

Abstract
When exposed to a hypoxic environment, the body's first response is a reflex increase in ventilation, termed the hypoxic ventilatory response (HVR). With chronic sustained hypoxia (CSH), such as during acclimatization to high altitude, an additional time-dependent increase in ventilation occurs, which increases the HVR and is termed ventilatory acclimatization to hypoxia (VAH). This secondary increase persists after exposure to CSH and involves plasticity within the circuits in the central nervous system that control breathing. The mechanisms of HVR plasticity are currently poorly understood. We hypothesized that changes in neuronal nitric oxide synthase (nNOS) activity or expression in the nucleus tractus solitarius contribute to this plasticity and underlie VAH in rats. To test this, we treated rats held in normoxia or 10% O2 (CSH, PiO2 = 70 Torr) for 7–9 days and measured ventilation in conscious, unrestrained animals before and after microinjecting the general NOS antagonist L-NG-Nitroarginine methyl ester into the nucleus tractus solitarius (NTS) or systemically injecting the nNOS-specific antagonist S-methyl-l-thiocitrulline. Localization of injection sites in the NTS was confirmed by histology following the experiment. We found that 1) neither NTS-specific nor systemic nNOS antagonism had any effect on hypoxia-mediated changes in breathing or metabolism (P > 0.05), but 2) nNOS protein expression was increased in the middle and caudal NTS by CSH. A persistent HVR after nNOS blockade in the NTS contrasts with results in awake mice, and our findings do not support the hypotheses that nNOS in the NTS contribute to the HVR or VAH in awake rats.
Keywords: synaptic plasticity, hypoxic ventilatory response, plethysmography, chronic sustained hypoxia
the hypoxic ventilatory response (HVR) is a reflex increase in ventilation that occurs in response to decreased arterial oxygen tension (PaO2). With chronic sustained hypoxia (CSH) of days to months (e.g., during acclimatization to altitude), additional time-dependent increases in ventilation occur that further improve PaO2 (44). This secondary increase is termed ventilatory acclimatization to hypoxia (VAH) and persists after the removal of hypoxic stimulation, indicating the occurrence of plasticity within the ventilatory control circuits (1, 22). Two mechanisms have been identified that contribute to VAH: 1) the sensitivity of carotid body arterial chemoreceptors to O2 increases and 2) the central nervous system (CNS) responsiveness to afferent inputs from carotid bodies also increases (3, 13, 29, 51). Considerable progress has been made toward elucidating mechanisms of enhanced carotid body sensitivity to O2 during CSH, but mechanisms that mediate increased CNS gain of the HVR are not well understood.
In the CNS, carotid body afferents terminate within the nucleus of the solitary tract (NTS) (31). There is considerable evidence implicating glutamate as the main excitatory neurotransmitter at this synapse. For example, during the onset of hypoxia, glutamate is released into the NTS coincident with the HVR, glutamate injection into the NTS mimics the HVR, and both the HVR and glutamate release are abrogated by carotid body denervation (34, 47). Furthermore, simultaneous antagonism of glutamate receptors within the NTS reduces ventilatory and cardiac responses to chemical or CO2-mediated carotid body stimulation in anesthetized and awake rats (5, 18, 49, 54). As the primary communication link between the carotid bodies and the NTS, activity-dependent alterations of this glutamatergic synapse is a likely candidate mechanism to underlie the CNS component of ventilatory plasticity to chronic hypoxia.
Long-term modification of glutamatergic synapses typically occurs via the activation of a canonical synaptic plasticity pathway, wherein 1) presynaptic glutamate release activates postsynaptic glutamate receptors, 2) Ca2+ influx through glutamatergic N-methyl-D-aspartate receptors (NMDARs) leads to dephosphorylation of neuronal nitric oxide synthases (nNOS), 3) nitric oxide (NO) production increases, and 4) retrograde diffusion of NO back across the synapse induces further glutamate release from the presynaptic neuron. Additionally, increased neuronal [Ca2+] induces the translation of glutamate receptors and nNOS proteins in the postsynaptic cell, further sensitizing the system to presynaptic inputs (6, 25, 28, 37).
