

Abstract
Imposed length changes of only a small percent produce transient reductions in active force in strips of airway smooth muscle (ASM) due to the temporary detachment of bound cross-bridges caused by the relative motion of the actin and myosin fibers. More dramatic and sustained reductions in active force occur following large changes in length. The Huxley two-state model of skeletal muscle originally proposed in 1957 and later adapted to include a four-state description of cross-bridge kinetics has been widely used to model the former phenomenon, but is unable to account for the latter unless modified to include mechanisms by which the contractile machinery in the ASM cell becomes appropriately rearranged. Even so, the Huxley model itself is based on the assumption that the contractile proteins are all aligned precisely in the direction of bulk force generation, which is not true for ASM. The present study derives a coarse-grained version of the Huxley model that is free of inherent assumptions about cross-bridge orientation. This simplified model recapitulates the key features observed in the force-length behavior of activated strips of ASM and, in addition, provides a mechanistically based way of accounting for the sustained force reductions that occur following large stretch.
Keywords: Huxley model, force-velocity relationship, cross-bridge kinetics
imposed length changes of only a small percent cause transient reductions in active force in strips of airway smooth muscle (ASM) (1, 9). These force reductions are due to the temporary detachment of bound cross-bridges by the relative motion of the actin and myosin fibers (1, 23) and are generally modeled using adaptations of the Huxley model of skeletal muscle (4, 23, 26), which is modified to include the Hai-Murphy four-state description of cross-bridge kinetics (13). A more sustained reduction in active force reduction, however, can be shown to occur following dramatic changes in length. Evidence for this phenomenon in vivo requires rather severe lung inflations during the development of bronchoconstriction (2), but it is readily demonstrated in vitro by subjecting activated strips of ASM to sudden stretches of sufficient amplitude (1). Recovery from this kind of force reduction is much slower than would be expected of normally cycling cross-bridges, suggesting that the mechanism responsible represents a more profound disruption than myosin cross-bridges merely becoming detached from their actin attachment sites.
We previously developed a computational model of ASM dynamics that takes into account the passive rheological properties of the connective tissue component of ASM as well as its cross-bridge kinetics (1). This model accurately accounts for the dynamic force-length behavior of activated ASM strips when length changes are small, but it contains no mechanism to account for the sustained decrements in active force produced by sudden, brief increases in length of more than about 5% (1). Models based solely on the Huxley model of skeletal muscle contraction (16, 17, 34) are also not able to account for this protracted decrement in stretch-induced force reduction (23, 24, 26). However, Silveira et al. (32), Brook (4), and Donovan (7) developed models that include remodeling of the contractile and force-bearing structures within the ASM cell, and which are able to mimic the adaptation of force generation caused by changes in ASM length, in line with experimental evidence that intracellular structural alterations are required to account for these phenomena (27).
The models of Silveira et al. (32), Brook (4), and Donovan (7) represent important advances in our understanding of ASM dynamics, but they highlight a conceptual conflict between the representation of structural remodeling and the use of the Huxley model in describing ASM dynamics. Specifically, structural remodeling requires that the contractile proteins in ASM be labile and thus relatively unconstrained in their spatial organization, whereas the Huxley model is based on the assumption that the contractile protein fibers are all oriented precisely in the direction of overall force generation (16, 19, 34). Although this assumption is arguably appropriate for skeletal muscle, the contractile machinery inside an ASM cell is oriented over a range of angles (33) and it is difficult to imagine that the active reorganization of this machinery during muscle contraction would result in perfect alignment. In an attempt to reconcile the advantages of the Huxley model with the difficulty just alluded to, we derive here a coarse-grained version of the Huxley model that is free of assumptions about contractile fiber orientation. We further show that the modified model provides a mechanistically motivated way of accounting for the sustained reductions in active ASM force that have been recorded following large stretch.
METHODS
Model derivation.
The Huxley model accounts for the macroscopic force and velocity of muscle contraction as the summed effects of a very large number of myosin cross-bridges that bind to and unbind from their target actin binding sites. It is assumed that at any point in time each cross-bridge is either unbound, in which case it does not contribute to force and velocity, or it is bound to an actin site located a distance, x, from the elastic equilibrium length of the myosin head. The entire population of cross-bridges thus defines a density distribution, n(x, t), where n(x, t)dx is the number of bound cross-bridges stretched between the distance values x and x + dx at time t, and the distance parameter, x, may have any value in the range [−X, X]. The partial differential equation of n that defines the Huxley model is (19, 34):
| (1) |
where F(x, t) and G(x, t) represent, respectively, the rates at which cross-bridges bind to and unbind from the actin binding sites, and v(t) is the velocity of movement of the actin and myosin fibers with respect to each other. The Huxley model further assumes that the elastic stresses in the stretched cross-bridges supply the force for muscle contraction (16, 19, 34), so that total muscle force is simply the elastic force in each cross-bridge integrated over the entire population. Specifically, if ε(x) is the force-extension function for a single cross-bridge, then the contribution to muscle force from all the cross-bridges at extension x at time t is ε(x)n(x,t). This gives the total muscle force P(t) as:
| (2) |
Huxley assumed (16, 19, 34) that there exists a maximum possible density, nmax(x), of bound cross-bridges at each value of x, and that both binding and unbinding follow first-order kinetics according to
| (3) |
and
| (4) |
for some suitable functions f(x) and g(x). Huxley originally chose functional forms for f(x) and g(x) that permitted an analytical solution to Eq. 1 (34), but many are possible (19). The one key constraint of the model is that unbound cross-bridges bind to actin sites located at x > 0, which means their binding is designed to contribute to muscle shortening and not lengthening. Once bound, however, some cross-bridges may be dragged into regions for which x < 0 as the muscle shortens.
