growth (8–10). EVs may bridge the chromosomal mutations (oncogenic), downstream epigenetic events, and pathways of intercellular communication in cancer (11) (Fig. 1). Indeed, oncogenic lesions, such as mutant epidermal growth factor receptor (EGFR), HER2, and RAS, impact cellular vesiculation processes, leading to release and intercellular exchange of oncogenic proteins, transcripts and DNA sequences (12–15). Vesiculation is also implicated as a removal mechanism for growth inhibitory factors, tumour suppressors (16–18), and non-coding RNA (13,19,20), which will all promote tumour growth.
Fig. 1.

The diverse roles of vesiculation in cancer.
While the ability of cells to release EVs has been known since the 1960s (21) and cancer cells have been studied in this regard since the 1970s (22) and 1980s (23), the biogenetic pathways leading to formation of different EV subsets in cancer remain obscure and controversial (24–27). So are the mechanisms of selective packaging of their molecular cargo, properties, mechanisms of intercellular trafficking (28,29) and their biological role in various disease contexts (11). Hence there is a growing interest in understanding the involvement of EVs in cancer progression (11,25,30–33). Moreover, usage of EVs for drug delivery (20,34) and vaccination (35) has attracted considerable interest, and intense efforts are underway to analyze and remotely monitor cancer-specific molecular events by taking advantage of their encapsulation in the cargo of EVs circulating in blood and other biofluids (36–40).
Horizontal gene transfer and vesiculation
Perhaps one of the most tantalizing aspects of cancer cell vesiculation is its role in extracellular emission and horizontal transfer of transforming oncogenes (11). Holmgren and colleagues showed the ability of apoptotic vesicles (bodies) to exchange functional oncogenic DNA between cells (41). Vesicular transport of transposons and single-stranded DNA amplicons containing MYC sequences was subsequently described by Balaj et al. and postulated to be a mechanism causing genomic instability in cells exposed to this material (14). More recently, genomic DNA sequences containing mutant H-Ras gene were found in the cargo of EVs produced by viable cancer cells in vitro (15), and this finding was confirmed in several other systems. EVs isolated from plasma of cancer patients carry cell-free DNA (cfDNA) (42,43), and material previously implicated in cellular transformation (44). While it still remains unclear whether EVs containing fragmented oncogenic DNA mediate horizontal gene transfer (15), evidence exists with regards to oncogenic transcripts (13) and proteins contributing to clonogenic growth, angiogenesis, invasion and metastasis (12,32,45–47). In addition, horizontal transformation may also be a function of stimuli unrelated to the passage of genetic cargo, but instead tumour-related EVs can carry also other growth altering activities, such as transglutaminase, with lasting effects on recipient cells (48).
Other factors influencing vesiculation
Oncogenic transformation represents but one mechanism implicated in cancer cell vesiculation. Indeed, the release of EVs is also influenced by several other processes, such as cellular adhesion, migration, differentiation, epithelial-to-mesenchymal transition (EMT) and deregulation of stemness (20,49–54). Moreover, metabolic states and exposure to hypoxia are reflected in the cargo of EVs produced by cancer cells (55). These observations suggest that EV cargo may not only serve as a source of information regarding the genetic evolution of the malignant cell population, but also reflect the functional and metabolic state of these cells. It is also of interest that hypoxia may alter the biological activity of tumour-related EVs and changes in cellular energy metabolism could be expected to have analogous effects (56).
Functions of cancer-derived EVs
Among many normal and abnormal cell populations that may be exposed to cancer-derived EVs. Endothelial cells, inflammatory cells and blood cells are especially interesting in this regard, because these cells are immediately and continually exposed to tumour EVs in the circulation, and because these EVs may be responsible for the systemic pathological impact of even localized forms of cancer. The emerging understanding of the consequences of cancer-derived EV exposure includes evidence for deregulation of angiogenesis (45,55–57), the inflammatory response, metastasis (32,58) and cancer-related coagulopathy. In the latter case, EVs constitute an attractive, albeit controversial, mechanism of spreading the procoagulant state from cancer cells into periphery leading to related morbidities, such as VTE (3,30,31,59–62). While EVs are often regarded as “carriers” of procoagulant TF activity in this setting, it is also possible that cells that have taken up tumour-related EVs may either expose or de novo express TF still resulting in haemostatic perturbations (11).
Potential clinical relevance of cancer-derived EVs
The diversity and intrinsic heterogeneity of human cancers may make the EV landscape associated with malignancy exceedingly complex. However, therein exist opportunities to exploit the vesiculation mechanisms in precision medicine for tumour diagnosis, prognosis, prediction of, for example, VTE or metastasis, monitoring of therapy, and dedicated treatment, such as administration of autologous EVs to increase the body's immune response and to eradicate the tumour.
With regard to prognosis of VTE, several EV-based assays are now being studied to identify cancer patients at high risk for developing VTE. To understand this development, one of the goals of the ISEV 2014 Educational Day was to discuss the related properties of TF, as the primary cellular trigger of coagulation, followed by a section on the currently available information on the putative contribution of EV-associated TF to VTE.
EVs in coagulation
The importance of EVs or “microparticles” to blood coagulation has long been recognized. Indeed the first described property of EVs was the ability to support thrombin generation (TG) (21). The formation of the principle complexes of the coagulation process, the intrinsic and extrinsic tenases and prothrombinase, requires a negatively charged phospholipid surface for the calcium-dependent binding of the vitamin K-dependent clotting proteins factors II (prothrombin), VII, IX and X (Fig. 2), which increases their biological activity by several orders of magnitude. During normal haemostasis, the negatively charged surface is provided by aminophospholipid (APL) externalization (predominantly phosphatidylserine) on the platelet membrane and platelet-derived EVs. The inability to externalize APL causes Scott syndrome, a rare bleeding disorder (63). Following vascular damage, blood is exposed to the subendothelium causing platelet activation and initiation of coagulation by perivascular TF. Only in pathological conditions, for example, sepsis, sickle cell anaemia and cancer, is there a significant amount of circulating TF, much of which is expressed by EVs.
Fig. 2.

Tissue factor initiates coagulation by binding factor VIIa to form membrane bound complex which activates factor X. This forms a complex with factor Va which activates prothrombin to thrombin. Thrombin activates factor XI which creates an amplification loop through the factor IXa/VIIIa activation of factor X. The resulting burst of thrombin generation causes platelet activation and conversion of insoluble fibrinogen to an insoluble thrombin clot.
Tissue factor
Background
TF (CD142) is a 263 amino acid integral transmembrane protein. Full length TF (flTF) has 3 domains: an extracellular domain (residues 1 – 219), a 23 amino acids transmembrane domain, and a short 21 amino acids intracellular domain, which is involved in signalling. The extracellular domain of flTF binds coagulation factor VII or its activated form (VIIa) with high affinity, thereby initiating blood coagulation and mediating haemostasis (Fig. 2). TF also plays roles in embryogenesis, angiogenesis, and inflammation (64). There is also a second form of TF known, alternatively spliced TF (asTF). This is a soluble version that lacks the transmembrane domain. Whereas soluble asTF has little or no procoagulant activity, asTF can stimulate angiogenesis independently of FVIIa and when bound to integrins on the cell surface acquires procoagulant activity (65).
TF is constitutively expressed by extravascular cells, such as adventitial fibroblasts, thereby providing a haemostatic envelope that can initiate haemostasis upon vessel wall damage. In fact, under normal conditions all components required for coagulation, such as coagulation factors and calcium ions, are sufficiently present within the blood, but coagulant TF, the initiator of coagulation, and is present only outside the blood vessel. Upon vascular damage, blood will contact the extravascular TF and coagulation will be initiated. Only under exceptional pathological conditions, can circulating cells, such as monocytes and cells lining the vessel wall (endothelial cells) express and produce coagulant TF. TF has also been detected in other types of circulating cells, such as platelets, but this may be due to binding to TF-positive EVs (66).
Although the molecular mechanisms regulating the coagulant activity of TF are still incompletely understood, this activity is increased in damaged cells due to the presence of the negatively charged phospholipid phosphatidylserine, and by thiol-disulphide bond modifications of the TF molecule. This concept of circulating blood-borne “thrombotic” TF on EVs (TF-EVs) is therefore of great interest, not only given their potential as biomarkers of major diseases, such as cancer, but also on their putative contribution to the development of thrombosis.
TF-exposing vesicles and coagulation activation in animal studies
Many lines of evidence suggest that TF-EVs are involved in coagulation activation in vivo, either directly or indirectly by adhesion to the damaged vessel wall.
