

Abstract
NF-κB induces transcriptional expression of proinflammatory genes and antiapoptotic genes. The two activities of NF-κB remain to be characterized in the mechanism of chronic inflammation in obesity. To address this issue, we inactivated NF-κB in adipose tissue by knocking out p65 (RelA) in mice (F-p65-KO) and examined the inflammation in lean and obese conditions. In the lean condition, KO mice exhibited a reduced inflammation in adipose tissue with a decrease in macrophage infiltration, M1 polarization, and proinflammatory cytokine expression. In the obese condition, KO mice had elevated inflammation with more macrophage infiltration, M1 polarization, and cytokine expression. In the mechanism of enhanced inflammation, adipocytes and macrophages exhibited an increase in cellular apoptosis, which was observed with more formation of crown-like structures (CLS) in fat tissue of KO mice. Body weight, glucose metabolism, and insulin sensitivity were not significantly altered in KO mice under the lean and obese conditions. A modest but significant reduction in body fat mass was observed in KO mice on HFD with an elevation in energy expenditure. The data suggest that in the control of adipose inflammation, NF-κB exhibits different activities in the lean vs. obese condition. NF-κB is required for expression of proinflammatory genes in the lean but not in the obese condition. NF-κB is required for inhibition of apoptosis in the obese condition, in which proinflammation is enhanced by NF-κB inactivation.
Keywords: inflammation, crown-like structure, macrophage polarization, apoptosis, nuclear factor-κB
adipocytes and macrophages are the major sources of proinflammatory cytokines in adipose tissue (49, 54). They are activated by hypoxia (20, 56), oxidative stress (11, 34), endoplasmic reticulum (ER) stress (35), Toll-like receptor 4 activation (25), cell death (7), etc., in the inflammatory response in obesity. It is generally believed that activation of the IKKβ/NF-κB signaling pathway in those cells occurs in obesity to promote the inflammatory response (42). In agreement, gain-of-function of NF-κB in adipose tissue promotes chronic inflammation in mice with overexpression of p65 subunit (47) or IKKβ (21). Inhibition of the pathway is proposed as an approach in the control of obesity-associated inflammation for improvement of insulin resistance (8). However, the view remains to be tested by loss-of-function of NF-κB in adipose tissue. Of the two subunits (p65 and p50) of NF-κB, the p65 subunit contains the activation domain and determines the transcriptional activity of NF-κB (4). Knockout of p65 gene is an approach to inactivate NF-κB activity in vivo. To address the issue, we made fat-selective p65-knockout (F-p65-KO) mice and examined the inflammatory status of adipose tissue in this study.
NF-κB regulates gene transcription through direct binding to the target gene promoter DNA. NF-κB induces expression of proinflammatory cytokines [such as TNFα, IL-1, IL-6, monocyte chemotactic protein-1 (MCP-1), etc.] in response to stress signals (4). However, NF-κB also controls cell survival by induction of antiapoptotic genes (22). NF-κB induces expression of multiple genes to block the apoptotic programs. The genes include those that 1) inhibit caspase function, such as the cellular inhibitor of apoptosis proteins, X-chromosome-linked IAP (XIAP), and caspase-8-c-FLIP (FLICE inhibitory protein) (46); 2) inhibit the apoptotic signaling downstream of TNF receptor 1 (TNF-R1), such as TNF-R-associated factors 1 and 2 (22); 3) preserve mitochondrial function, such as Bcl-xL and A1/Bf1 (22); and 4) inhibit proapoptotic signaling by the c-Jun kinase (JNK) pathway, such as XIAP and Gadd45β (37, 46, 60). These genes eventually block activation of the caspase pathways to control apoptosis. Although inhibition of NF-κB may reduce expression of proinflammatory cytokines, it may also increase the risk of apoptosis. Adipocyte apoptosis is a risk factor of chronic inflammation in adipose tissue (1, 7, 52). The relative significance of two possibilities remains to be determined in obesity after NF-κB inhibition.
To test the NF-κB activities in adipose tissue, we inactivated NF-κB activity by knocking out the p65 gene in adipocytes and macrophages using the FABP4-Cre (fatty acid-binding protein 4 Cre recombinase, aP2-Cre) system. The knockout was a consequence of exon 2–4 deletion in p65 gene. The results suggest that NF-κB inactivation promotes adipose tissue inflammation in obesity through induction of cellular apoptosis.
EXPERIMENTAL PROCEDURES
Generation of floxed-p65 mice.
The loxP p65 mice were made by inserting loxP sites in introns between exons 1 and 5 of the p65 gene, which was designed to remove exons 2–4 after Cre-mediated recombination to inactivate the p65 gene (Fig. 1). FABP4-Cre mice on the C57BL/6 genetic background (aP2-Cre mice, stock no. 005069) were purchased from The Jackson Laboratory (Bar Harbor, ME). F-p65-KO (p65f/f Cre+/−) mice were generated by crossing the floxed-p65 mice with aP2-Cre mice. Floxed p65 littermates (p65f/f) of F-p65-KO (p65−/−) were used as wild-type control in this study. The mice were on the C57BL/6 genetic background. The study was conducted in male mice in the animal facility of the Pennington Biomedical Research Center. The mouse housing environment included a 12:12-h light-dark cycle, constant room temperature (22–24°C), and free access to water and diet. Chow diet (cat. no. 5001, containing 5% wt/wt or 11% calories in fat; Labdiet, St. Louis, MO) and the high-fat diet (HFD; D12331, 36% wt/wt or 58% calories in fat; Research Diets, New Brunswick, NJ) were used. Mice were fed HFD at 8 wk of age to generate a diet-induced obese model. All procedures were performed in accordance with the National Institutes of Health guidelines for the care and use of animals and were approved by the Institutional Animal Care and Use Committee at the Pennington Biomedical Research Center.
Fig. 1.