We now derive, starting from the Huxley model, a model for ASM that avoids making assumptions about the distribution of cross-bridge extension, n(x, t). We begin by assuming that nmax is a constant and thus independent of x (19, 34). The time-derivative of Eq. 2 provides an expression for the rate of change of active muscle force as
| (5) |
where the second line was obtained by substitution of Eqs. 1, 3, and 4. Applying integration by parts to the second integral on the right-hand side of Eq. 5 gives
| (6) |
We can assume without loss of generality that no cross-bridges are bound at the extremes of their ranges of extension by making the magnitude of X large enough to ensure this is the case, so that n(−X, t) = n(X, t) = 0. If we further assume that the cross-bridges behave as Hookean springs such that
| (7) |
then Eq. 6 becomes
| (8) |
where N(t) is the total number of cross-bridges bound at time t.
To deal with the first integral on the right-hand side of Eq. 5 it is necessary to know the functions f(x) and g(x). As mentioned above, these functions remain a matter of some debate even for skeletal muscle (19, 34), and in the case of ASM the variations in cross-bridge orientation mean that we really have no idea what f(x) and g(x) should be. Accordingly, in the absence of any particular reason to the contrary we will invoke Occam's Razor and assume that, to a first approximation, f(x) and g(x) can be considered constant for x ∈ [0, X] and x ∈ [−X, X], respectively, and zero elsewhere. Furthermore, f(x) is nonzero only for positive allowable values of x where it equals a constant, kon, which reflects the rate at which unbound cross-bridges bind onto actin. This condition ensures that cross-bridge binding acts only to shorten the muscle, in contrast to mechanistically similar models of passive tissue rheology in which binding can be symmetric about the elastic equilibrium position (8). Thus
| (9) |
In the case of g(x) it is necessary to allow for bound cross-bridges to detach after they have been dragged into regions of negative x as a result of muscle shortening. Furthermore, it seems reasonable to suppose that relative movement of the actin and myosin fibers will make cross-bridges more likely to detach. Indeed, we have previously invoked this assumption (1), as has Seow (28). Furthermore, Piazzesi et al. (25) found experimentally that detachment rate was close to being linearly related to shortening velocity. Accordingly, we will let g(x) be a linear function of v(t) such that
| (10) |
where g0 is a constant that ensures a finite detachment rate under isometric conditions, koff is a constant, and we have normalized v(t) by the maximum unloaded shortening velocity of the muscle, vmax, so that koff has the same units as kon. Equation 10 provides a reasonable description of experimental measurements of the detachment rate constant in skeletal muscle, but the contribution of g0 to g(x) over most of the velocity range is minimal [see Fig. 4D in (25)] so we will ignore g0 from now on.
Fig. 4.
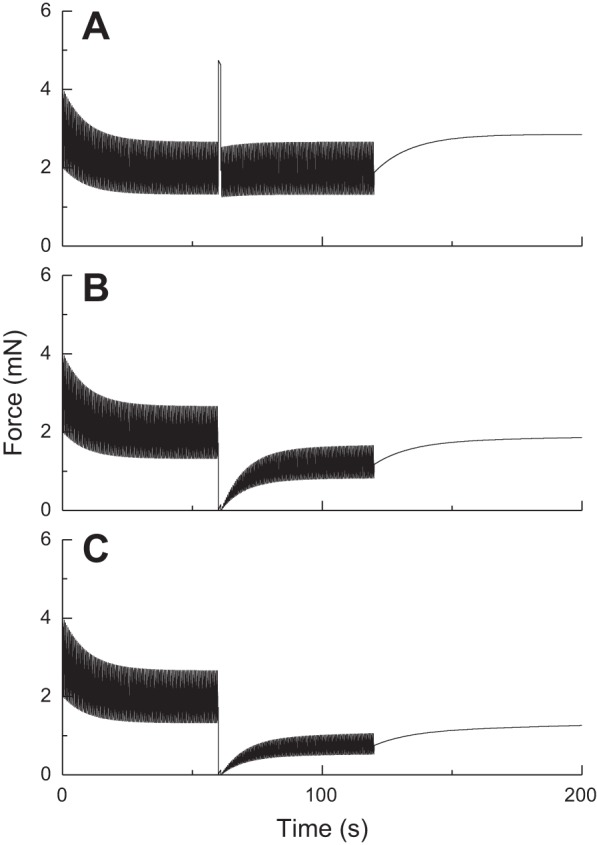
Effect of brief stretches, applied at 60 s on the oscillatory force-length behavior of ASM as determined by Eq. 14 with Nmax being modulated according to Eq. 15. The stretch amplitudes are A: 5% lref (A), 15% lref (B), and 25% lref (C). (Compare with experimental data in Fig. 1B.)