Animal studies have shown that tumour-derived EVs are released from a variety of tumours in vivo, and that these EVs expose procoagulant TF as demonstrated in vitro. In the circulation of mice bearing an orthotopic tumour derived from a human pancreatic cancer cell line, the human TF coagulant activity of the tumour-derived EVs was paralleled by in vivo coagulation activation. Moreover, plasma from the tumour-bearing mice increased TG in vitro in a human TF-dependent manner, and the inhibition of human TF reduced thrombin–antithrombin (TAT) levels in vivo, an indication of coagulation system activation (6,67).
In addition, tumour-derived EVs enhance the development of thrombosis in mice in vivo. Thomas and colleagues found that cancer cell–derived EVs, after infusion into the circulation of a living mouse, accumulated at the site of vessel injury by binding to activated platelets, suggesting that tumour-derived EVs participate in thrombus formation in vivo (68).
Retrospective studies on TF-exposing vesicles, coagulation activation, and venous thrombosis in cancer patients: cause or consequence?
Several retrospective studies have analyzed the levels of circulating TF-EVs in patients with cancer. In general, elevated levels of circulating TF-EVs have been reported compared to healthy controls, and these increased levels are associated with in vivo coagulation activation, for example, in colorectal cancer patients (69) and in patients with early prostate cancer (70). However, one cannot conclude from these studies that high levels of circulating TF-EVs in blood of cancer patients are a causal factor for the development of VTE.
Tesselaar and colleagues were the first to report that the TF-EV coagulant activity is higher in pancreatic and metastatic breast cancer patients who presented with VTE compared to cancer patients without VTE (71). This finding was confirmed in later case-control studies (72,73). Similarly, Zwicker and colleagues (74) observed TF-EVs in plasma of 60% of the cancer patients with VTE, compared with 27% of patients with cancer without VTE. Moreover, in 3 patients undergoing pancreatectomy, the TF-EV level was markedly reduced after surgery. In a 1-year follow-up, 25% (4 out of 16) patients with detectable TF-EV levels developed VTE, compared with 0% (0 out of 44) in patients without detectable TF-EVs, a finding confirmed recently by Campello et al. (75). One has to bear in mind, however, that these studies do not provide an answer to the question whether the increased presence of coagulant TF-EVs is either cause or consequence of cancer-associated VTE.
Prospective studies on TF-exposing vesicles, coagulation activation, and venous thrombosis in cancer patients
The most extensive prospective study measured TF-EV activity in plasma samples from the Vienna Cancer and Thrombosis Study (CATS) (76). A total of 348 patients suffering from pancreatic, gastric, colorectal or brain cancer, had a 2-year follow-up for the occurrence of symptomatic VTE. Patients with pancreatic and gastric cancer expressed higher levels of coagulant TF-EV activity in comparison to patients with brain and colorectal cancer. Moreover, patients with pancreatic cancer demonstrated borderline significance for an association between TF-EV activity and development of VTE, whereas this association was not found for 3 other tumour types. This is in line with findings of a later study which demonstrated an association between elevated plasma TF-EV activity and development of VTE in patients with newly diagnosed cancer of pancreaticobiliary origin (77). Recently, the interim results of a prospective study which evaluated the predictive value of TF-EV activity for VTE in 88 patients with pancreatic cancer were reported (78). Patients with a high TF-EV activity at baseline were 6 times more likely to develop deep vein thrombosis (DVT) or pulmonary embolism (PE) during 6 months follow-up compared to patients with low EV-TF activity, although statistical significance was not reached (HR 6.0; 95% CI 0.74–48.99). Nevertheless, taken together with the findings of other studies, these results support a role for TF-EV activity as a predictive biomarker for cancer-associated VTE in pancreatic cancer patients.
Also in patients with glioblastoma multiforme (GBM), levels of circulating TF-EVs are increased compared to healthy controls (60). Furthermore, patients who developed VTE during 7 months follow-up had significantly higher levels of TF-EVs at baseline than the non-VTE patients. This study was expanded in order to confirm the results (unpublished data). Twenty-five GBM patients and 20 patients with meningioma, a prothrombotic benign neoplasm, were included and followed for up to 7 months. Both these brain neoplasms had higher levels of TF-EVs compared to healthy subjects, as shown in Fig. 3, Panel a. Seven glioma patients developed VTE during follow-up, and these patients presented with significantly higher levels of TF-EVs at baseline and 1 month after surgery. In contrast, 2 meningioma patients developed VTE but they did not present with higher levels of TF-EVs compared with patients without VTE. A possible explanation is that circulating TF-EVs in patients with GBM are more procoagulant than those TF-EVs in patients with meningioma. However, since only a minority of TF-EVs in GBM patients also stained for GFAP (10%) (Fig. 3, Panel b), a marker for glial cells, the true cellular origin of coagulant EV-associated TF in blood still remains to be determined.
Fig. 3.

(a) TF-bearing EVs in glioma and meningioma patients and in healthy controls. (b) Origin of TF-bearing EVs in glioma patients and controls.
The main question obviously is, whether coagulant TF-EV activity can be used to predict who will develop VTE, so that prevention of VTE becomes a realistic option. In a recent, small phase II study (the Microtec study) 66 patients with advanced cancer (30 pancreatic, 21 non-small cell lung and 15 colorectal) were assigned into 1 of 2 groups on the basis of the number of circulating TF-EV: low TF-EVs (≤3.5×104/µl) or high TF-EVs (>3.5×104/µL) as measured with impedance-based flow cytometry. The low TF-EV patients were followed without treatment. The high TF-EV patients were randomized to either enoxaparin or observation without treatment. The untreated high TF-EV patients had a higher 2-month cumulative incidence of mostly asymptomatic venous thrombosis compared with low TF-EV patients, 27% versus 7%, respectively. High TF-EV patients randomized to enoxaparin had a low cumulative incidence of venous thrombosis (6%). Thus, it may be feasible to use the number of circulating TF-EVs to identify patients at high risk for developing VTE, and thus patients eligible for prophylactic anticoagulant therapy (79).
Is there a role for EVs exposing coagulant TF in cancer-related thrombosis?
As summarized in the previous sections, experimental and cross-sectional clinical studies provide evidence that TF-EVs in cancer patients represent a coagulant EV subpopulation that may play a role in the prothrombotic state found in different malignancies (3). At present, however, results from prospective studies that investigated the potential of TF-EV coagulant activity as a biomarker for prediction of future VTE in cancer patients are inconsistent. It needs to be kept in mind that ambiguous results can result from inclusion of different types and stages of cancer and the presence of only limited data from large prospective studies, but also methodological discrepancies and a lack of standardization. For instance, the sensitivity of conventional flow cytometry is too low to detect the majority of TF-EVs which hampers interpretation of the results. Hence, functional assays are in general preferred to measure TF-EVs, but it is yet unknown which is the best assay for measuring the coagulant activity of the TF-EV population. Also pre-analytical variables, such as blood withdrawal, EV isolation, and EV preparation are not consistent across the studies. However, despite these differences, evidence primarily in patients with pancreatic cancer indicates that TF-bearing EVs might play an important role in the pathogenesis of VTE.
Unanswered questions
Although cancer chemotherapy is associated with increased risk of VTE (80,81), Tesselaar found no elevated plasma TF-EV activity in patients treated with chemotherapy (73). More recently, also no increase in TF-EV activity was observed in response to chemotherapy in breast cancer patients (82), or in GBM patients (60). These findings suggest that mechanisms other than increased levels of circulating and coagulant TF-EVs are responsible for thrombosis during in response to chemotherapy, for example, increased neutrophil extracellular trap formation or increased apoptosis and/or necrosis. Again, most studies are small and effects of chemotherapy may differ between various types of cancer. Clearly, larger and more mechanistic studies are necessary to address this question.
Although an association between the level of TF-EV release and thrombosis has been established for some types of cancer, causality has not yet been firmly established. The results of future prospective clinical trials should aim to identify any causative relationship.
Another issue is the optimum timing of sampling. TF-EV coagulant activity progressively increased in the months prior to the development of thrombosis, but baseline levels may be similar to those patients who did not develop thrombosis (3). Thus, a single baseline sample may not be sufficient to predict future thrombotic events. Furthermore, basal TF-EV determination was found to be correlated with VTE in studies over a 6-month period but only weakly in studies with a 2-year follow-up (3).
Mechanisms for the selective incorporation of TF into EV
One important aspect in the formation of TF-containing EVs involves understanding the mechanisms by which TF is incorporated into EVs.
Role of serine phosphorylation
The induction and termination of the incorporation of TF into EVs seems regulated by phosphorylation of 2 serine residues within the cytoplasmic domain of TF (83). The phosphorylation of Ser253 is involved in the incorporation and release of TF within EVs (Fig. 4a). Upon cellular activation, protein kinase C-α phosphorylates Ser253 (84,85), resulting in the interaction of phosphorylated TF with the cytoskeletal protein filamin-A (86,87) (Fig. 4b). The termination of TF incorporation in EVs may be regulated by phosphorylation of Ser258 (83,88) (Fig. 4a). This phosphorylation occurs only after phosphorylation of Ser253 (83,84) and is mediated by p38α (88).