There is some evidence in support of a role for this glutamate-NO synaptic plasticity mechanism in the HVR; for example, NOS have been identified in the NTS of rats and cats (19), and NO donors injected into the NTS of rats increase spontaneous discharge rates of neurons and also ventilation (38, 48). There is also some experimental support for a role for NOS in mediating VAH. For example, nNOS and NMDAR mRNA and protein expression increase following 2 wk of CSH in murine medulla (15), and systemic nNOS blockade reduces VAH in CSH acclimated mice (15) but increases ventilation in hypoxia-adapted plateau pika (42). Unfortunately, the impact of studies examining the role of NOS specifically in VAH is limited because these studies employed systemic methods of drug delivery instead of specific examinations targeted to the NTS. Indeed, NO has opposing effects at the carotid body (inhibitory) vs. the NTS (excitatory), which makes these systemic manipulations difficult to interpret (8, 25, 38). Determining the role of nNOS specifically in the NTS during chronic hypoxia should help unravel synaptic mechanisms of plasticity in the CNS that underlie VAH and improve our understanding of systemic physiological effects of chronic hypoxemia. The goal of the present paper was to determine if synaptic plasticity mediated by nNOS in the NTS contributes to the CNS component of VAH.
MATERIALS AND METHODOLOGY
Experimental animals and surgery.
All surgical procedures and protocols were performed in accordance with the relevant guidelines of the University of California San Diego Institutional Animal Care and Use Committee. Male Sprague-Dawley rats (Charles River) weighing 200–250 g were housed under a 12:12 h light-dark cycle and fed a standard diet ad libitum.
Animals were divided into two groups treated with 1) systemic intraperitonial injection of the specific nNOS antagonist S-methyl-l-thiocitrulline (SMTC) (10 mg/kg; n = 18) or 2) NTS-specific bilateral microinjection of 50 nl of the general NOS antagonist L-NG-Nitroarginine methyl ester (L-NAME) (500 mM; n = 20). Within these treatment groups, animals were further divided into two experimental groups: 1) normoxic sea level controls (CON, n = 9 for SMTC, 10 for L-NAME) and 2) CSH rats (n = 9 for SMTC, 10 for L-NAME). In this fashion we examined separate groups of animals treated with CON or CSH, and all animals within each of these groups were tested for responses to acute hypoxia and the effect of a single drug. The CON rats were kept in normoxia for 7 days, whereas the CSH rats were acclimatized to a simulated altitude of 5,500 m in a hypobaric chamber at 380 Torr (PiO2= 70 Torr, equivalent to ≈10% O2 at sea level barometric pressure) for 7 days. For CSH animals, the chamber was returned to sea level for 15 min every 3 to 4 days for general cage maintenance, or when it was necessary to remove animals for experimentation. At least 2 days prior to acclimatization in chronic sustained normoxia or hypoxia all animals in the NTS-specific microinjection group underwent surgery for implantation of guide cannulae and body temperature telemetry probes. At the same time point all animals in the systemic injection group underwent surgery for implantation of body temperature telemetry probes only.
All surgeries were performed under isoflurane anesthesia (initially 5% isoflurane in 100% O2 and maintained at 2 to 3% isoflurane). Stereotaxic surgery (Kopf Instruments, Tujunga, CA) was used to implant a stainless steel guide cannula (Plastics One, Roanoke, VA) bilaterally into the NTS [coordinates: AP −0.3 mm (from obex), ML 0.7 mm, DV 0.5 mm] to deliver pharmacological agents. Two holes were drilled into the cranium into which screws would fit firmly, and the guide cannula was secured to the skull with acrylic resin that fixed the guide cannula to these screws. The microinjection needle was 1 mm longer than the guide cannula and projected into the NTS.
A telemetry thermometer probe (Emitter, Respironics, Bend, OR) was implanted to monitor body temperature. The body temperature is required for an accurate calculation of the tidal volume (Vt) (see below). The emitter was implanted into the abdominal cavity and sutured in place to the interior wall of the abdomen. Postsurgery recovery was assessed in rats 2 days after surgery. Animals were considered to have recovered from surgery if they were behaving normally (i.e., rats were grooming, eating, sleeping, and exploring normally, displayed normal appetite, and displayed no signs of infection, pain, or stress).