Inflammation of fat-selective p65-knockout (F-p65-KO) mice on chow diet. A: floxed-p65 (RelaΔallele) gene. Exons (■) and loxP sites (△ and ▲) are indicated in the gene structure. B: genotyping. Primers L1F and P4 were used in genotyping of floxed p65 mice. C: p65 protein in epididymal fat. Proteins of p65, Cre, and IκBα were determined by Western blot. D: mRNA of NF-κB target genes. The test was conducted in epididymal of mice at 8 wk of age by quantitative (q)RT-PCR. In the bars, values are means ± SE (n = 8). *P < 0.05 by Student's t-test. WT, wild type; KO, knockout; MCP-1, monocyte chemotactic protein-1.
Body weight and composition.
Body weight and composition were measured every 2 wk. Body composition was measured using quantitative nuclear magnetic resonance (NMR; Minispec Mn10 NMR scanner; Brucker Canada, Milton, ON, Canada), as described previously (47).
Food intake.
Food intake was determined manually in individually housed mice on chow diet at 16 wk of age. The average daily food intake was determined over 3 days by the net reduction in diet weight with exclusion of spilled diet. Food intake in grams was converted to kilocalories and adjusted by lean body mass. Food intake on HFD was monitored in mice using the metabolic chamber, as described below.
Energy expenditure.
Indirect calorimetry was performed in mice at 6 wk on HFD within the Comprehensive Laboratory Animal Monitoring System (Columbus Instruments, Columbus, OH). Mice were kept in the metabolic chamber for 6 days. Oxygen consumption (V̇o2), carbon dioxide production (V̇co2), spontaneous physical activity, and food intake were recorded daily. Energy expenditure (EE; kcal·kg−1·h−1) was calculated with data on day 5 using the formula EE = [3.815 + 1.232 × V̇co2/V̇o2] × V̇o2 × 0.001 (19). EE data were normalized with body lean mass, and the average daily food intake was derived over 3 days (days 4–6).
Insulin tolerance test and glucose tolerance test.
Insulin tolerance test (ITT) was conducted with peritoneal injection of insulin (0.75 U/kg) after 4-h fasting in mice at 9 wk on HFD. Glucose tolerance test (GTT) was performed with peritoneal injection of glucose (2 g/kg) after overnight fasting in mice at 10 wk on HFD. Blood glucose was monitored in the tail vein blood using the FreeStyle OneTouch blood glucose-monitoring system (TheraSense, Phoenix, AZ).
Western blot.
Whole cell lysates were prepared from epididymal white adipose tissue and used in Western blotting according to the protocols described elsewhere (47). Antibodies to NF-κB p65 (sc-8008), IκBα (sc-371), phospho-c-JUN (sc-822), and c-JUN (sc-1694) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies to β-actin (ab6276), Cre (ab24608), and tubulin (ab7291) were obtained from Abcam (Cambridge, UK). Antibodies to cleaved caspase 7 (no. 8438) and caspase 7 (no. 12827) were obtained from Cell Signaling Technologies (Danvers, MA). Tubulin and β-actin were used as internal controls.
Quantitative real time RT-PCR.
Total mRNA was extracted using TRIzol reagent, following the manufacturer's protocol (Invitrogen, Carlsbad, CA). The ABI 7900 platform with TaqMan Universal PCR Master Mix (4304437; Applied Biosystems, Carlsbad, CA) was used to quantify mRNA in total RNA extracts. Target gene expression was normalized to ribosome 18S RNA as the endogenous control. Primers and probes were purchased from Applied Biosystems (Carlsbad, CA). These included F4/80 (Mm00802530-m1), integrin αM (CD11b; Mm00434455-m1), integrin αX (CD11c; Mm00498698-m1), arginase 1 (Mm00475988-m1), sterol regulatory element-binding protein (SREBP; Mm00550338-m1), PPARγ (Mm00440945_m1), fatty acid synthase (FAS; Mm00662319-m1), hormone-sensitive lipase (HSL; Mm00495359-m1), lipoprotein lipase (LPL; Mm00484770-m1), p65 (Mm00501346_m1), adiponectin (ACDC; Mm00456425_m1), aP2 (Mm00445880_m1), leptin (Mm00434759_m1), IL-1β (Mm00434228_ml), IL-6 (Mm00446190_m1), TNFα (Mm00443258_m1), MCP-1 (Mm00441242_m1), and IκBα (Mm00477798_m1). For adipokine expression, macrophage markers, lipogenic genes, inflammatory cytokines, and angiogenic genes, RNA was extracted from epididymal white adipose tissue (WAT) at 8 wk of age from mice on chow diet and 26 wk of age from mice on HFD.
Apoptosis in adipose tissue.
After 18 wk of HFD feeding, epididymal WAT was isolated from mice. In the assay, formalin-fixed epididymal WAT was processed, embedded in paraffin, and sectioned at 5 μm. Viable cells were blotted with a perilipin antibody (ab3526; Abcam, Cambridge, MA) at 1:200 and a FITC-conjugated secondary antibody (F2765; Invitrogen) at 1:400. Apoptotic cells were detected using the In situ Cell Death Detection Kit (TUNEL assay) according to the manufacturer's protocol (12156792910; Roche Applied Science, Indianapolis, IN). Cleaved caspase 3 was determined in the immunohistostaining using a primary antibody (no. 9661; Cell Signaling Technology, Danvers, MA). Cleaved caspase 7 was determined in the tissue in Western blot to facilitate the apoptosis assay. Inflorescent images were captured with an upright microscope (Zeiss Axioplan 2, Intelligent Imaging Innovations, Denver, CO) and a confocal microscope (Leica TCS SP5; Leica Microsystems, Wetzlar, Germany) equipped with an AOBS system and tandem scanner (resonant scanner). Slidebook Software version 2.0 (Intelligent Imaging Innovations) and Leica TCS SP5 LASAF software (Leica Microsystems) were used in image analysis.
Apoptosis in vitro.
Primary adipocytes and macrophages were treated with TNFα (10 ng/ml) for different times, as indicated. Apoptosis was determined in the cells by measuring cleaved caspase 7 in Western blot. Adipocytes were differentiated from preadipocytes of inguinal fat, as described elsewhere (47). Macrophages were prepared as described below.