Fig. 4.

(a) The phosphorylation of Ser253 within the cytoplasmic domain acts to initiate the incorporation and release of TF within EVs. (b) The interaction between TF and filamin-A is required for the active incorporation of TF into EVs.
Interestingly, the incorporation of TF into EVs seems to occur via different mechanisms to that of the formation of the EVs themselves. Firstly, inhibition of the incorporation of TF into EVs does not interfere with the release of EVs (83,87). Secondly, preliminary studies suggest the involvement of caspases but not calpains in the incorporation of TF into EVs (Collier et al., unpublished data), which both are thought to contribute to the release of EVs (89).
In conclusion, the regulation of the release of TF into EVs is mediated through mechanisms regulated by phosphorylation of the cytoplasmic domain of TF and may involve the cytoskeletal protein filamin-A. The phosphorylation of TF in turn also induces feedback-signalling mechanisms which promote cell proliferation or apoptosis, depending on the level of TF within the cell. These mechanisms may constitute part of the process used by cells to gauge the level of surrounding injury and trauma, and contribute to how cells determine the appropriate response to injury (88,90–98).
Role of the microenvironment
TF can be present in a coagulant or a non-coagulant form. The underlying mechanisms to switch between both forms, however, are still unclear but may involve (a) dimerization (99), (b) exposure of anionic phospholipids (100), (c) oxidation and reduction of the disulphide bonds (Cys186–Cys209) of TF (101), and/or (d) the association with lipid rafts (102).
The role of rafts has remained especially obscure, particularly for TF present in plasma membranes, in other words cell-exposed TF. In previous studies, the association between TF and lipid rafts, also known as detergent-resistant membranes (DRMs), was determined in whole cell lysates. Because in whole cell lysates most TF originates from intracellular membranes (103), the role of DRMs in regulation of TF coagulant activity has remained controversial (103–105). Therefore, the association of TF with DRMs was studied in purified plasma membranes and in EVs.
Plasma membranes of various human TF producing cells and cell lines contain 2 different types of DRMs based on density, DMR-H with high density and DRM-L with low density. DRM-H contains the bulk of TF, but lacks any detectable coagulant activity. In contrast, DRM-L contains only minute amounts of TF, but this TF initiates coagulation. So, different forms of coagulant and non-coagulant TF seem to co-exist in plasma membranes. The lack of coagulant activity was not due to the presence of the endogenous plasma inhibitor of TF, tissue factor pathway inhibitor (TFPI). In contrast, the regulator of oxidation and reduction of disulphide bonds, protein disulphide isomerase (PDI), was present in DRM-H, suggesting that PDI may be involved in keeping TF in a dormant, non-coagulant form. Similarly to plasma membranes, vesicles from smooth muscle cells and human saliva, both known to contain coagulant TF (106), also contain DRM-H and DRM-L. Again, the non-coagulant form of TF and PDI were associated with DRM-H, whereas the coagulant form was associated with DRM-L.
With this knowledge, a model for the release of TF-EVs can be proposed, shown in Fig. 5, which may help to explain the observed discrepancy between the presence of detectable levels of TF-EVs and the concurrent lack of TF coagulant activity, both in mouse models (67) and cancer patients (107). With our model, we hypothesize the existence of TF-EVs originating from different membrane compartments. In this model, the non-coagulant TF-EVs are exclusively released from the DRM-H compartments of the plasma membrane (option 1, Fig. 5), whereas the coagulant TF-EVs are released from DRM-L compartments of the plasma membrane only (option 2, Fig. 5). Although not shown in Fig. 5 for clarity, it seems likely that hybrid vesicles may occur which contain both forms of TF.
Fig. 5.

Model for the release of TF-exposing vesicles. TF-exposing vesicles originating from different membrane compartments.
TF in tumour angiogenesis
Cancer cells thrive in the tumour microenvironment. Several stress-related phenomena of this microenvironment such as hypoxia, acidosis, starvation, and coagulation (108,109), are major drivers of tumour development and aggressiveness, and thus select for tumour cells that successfully adapt to microenvironmental stress. This adaptive response of cancer cells is strongly associated with resistance to oncological treatment. However, such mechanisms also represent potential Achilles’ heels of the cancer cell machinery and thus may offer alternative treatment targets of cancer (110).
It was previously shown that the coagulant TF–FVIIa complex promotes retinal angiogenesis through protease-activated receptor-2 (PAR-2) (91). For example, hypoxic cancer cells release EVs exposing the TF–FVIIa complex that induce PAR-2-mediated angiogenesis (55) (Fig. 6). In addition, hypoxia also affects the sorting, of mRNAs and proteins involved in tumour development, to EVs from glioma cells and these EVs are present in plasma of glioma patients (56). It may be concluded that EVs represent novel players in hypoxia-dependent cross talk between malignant cells and stromal endothelial cells during tumour formation. Further, the EV molecular composition may reflect the oxygenation status and aggressiveness of malignant tumours.
Fig. 6.
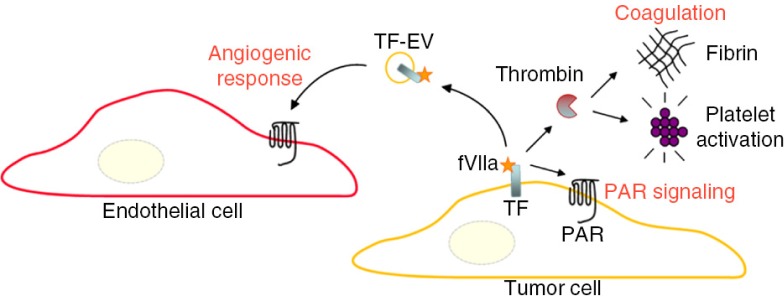
TF-dependent coagulation activation is of relevance for metastasis, tumour growth, and angiogenesis through several distinct mechanisms, including the activation of thrombin further resulting in the generation of a fibrin network, and platelet activation. These end stages of coagulation can contribute to tumour progression and metastasis by, for example, protection of circulating cancer cells from immune cell attack. In addition, TF and associated coagulation proteases up-stream of thrombin can cleave and activate protease-activated receptors (PARs) to induce pro-migratory and survival signalling in cancer cells. Endothelial cells, however, are normally devoid in TF. At hypoxic conditions, cancer cells may release TF/FVIIa-bearing EVs that in a paracrine manner trigger an angiogenic response through activation of PAR-2 in endothelial cells. Further, it may be hypothesized that systemic release of TF-EVs from hypoxic tumour regions contributes to the hypercoagulable state of cancer patients.
Techniques for analyzing EVs
Measurement of EVs in suspension
EVs are heterogeneous in origin, size and function (111). To determine their origin and number, measurements of size, concentration and antigens at the level of single EV is required. At this time, however, only several imperfect EV detection technologies are available. Here, only methods for the detection of EVs in suspension will be discussed.
Flow cytometry is the most applied technique for EV detection. A sensitive system can detect the scatter and fluorescence phenotype of more than 100,000 EVs per minute, with fluorescence sensitivity of 50 antigens on a single EV. Due to their small size, low refractive index and low number of antigens, the fluorescence and scatter signals from EVs are difficult to distinguish from the noise, causing the smallest EV to pass undetected. Due to the large number of small (112), co-incidence or swarm detection may occur (113). Due to differences in refractive index, gating using polystyrene beads does not result in gating of the same size for EVs.
In a typical flow cytometer, one of the detector channels is used for triggering. If the signal in the trigger channel exceeds a threshold level, any potential signals in all the detection channels are also then recorded. Because the fluorescence sensitivity of a typical flow cytometer is higher than the scatter sensitivity, triggering on fluorescence may result in detection of smaller EVs. Fluorescence triggering will only detect fluorescent EVs, which still means either a subset of EVs is detected or a complex protocol is needed to label all EVs (114). To date, a suitable pan-EV marker has not been identified.
In resistive pulse sensing, a single EV is pushed through a pore. An electrical circuit measures the resistance of the pore. When an EV moves through the pore, the resistance increases, and this increase is proportional to the volume of the EV (115). To date, the major limitation of resistive pulse sensing is that it lacks the possibility to determine antigen expression on EVs.
Nanoparticle tracking analysis relates the rate of Brownian motion to EV size. This relation requires knowledge of the medium viscosity and temperature. The refractive index of EVs does not affect the size that is measured, but does affect the minimum detectable size. Although nanoparticle tracking analysis can measure fluorescence, the sensitivity needs to be improved (112). The system requires careful calibration (116), and determined size and concentration have limited accuracy and precision (115).
In cryo-electron microscopy (EM), the sample is labelled with the immune-gold method, and snap frozen before analysis (117). This approach has several major advantages. The minimum detectable size is in the order of nanometres, and this method allows immune-phenotyping of all EVs. The major disadvantages, however, are that concentration determination is in its infancy, and that the time needed for a measurement precludes clinical application.