Employing Eqs. 7, 9, and 10 (ignoring g0), the first integral on the right-hand side of Eq. 5 becomes
| (11) |
In analogy with Eq. 2, the middle term on the right-hand side of Eq. 11 is proportional to the component of P(t) arising from those bound cross-bridges for which x is positive. However, because our fundamental premise is that Eq. 2 is inappropriate for ASM, we cannot use it to evaluate n(x, t) for x ∈ [0, X] and thus calculate the middle term of Eq. 11 directly. Instead, we note that this term describes the component of muscle force that acts in the direction of muscle shortening, which is counteracted by the cross-bridges that are bound at values of x that are negative enough to oppose muscle shortening. The greater the velocity of shortening, the greater will be the fraction of bound cross-bridges for which x is negative until eventually the force opposing contraction equals that producing it. At this point the muscle reaches a limiting shortening velocity, vmax, and generates no force. There is also evidence in skeletal muscle that the total number of attached cross-bridges decreases as velocity increases (25). Both mechanisms will cause the second term in Eq. 11 to make a contribution to P(t) that decreases with the velocity of shortening until velocity reaches vmax. Assuming a linear relationship for this effect gives
| (12) |
Substituting Eqs. 8, 11, and 12 into Eq. 5 gives
| (13) |
The first term on the right-hand side can be written kon(nmaxX)(EX/2), which permits the following observations. The factor nmaxX is the maximum number, Nmax, of cross-bridges that can be bound throughout the entire muscle. The factor EX/2 is another constant of the model, and is the force in a single cross-bridge stretched to half its possible length. This factor can thus be viewed as a measure of the characteristic force, Pbr, in a single cross-bridge. Of course not all cross-bridges experience the same force, and the mean cross-bridge force decreases with increasing v(t) as increasing numbers of cross-bridges become dragged into regions of negative x. Therefore, we cannot simply equate N(t) in the last term of Eq. 13 to the ratio P(t)/Pbr. Rather, we again invoke the notion, used to derive Eq. 12, that the number of attached cross-bridges decreases with increasing v(t). Assuming direct inverse proportionality between v(t) and N(t) makes this last term a constant, α.
With the above observations Eq. 13 becomes
| (14) |
We show in appendix a that Eq. 14 also predicts a hyperbolic force-velocity relationship for ASM of the type that has been measured experimentally (14). Equation 14 is an ordinary, rather than a partial, differential equation because it simply assumes that cross-bridges are either bound or unbound at any point in time. It thus represents a cruder description of cross-bridge kinetics than the classic Huxley model (Eq. 1), but it achieves this description without making assumptions about distributions of cross-bridge extension that are arguably inappropriate for the relatively amorphous internal structure of ASM.
What we were unable to find a convincing explanation for in our previous study (1) is the large reduction in force-generating capacity observed in ASM strips immediately following a large, brief stretch. This phenomenon is illustrated in Fig. 6A in (1), which shows a stretch-dependent decrement in the amplitude of the force oscillations. This phenomenon cannot be accounted for simply in terms of temporarily detached cross-bridges reattaching with kinetics governed by the parameters koff and kon because if this were the case, then the recovery in force would be much more rapid than is actually observed. The dramatic and sustained reductions in oscillatory force observed experimentally (1) must therefore reflect a more profound mechanical disruption of the contractile apparatus. What such disruptions precisely entail is not clear, but one could imagine that a sudden, violent stretch could rupture the actin and myosin filaments that interact via cross-bridges to generate ASM force. Qi et al. (27) have demonstrated partial dissolution and then reassembly of myosin filaments caused by mechanical perturbation of ASM, although the reassembly process may too rapid to explain the protracted force reduction evident in Fig. 1B. Alternatively, a large enough stretch might even break elements of the cytoskeleton that play a key role in transmitting the force generated by cross-bridge cycling from the ASM cell to the tissue in which it is embedded (10, 12, 35). This would decouple some of the internal contractile filaments from the cell as a whole, preventing their shortening from contributing to overall force generation. In any case, it appears that there is a level of mechanical stretch above which the contracted ASM tissue sustains some kind of internal damage, as we previously suggested (1), with a commensurate reduction in its capacity to generate active force.
Fig. 1.

A: previously observed experimental force-time behavior in a strip of airway smooth muscle (ASM) that was stimulated to contract at t = 0 and then subjected to 2-Hz oscillations about the initial length, with the amplitudes indicated in the figure, beginning at t = 120 s and ending at t = 700 s. B: oscillatory ASM force data (2% amplitude) obtained with a 1-s stretch applied at t = 60 s. The stretch amplitudes are indicated in the figure. [Adapted from (1)].
Inspection of Eq. 14 immediately suggests a mechanistically motivated way in which to account for this effect. The factor Nmax in the first term of Eq. 14 specifies the maximum number of possible cross-bridges that can be bound at any point in time and thus defines the maximum force the muscle can generate. The damage to the contractile machinery caused by sudden stretch presumably reduces Nmax. To model this, we define a threshold tissue length, lcrit, above which damage occurs, and assume that the damage incurred is proportional to the amount by which tissue strain, l(t), exceeds lcrit. We also assume that recovery from damage occurs exponentially with time, so the muscle eventually regains its full force-generating capacity as the disrupted contractile components are able to repolymerize. Accordingly, we make Nmax a function of stress and time, thus:
| (15) |
where kbreak is a constant governing the rate at which stress disrupts the contractile machinery, kfix is a constant governing the rate at which the machinery is reassembled, and Nmax,0 is the maximum value of Nmax that is achieved when the machinery is fully assembled.