Crown-like structure.
Epididymal WAT was isolated from mice at 16 wk on HFD. Hematoxylin and eosin (H & E) staining of tissue sections and fluorescent staining of living tissue were performed as described elsewhere (53). In the fluorescent staining, griffonia simplicifolia IB4 isolectin Alexa Fluor 488 (40 μg/ml in PBS) was used to stain macrophages and endothelial cells. BODIPY 558/568 (5 μM in PBS) was used in the staining of adipocytes. Images were captured using the confocal microscope (Leica Microsystems).
Triglyceride analysis.
Triglyceride content in the feces was measured in mice after 8 wk on chow diet or 16 wk of HFD feeding.
Macrophage function.
Primary peritoneal macrophages were isolated from mice 48–72 h after intraperitoneal injection of a sterilized starch solution (2% wt/wt, 2 ml; Sigma 85643), as described elsewhere (47). LPS (2 ng/ml; Sigma) was used to induce cytokine expression in serum-free RPMI 1640 medium. Total RNA was extracted from cells, and mRNA expression was determined for IL-1β, IL-6, and TNFα.
Serum cytokine.
Serum was collected through retro-optical bleeding at 18 wk on HFD. The collection was made after overnight fasting. Cytokines IL-1β, IL-6, and TNFα were tested using a multiplex kit (MCYTOMAG-70K; Millipore).
Statistical analysis.
Statistical analysis was performed using two-tailed, unpaired Student's t-test for the WT littermates vs. KO mice. P < 0.05 was considered significant. Results are presented as means ± SE.
RESULTS
Inflammation in F-p65-KO mice on chow diet.
Our earlier study suggested that activation of NF-κB in adipose tissue leads to increased chronic inflammation in aP2-p65 mice (47), which suggests that inhibition of NF-κB may attenuate the adipose inflammation. To test this possibility, we inactivated the p65 gene to suppress NF-κB activity in adipocytes using the Cre-LoxP strategy. Floxed-p65 mice were generated by flanking the exons 2–4 with a pair of LoxP sites, and the flox result was confirmed by PCR genotyping (Fig. 1, A and B). The selection marker Neo (neomycin resistance gene) was removed from the p65 gene in floxed-p65 mice. Fat-selective inactivation was made by crossing the floxed-p65 mice with aP2-Cre mice. In the homozygous F-p65-KO mice (p65−/−, aP2-Cre+/−), P65 protein was reduced by 90% in the fat tissue relative to the wild-type (WT) littermates of floxed p65 (p65f/f) (Fig. 1C). Expression of NF-κB target genes was also decreased in the fat tissue, as indicated by IκBα (protein and mRNA), TNFα, IL-6, IL-1β, and MCP-1 (Fig. 1D). Expression of Cre protein was detected in the adipose tissue of KO mice (Fig. 1C). The results suggest that NF-κB is inactivated in the adipose tissue of F-p65-KO mice. The inactivation reduced expression of proinflammatory cytokines.
Metabolic phenotype was examined in F-p65-KO mice on chow diet. The data were collected on body weight, body fat mass, lean body mass, food intake, nutrient absorption, energy expenditure, physical activity, substrate utilization, insulin tolerance, and glucose tolerance (data not shown). These data demonstrate that KO mice exhibit no difference from WT mice in energy balance and insulin sensitivity, suggesting that NF-κB inactivation has no impact on metabolism in the lean mice.
Macrophage in KO mice on chow diet.
Macrophage infiltration is a marker of chronic inflammation in the white adipose tissue during obesity (49, 54). The aP2 promoter exhibits an activity in macrophages (47, 50). Macrophages were examined in F-p65-KO mice on the chow diet. P65 protein was decreased, and expression of NF-κB target genes (IL-1, IL-6, and TNFα) was reduced in macrophages of KO mice (Fig. 2, A–D). The reduction was especially significant in response to LPS (Fig. 2, B–D). Macrophage infiltration and polarization were examined in epididymal fat. Compared with WT mice (floxed-p65 mice), KO mice exhibited a significant reduction in macrophage markers (F4/80, CD11b, and CD11c) (Fig. 2E). The reduction in CD11c (not detectable) suggests a decrease in type 1 macrophages (30) in KO mice. Expression of arginase 1 (a marker of type 2 macrophages) was enhanced (Fig. 2E), suggesting more type 2 macrophages in KO mice. The data suggest that macrophage infiltration, type 1 polarization, and expression of proinflammatory mediators are decreased in adipose tissue of F-p65-KO mice.
Fig. 2.

Macrophages in F-p65-KO mice on chow diet. A: p65 protein in macrophages of KO mice. Western blot was conducted with cell lysate of peritoneal macrophages. B: IL-1. mRNA of NF-κB target gene was expressed in fold change in peritoneal macrophages. LPS (2 ng/ml) was used to induce the target gene expression in macrophages. C: IL-6. D: TNFα. E: macrophages in adipose tissue. mRNA was determined in epididymal fat of mice at 8 wk of age on chow diet (n = 8). *P < 0.05, **P < 0.001 by Student's t-test. CNT, control.
Reduced fat content in F-p65-KO mice on HFD.
The data in lean mice suggest that p65 inactivation may reduce adipose inflammation in the obese condition. To test this possibility, KO mice were fed HFD (58% calories in fat) for 18 wk to induce obesity. Compared with WT mice, KO mice gained a little less weight on HFD. However, the difference was not significant in the 18-wk study (14-wk data are presented in Fig. 3A). Despite the lack of change in body weight, body fat mass was significantly reduced in KO mice, and the lean body mass was not altered (Fig. 3, B and C), suggesting that the modest body weight reduction is from fat decrease. Food intake was not different between the two groups (Fig. 3D). However, EE was slightly elevated in KO mice compared with WT (Fig. 3E). The increase was observed in both day and night times. The physical activity, insulin, and glucose tolerance were not significantly different between KO and WT mice (Fig. 3, F–H). ITT and GTT were tested at 9–10 and 16–18 wk in KO mice on HFD. The data are consistent at the two time points, and the data for 9–10 wk are presented. The data suggest that NF-κB inactivation may modestly reduce fat mass through increasing EE without a significant impact on lean body mass and insulin sensitivity.