In summary, at this time no technique capable of measuring size, concentration and antigen expression of single EV exists. However, each of the techniques discussed is evolving rapidly, and we expect suitable detection techniques to become available in the near future.
Measuring EV-associated TF
Measurement methodologies for (full length) TF can be classified into antigenic and functional – mostly coagulant – assays. At present, there is a lack of standardization in the measurement of TF-EVs. Therefore, measuring EV-associated TF antigen or TF coagulant activity presents even more problems than one would normally encounter (see Table I). In general, the TF antigen-based assays lack the sensitivity necessary to detect the low levels of coagulant TF-EVs in blood (118).
Table I.
Issues with measurement of MV-associated TF.
| Pre-analytical variables – cell-free plasma, anticoagulant, g-values for centrifugation, discard tube in blood draw, freeze thawing |
| TF activity can be increased by post-translational modification and decreased by its inhibitor tissue factor pathway inhibitor |
| Higher concentrations of Factor VIIa can activate FX in a TF-independent manner in the presence of phospholipids |
| Binding of Factor VII/VIIa to TF blocks access of some anti-TF antibodies |
| The contact pathway may activate coagulation in some clotting-based assays with low TF as initiator. This pathway can be inhibited |
| Presence of antibody microaggregates causing false positive signals in flow cytometry studies |
| Detection limit of most flow cytometers above the size of small TF-positive vesicles |
| Few studies simultaneously compare more than one assay |
| No accepted international standard |
| No gold standard assay – the MP TF activity assay is the best available assay |
| Positive (plasma from whole blood treated with LPS) and Negative Controls (use of inhibitory antibodies are important to confirm that the procoagulant activity measured is due to TF) |
EV-TF coagulant activity
Despite limitations, however, it is possible to measure a true TF-EV coagulant activity by using appropriate positive controls such as monocyte-EV-TF and specific inhibitory antibodies against TF or FVIIa. One approach to detect TF-EV coagulant activity is to isolate EVs by ultracentrifugation, which are washed and then incubated with purified human coagulation factors to generate coagulation factor Xa, which activity is measured using a chromogenic substrate. Alternatively, the TF-EV coagulant activity can also be measured directly in the plasma after recalcification. Also several commercial assays are available to measure TF-EV coagulant activity, but the sensitivity of such assays is relatively low (118). So far, measuring TF-EV coagulant activities have been insufficiently compared or standardized between laboratories.
EV-TF antigen detection
The most popular method to measure TF-EVs is flow cytometry. Measurement of cell-associated TF has created controversy in the past, so one can appreciate that these difficulties are multiplied when measuring EVs. Again the use of appropriate controls is a prerequisite. Recently, commercially available clones of TF antibodies were compared (119). This study revealed a great deal of variability (up to 2 or 3 orders of magnitude) between clones in non-specific binding, avidity and the ability to inhibit TF activity. In addition to selecting the appropriate antibody clone, the need for target-specific titration of the antibodies and selection of the brightest fluorophores were also identified as critical factors in achieving adequate sensitivity and specificity when measuring TF by flow cytometry.
In summary, measurement of TF-EVs is still in its infancy and requires larger and comparative studies to pave the road towards more standardized measurements and inter-laboratory acceptance of applied methods and obtained results.
High-grade gliomas as a model disease for EV studies
Primary high-grade gliomas, particularly glioblastomas (GBM), arise from progenitor and supporting glial cells in the brain. Patients experience poor survival despite advances in surgical and chemotherapeutic approaches (120). A major contribution to mortality and recurrent hospitalization emerges from a high risk of VTE. These complications profoundly impact the cost of GBM treatment. Our limited understanding of the pathophysiology of GBM-related hypercoagulability, combined with the haemorrhage risk of anticoagulation therapy, has prevented the establishment of a consensus on the role of anticoagulation in patients with GBM. This population represents an appealing model upon which to base the validation of novel markers of coagulation risk and to design studies to identify patients most likely to benefit from prophylactic and therapeutic anticoagulation.
Identification of clinically relevant GBM subtypes
Molecular subtyping of GBM has increased our understanding of these tumours and disclosed heterogeneity that demands individualized molecularly-targeted treatment approaches (120). Specifically, integrated genomic analysis has identified 4 clinically relevant GBM subtypes, which differ in clinical prognosis, response to therapy, survival, and the risk of developing VTE (120).
Venous thromboembolism in GBM: incidence, cost and relationship to molecular subtype
The incidence of VTE is of particular concern in patients with brain tumours (121,122). In GBM, VTE affects between 3 and 20% of patients in the post-operative period (122), with a cumulative lifetime risk of up to 32% (123). While the cost of treating VTE in patients with GBM has not been specifically examined, the estimated annual provider payments associated with a DVT diagnosis are $7,000 to $10,000 (124). Similarly, a PE diagnosis is estimated to cost between $13,000 and $16,000 (124). Re-admissions, a particularly relevant concern in GBM patients with VTEs given their persistently increased thrombotic risk and the unclear long-term anticoagulation guidelines for this group, can occur in up to 14.3% of patients with VTE and are associated with similar or higher treatment costs (124).
Mirroring the survival heterogeneity of GBMs, the risk of VTE is variable across high-grade gliomas (125). There is also indirect evidence that GBM molecular subtype can affect VTE risk (52,126), making this GBM classification scheme appealing as a predictor of thrombotic risk. Current GBM subtyping nonetheless requires primary tissue samples for genetic analysis, limiting its utility to the post-operative setting. Less invasive biomarkers, such as diagnostic biofluid analyses of tumour-specific exosome amplifications and mutations of GBM subtypes are needed in order to fully characterize their impact on thrombotic risk.
Pathophysiology of VTE in GBM
Understanding the pathophysiology of VTE in GBM is critical to risk stratification and can provide insight into potential GBM subtype biomarkers. In addition to demographic thrombotic risk factors, there are both treatment- and glioma-associated mechanisms that can affect patient risk (122). Gliomas themselves can directly affect VTE risk through the upregulation or ectopic expression of procoagulant molecules, including TF (122,127). Underscoring this direct causal relationship, the extent of tumour resection demonstrates an inverse thrombotic risk correlation. Expression of glioma-specific pro-coagulants can also be stratified based on GBM molecular profile (126), highlighting the potential utility of GBM subtype defining biomarkers as prospective indicators of VTE risk.
Thrombotic management and risk avoidance in GBM
Effective management and prophylaxis of VTE in patients with GBM involves balancing the risk of VTE with the risk of intracranial bleeding due to anticoagulation.
Given the risks of VTE in GBM, prophylactic anticoagulation is appealing but a large multicentre, phase III, placebo-controlled trial assessing prophylactic anticoagulation was terminated early by the sponsor (128). Therefore, the safety and efficacy of this approach remains unknown. Biomarkers identifying those patients at increased risk for VTE would significantly aid clinical decision making in this population.
EVs as biomarkers of GBM thrombotic risk
GBM-secreted EVs are appealing diagnostic biomarkers. GBM-specific proteins or genetic material, such as the mutated epidermal growth factor receptor EGFRvIII and enzymes IDH1/IDH2), are detectable in both the plasma and CSF at levels significantly above healthy controls (129). Because the levels of these markers decrease with surgical resection (129), they are appealing as markers for tumour recurrence. EVs have also shown prognostic utility in GBM, as patients with higher levels of GBM-derived EVs are more likely to fail standard treatment protocols (129).
EVs are potentially ideal biomarkers of both GBM molecular subtype and thrombotic risk, especially given the likely association of these 2 phenomena (126). Thus, overexpression of EGFRvIII correlated with classical type of GBM, has been found to drive prothrombotic TF production and is likely responsible for the increased coagulant profile associated with this subtype (120,126). Further, cancer cell–derived TF-EVs induce procoagulant activity in the endothelium (52). These data suggest a causative relationship between the classical GBM oncogenic pathway and increased thrombotic risk, with TF-EVs serving as a detectable intermediary. Validation of this relationship in the clinical setting demands further exploration, as does the identification of analogous biomarkers for the remaining GBM subtypes.
The recent clinical trial exploring other biomarkers of cancer-related thrombotic risk provides a roadmap for this work (121). Similar trials investigating the utility of exosomes as GBM biomarkers are needed to: (a) prospectively correlate GBM-specific EVs with molecular subtype and risk of VTE, and (b) test the role of preventative anticoagulation in those GBM subtypes with increased thrombotic risk. TF-EVs, identified by either analysis or functional assay, are suited for trials assessing their predictive value as markers for both the classical GBM subtype and for increased thrombotic risk. Combined with identification and testing of additional GBM subtype–specific exosomes, this approach could facilitate individualization of GBM treatment plans based on tumour-specific risk profiles.