Numerical simulations.
The model was driven by an imposed oscillatory length change, the time-derivative of which gives v(t). Solving Eqs. 14 and 15 for an arbitrary v(t), however, requires numerical integration. We integrated Eq. 15 using forward Euler integration, but we found that integrating Eq. 14 in this manner required an extremely small step size, δt, to obtain solutions that were independent of δt. We found that convergent solutions could be obtained with a much larger δt using a combination of forward and implicit Euler integration (see appendix b). The initial conditions were P(0) = 0 with an arbitrary initial length of 100%.
We used δt = 0.002 s to obtain the results shown below. We also did not allow P(t) to become negative [whenever this happened during the integration of the above equations we simply set P(t) = 0] because soft ASM tissue is not capable of sustaining a compressive force.
Experimental comparison data.
We evaluate the performance of the model described above by comparing its ability to predict the key features of experimental data collected in a previous study (1). These data sets are provided in Fig. 1.
RESULTS
Figure 2 shows the predictions of P(t) obtained by first activating the muscle at constant length for 120 s, then imposing constant-amplitude 2-Hz oscillations in length of the indicated amplitudes about the original length until 700 s, and then maintain the muscle at its original length for the remaining time. The parameters in Eq. 14 were assigned, on the basis of trial and error, the following values: kon = 0.07 s−1, koff = 0.001 s−1, vmax = 0.4 lref s−1 (lref is the reference length of the ASM measured at baseline), and PmaxNmaxkon − α = 0.2 s−1. The simulated P(t) profiles show transient and steady-state oscillatory behavior that is qualitatively and quantitatively similar, in terms of its key features, to previous experimental observations obtained under corresponding experimental conditions (Fig. 1A). These key features include a transient rise in force upon ASM stimulation that reaches a plateau, a dramatic and rapid reduction in peak force immediately following instigation of length oscillations that achieves a level almost independent of the amplitude of the oscillations, and a transient recovery in force when the oscillations stop.
Fig. 2.
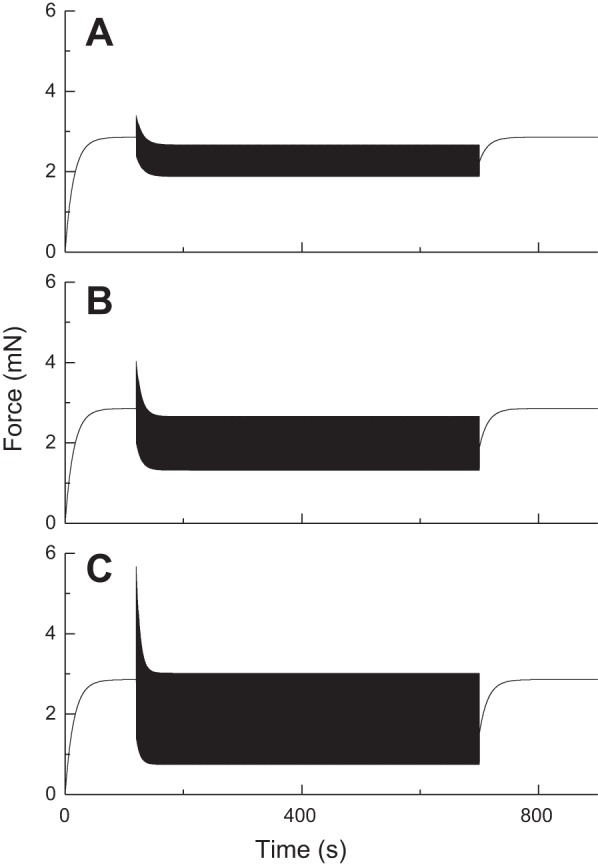
Transient and steady-state force-length loops for activated ASM predicted by Eq. 14. The amplitudes of length oscillation, beginning at 120 s, are 1% lref (A), 2% lref (B), and 4% lref (C). (Compare with experimental data in Fig. 1A.)
Figure 3 shows the predictions of P(t) obtained with Eq. 14 using the same parameters values when the length oscillations (amplitude 2% lref) are interrupted by sudden, brief (1 s duration) stretches of amplitudes 5%, 15%, and 25% lref. The amplitude of P(t) is briefly affected by stretch in each case (although barely so with the smallest amplitude of 5% lref) but recovers quickly to its prestretch level with a time course that resembles that of initial force generation from the relaxed state (see Fig. 2). Furthermore, the nature of this recovery is independent of the amplitude of the sudden stretch, in marked contradiction to the experimental observations reproduced in Fig. 1B. However, when Eq. 14 is combined with Eq. 15 to produce a strain-related decrement in Nmax, the predicted effects of a sudden stretch are much more like the experimental observations, as shown in Fig. 4 (kfix = 0.001 s−1, kbreak = 0.05 s−1, lcrit = 5% lref). That is, the amplitude of P(t) is reduced in a dose-dependent manner after the stretch, and its recovery is markedly retarded.