Fig. 3.

Reduced fat content in KO mice on high-fat diet (HFD). Mice were subjected to HFD feeding for 14 wk at 8 wk of age. Body weight and body composition were monitored biweekly. A: body weight. B: body fat. %Fat mass in body weight is presented. C: body lean mass. The interruption in weight and fat gain at 6–8 wk was due to metabolic cage study for energy expenditure that induced a stress response from cage change. D: food intake. Data represent average energy intake over 3 days in the metabolic cage at 6 wk on HFD. The food intake was expressed in kcal and normalized with the lean body mass (kg) and time (h). E: energy expenditure. Data were normalized with lean body mass and expressed in kcal·kg lean body mass−1·h−1. F: physical activity. The data represents mean value over 72 h after a 48-h acclimation to the metabolic cage. G: insulin tolerance test (ITT). H: glucose tolerance test (GTT). ITT and GTT were performed in mice at 9 and 10 wk on HFD. Data are expressed as means ± SE (n = 8–9). *P < 0.05, **P < 0.001 by Student's t-test.
Enhanced inflammation in fat of F-p65-KO mice on HFD.
Inflammatory status was determined in the epididymal fat of F-p65-KO mice on HFD through macrophage markers and proinflammatory cytokines. Compared with WT mice, KO mice exhibited significant increases in macrophage infiltration, macrophage activation, and cytokine expression. In the KO mice, expression of F4/80, CD11b, and CD11c was increased significantly (Fig. 4A). Arginase 1 expression was not altered (Fig. 4A). Expression of proinflammatory cytokines (TNFα, MCP-1, and IL-6) was significantly enhanced, but IL-1β was not altered (Fig. 4B). However, the cytokine proteins were not elevated in the circulation (Fig. 4C). Expression of lipogenesis-related genes was not significantly altered in the fat tissue, except for an increase in SREBP (Fig. 4D). ACDC was not changed, but leptin was reduced in KO compared with WT mice (Fig. 4E). The data suggest that the inflammatory response is enhanced in the adipose tissue of F-p65-KO mice in the obese condition.
Fig. 4.

Adipose inflammation and gene expression on HFD. Epididymal white adipose tissue (WAT) was collected from mice on HFD for 18 wk, and mRNA expression was analyzed in qRT-PCR. A: macrophage markers. B: proinflammatory genes. C: serum cytokines. D: lipogenic genes. E: adipocyte genes. All bar graphs represent means ± SE (n = 6). *P < 0.05 by Student's t-test. F4/80, F4/80 antigen, a glycoprotein expressed by macrophages; CD11b, integrin αM; CD11c, integrin αX; SREBP, sterol regulatory element-binding protein; FAS, fatty acid synthase; HSL, hormone-sensitive lipase; LPL, lipoprotein lipase; ACDC, adiponectin.
Cell apoptosis in fat tissue of KO mice.
It was surprising that the inflammation was enhanced in KO mice on HFD. To understand the mechanism, we examined cell apoptosis in the fat tissue of F-p65-KO mice on chow and HFD. NF-κB controls cellular apoptosis, but the activity remains unknown in adipose tissue. Adipocyte death contributes to the adipose inflammation in obesity and is characterized by “crown-like structure” (CLS) in adipose tissue (7). Viable cells and apoptotic cells were determined with perilipin (viable) and TUNEL (apoptotic) assays in the adipose tissue. DAPI was used to determine cell density for nuclear DNA staining. On chow diet, KO mice exhibited no difference from WT mice in viable and apoptotic cells (chow; Fig. 5A). On HFD, KO mice had less viable cells and more apoptotic cells (HFD; Fig. 5A). Reduced viable cells were observed by decreased perilipin signal, and increased apoptotic cells were observed with enhanced TUNEL signal in KO mice (Fig. 5A). The apoptosis was confirmed with caspase assays. Cleavage of caspase 7 was increased in KO mice (Fig. 5B). In vitro, adipocytes and macrophages of KO mice exhibited more cleaved caspase 7 in response to TNFα stimulation (Fig. 5, C and D). The data suggest that NF-κB inactivation increased apoptosis in adipocytes and macrophages in adipose tissue of F-p65-KO mice under obesity.
Fig. 5.

Apoptosis in WAT. A: TUNEL test. Formalin-fixed epididymal WAT was blotted with a monoclonal primary antibody to perilipin (FITC, green fluorescence), followed by TUNEL (red fluorescence) and DAPI staining (total DNA). Pictures were taken under a microscope with a ×40 lens. B: caspase assay. Cleaved caspase 7 was determined over caspase 7 in a Western blot. β-Actin was used as a protein loading control. C: adipocyte apoptosis. Cleaved and full-length caspase 7 were determined in Western blot. D: macrophage apoptosis. Primary macrophages were collected from peritoneal cavity and treated with TNFα for apoptosis.
Increased CLS in KO mice.
CLS are formed by accumulated macrophages around a dead adipocyte (7). An increase in CLS was observed in KO mice with hematoxylin and eosin staining, in which macrophages were in heavy purple color in the extracellular matrix of adipose tissue (Fig. 6A, arrows). CLS was further examined by analysis of macrophages and adipocytes using fluorescent staining, in which macrophages were stained by isolectin (green) and adipocytes were stained by Bodipy (red) (Fig. 6B). Macrophages were located in groups as indicated by green dots in the extracellular matrix (Fig. 6B, white arrows). Adipocytes were in red color from Bodipy staining. Apoptotic cells in CLS were observed by double staining with TUNEL signal (red) and cleaved caspase 3 (green) (Fig. 6, C and D). The apoptotic adipocytes are in orange color due to colocalization of the red and green signals (Fig. 6, C and D). Apoptotic macrophages are small cells double stained by red and green colors in the extracellular metrics (Fig. 6D). The data suggest that, in obesity, NF-κB inactivation leads to an increase in CLS in adipose tissue of F-p65-KO mice through adipocyte and macrophage apoptosis.