Taken together, a role for EVs in complex processes such as cellular transformation, tumour growth and development, and the risk of VTE is emerging. Although there is a growing clinical interest in the application of EVs as (a) biomarkers for diagnosis and prognosis, (b) therapeutic application, (c) usefulness to monitor therapeutic efficacy, (d) patient risk stratification to enable personalized medicine, more and much larger studies will be essential to firmly establish the clinical relevance of EVs. Nevertheless, we hope that the present review will increase the understanding of EV investigators from different fields.
Conflict of interest and funding
The authors have not received any funding or benefits from industry or elsewhere to conduct this study.
References
- 1.Bouillaud S, Bouillaud J. De l'Obliteration des veines et de son influence sur la formation des hydropisies partielles: consideration sur la hydropisies passive et general. Archiv Gen Med. 1823;1:188–204. [Google Scholar]
- 2.Buller HR, van Doormaal FF, van Sluis GL, Kamphuisen PW. Cancer and thrombosis: from molecular mechanisms to clinical presentations. J Thromb Haemost. 2007;5:246–54. doi: 10.1111/j.1538-7836.2007.02497.x. [DOI] [PubMed] [Google Scholar]
- 3.Geddings JE, Mackman N. Tumor-derived tissue factor-positive microparticles and venous thrombosis in cancer patients. Blood. 2013;122:1873–80. doi: 10.1182/blood-2013-04-460139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dvorak HF, Van DeWater L, Bitzer AM, Dvorak AM, Anderson D, Harvey VS, et al. Procoagulant activity associated with plasma membrane vesicles shed by cultured tumour cells. Cancer Res. 1983;43:4434–42. [PubMed] [Google Scholar]
- 5.Bastida E, Ordinas A, Escolar G, Jamieson GA. Tissue factor in microvesicles shed from U87MG human glioblastoma cells induces coagulation, platelet aggregation, and thrombogenesis. Blood. 1984;64:177–84. [PubMed] [Google Scholar]
- 6.Davila M, Amirkhosravi A, Coll E, Desai H, Robles L, Colon J, et al. Tissue factor-bearing microparticles derived from tumour cells: impact on coagulation activation. J Thromb Haemost. 2008;6:1517–24. doi: 10.1111/j.1538-7836.2008.02987.x. [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Heppner GH. Tumor cell societies. J Natl Cancer Inst. 1989;81:648–9. doi: 10.1093/jnci/81.9.648. [DOI] [PubMed] [Google Scholar]
- 9.Wu M, Pastor-Pareja JC, Xu T. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature. 2010;463:545–8. doi: 10.1038/nature08702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, et al. Reconstructing and reprogramming the tumour-propagating potential of glioblastoma stem-like cells. Cell. 2014;157:580–94. doi: 10.1016/j.cell.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rak J. Extracellular vesicles - biomarkers and effectors of the cellular interactome in cancer. Front Pharmacol. 2013;4:21. doi: 10.3389/fphar.2013.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008;10:619–24. doi: 10.1038/ncb1725. [DOI] [PubMed] [Google Scholar]
- 13.Skog J, Wurdinger T, van RS, Meijer DH, Gainche L, Sena-Esteves M, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–6. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Balaj L, Lessard R, Dai L, Cho YJ, Pomeroy SL, Breakefield XO, et al. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat Commun. 2011;2:180. doi: 10.1038/ncomms1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee TH, Chennakrishnaiah S, Audemard E, Montermini L, Meehan B, Rak J. Oncogenic ras-driven cancer cell vesiculation leads to emission of double-stranded DNA capable of interacting with target cells. Biochem Biophys Res Commun. 2014;451:295–301. doi: 10.1016/j.bbrc.2014.07.109. [DOI] [PubMed] [Google Scholar]
- 16.Yu X, Harris SL, Levine AJ. The regulation of exosome secretion: a novel function of the p53 protein. Cancer Res. 2006;66:4795–801. doi: 10.1158/0008-5472.CAN-05-4579. [DOI] [PubMed] [Google Scholar]
- 17.Putz U, Howitt J, Doan A, Goh CP, Low LH, Silke J, et al. The tumour suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci Signal. 2012;5 doi: 10.1126/scisignal.2003084. ra70. [DOI] [PubMed] [Google Scholar]
- 18.Gabriel K, Ingram A, Austin R, Kapoor A, Tang D, Majeed F, et al. Regulation of the tumour suppressor PTEN through exosomes: a diagnostic potential for prostate cancer. PLoS One. 2013;8:e70047. doi: 10.1371/journal.pone.0070047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–9. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 20.Quesenberry PJ, Goldberg LR, Aliotta JM, Dooner MS, Pereira MG, Wen S, et al. Cellular phenotype and extracellular vesicles: basic and clinical considerations. Stem Cells Dev. 2014;23:1429–36. doi: 10.1089/scd.2013.0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolf P. The nature and significance of platelet products in human plasma. Br J Haematol. 1967;13:269–88. doi: 10.1111/j.1365-2141.1967.tb08741.x. [DOI] [PubMed] [Google Scholar]
- 22.Taylor DD, Doellgast GJ. Quantitation of peroxidase-antibody binding to membrane fragments using column chromatography. Anal Biochem. 1979;98:53–9. doi: 10.1016/0003-2697(79)90704-8. [DOI] [PubMed] [Google Scholar]
- 23.Poste G, Nicolson GL. Arrest and metastasis of blood-borne tumour cells are modified by fusion of plasma membrane vesicles from highly metastatic cells. Proc Natl Acad Sci USA. 1980;77:399–403. doi: 10.1073/pnas.77.1.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnstone RM. Exosomes biological significance: a concise review. Blood cells Mol Dis. 2006;36:315–21. doi: 10.1016/j.bcmd.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 25.Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–93. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 26.Bobrie A, Thery C. Exosomes and communication between tumours and the immune system: are all exosomes equal? Biochem Soc Trans. 2013;41:263–7. doi: 10.1042/BST20120245. [DOI] [PubMed] [Google Scholar]
- 27.Shen B, Wu N, Yang JM, Gould SJ. Protein targeting to exosomes/microvesicles by plasma membrane anchors. J Biol Chem. 2011;286:14383–95. doi: 10.1074/jbc.M110.208660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bolukbasi MF, Mizrak A, Ozdener GB, Madlener S, Strobel T, Erkan EP, et al. miR-1289 and “Zipcode”-like Sequence Enrich mRNAs in Microvesicles. Mol Ther Nucleic Acids. 2012;1:e10. doi: 10.1038/mtna.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gibbings DJ, Ciaudo C, Erhardt M, Voinnet O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat Cell Biol. 2009;11:1143–9. doi: 10.1038/ncb1929. [DOI] [PubMed] [Google Scholar]
- 30.Zwicker JI, Trenor CC, 3rd, Furie BC, Furie B. Tissue factor-bearing microparticles and thrombus formation. Arterioscler Thromb Vasc Biol. 2011;31:728–33. doi: 10.1161/ATVBAHA.109.200964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nieuwland R. Cellular origin of microparticles exposing tissue factor in cancer: a mixed double? J Thromb Haemost. 2008;6:1514–6. doi: 10.1111/j.1538-7836.2008.03069.x. [DOI] [PubMed] [Google Scholar]
- 32.Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883–91. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janowska-Wieczorek A, Marquez-Curtis LA, Wysoczynski M, Ratajczak MZ. Enhancing effect of platelet-derived microvesicles on the invasive potential of breast cancer cells. Transfusion. 2006;46:1199–209. doi: 10.1111/j.1537-2995.2006.00871.x. [DOI] [PubMed] [Google Scholar]
- 34.El-Andaloussi S, Lee Y, Lakhal-Littleton S, Li J, Seow Y, Gardiner C, et al. Exosome-mediated delivery of siRNA in vitro and in vivo . Nat Protoc. 2012;7:2112–26. doi: 10.1038/nprot.2012.131. [DOI] [PubMed] [Google Scholar]
- 35.Mignot G, Roux S, Thery C, Segura E, Zitvogel L. Prospects for exosomes in immunotherapy of cancer. J Cell Mol Med. 2006;10:376–88. doi: 10.1111/j.1582-4934.2006.tb00406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Nedawi K, Meehan B, Rak J. Microvesicles: messengers and mediators of tumour progression. Cell Cycle. 2009;8:2014–8. doi: 10.4161/cc.8.13.8988. [DOI] [PubMed] [Google Scholar]