Fig. 3.
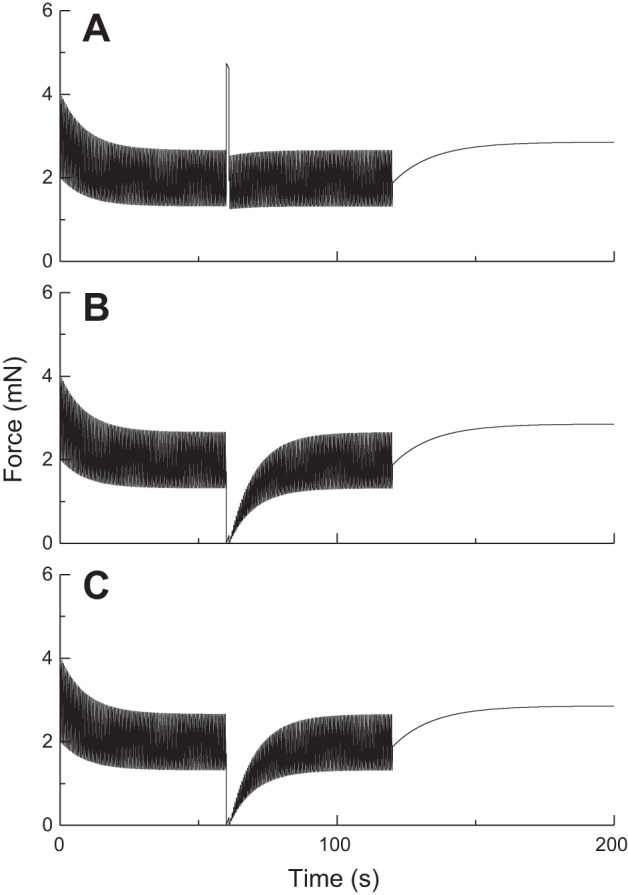
Effect of brief stretches applied at 60 s on the oscillatory force-length behavior of ASM as determined by Eq. 14. The stretch amplitudes are 5% lref (A), 15% lref (B), and 25% lref. (C). (Compare with experimental data in Fig. 1A.)
DISCUSSION
The force-length behavior of strips of activated ASM has been the subject of intense investigation because of its relevance to the modulation of bronchoconstriction by both tidal breathing and deep lung inflation (1, 3, 5, 11, 15, 23, 30, 31). These investigations show that imposed length changes can substantially affect active ASM force, but the mechanisms involved still remain a matter of some debate. Several recent modeling efforts have attempted to place ASM force adaptation on a mechanistic basis (4, 7, 32), all invoking the entirely reasonable idea that some kind of rearrangement of the structural components of the ASM cell is responsible for the response to stretch. These models can become quite complicated in all their details, however, which can be problematic if they are to be uniquely fit to experimental data sets. Also, although these recent models represent notable advances in the field of ASM physiology, their increasing complexity can make it difficult to appreciate the relative roles of all the disparate mechanisms they embody. Our goal in the present study, therefore, was to derive a mechanistically based model of ASM dynamics that recapitulates the essential features of ASM force-length behavior but without becoming burdened by second-order details that, while perhaps affecting the precision of model predictions, do not add to these predictions in an important qualitative sense.
The above considerations caused us to question the widespread use of the Huxley model (16) in combination with the four-state cross-bridge scheme of Hai and Murphy (13) as a description of ASM. Certainly, the Huxley model (Eq. 1) has a venerable history in the annals of muscle physiology, linking structure to function in a very elegant manner that seems entirely appropriate for the ordered sarcomeres of skeletal muscle (16, 19, 34). In particular, Eqs. 1 and 2 are based on the notion that v(t) must be constant along the lengths of every pair of interacting actin and myosin fibers in the muscle, which means that the fibers must all be aligned in the same direction so that each cross-bridge pulls precisely in this direction (Fig. 5A). This is significantly more difficult to justify in the case of ASM, however, which has a somewhat amorphous structure in which the actin and myosin filaments are aligned at a variety of angles (33) (Fig. 5B). There are two approaches to addressing this difficulty. One is to take fiber orientation specifically into account, as in the model of Kroon (20), but this leads to significant additional complexities, particularly as fiber orientation is likely to change as the muscle changes length (21, 32). Therefore, in line with our goals of seeking simplicity where possible, we took the alternative approach of attempting to coarse-grain the microlevel details of the ASM cell to account for the average effects of these details without explicitly taking the details themselves into account.
Fig. 5.

A: schematic of the arrangement of contractile proteins in skeletal muscle showing three bound and three unbound myosin cross-bridges. The extension of each bound cross-bridge past its unstressed position (shown by the vertical dashed lines) is represented by x. X and −X denote the limits of x for the third cross-bridge from the left. B: schematic of a smooth muscle cell illustrating actin and myosin filaments arranged at a variety of angles to the direction of overall muscle shortening.