Plethysmography.
Inspired ventilation (V̇I) was measured by barometric pressure method of plethysmography modified for continuous flow (23). On the day of experimentation, individual animals were sealed into a 7-liter Plexiglas chamber. An electronic gas mixer (MFC-4, Sable Systems, Las Vegas, NV) was used to supply the animal with an inflowing gas mixture (3 liters/min) of controlled O2 and CO2 (balance N2). Inflowing gas entered the chamber through a tube (7 cm long and 1 cm in diameter) that was filled with smaller PE-50 tubing of similar length to create a high-impedance input and reduce the loss of pressure signals. Gas exited the plethysmograph chamber through a high-impedance valve to a vacuum pump (Dayton Electric, Chicago, IL). Pressure inside the box was referenced to atmospheric pressure with a water manometer. Atmospheric pressure corrected for standard gravity and room temperature was measured on each experimental day. To ensure a controlled gas mixture in the chamber, the pressure inside the chamber was positive (<0.5 cmH20) with a small leak from the chamber (20-ga needle) that also prevented slow changes in baseline with temperature fluctuations. Chamber gas concentrations were measured with a mass spectrometer (Perkin-Elmer 1100 Medical Gas Analyzer, Pomona, CA) that was calibrated for O2 and CO2 on each experimental day. A chamber temperature probe (Thermalert TH-5, Physitemp, Clifton, NJ) was sealed inside the box and a humidity probe was placed into the box through a hole that was cut and sealed for this purpose. Inspiration produces humidity-related changes in pressure that can be monitored with a differential pressure transducer (DP45, Validyne, Northridge, CA) referenced to atmosphere. Output from the transducer was recorded on a digital data acquisition system (Labdat, see below). Respiratory frequency (fr) was calculated directly from the ventilation-induced pressure swings. Tidal Volume (Vt) was calculated from the ventilation-induced pressure changes by an equation from Drorbaugh and Fenn (1955) and modified for flow-through plethysmography by Jacky (12, 23). Prior to each experiment, calibration pulses (0.2, 0.5, and 1.0 ml) were injected into the chamber at a rate similar to the rats' inspiratory time. The amplitude of calibration pulses varied <10% for injections lasting between 100 and 350 ms (corresponding to inspiratory times).
Ventilation was determined under poikilocapnic conditions. The animals were given at least 40 min to habituate to the plethysmograph at their chronic inspired O2 levels before study (i.e., CSH animals were kept in 10% O2 while adjusting to the chamber; CON animals were kept in 21%). Two inspired O2 levels (FIO2 = 10 and 21%) were used in the study. Following acclimatization, baseline V̇i measurements were obtained for 5–10 min, and then animals were injected with the sham treatment and then exposed to the opposite gas mixture for 30 min (i.e., CSH animals were exposed to room air; CON animals were exposed to hypoxia). Animals were then returned to their control gas mixture for 15 min and then given a hypercapnic challenge of 7% CO2 for 5 min. Animals then received the drug treatment and were reexamined as before. V̇i was measured for 30 min after the rats were exposed to an acute inspired gas concentration or drug injections. Results were analyzed at 1, 2, 5, 10, 15, and 30 min following a treatment change; however, no differences were found between data at these time points, and therefore V̇i is reported as the average value obtained between 10–15 min after the rats were exposed to an acute inspired gas concentration or drug injection unless otherwise noted. This time point was chosen as the representative time point as it is consistent with previous related studies performed in our laboratory and others, and thus allows for accurate comparison between studies.
Pharmacology and microinjections.
L-NAME and SMTC were prepared in artificial cerebrospinal fluid (ACSF) (in mM: 115 NaCl, 2.0 KCl, 2.2 KH2PO4, 25 NaHCO3, 10 D-glucose, 1.2 MgSO4, and 2.5 CaCl2, adjusted to pH 7.4 with HCl) (52). The ACSF was microinjected as a sham in all experimental groups studied. All drugs were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated.