- 37.Garnier D, Jabado N, Rak J. Extracellular vesicles as prospective carriers of oncogenic protein signatures in adult and paediatric brain tumours. Proteomics. 2013;13:1595–607. doi: 10.1002/pmic.201200360. [DOI] [PubMed] [Google Scholar]
- 38.Chen WW, Balaj L, Liau LM, Samuels ML, Kotsopoulos SK, Maguire CA, et al. BEAMing and droplet digital PCR analysis of mutant IDH1 mRNA in glioma patient serum and cerebrospinal fluid extracellular vesicles. Mol Ther Nucleic Acids. 2013;2:e109. doi: 10.1038/mtna.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noerholm M, Balaj L, Limperg T, Salehi A, Zhu LD, Hochberg FH, et al. RNA expression patterns in serum microvesicles from patients with glioblastoma multiforme and controls. BMC Cancer. 2012;12:22. doi: 10.1186/1471-2407-12-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shao H, Chung J, Balaj L, Charest A, Bigner DD, Carter BS, et al. Protein typing of circulating microvesicles allows real-time monitoring of glioblastoma therapy. Nat Med. 2012;18:1835–40. doi: 10.1038/nm.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bergsmedh A, Szeles A, Henriksson M, Bratt A, Folkman MJ, Spetz AL, et al. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc Natl Acad Sci USA. 2001;98:6407–11. doi: 10.1073/pnas.101129998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kahlert C, Melo SA, Protopopov A, Tang J, Seth S, Koch M, et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J Biol Chem. 2014;289:3869–75. doi: 10.1074/jbc.C113.532267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014;24:766–9. doi: 10.1038/cr.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garcia-Olmo DC, Dominguez C, Garcia-Arranz M, Anker P, Stroun M, Garcia-Verdugo JM, et al. Cell-free nucleic acids circulating in the plasma of colorectal cancer patients induce the oncogenic transformation of susceptible cultured cells. Cancer Res. 2010;70:560–7. doi: 10.1158/0008-5472.CAN-09-3513. [DOI] [PubMed] [Google Scholar]
- 45.Al-Nedawi K, Meehan B, Kerbel RS, Allison AC, Rak J. Endothelial expression of autocrine VEGF upon the uptake of tumour-derived microvesicles containing oncogenic EGFR. Proc Natl Acad Sci USA. 2009;106:3794–9. doi: 10.1073/pnas.0804543106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Demory Beckler M, Higginbotham JN, Franklin JL, Ham AJ, Halvey PJ, Imasuen IE, et al. Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Mol Cell Proteomics. 2013;12:343–55. doi: 10.1074/mcp.M112.022806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luga V, Zhang L, Viloria-Petit AM, Ogunjimi AA, Inanlou MR, Chiu E, et al. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell. 2012;151:1542–56. doi: 10.1016/j.cell.2012.11.024. [DOI] [PubMed] [Google Scholar]
- 48.Antonyak MA, Li B, Boroughs LK, Johnson JL, Druso JE, Bryant KL, et al. Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc Natl Acad Sci USA. 2011;108:4852–7. doi: 10.1073/pnas.1017667108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Di Vizio D, Kim J, Hager MH, Morello M, Yang W, Lafargue CJ, et al. Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 2009;69:5601–9. doi: 10.1158/0008-5472.CAN-08-3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koumangoye RB, Sakwe AM, Goodwin JS, Patel T, Ochieng J. Detachment of breast tumour cells induces rapid secretion of exosomes which subsequently mediate cellular adhesion and spreading. PLoS One. 2011;6:e24234. doi: 10.1371/journal.pone.0024234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tauro BJ, Mathias RA, Greening DW, Gopal SK, Ji H, Kapp EA, et al. Oncogenic H-ras reprograms Madin-Darby canine kidney (MDCK) cell-derived exosomal proteins following epithelial-mesenchymal transition. Mol Cell Proteomics. 2013;12:2148–59. doi: 10.1074/mcp.M112.027086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garnier D, Magnus N, Lee TH, Bentley V, Meehan B, Milsom C, et al. Cancer cells induced to express mesenchymal phenotype release exosome-like extracellular vesicles carrying tissue factor. J Biol Chem. 2012;287:43565–72. doi: 10.1074/jbc.M112.401760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garnier D, Magnus N, Meehan B, Kislinger T, Rak J. Qualitative changes in the proteome of extracellular vesicles accompanying cancer cell transition to mesenchymal state. Exp Cell Res. 2013;319:2747–57. doi: 10.1016/j.yexcr.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 54.Gesierich S, Berezovskiy I, Ryschich E, Zoller M. Systemic induction of the angiogenesis switch by the tetraspanin D6.1A/CO-029. Cancer Res. 2006;66:7083–94. doi: 10.1158/0008-5472.CAN-06-0391. [DOI] [PubMed] [Google Scholar]
- 55.Svensson KJ, Kucharzewska P, Christianson HC, Skold S, Lofstedt T, Johansson MC, et al. Hypoxia triggers a proangiogenic pathway involving cancer cell microvesicles and PAR-2-mediated heparin-binding EGF signaling in endothelial cells. Proc Natl Acad Sci USA. 2011;108:13147–52. doi: 10.1073/pnas.1104261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kucharzewska P, Christianson HC, Welch JE, Svensson KJ, Fredlund E, Ringner M, et al. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumour development. Proc Natal Acad Sci USA. 2013;110:7312–7. doi: 10.1073/pnas.1220998110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bussolati B, Grange C, Camussi G. Tumor exploits alternative strategies to achieve vascularization. FASEB J. 2011;25:2874–82. doi: 10.1096/fj.10-180323. [DOI] [PubMed] [Google Scholar]
- 58.Janowska-Wieczorek A, Wysoczynski M, Kijowski J, Marquez-Curtis L, Machalinski B, Ratajczak J, et al. Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int J Cancer. 2005;113:752–60. doi: 10.1002/ijc.20657. [DOI] [PubMed] [Google Scholar]
- 59.Thaler J, Preusser M, Ay C, Kaider A, Marosi C, Zielinski C, et al. Intratumoral tissue factor expression and risk of venous thromboembolism in brain tumour patients. Thromb Res. 2013;131:162–5. doi: 10.1016/j.thromres.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 60.Sartori MT, Della Puppa A, Ballin A, Campello E, Radu CM, Saggiorato G, et al. Circulating microparticles of glial origin and tissue factor bearing in high-grade glioma: a potential prothrombotic role. Thromb Haemost. 2013;110:378–85. doi: 10.1160/TH12-12-0957. [DOI] [PubMed] [Google Scholar]
- 61.Milsom CC, Yu JL, Mackman N, Micallef J, Anderson GM, Guha A, et al. Tissue factor regulation by epidermal growth factor receptor and epithelial-to-mesenchymal transitions: effect on tumour initiation and angiogenesis. Cancer Res. 2008;68:10068–76. doi: 10.1158/0008-5472.CAN-08-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu JL, May L, Lhotak V, Shahrzad S, Shirasawa S, Weitz JI, et al. Oncogenic events regulate tissue factor expression in colorectal cancer cells: implications for tumour progression and angiogenesis. Blood. 2005;105:1734–41. doi: 10.1182/blood-2004-05-2042. [DOI] [PubMed] [Google Scholar]
- 63.Clark SR, Thomas CP, Hammond VJ, Aldrovandi M, Wilkinson GW, Hart KW, et al. Characterization of platelet aminophospholipid externalization reveals fatty acids as molecular determinants that regulate coagulationm. Proc Natl Acad Sci USA. 2013;110:5875–80. doi: 10.1073/pnas.1222419110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mackman N. The many faces of tissue factor. J Thromb Haemost. 2009;7:136–9. doi: 10.1111/j.1538-7836.2009.03368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van den Berg YW, Osanto S, Reitsma PH, Versteeg HH. The relationship between tissue factor and cancer progression: insights from bench and bedside. Blood. 2012;119:924–32. doi: 10.1182/blood-2011-06-317685. [DOI] [PubMed] [Google Scholar]