Achieving our aim while still retaining a valid approximation of reality requires careful consideration of what is of primary importance, and what can be relegated to the level of second-order detail. One of the main justifications for the four-state version of the Huxley model has been its ability to recapitulate the steady-state behavior of activated strips of ASM (4, 23, 24). On the other hand, models of a Hill force-velocity generator in series with a passive elastic element can do just as well, if not better in this regard (3). Nevertheless, it is also clear that the transient force-length behavior of ASM cannot be accounted for without including the kinetics of cross-bridge binding to, and unbinding from, actin (1). Thus there are clearly aspects of the Huxley model that are entirely appropriate for, indeed demanded by, a valid description of ASM dynamics.
Accordingly, we took the view that an alternative model of ASM behavior should bear some definable mathematical relationship to the Huxley model. We found that it is possible to transform the partial differential equation of the Huxley model (Eq. 1) into a simpler, ordinary differential equation (Eq. 14) that still embodies the key notions of force production by cross-bridge cycling as well as the manner in which the velocity of shortening affects the kinetics of cross-bridge binding. This simpler model is also consistent with the classic Hill force-velocity relationship (appendix a) as a result of the assumption of a maximum shortening velocity, vmax, via Eq. 12, following the recent analysis of Seow (28). This model also recapitulates the key features of the transient and steady-state behavior of strips of ASM subjected to small-amplitude sinusoidal oscillations, as evidenced by the comparison of the simulations in Fig. 2 to the experimental data shown in Fig. 1A. In particular, the model produces marked and rapid decreases in peak oscillatory force once oscillations commence, with peak steady-state force being virtually independent of the amplitude of length oscillation. Also, when oscillations stop, the model predicts a transient recovery of active force to its previous isometric level. These features are all reflective of the tendency for cross-bridges to fall off their binding sites when the actin and myosin fibers move relative to each other, and to then reattach when movement stops, just as predicted by the Huxley model (Eq. 1).
What our previous modeling efforts were not able to achieve, however, was a satisfactory recapitulation of the dramatic and slowly recovering degradations in force-generating capacity observed when activated ASM strips are subjected to sufficiently vigorous stretches (1). With the new model (Eq. 14) we are able to achieve this in a mechanistically motivated way simply by including a stretch-based reduction in the number of available cross-bridges specified by the parameter Nmax. This represents the temporary disruption of the contractile apparatus induced by sudden stretch above a specified threshold. The precise mechanisms by which this disruption occurs remain uncertain, but the actin and myosin filaments themselves could well be involved because both are known to polymerize upon muscle activation and to be disrupted by mechanical perturbations (15, 18, 27). The same mechanism could, in principle, be implemented in the classic Huxley model (Eq. 1), although then it would be necessary to specify stretch-induced alterations to nmax as a function of x; in our simpler model we avoid having to decide what this x-dependence might be and simply focus on the total number, Nmax, of cross-bridges in the muscle.
Also, in the interests of simplicity, we assume here that Nmax has an inverse dependence on strain above the damage threshold, lcrit, and that disruption and recovery of the contractile apparatus follow first-order kinetics (Eq. 15). It could well be that damage also depends on the stress generated in the tissue by stretch. Furthermore, although our choice of the parameter kbreak in Eq. 15 gives results (Fig. 4) that agree reasonably well with experimental data (Fig. 1B), the experimental data do not extend out far enough in time beyond the stretch to define the nature of kfix, so the value we use here is merely a guess. Indeed, there is evidence that full recovery of force-generating capacity after a stretch requires repeated cycles of ASM stimulation and relaxation (5). The nature of the recovery phase also raises questions about the possible role of latch-bridges in the generation of force, because these have been posited to progressively take over from conventional cross-bridges as more energetically efficient load bearers in ASM (6). Such considerations are beyond the scope of the present study, but may need to be incorporated into the model if it is to accurately account for force recovery over extended time periods.
The models of Silveira et al. (32), Brook (4), and Donovan (7) also invoke structural remodeling within the ASM cell in response to length change. These models incorporate details of how the remodeling takes place, whereas we have taken the approach here of simply accounting for their net effect by adjusting the value of Nmax. In so doing, we are not passing judgment on the validity of these more complicated models. Obviously they require a finer level of validation than our simpler model, but on the other hand they offer the possibility of greater mechanistic elucidation. By the same token, we believe that our simpler model highlights the principle phenomena more clearly. These various modeling approaches should thus be viewed as complementary rather than competitive.
Finally, it goes without saying that Eqs. 14 and 15 do not constitute a perfect representation of ASM behavior. For example, the simulated force-length loops (not shown) are significantly narrower than those observed experimentally (1, 9, 31), but this discrepancy may be explained by the viscoelastic behavior of the connective tissue present in strips of ASM (1, 3), or by the presence of different populations of cross-bridges (23), or by both of these explanations, neither of which have been accounted for in the present model. Also, the variations in minimum and maximum force during the steady-state portion of the P(t) oscillations in Fig. 2 are not exactly the same as in the experimental observations in Fig. 1A. There are numerous reasons one might invoke to explain a less than perfect match between simulations and data, such as the assumption of a linear dependence of cross-bridge detachment on velocity (Eq. 10); it could easily be that a more complicated relationship pertains in reality. Nevertheless, the general dependencies are the same, showing that Eqs. 14 and 15 together account for many of the key dynamic mechanical features observed in activated strips of ASM. Furthermore, Eq. 15 embodies a mechanistic basis for the stretch-induced reductions in force that have been observed experimentally (1). Accordingly, we conclude that Eqs. 14 and 15 together constitute a simple yet biophysically justifiable model of ASM contraction dynamics that is consistent with ASM structure. In fact, there is no reason why these two equations should be limited to the description of ASM, because the Huxley model has been applied to other forms of smooth muscle (22) for which the same reservations presumably apply.