Fig. 6.

Crown-like structure (CLS). A: hematoxylin and eosin staining of adipose tissue for CLS. Images were captured at ×40. B: isolectin staining of CLS. Macrophages and endothelial cells were stained with isolectin. Adipocytes were stained with Bodipy. The staining was performed in fresh living tissue, and the picture was taken under the confocal microscope with a ×40 lens. Capillaries are also in green color, as isolectin reacts with endothelial cells (highlighted by yellow arrows). C: adipocyte apoptosis. Cleaved caspase 3 (green) and TUNEL (red) signals were determined in fixed epididymal fat. Picture was taken under the confocal microscope with a ×40 lens to show apoptotic adipocytes in the CLS. D: macrophage apoptosis. Apoptosis was determined by cleaved caspase 3 and TUNEL signals in macrophages in the CLS by colocalization. The picture was taken with a confocal microscope with a ×60 lens.
Enhanced JNK activity.
NF-κB inhibits JNK (c-Jun NH2-terminal kinase) activity through induction of gene expression. The increased apoptosis may be a result of enhanced JNK activity, which is known to induce cellular apoptosis in NF-κB-deficient cells. To test this possibility, JNK activity was determined with the phosphorylation status of c-Jun in the adipose tissue of KO mice on HFD. The phosphorylation was significantly enhanced in Western blots of KO mice (Fig. 7), suggesting enhanced JNK activity in KO mice.
Fig. 7.
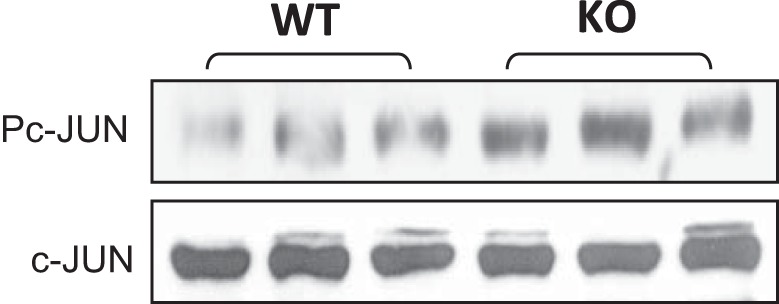
JNK activity. Phosphorylation of c-Jun was determined in epididymal fat of F-p65-KO mice on HFD (18 wk) in Western blot.
DISCUSSION
This study provides evidence that NF-κB may control adipose tissue inflammation through apoptosis. It is generally believed that NF-κB promotes inflammation through induction of transcriptional expression of proinflammatory cytokines. This activity is supported by the reduced adipose inflammation of F-p65-KO mice on chow diet in this study and increased adipose inflammation in mice with p65 or IKK overexpression in other studies (21, 47). However, p65 inactivation did not inhibit the inflammation in F-p65-KO mice on HFD, suggesting that NF-κB is not necessary for chronic inflammation in the obese condition. Apoptosis was enhanced in adipocytes and macrophages in adipose tissue of F-p65-KO mice on HFD but not on chow diet, suggesting that NF-κB is required for the inhibition of apoptosis in the obese condition. The association of increased inflammation and apoptosis suggests that the antiapoptotic activity of NF-κB is more important in the control of inflammation in the obese condition. The apoptosis may block expression of cytokine proteins from the elevated mRNA in macrophages. As such, plasma cytokines were not upregulated, and adiponectin expression was not downregulated in KO mice relative to WT mice.
Apoptosis is a mechanism of removing aging or damaged adipocytes in adipose tissue, which is elevated in obese mice and patients (5, 7). Average lifespan of adipocytes is ∼10 yr in humans (3). The adipocyte lifespan is reduced in obesity with acceleration in adipocyte turnover rate (43, 44). Adipocytes have less activity in apoptosis than preadipocytes, as indicated by expression of apoptotic genes. Adipocyte differentiation reduces expression of proapoptotic molecule (TRAIL-R) (23) and increases expression of antiapoptotic molecule (bcl-2) (33). Although adipocytes are more tolerant of apoptosis, adipocyte apoptosis is elevated by obesity, with increased expression of proapoptotic molecule FAS (CD95) (5, 52). The expression may mediate signals of inflammatory cytokines (38) or hypoxia (58) in the induction of apoptosis. Strong apoptosis reduces adipose tissue mass through decreasing adipocyte number, which is reported in transgenic mice with caspase 8 overexpression (36). Adipocyte apoptosis contributes to adipose inflammation in obesity (5, 7). Reduction of adipocyte apoptosis leads to a decrease in adipose inflammation in obese mice with Bid or FAS inactivation (1, 52). The antiapoptosis activity of NF-κB has never been tested in the context of adipose inflammation. The current study provides evidence that apoptosis from NF-κB inactivation promotes adipose inflammation in F-p65-KO mice on HFD. The apoptosis was not altered in the KO mice on chow diet, which is likely a result of less apoptotic stimuli in the lean condition. The data suggest that NF-κB has an anti-inflammatory effect on the adipose tissue in obesity through inhibition of cell apoptosis.
The reduced adipose tissue mass is related to the adipocyte apoptosis and energy expenditure without an alteration in food intake in F-p65-KO mice. The apoptosis modestly reduced fat mass, which was not strong enough to make a significant reduction in the body weight. An increase in adipogenesis may compensate for the apoptosis to avoid a huge fat reduction in F-p65-KO mice. The increased CLS suggests more adipogenic activities. CLS serves as an adipogenic niche in the fat tissue (27). NF-κB inhibition by IκBα overexpression in adipocytes does not significantly change body weight in mice (50). The energy expenditure was elevated in F-p65-KO mice, which may contribute to the reduced fat mass. The food intake was not significantly altered in KO mice. The energy expenditure is likely a result of elevated inflammation in the fat tissues, which promotes lipolysis and fat mobilization.