- 66.Mackman N, Luther T. Platelet tissue factor: to be or not to be. Thromb Res. 2013;132:3–5. doi: 10.1016/j.thromres.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 67.Wang JG, Geddings JE, Aleman MM, Cardenas JC, Chantrathammachart P, Williams JC, et al. Tumor-derived tissue factor activates coagulation and enhances thrombosis in a mouse xenograft model of human pancreatic cancer. Blood. 2012;119:5543–52. doi: 10.1182/blood-2012-01-402156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thomas GM, Panicot-Dubois L, Lacroix R, Dignat-George F, Lombardo D, Dubois C. Cancer cell-derived microparticles bearing P-selectin glycoprotein ligand 1 accelerate thrombus formation in vivo . J Exp Med. 2009;206:1913–27. doi: 10.1084/jem.20082297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hron G, Kollars M, Weber H, Sagaster V, Quehenberger P, Eichinger S, et al. Tissue factor-positive microparticles: cellular origin and association with coagulation activation in patients with colorectal cancer. Thromb Haemost. 2007;97:119–23. [PubMed] [Google Scholar]
- 70.Haubold K, Rink M, Spath B, Friedrich M, Chun FK, Marx G, et al. Tissue factor procoagulant activity of plasma microparticles is increased in patients with early-stage prostate cancer. Thromb Haemost. 2009;101:1147–55. [PubMed] [Google Scholar]
- 71.Tesselaar ME, Romijn FP, van der Linden IK, Prins FA, Bertina RM, Osanto S. Microparticle-associated tissue factor activity: a link between cancer and thrombosis? J Thromb Haemost. 2007;5:520–7. doi: 10.1111/j.1538-7836.2007.02369.x. [DOI] [PubMed] [Google Scholar]
- 72.Manly DA, Wang J, Glover SL, Kasthuri R, Liebman HA, Key NS, et al. Increased microparticle tissue factor activity in cancer patients with Venous Thromboembolism. Thromb Res. 2010;125:511–2. doi: 10.1016/j.thromres.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tesselaar ME, Romijn FP, van der Linden IK, Bertina RM, Osanto S. Microparticle-associated tissue factor activity in cancer patients with and without thrombosis. J Thromb Haemost. 2009;7:1421–3. doi: 10.1111/j.1538-7836.2009.03504.x. [DOI] [PubMed] [Google Scholar]
- 74.Zwicker JI, Liebman HA, Neuberg D, Lacroix R, Bauer KA, Furie BC, et al. Tumor-derived tissue factor-bearing microparticles are associated with venous thromboembolic events in malignancy. Clin Cancer Res. 2009;15:6830–40. doi: 10.1158/1078-0432.CCR-09-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Campello E, Spiezia L, Radu CM, Bulato C, Castelli M, Gavasso S, et al. Endothelial, platelet, and tissue factor-bearing microparticles in cancer patients with and without venous thromboembolism. Thromb Res. 2011;127:473–7. doi: 10.1016/j.thromres.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 76.Thaler J, Ay C, Mackman N, Bertina RM, Kaider A, Marosi C, et al. Microparticle-associated tissue factor activity, venous thromboembolism and mortality in pancreatic, gastric, colorectal and brain cancer patients. J Thromb Haemost. 2012;10:1363–70. doi: 10.1111/j.1538-7836.2012.04754.x. [DOI] [PubMed] [Google Scholar]
- 77.Bharthuar A, Khorana AA, Hutson A, Wang JG, Key NS, Mackman N, et al. Circulating microparticle tissue factor, thromboembolism and survival in pancreaticobiliary cancers. Thromb Res. 2013;132:180–4. doi: 10.1016/j.thromres.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 78.van Es N, Bleker S, Kleinjan A, Berckmans RJ, Wilmink JW, Nieuwland R, et al. A new microparticle coagulant activity assay to predict venous thromboembolism in patients with pancreatic cancer. San Francisco, CA: The American Society of Hematology; 2014. Dec 4–9, Abstract 4250. [Google Scholar]
- 79.Zwicker JI, Liebman HA, Bauer KA, Caughey T, Campigotto F, Rosovsky R, et al. Prediction and prevention of thromboembolic events with enoxaparin in cancer patients with elevated tissue factor-bearing microparticles: a randomized-controlled phase II trial (the Microtec study) Br J Haematol. 2013;160:530–7. doi: 10.1111/bjh.12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aalberts M, van Dissel-Emiliani FM, van Adrichem NP, van WM, Wauben MH, Stout TA, et al. Identification of distinct populations of prostasomes that differentially express prostate stem cell antigen, annexin A1, and GLIPR2 in humans. Biol Reprod. 2012;86:82. doi: 10.1095/biolreprod.111.095760. [DOI] [PubMed] [Google Scholar]
- 81.Khorana AA, Streiff MB, Farge D, Mandala M, Debourdeau P, Cajfinger F, et al. Venous thromboembolism prophylaxis and treatment in cancer: a consensus statement of major guidelines panels and call to action. J Clin Oncol. 2009;27:4919–26. doi: 10.1200/JCO.2009.22.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mukherjee SD, Swystun LL, Mackman N, Wang JG, Pond G, Levine MN, et al. Impact of chemotherapy on thrombin generation and on the protein C pathway in breast cancer patients. Pathophysiol Haemost Thromb. 2010;37:88–97. doi: 10.1159/000324166. [DOI] [PubMed] [Google Scholar]
- 83.Collier ME, Ettelaie C. Regulation of the incorporation of tissue factor into microparticles by serine phosphorylation of the cytoplasmic domain of tissue factor. J Biol Chem. 2011;286:11977–84. doi: 10.1074/jbc.M110.195214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dorfleutner A, Ruf W. Regulation of tissue factor cytoplasmic domain phosphorylation by palmitoylation. Blood. 2003;102:3998–4005. doi: 10.1182/blood-2003-04-1149. [DOI] [PubMed] [Google Scholar]
- 85.Zioncheck TF, Roy S, Vehar GA. The cytoplasmic domain of tissue factor is phosphorylated by a protein kinase C-dependent mechanism. J Biol Chem. 1992;267:3561–4. [PubMed] [Google Scholar]
- 86.Ott I, Fischer EG, Miyagi Y, Mueller BM, Ruf W. A role for tissue factor in cell adhesion and migration mediated by interaction with actin-binding protein 280. J Cell Biol. 1998;140:1241–53. doi: 10.1083/jcb.140.5.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Collier ME, Maraveyas A, Ettelaie C. Filamin-A is required for the incorporation of tissue factor into cell-derived microvesicles. Thromb Haemost. 2014;111:647–55. doi: 10.1160/TH13-09-0769. [DOI] [PubMed] [Google Scholar]
- 88.Ettelaie C, Elkeeb AM, Maraveyas A, Collier ME. p38alpha phosphorylates serine 258 within the cytoplasmic domain of tissue factor and prevents its incorporation into cell-derived microparticles. Biochim Biophys Acta. 2013;1833:613–21. doi: 10.1016/j.bbamcr.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 89.Vion AC, Birukova AA, Boulanger CM, Birukov KG. Mechanical forces stimulate endothelial microparticle generation via caspase-dependent apoptosis-independent mechanism. Pulm Circ. 2013;3:95–9. doi: 10.4103/2045-8932.109921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Camerer E, Gjernes E, Wiiger M, Pringle S, Prydz H. Binding of factor VIIa to tissue factor on keratinocytes induces gene expression. J Biol Chem. 2000;275:6580–5. doi: 10.1074/jbc.275.9.6580. [DOI] [PubMed] [Google Scholar]
- 91.Belting M, Dorrell MI, Sandgren S, Aguilar E, Ahamed J, Dorfleutner A, et al. Regulation of angiogenesis by tissue factor cytoplasmic domain signaling. Nat Med. 2004;10:502–9. doi: 10.1038/nm1037. [DOI] [PubMed] [Google Scholar]
- 92.Collier ME, Ettelaie C. Induction of endothelial cell proliferation by recombinant and microparticle-tissue factor involves beta1-integrin and extracellular signal regulated kinase activation. Arterioscler Thromb Vasc Biol. 2010;30:1810–7. doi: 10.1161/ATVBAHA.110.211854. [DOI] [PubMed] [Google Scholar]
- 93.Kocaturk B, Van den Berg YW, Tieken C, Mieog JS, de Kruijf EM, Engels CC, et al. Alternatively spliced tissue factor promotes breast cancer growth in a beta1 integrin-dependent manner. Proc Natl Acad Sci USA. 2013;110:11517–22. doi: 10.1073/pnas.1307100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pradier A, Ettelaie C. The influence of exogenous tissue factor on the regulators of proliferation and apoptosis in endothelial cells. J Vasc Res. 2008;45:19–32. doi: 10.1159/000109074. [DOI] [PubMed] [Google Scholar]
- 95.Frentzou GA, Collier ME, Seymour AM, Ettelaie C. Differential induction of cellular proliferation, hypertrophy and apoptosis in H9c2 cardiomyocytes by exogenous tissue factor. Mol Cell Biochem. 2010;345:119–30. doi: 10.1007/s11010-010-0565-8. [DOI] [PubMed] [Google Scholar]
- 96.Aharon A, Tamari T, Brenner B. Monocyte-derived microparticles and exosomes induce procoagulant and apoptotic effects on endothelial cells. Thromb Haemost. 2008;100:878–85. doi: 10.1160/th07-11-0691. [DOI] [PubMed] [Google Scholar]