GRANTS
This study was supported by National Institutes of Health Grants R01 HL-087788 and P20 GM-103532.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author.
AUTHOR CONTRIBUTIONS
J.H.B. conception and design of research; J.H.B. performed experiments; J.H.B. analyzed data; J.H.B. interpreted results of experiments; J.H.B. prepared figures; J.H.B. drafted manuscript; J.H.B. edited and revised manuscript; J.H.B. approved final version of manuscript.
Appendix A: Force-Velocity Relationship
The solution to Eq. 14 depends on the forcing function, v(t), but it is instructive to consider when v(t) is a positive constant, v. In this case the muscle is contracting at a constant rate, in which case |v| = v. Note that we also make the approximation, in the spirit of coarse-graining, that v(t) = dx(t)/dt, even though this is not strictly correct unless the contractile fibers are precisely lined up in the direction of muscle shortening. This mirrors the measurement of the force-velocity characteristics of a strip of activated muscle tissue where, in the laboratory, the muscle is exposed to a fixed load while its steady-state velocity of shortening is measured after allowing for a brief transient (28, 29). Equation 14 thus becomes
| (A-1) |
where
| (A-2) |
The solution to Eq. A-1 is
| (A-3) |
The constant, M, has a value determined by the initial conditions, but the steady-state force, P, is simply the ratio
| (A-4) |
Now, there is always some internal load, Pi, that the muscle must overcome to shorten, even when the external load, Pe, is zero. Because P is the sum of Pi and Pe, we have
| (A-5) |
which can be combined with Eq. A-4 to give an expression having the same functional form as the classic Hill force-velocity relationship (1, 28). That is,
| (A-6) |
where a and b are constants. P0 is the isometric force produced when v = 0, which is found by substituting Eqs. A-2 into A-6 to be
| (A-7) |
Equation A-7 predicts that isometric force increases with the number of cross-bridges that can potentially bind to actin (Nmax) and the elastic force in each bound cross-bridge (Pbr). Equation A-6 also predicts that isometric force decreases with the magnitude of the internal load, Pi, which must be overcome before the muscle can perform useful external work. All of these dependencies make intuitive sense.
Finally, substituting Eq. A-2 into Eq. A-6, setting v = vmax corresponding to Pe = 0, and solving for Pi gives
| (A-8) |
Appendix B
The composite forward/implicit Euler integration scheme is
| (B-1) |
where from Eq. 14 we have
Expressing this equation explicitly in terms of P(t + δt) gives the P(t) updated formula:
| (B-2) |
REFERENCES
- 1.Bates JH, Bullimore SR, Politi AZ, Sneyd J, Anafi RC, Lauzon AM. Transient oscillatory force-length behavior of activated airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 297: L362–L372, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bates JH, Cojocaru A, Lundblad LK. Bronchodilatory effect of deep inspiration on the dynamics of bronchoconstriction in mice. J Appl Physiol 103: 1696–1705, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Bates JH, Lauzon AM. Modeling the oscillation dynamics of activated airway smooth muscle strips. Am J Physiol Lung Cell Mol Physiol 289: L849–L855, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Brook BS. Emergence of airway smooth muscle mechanical behavior through dynamic reorganization of contractile units and force transmission pathways. J Appl Physiol 116: 980–997, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bullimore SR, Siddiqui S, Donovan GM, Martin JG, Sneyd J, Bates JH, Lauzon AM. Could an increase in airway smooth muscle shortening velocity cause airway hyperresponsiveness? Am J Physiol Lung Cell Mol Physiol 300: L121–L131, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dillon PF, Aksoy MO, Driska SP, Murphy RA. Myosin phosphorylation and the cross-bridge cycle in arterial smooth muscle. Science 211: 495–497, 1981. [DOI] [PubMed] [Google Scholar]
- 7.Donovan GM. Modelling airway smooth muscle passive length adaptation via thick filament length distributions. J Theor Biol 333: 102–108, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donovan GM, Bullimore SR, Elvin AJ, Tawhai MH, Bates JH, Lauzon AM, Sneyd J. A continuous-binding cross-linker model for passive airway smooth muscle. Biophys J 99: 3164–3171, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fredberg JJ, Inouye D, Miller B, Nathan M, Jafari S, Raboudi SH, Butler JP, Shore SA. Airway smooth muscle, tidal stretches, and dynamically determined contractile states. Am J Respir Crit Care Med 156: 1752–1759, 1997. [DOI] [PubMed] [Google Scholar]
- 10.Gunst SJ, Meiss RA, Wu MF, Rowe M. Mechanisms for the mechanical plasticity of tracheal smooth muscle. Am J Physiol Cell Physiol 268: C1267–C1276, 1995. [DOI] [PubMed] [Google Scholar]