A microinjection needle made to fit the guide cannula was connected to a 500-nl Hamilton microsyringe through a polyethylene tube. The polyethylene tube ran through the lid of the plethysmograph and was sealed. After control ventilatory measurements were made at 10 and 21% O2, 50 nl of L-NAME (100 nM) was injected into the NTS. The pharmacological doses of L-NAME used in the present study are sufficient to inhibit physiological responses to NTS-mediated stimulation throughout the time frame of our experiments in awake rats, in accordance with studies conducted in similar preparations from other laboratories (9–11). The concentration of SMTC used in our study is consistent with that used in previous studies examining the role of nNOS in ventilation that employed systemic SMTC administration in rodents and lagomorphs (15, 16, 42). Following drug administration, ventilatory measurements were again collected in each gas mixture. Ventilatory effects of the drugs were determined by comparing the data obtained following the control (ACSF injection) measurements with those collected following the ACSF + drug injection. Animals were permitted at least 1 h to rest in their home cages between the completion of sampling following the first injection and beginning the second experimental trial. Animals received ACSF injections before drug injections, and experimental protocols were not randomized because of long drug wash off times. In some experiments, animals received a second injection of ACSF in place of cocktail microinjection to assess the impact of the experimental time course. No differences were observed between the effects of ACSF in multiple trials in the same animal (data not shown).
Localization of microinjections.
Evan's blue microinjections were used to localize the microinjection sites (Fig. 1). At the end of the experiment, Evan's blue (50 nl) was microinjected through the guide cannulae into the NTS at the same site as the drug delivery. The animals were anaesthetized with an overdose of sodium pentobarbital and transcardially perfused with ice-cold ACSF followed by 4% paraformaldehyde. The brainstem was removed and postfixed in 2% paraformaldehyde for 1 day and then stored in sucrose (30%). The brainstems were frozen in isopentane (−140°C) and sectioned (slice thickness = 30–50 μm) on a cryostat (Cryocut 1800, Leica Biosystems, Wetzlar, Germany). Fig. 1A shows that the spread of a typical 50-nl microinjection is ∼250 μm, although it is not known exactly how the pharmacological effects of nNOS antagonists spread compares with a histological marker. On a day before they were studied with the full protocol, rats received test glutamate microinjections (data not shown). For data analysis, we only used animals that had a positive response to glutamate and in which the microinjection site was located within 500 μm of the NTS target. We have shown previously that no ventilatory responses are observed if the histological data shows a microinjection that is more than 500 μm away from the commissural caudal NTS target site, but similar responses are obtained if microinjections are localized within 500 μm of the target (39).
Fig. 1.
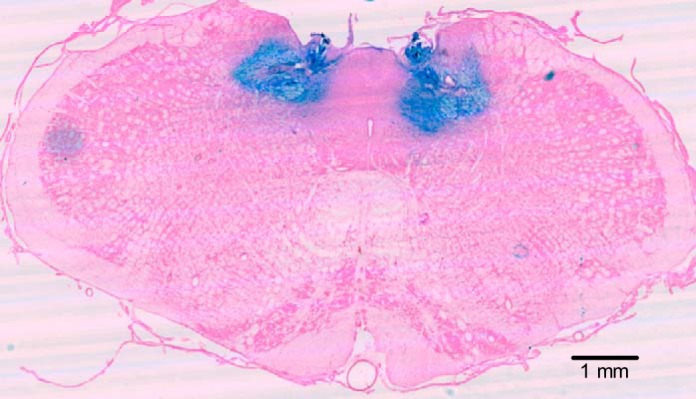
Location of nucleus tractus solitarius (NTS) microinjections. Representative image of a coronal section of the brainstem of a rat showing the sites of successful microinjection with Evan's blue in the caudal region of the NTS (−14.0 bregma; −4.8 interautal). Magnification to ×6.3.
Protein extraction and Western blots.
Rats exposed to 7 days of room air or CSH (n = 3 each) were euthanized with a pentobarbital overdose. The skull was rapidly opened and the brainstem extracted and immediately placed in liquid nitrogen. The NTS was extracted under liquid nitrogen to prevent protein denaturation. Briefly, the obex was visually identified and a 1-mm-thick coronal section was obtained 1.5 mm caudal to 1.5 mm rostral to the obex. The dorsal half of this brainstem section was carefully removed under liquid nitrogen, weighed, and stored at −80°C until analysis. To extract proteins, tissues were homogenized on ice with a tissue blender in RIPA buffer (Cell Signaling, Boston, MA) supplemented with Complete protease inhibitor cocktail (Roche Applied Sciences) and Halt phosphatases inhibitor cocktail (Thermo Sci, Rockford, IL). The resulting homogenate was centrifuged for 10 min at 10,000 g at 4°C to remove cell debris. Supernatants were taken as whole cell lysates and protein concentration was measured with a bicinchoninic acid kit, according to the manufacturer's instructions (Sigma).