Our data suggest that NF-κB may regulate macrophage polarization in adipose tissue. Differentiation of M2 to M1 macrophages (macrophage polarization) is increased in adipose tissue in obesity (30). In F-p65-KO mice, the decreased M1 in the lean and increased M1 in the obese conditions suggest that the NF-κB activity is dependent on fat expansion. The increased M1 is likely a result of macrophage uptaking of adipocyte debris from apoptosis in the obese condition. The engulf activity may promote macrophage apoptosis in F-p65-KO mice. Our result is different from myeloid IKKβ-knockout mice, in which proinflammation is reduced in obese mice (2). The difference may be related to the fact that myeloid cells include neutrophils, basophils, and eosinophils in addition to macrophages. The multinucleate cells play a role in the chronic inflammation (39, 45, 51). The difference in IKKβ vs. NF-κB and aP2-Cre vs. Lys-Cre may contribute to the discrepancy as well.
Activation of JNK may contribute to the enhanced inflammatory response in F-p65-KO mice in the obese condition. JNK induces expression of inflammatory genes through activation of transcription factor AP-1. JNK is activated in adipose tissue to promote chronic inflammation in obesity (18). The JNK activity is supported by fat-selective knockout using the aP2-Cre system (40) and myeloid cell-selective knockout using the Lyz-Cre system (15). Alternatively, JNK induces cellular apoptosis to promote inflammation. JNK activity was enhanced in the fat tissue of KO mice on HFD, suggesting a role for JNK in promotion of inflammation and cellular apoptosis.
Chronic inflammation of adipose tissue is disassociated with systemic insulin resistance in F-p65-KO mice on HFD. Although adipose inflammation was elevated in KO mice, insulin sensitivity was not altered by the inflammation. ITT and GTT were not impaired in KO mice on HFD, as suggested by data at two time points (9–10 and 16–18 wk). Energy expenditure was elevated in F-p65-KO mice. The increased energy expenditure may be a mechanism for disassociation of inflammation and insulin sensitivity. Energy expenditure improves insulin sensitivity in many mouse models, including the inflammation model (21, 41, 47, 55). Energy expenditure is a positive impact of inflammation on metabolism (57). The positive effect provides a potential answer to the unaltered insulin sensitivity in our KO mice. It also explains the disappointing results of anti-inflammation therapies in clinical trials for insulin sensitization. Various anti-inflammatory medicines have been tested to improve insulin sensitivity in human studies, in which the medicines yielded negative results in most small clinical trials (8, 57). In the best case, salsalate yielded positive results in the small clinical trials (10, 14) but gave a negative result in a large clinical trial (12, 13). The anti-inflammatory therapies induce body weight in patients in the large clinical trial with 1-kg gain on average (12, 13). The mechanism of weight gain is unknown, but a reduction in energy expenditure may play a role. In animals, salsalate does not improve insulin sensitivity in lipodystrophic mice (17). In current study, the increased energy expenditure may protect insulin sensitivity from the detrimental effects of elevated inflammation in our KO mice.
In the present study, the conditional p65 gene knockout was made with aP2-Cre mic from The Jackson laboratory (stock no. 005069) (16). This aP2-Cre system exhibited recombination efficiency in adipocytes and macrophages in gene deletion. The observation is consistent with aP2 promoter activity in both adipocytes and macrophages in many studies (6, 24, 31, 47, 50, 52). The macrophage activity provides an answer to the aP2-promoter activity in multiple tissues such as brain, liver, and muscle in embryo or adult mice (26, 32, 59). However, in some studies, the macrophage activity was not observed in transgenic mice (28, 29). The aP2-promote does not exhibit leakage in myeloid cells (52). The reason for this discrepancy remains unknown and may be related to floxed genes. Although the aP2-Cre system is limited in specificity to adipocytes, the system remains a powerful tool in the study of adipocytes and macrophages together, according to recent reports (28, 50). Caution is recommended in the data interpretation for systemic insulin resistance in this study due to the involvement of multiple organs. In this regard, our conclusion remains to be verified using a more sophisticated Cre system, such as adiponectin-Cre mice (9, 26), or an inducible adiponectin Cre system (48).
In summary, this study suggests that NF-κB regulates proinflammation in adipose tissue using different mechanisms according to the adiposity conditions. In the lean condition, NF-κB contributes to inflammation through induction of proinflammatory cytokines. NF-κB inactivation leads to reduced inflammation, as suggested by expression of proinflammatory genes, macrophage infiltration, and M1 polarization in F-p65-KO mice. In the obese condition, NF-κB exhibits an anti-inflammation effect through inhibition of apoptosis. NF-κB inactivation leads to elevated inflammation, as suggested by increased expression of proinflammatory genes, M1 macrophages, and CLS in the white adipose tissue. This study suggests that NF-κB may not be an ideal drug target in the control of chronic inflammation in obesity.
GRANTS
This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) funds (DK-085495 and DK-068036) to J. Ye, the National Natural Science Foundation of China (grant no. 81370915) to Z. Gao, and a National Institutes of Health (NIH) T32 Award to T. M. Henagan (5T32-DK-064584-08). The quantitative RT-PCR test, metabolic phenotyping, and imaging studies were conducted in the genomic core, phenotyping core, and imaging core, which were supported by NIH Grants (P30-DK-072476 and P20-RR-021945).
DISCLOSURES
The authors have no conflicts of interest, financial or otherwise, in the publication of this study.
AUTHOR CONTRIBUTIONS
Z.G., J.Z., T.M.H., J.H.L., and X.Y. performed experiments; Z.G., J.Z., T.M.H., J.H.L., H.W., X.Y., and J.Y. analyzed data; Z.G., J.Z., T.M.H., J.H.L., and J.Y. interpreted results of experiments; Z.G., J.Z., T.M.H., J.H.L., H.W., X.Y., and J.Y. prepared figures; Z.G., J.Z., T.M.H., J.H.L., H.W., and J.Y. edited and revised manuscript; Z.G., J.Z., T.M.H., J.H.L., H.W., X.Y., and J.Y. approved final version of manuscript; H.W. and J.Y. conception and design of research; J.Y. drafted manuscript.