- 11.Gunst SJ, Wu MF. Selected contribution: plasticity of airway smooth muscle stiffness and extensibility: role of length-adaptive mechanisms. J Appl Physiol 90: 741–749, 2001. [DOI] [PubMed] [Google Scholar]
- 12.Gunst SJ, Zhang W. Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am J Physiol Cell Physiol 295: C576–C587, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hai CM, Murphy RA. Cross-bridge phosphorylation and regulation of latch state in smooth muscle. Am J Physiol Cell Physiol 254: C99–C106, 1988. [DOI] [PubMed] [Google Scholar]
- 14.Hanks BS, Stephens NL. Mechanics and energetics of lengthening of active airway smooth muscle. Am J Physiol Cell Physiol 241: C42–C46, 1981. [DOI] [PubMed] [Google Scholar]
- 15.Herrera AM, Martinez EC, Seow CY. Electron microscopic study of actin polymerization in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 286: L1161–L1168, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Huxley AF. Muscle structure and theories of contraction. Progr Biophys Biophys Chem 7: 255–318, 1957. [PubMed] [Google Scholar]
- 17.Huxley HE. Memories of early work on muscle contraction and regulation in the 1950’s and 1960’s. Biochem Biophys Res Commun 369: 34–42, 2008. [DOI] [PubMed] [Google Scholar]
- 18.Ijpma G, Al-Jumaily AM, Cairns SP, Sieck GC. Myosin filament polymerization and depolymerization in a model of partial length adaptation in airway smooth muscle. J Appl Physiol 111: 734–742, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keener J, Sneyd J. Mathematical Physiology. New York: Springer, 1998. [Google Scholar]
- 20.Kroon M. Influence of dispersion in myosin filament orientation and anisotropic filament contractions in smooth muscle. J Theor Biol 272: 72–82, 2011. [DOI] [PubMed] [Google Scholar]
- 21.Kuo KH, Herrera AM, Seow CY. Ultrastructure of airway smooth muscle. Respir Physiol Neurobiol 137: 197–208, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Laforet J, Guiraud D, Andreu D, Taillades H, Coste CA. Smooth muscle modeling and experimental identification: application to bladder isometric contraction. J Neural Eng 8: 036024, 2011. [DOI] [PubMed] [Google Scholar]
- 23.Mijailovich SM, Butler JP, Fredberg JJ. Perturbed equilibria of myosin binding in airway smooth muscle: bond-length distributions, mechanics, and ATP metabolism. Biophys J 79: 2667–2681, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mijailovich SM, Fredberg JJ, Butler JP. On the theory of muscle contraction: filament extensibility and the development of isometric force and stiffness. Biophys J 71: 1475–1484, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piazzesi G, Reconditi M, Linari M, Lucii L, Bianco P, Brunello E, Decostre V, Stewart A, Gore DB, Irving TC, Irving M, Lombardi V. Skeletal muscle performance determined by modulation of number of myosin motors rather than motor force or stroke size. Cell 131: 784–795, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Politi AZ, Donovan GM, Tawhai MH, Sanderson MJ, Lauzon AM, Bates JH, Sneyd J. A multiscale, spatially distributed model of asthmatic airway hyper-responsiveness. J Theor Biol 266: 614–624, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qi D, Mitchell RW, Burdyga T, Ford LE, Kuo KH, Seow CY. Myosin light chain phosphorylation facilitates in vivo myosin filament reassembly after mechanical perturbation. Am J Physiol Cell Physiol 282: C1298–C1305, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Seow CY. Hill's equation of muscle performance and its hidden insight on molecular mechanisms. J Gen Physiol 142: 561–573, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seow CY, Ford LE. Contribution of damped passive recoil to the measured shortening velocity of skinned rabbit and sheep muscle fibres. J Muscle Res Cell Motil 13: 295–307, 1992. [DOI] [PubMed] [Google Scholar]
- 30.Shen X, Gunst SJ, Tepper RS. Effect of tidal volume and frequency on airway responsiveness in mechanically ventilated rabbits. J Appl Physiol 83: 1202–1208, 1997. [DOI] [PubMed] [Google Scholar]
- 31.Shen X, Wu MF, Tepper RS, Gunst SJ. Mechanisms for the mechanical response of airway smooth muscle to length oscillation. J Appl Physiol 83: 731–738, 1997. [DOI] [PubMed] [Google Scholar]
- 32.Silveira PS, Butler JP, Fredberg JJ. Length adaptation of airway smooth muscle: a stochastic model of cytoskeletal dynamics. J Appl Physiol 99: 2087–2098, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Walmsley JG, Murphy RA. Force-length dependence of arterial lamellar, smooth muscle, and myofilament orientations. Am J Physiol Heart Circ Physiol 253: H1141–H1147, 1987. [DOI] [PubMed] [Google Scholar]
- 34.Williams WO. Huxley's model of muscle contraction with compliance. J Elast 105: 365–380, 2011. [Google Scholar]
- 35.Zhang W, Gunst SJ. Interactions of airway smooth muscle cells with their tissue matrix: implications for contraction. Proc Am Thorac Soc 5: 32–39, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]