For Western blot analysis, equal amounts of protein (75 μg/well) were separated at 80 V for 3 h on 4–12% Bolt Mini Bis-Tris Plus gels (Invitrogen) and transferred onto a PVDF membrane (Millipore, Billerica, MD). Western blots were performed with antibodies against nNOS (1:1,000; Millipore) and GAPDH (1:1,000; Millipore). Membranes were incubated in primary antibodies overnight at 4°C, rinsed 3 × 15 min in TBS + 0.1% Tween20 and then blocked in Odyssey blocking buffer (LI-COR, Lincoln, NE). Specific bands were visualized after 2 h incubation with the respective fluorescent secondary antibodies (1:15,000) with an Odyssey fluorescent scanner (LI-COR). Densitometry of Western blots from each experimental group were obtained using ImageJ (National Institutes of Health, Bethesda, MD), and absolute values were normalized to GAPDH expression on the same blot. Results were analyzed in arbitrary units, comparing each value with that obtained from the paired GAPDH measurement on each blot, and results are expressed as fold change relative to normoxic controls run simultaneously. Gels were repeated 3 times.
Immunohistochemistry.
Rats exposed to 7 days of room air or CSH (n = 6 each) were euthanized with a pentobarbital overdose and then perfusion fixed with 4% paraformaldehyde in PBS. Following fixation, the skull was rapidly opened and the brainstem extracted and immediately placed in 4% paraformaldehyde in PBS and fixed overnight at 4°C. Brainstems were then cryoprotected in 30% sucrose for 48 h at 4°C and then embedded in OCT on a piece of cork. The NTS was identified as described above, and six frozen 20-μm sections were cut with a microtome. These six sections included bilateral sections from the left and right hemisphere of the brainstem that encompassed the caudal (−14.1 mm from bregma), middle (−13.8 mm from bregma), and rostral (−13.3 mm from bregma) regions of the NTS. Sections were then incubated in an nNOS antibody (BD Biosciences, San Jose, CA) at 1:2,000 overnight at 4°C. Following incubation, samples were counter-stained with DAPI and fixed. Samples were imaged with a ×40 objective and a Keyence fluorescent microscope with 405 (DAPI) and 488 nm (FITC) fluorescence cubes (Keyence, Mississauga, ON, Canada). For data collection, the parameters of the microscope such as light intensity, exposure time, camera gain, etc., were determined for the brightest fluorescing sample and standardized for subsequent samples. Six images were taken from each rat, including two each from the caudal, middle and rostral NTS. ImageJ image analysis software was used to determine 1) the percentage of nNOS-positive cells relative to the total number of cells and 2) the average fluorescence intensity of nNOS-positive cells in each sample.
Data collection and analysis.
Ventilatory data from the plethysmograph was digitized with an analog-digital converter (National Instruments E-Series 16 channel with 16-bit multiplexing) and custom software (National Instruments, LabVIEW SignalExpress). Digitized data was analyzed with custom software (UCSD Division of Physiology Plethysmography Toolbox running on The Mathworks MATLAB). The pressure signal was analyzed for amplitude [with corrections for changes in the baseline during a breath to calculate Vt as described above], timing to calculate fr, and the duration of inspiration and expiration (Ti and Te, respectively). At least 10 sets of breaths comprising at least 10 consecutive breaths were selected for analysis from the final 5 min of each experimental treatment epoch and were analyzed for each period. Breaths were chosen from periods where the animal was awake but not active (i.e., not actively grooming or exploring but also not asleep, as determined by visual examination).