ACKNOWLEDGMENTS
J. Ye is the guarantor of this work.
REFERENCES
- 1.Alkhouri N, Gornicka A, Berk MP, Thapaliya S, Dixon LJ, Kashyap S, Schauer PR, Feldstein AE. Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J Biol Chem 285: 3428–3438, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 11: 191–198, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Arner E, Westermark PO, Spalding KL, Britton T, Ryden M, Frisen J, Bernard S, Arner P. Adipocyte turnover: relevance to human adipose tissue morphology. Diabetes 59: 105–109, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic Disease. Cell Metab 13: 11–22, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bluher M, Klöting N, Wueest S, Schoenle EJ, Schön MR, Dietrich A, Fasshauer M, Stumvoll M, Konrad D. Fas and FasL expression in human adipose tissue is related to obesity, insulin resistance, and type 2 diabetes. J Clin Endocrinol Metab 99: E36–E44, 2014. [DOI] [PubMed] [Google Scholar]
- 6.Boord JB, Maeda K, Makowski L, Babaev VR, Fazio S, Linton MF, Hotamisligil GS. Combined adipocyte-macrophage fatty acid-binding protein deficiency improves metabolism, atherosclerosis, and survival in apolipoprotein E-deficient mice. Circulation 110: 1492–1498, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, Wang S, Fortier M, Greenberg AS, Obin MS. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res 46: 2347–2355, 2005. [DOI] [PubMed] [Google Scholar]
- 8.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 11: 98–107, 2011. [DOI] [PubMed] [Google Scholar]
- 9.Eguchi J, Wang X, Yu S, Kershaw EE, Chiu PC, Dushay J, Estall JL, Klein U, Maratos-Flier E, Rosen ED. Transcriptional control of adipose lipid handling by IRF4. Cell Metab 13: 249–259, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fleischman A, Shoelson SE, Bernier R, Goldfine AB. Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care 31: 289–294, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 114: 1752–1761, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldfine AB, Conlin PR, Halperin F, Koska J, Permana P, Schwenke D, Shoelson SE, Reaven PD. A randomised trial of salsalate for insulin resistance and cardiovascular risk factors in persons with abnormal glucose tolerance. Diabetologia 56: 714–723, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldfine AB, Fonseca V, Jablonski KA, Chen YD, Tipton L, Staten MA, Shoelson SE; Targeting Inflammation Using Salsalate in Type 2 Diabetes Study Team. Salicylate (salsalate) in patients with type 2 diabetes: a randomized trial. Ann Intern Med 159: 1–12, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldfine AB, Silver R, Aldhahi W, Cai D, Tatro E, Lee J, Shoelson SE. Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin Transl Sci 1: 36–43, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han MS, Jung DY, Morel C, Lakhani SA, Kim JK, Flavell RA, Davis RJ. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 339: 218–222, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He W, Barak Y, Hevener A, Olson P, Liao D, Le J, Nelson M, Ong E, Olefsky JM, Evans RM. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci USA 100: 15712–15717, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herrero L, Shapiro H, Nayer A, Lee J, Shoelson SE. Inflammation and adipose tissue macrophages in lipodystrophic mice. Proc Natl Acad Sci USA 107: 240–245, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirosumi J, Tuncman G, Chang L, Gorgun C, Uysal K, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature 420: 333–336, 2002. [DOI] [PubMed] [Google Scholar]
- 19.Hofmann SM, Zhou L, Perez-Tilve D, Greer T, Grant E, Wancata L, Thomas A, Pfluger PT, Basford JE, Gilham D, Herz J, Tschop MH, Hui DY. Adipocyte LDL receptor-related protein-1 expression modulates postprandial lipid transport and glucose homeostasis in mice. J Clin Invest 117: 3271–3282, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M, Shimomura I. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 56: 901–911, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Jiao P, Feng B, Ma J, Nie Y, Paul E, Li Y, Xu H. Constitutive activation of IKKβ in adipose tissue prevents diet-induced obesity in mice. Endocrinology 153: 154–165, 2012. [DOI] [PubMed] [Google Scholar]
- 22.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol 3: 221–227, 2002. [DOI] [PubMed] [Google Scholar]
- 23.Keuper M, Wernstedt Asterholm I, Scherer PE, Westhoff MA, Möller P, Debatin KM, Strauss G, Wabitsch M, Fischer-Posovszky P. TRAIL (TNF-related apoptosis-inducing ligand) regulates adipocyte metabolism by caspase-mediated cleavage of PPARgamma. Cell Death Dis 4: e474, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar A, Lawrence JC Jr, Jung DY, Ko HJ, Keller SR, Kim JK, Magnuson MA, Harris TE. Fat cell-specific ablation of rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes 59: 1397–1406, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, Sizemore N, Hwang DH. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem 278: 37041–37051, 2003. [DOI] [PubMed] [Google Scholar]
- 26.Lee KY, Russell SJ, Ussar S, Boucher J, Vernochet C, Mori MA, Smyth G, Rourk M, Cederquist C, Rosen E, Kahn B, Kahn CR. Lessons on conditional gene targeting in mouse adipose tissue. Diabetes 62: 864–874, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee YH, Petkova AP, Granneman JG. Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab 18: 355–367, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee Yun S, Kim JW, Osborne O, Oh Da Y, Sasik R, Schenk S, Chen A, Chung H, Murphy A, Watkins SM, Quehenberger O, Johnson RS, Olefsky JM. Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell 157: 1339–1352, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li P, Fan W, Xu J, Lu M, Yamamoto H, Auwerx J, Sears DD, Talukdar S, Oh D, Chen A, Bandyopadhyay G, Scadeng M, Ofrecio JM, Nalbandian S, Olefsky JM. Adipocyte NCoR knockout decreases PPARgamma phosphorylation and enhances PPARgamma activity and insulin sensitivity. Cell 147: 815–826, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117: 175–184, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Makowski L, Boord JB, Maeda K, Babaev VR, Uysal KT, Morgan MA, Parker RA, Suttles J, Fazio S, Hotamisligil GS, Linton MF. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat Med 7: 699–705, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mullican SE, Tomaru T, Gaddis CA, Peed LC, Sundaram A, Lazar MA. A novel adipose-specific gene deletion model demonstrates potential pitfalls of existing methods. Mol Endocrinol 27: 127–134, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagel SA, Keuper M, Zagotta I, Enlund E, Ruperez AI, Debatin KM, Wabitsch M, Fischer-Posovszky P. Up-regulation of Bcl-2 during adipogenesis mediates apoptosis resistance in human adipocytes. Mol Cell Endocrinol 382: 368–376, 2014. [DOI] [PubMed] [Google Scholar]