- 97.Collier ME, Mah PM, Xiao Y, Maraveyas A, Ettelaie C. Microparticle-associated tissue factor is recycled by endothelial cells resulting in enhanced surface tissue factor activity. Thromb Haemost. 2013;110:966–76. doi: 10.1160/TH13-01-0055. [DOI] [PubMed] [Google Scholar]
- 98.Liou YC, Zhou XZ, Lu KP. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem Scie. 2011;36:501–14. doi: 10.1016/j.tibs.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bach RR, Moldow CF. Mechanism of tissue factor activation on HL-60 cells. Blood. 1997;89:3270–6. [PubMed] [Google Scholar]
- 100.Wolberg AS, Monroe DM, Roberts HR, Hoffman MR. Tissue factor de-encryption: ionophore treatment induces changes in tissue factor activity by phosphatidylserine-dependent and -independent mechanisms. Blood Coagul Fibrinolysis. 1999;10:201–10. [PubMed] [Google Scholar]
- 101.Chen VM, Ahamed J, Versteeg HH, Berndt MC, Ruf W, Hogg PJ. Evidence for activation of tissue factor by an allosteric disulfide bond. Biochemistry. 2006;45:12020–8. doi: 10.1021/bi061271a. [DOI] [PubMed] [Google Scholar]
- 102.Mandal SK, Iakhiaev A, Pendurthi UR, Rao LV. Acute cholesterol depletion impairs functional expression of tissue factor in fibroblasts: modulation of tissue factor activity by membrane cholesterol. Blood. 2005;105:153–60. doi: 10.1182/blood-2004-03-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mandal SK, Pendurthi UR, Rao LV. Cellular localization and trafficking of tissue factor. Blood. 2006;107:4746–53. doi: 10.1182/blood-2005-11-4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dietzen DJ, Page KL, Tetzloff TA. Lipid rafts are necessary for tonic inhibition of cellular tissue factor procoagulant activity. Blood. 2004;103:3038–44. doi: 10.1182/blood-2003-07-2399. [DOI] [PubMed] [Google Scholar]
- 105.Sevinsky JR, Rao LV, Ruf W. Ligand-induced protease receptor translocation into caveolae: a mechanism for regulating cell surface proteolysis of the tissue factor-dependent coagulation pathway. J Cell Biol. 1996;133:293–304. doi: 10.1083/jcb.133.2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Berckmans RJ, Sturk A, van Tienen LM, Schaap MC, Nieuwland R. Cell-derived vesicles exposing coagulant tissue factor in saliva. Blood. 2011;117:3172–80. doi: 10.1182/blood-2010-06-290460. [DOI] [PubMed] [Google Scholar]
- 107.van Doormaal F, Kleinjan A, Berckmans RJ, Mackman N, Manly D, Kamphuisen PW, et al. Coagulation activation and microparticle-associated coagulant activity in cancer patients. An exploratory prospective study. Thromb Haemost. 2012;108:160–5. doi: 10.1160/TH12-02-0099. [DOI] [PubMed] [Google Scholar]
- 108.Milsom C, Yu J, May L, Magnus N, Rak J. Diverse roles of tissue factor-expressing cell subsets in tumour progression. Semin Thromb Hemost. 2008;34:170–81. doi: 10.1055/s-2008-1079257. [DOI] [PubMed] [Google Scholar]
- 109.Bailey KM, Wojtkowiak JW, Hashim AI, Gillies RJ. Targeting the metabolic microenvironment of tumors. Adv Pharmacol. 2012;65:63–107. doi: 10.1016/B978-0-12-397927-8.00004-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kucharzewska P, Belting M. Emerging roles of extracellular vesicles in the adaptive response of tumour cells to microenvironmental stress. J Extracell Vesicles. 2013;2 doi: 10.3402/jev.v2i0.20304. 20304, doi: http://dx.doi.org/10.3402/jev.v2i0.20304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.van der Pol E, Boing AN, Harrison P, Sturk A, Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev. 2012;64:676–705. doi: 10.1124/pr.112.005983. [DOI] [PubMed] [Google Scholar]
- 112.van der Pol E, Coumans F, Varga Z, Krumrey M, Nieuwland R. Innovation in detection of microparticles and exosomes. J Thromb Haemost. 2013;11:36–45. doi: 10.1111/jth.12254. [DOI] [PubMed] [Google Scholar]
- 113.van der Pol E, van Gemert MJ, Sturk A, Nieuwland R, van Leeuwen TG. Single vs. swarm detection of microparticles and exosomes by flow cytometry. J Thromb Haemost. 2012;10:919–30. doi: 10.1111/j.1538-7836.2012.04683.x. [DOI] [PubMed] [Google Scholar]
- 114.van der Vlist EJ, Nolte-‘t Hoen EN, Stoorvogel W, Arkesteijn GJ, Wauben MH. Fluorescent labeling of nano-sized vesicles released by cells and subsequent quantitative and qualitative analysis by high-resolution flow cytometry. Nat Protoc. 2012;7:1311–26. doi: 10.1038/nprot.2012.065. [DOI] [PubMed] [Google Scholar]
- 115.van der Pol E, Coumans FA, Grootemaat AE, Gardiner C, Sargent IL, Harrison P, et al. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. J Thromb Haemost. 2014;12:1182–92. doi: 10.1111/jth.12602. [DOI] [PubMed] [Google Scholar]
- 116.Gardiner C, Ferreira YJ, Dragovic RA, Redman CW, Sargent IL. Extracellular vesicle sizing and enumeration by nanoparticle tracking analysis. J Extracell Vesicles. 2013;2 doi: 10.3402/jev.v2i0.19671. 19671, doi: http://dx.doi.org/10.3402/jev.v2i0.19671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Arraud N, Linares R, Tan S, Gounou C, Pasquet JM, Mornet S, et al. Extracellular vesicles from blood plasma: determination of their morphology, size, phenotype and concentration. J Thromb Haemost. 2014;12:614–27. doi: 10.1111/jth.12554. [DOI] [PubMed] [Google Scholar]
- 118.Tatsumi K, Antoniak S, Monroe DM, 3rd, Khorana AA, Mackman N. Evaluation of a new commercial assay to measure microparticle tissue factor activity in plasma: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12:1932–4. doi: 10.1111/jth.12718. [DOI] [PubMed] [Google Scholar]
- 119.Basavaraj MG, Olsen JO, Osterud B, Hansen JB. Differential ability of tissue factor antibody clones on detection of tissue factor in blood cells and microparticles. Thromb Res. 2012;130:538–46. doi: 10.1016/j.thromres.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 120.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ay C, Dunkler D, Marosi C, Chiriac AL, Vormittag R, Simanek R, et al. Prediction of venous thromboembolism in cancer patients. Blood. 2010;116:5377–82. doi: 10.1182/blood-2010-02-270116. [DOI] [PubMed] [Google Scholar]
- 122.Perry JR. Thromboembolic disease in patients with high-grade glioma. Neuro Oncol. 2012;14:iv73–80. doi: 10.1093/neuonc/nos197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Blom JW, Vanderschoot JP, Oostindier MJ, Osanto S, van der Meer FJ, Rosendaal FR. Incidence of venous thrombosis in a large cohort of 66,329 cancer patients: results of a record linkage study. J Thromb Haemost. 2006;4:529–35. doi: 10.1111/j.1538-7836.2006.01804.x. [DOI] [PubMed] [Google Scholar]
- 124.Spyropoulos AC, Lin J. Direct medical costs of venous thromboembolism and subsequent hospital readmission rates: an administrative claims analysis from 30 managed care organizations. J Manag Care Pharm. 2007;13:475–86. doi: 10.18553/jmcp.2007.13.6.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brandes AA, Scelzi E, Salmistraro G, Ermani M, Carollo C, Berti F, et al. Incidence of risk of thromboembolism during treatment high-grade gliomas: a prospective study. Eur J Cancer. 1997;33:1592–6. doi: 10.1016/s0959-8049(97)00167-6. [DOI] [PubMed] [Google Scholar]
- 126.Magnus N, Gerges N, Jabado N, Rak J. Coagulation-related gene expression profile in glioblastoma is defined by molecular disease subtype. J Thromb Haemost. 2013;11:1197–200. doi: 10.1111/jth.12242. [DOI] [PubMed] [Google Scholar]
- 127.Magnus N, Garnier D, Rak J. Oncogenic epidermal growth factor receptor up-regulates multiple elements of the tissue factor signaling pathway in human glioma cells. Blood. 2010;116:815–8. doi: 10.1182/blood-2009-10-250639. [DOI] [PubMed] [Google Scholar]
- 128.Perry JR, Julian JA, Laperriere NJ, Geerts W, Agnelli G, Rogers LR, et al. PRODIGE: a randomized placebo-controlled trial of dalteparin low-molecular-weight heparin thromboprophylaxis in patients with newly diagnosed malignant glioma. J Thromb Haemost. 2010;8:1959–65. doi: 10.1111/j.1538-7836.2010.03973.x. [DOI] [PubMed] [Google Scholar]
- 129.Gonda DD, Akers JC, Kim R, Kalkanis SN, Hochberg FH, Chen CC, et al. Neuro-oncologic applications of exosomes, microvesicles, and other nano-sized extracellular particles. Neurosurgery. 2013;72:501–10. doi: 10.1227/NEU.0b013e3182846e63. [DOI] [PubMed] [Google Scholar]