- 34.Ogihara T, Asano T, Katagiri H, Sakoda H, Anai M, Shojima N, Ono H, Fujishiro M, Kushiyama A, Fukushima Y, Kikuchi M, Noguchi N, Aburatani H, Gotoh Y, Komuro I, Fujita T. Oxidative stress induces insulin resistance by activating the nuclear factor-kappaB pathway and disrupting normal subcellular distribution of phosphatidylinositol 3-kinase. Diabetologia 47: 794–805, 2004. [DOI] [PubMed] [Google Scholar]
- 35.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306: 457–461, 2004. [DOI] [PubMed] [Google Scholar]
- 36.Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, Kitsis RN, Scherer PE. Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat Med 11: 797–803, 2005. [DOI] [PubMed] [Google Scholar]
- 37.Papa S, Zazzeroni F, Bubici C, Jayawardena S, Alvarez K, Matsuda S, Nguyen DU, Pham CG, Nelsbach AH, Melis T, Smaele ED, Tang WJ, D'Adamio L, Franzoso G. Gadd45beta mediates the NF-kappaB suppression of JNK signalling by targeting MKK7/JNKK2. Nat Cell Biol 6: 146–153, 2004. [DOI] [PubMed] [Google Scholar]
- 38.Prins JB, Niesler CU, Winterford CM, Bright NA, Siddle K, O'Rahilly S, Walker NI, Cameron DP. Tumor necrosis factor-alpha induces apoptosis of human adipose cells. Diabetes 46: 1939–1944, 1997. [DOI] [PubMed] [Google Scholar]
- 39.Qiu Y, Nguyen KD, Odegaard JI, Cui X, Tian X, Locksley RM, Palmiter RD, Chawla A. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell 157: 1292–1308, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, Barrett T, Kim JK, Davis RJ. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 322: 1539–1543, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell 148: 852–871, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 116: 1793–1801, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, Blomqvist L, Hoffstedt J, Naslund E, Britton T, Concha H, Hassan M, Ryden M, Frisen J, Arner P. Dynamics of fat cell turnover in humans. Nature 453: 783–787, 2008. [DOI] [PubMed] [Google Scholar]
- 44.Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW 2nd, DeFuria J, Jick Z, Greenberg AS, Obin MS. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 56: 2910–2918, 2007. [DOI] [PubMed] [Google Scholar]
- 45.Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, Ofrecio J, Lin M, Brenner MB, Olefsky JM. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med 18: 1407–1412, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, Lin A. Inhibition of JNK activation through NF-kappaB target genes. Nature 414: 313–317, 2001. [DOI] [PubMed] [Google Scholar]
- 47.Tang T, Zhang J, Yin J, Staszkiewicz J, Gawronska-Kozak B, Jung DY, Ko HJ, Ong H, Kim JK, Mynatt R, Martin RJ, Keenan M, Gao Z, Ye J. Uncoupling of inflammation and insulin resistance by NF-kappaB in transgenic mice through elevated energy expenditure. J Biol Chem 285: 4637–4644, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang QA, Tao C, Gupta RK, Scherer PE. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med 19: 1338–1344, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796–1808, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wernstedt Asterholm I, Tao C, Morley TS, Wang QA, Delgado-Lopez F, Wang ZV, Scherer PE. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab 20: 103–118, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, Chawla A, Locksley RM. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 332: 243–247, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wueest S, Rapold RA, Schumann DM, Rytka JM, Schildknecht A, Nov O, Chervonsky AV, Rudich A, Schoenle EJ, Donath MY, Konrad D. Deletion of Fas in adipocytes relieves adipose tissue inflammation and hepatic manifestations of obesity in mice. J Clin Invest 120: 191–202, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu F, Burk D, Gao Z, Yin J, Zhang X, Weng J, Ye J. Angiogenic deficiency and adipose tissue dysfunction are associated with macrophage malfunction in SIRT1−/− mice. Endocrinology 1531706–1716, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112: 1821–1830, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ye J. Mechanisms of insulin resistance in obesity. Front Med 7: 14–24, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab 293: E1118–E1128, 2007. [DOI] [PubMed] [Google Scholar]
- 57.Ye J, McGuinness OP. Inflammation during obesity is not all bad: evidence from animal and human studies. Am J Physiol Endocrinol Metab 304: E466–E477, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yin J, Gao Z, He Q, Ye J. Role of hypoxia in obesity-induced disorders of glucose and lipid metabolism in adipose tissue. Am J Physiol Endocrinol Metab 296: E333–E342, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang J, Wang Y, Gao Z, Yun Z, Ye J. Hypoxia-inducible factor 1 activation from adipose protein 2-cre mediated knockout of von hippel-lindau gene leads to embryonic lethality. Clin Exp Pharmacol Physiol 39: 145–150, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang N, Ahsan MH, Zhu L, Sambucetti LC, Purchio AF, West DB. NF-kappaB and not the MAPK signaling pathway regulates GADD45beta expression during acute inflammation. J Biol Chem 280: 21400–21408, 2005. [DOI] [PubMed] [Google Scholar]