Statistical analysis was performed with commercial software (SPSS 15.0, SPSS, Chicago, IL). For all experiments, individual n values correspond to a single animal treated with CON or CSH acclimatization protocols and then treated with ACSF and then NOS antagonists as described above, before and after exposure to 21% O2 and then either acute hypoxia (10% O2) or acute hypercapnia (7% CO2). Values are presented as means ± SD. P < 0.05 was considered to achieve statistical significance. All data was normally distributed with equal variance (P > 0.05). For drug microinjection data, a 3-way repeated measures ANOVA was used to determine if there was a statistically significant difference between the three independent factors considered: 1) the acute ventilatory response (FiO2 of 0.10 vs. 0.21), 2) the chronic oxygen condition (CON vs. CSH), and 3) the treatment [control/sham (ACSF) vs. drug microinjection]. If there was a significant 3-way interaction, a 2-way ANOVA was applied to the CON and CSH groups independently to test for significant interactions between FiO2 and drug, along with Bonferonni post hoc tests to determine significance between the independent variables or the change in the HVRs (Δ's). Bonferonni post hoc multiple comparisons tests were also performed on each of the dependent variables to compare the single point means of interest. The dependent variables analyzed were V̇i, fr, Vt, and hypoxic and hypercapnic Δ's for V̇i, fr, and Vt, and metabolic rate.
Unpaired two-tailed t-tests were used to compare changes in the expression of nNOS protein between CON and CSH groups in Western blot and immunohistochemistry (IHC) experiments. For Western blot experiments, nNOS expression was normalized to the expression of the housekeeping protein GAPDH to account for loading differences between samples.
RESULTS
NTS-specific NOS antagonism did not effect changes in ventilation mediated by acute or chronic hypoxia.
Animals received bilateral ACSF microinjections into the NTS as a sham control. ACSF microinjections had no effect on any ventilatory parameter examined in either CON or CSH rats relative to preinjection controls (data not shown), consistent with our previous study (39); therefore, ACSF injections are presented as the control condition for all experiments to isolate the effect of drugs on ventilation. In sham-treated animals, acute and chronic hypoxia had the same effects on ventilation in all treatment groups (Fig. 2). In control experiments (ACSF microinjection), baseline normoxic ventilation in CSH-acclimatized animals was 88% greater than in CON animals (773 vs. 410 ml·min−1·kg−1; n = 10 for each; P < 0.05; Fig. 2A). This difference was due to a combination of significantly greater fr (122 vs. 102 breaths·min−1) and Vt (6.3 vs. 4.1 ml·min−1) in CSH vs. CON rats, respectively (Fig. 2, B and C). Exposure to acute hypoxia (10% O2) increased V̇i in CON and CSH rats to 817 and 1,085 ml·min−1·kg−1, respectively (Fig. 2A). These increases were due to significant increases in both fr (to 145 and 151 breaths·min−1; Fig. 2B) and Vt (to 5.7 and 7.1 ml·min−1; Fig. 2C) in CON and CSH groups, respectively. There was no significant difference between the HVRs of V̇i, fr, or Vt between CON and CSH groups (Fig. 2, A–C). The effects of acute inspired [O2] and CSH acclimatization (i.e., the HVR and VAH) on V̇i, fr, and Vt were within the normal physiological range for rats reported previously by our laboratory and others (1, 21, 39, 40, 43, 45, 46, 50, 51).
Fig. 2.

Effects of artificial cerebrospinal fluid (ACSF) or L-NG-Nitroarginine methyl ester (L-NAME) NTS microinjections on ventilation in normoxic control (CON) and chronic sustained hypoxia (CSH) rats. Effects of acute hypoxia on total minute ventilation (V̇i) (A), breathing frequency (fr) (B), and tidal volume (Vt) (C) in CON and CSH rats before and after bilateral microinjection of 50 nl ACSF or L-NAME (100 nM). Effects of acute hypercapnia on V̇i (D), fr (E), and Vt (F) in CON and CSH rats before and after bilateral microinjection of 50 nl ACSF or L-NAME. Data are means ± SD from n = 10 animals per group. Asterisks (*) indicate significant differences between 10 and 21% O2 values (A–C) and 0% and 7% CO2 values (D–F) (P < 0.05).
To determine the role of NOS specifically in the rat NTS on ventilatory responses to acute and chronic hypoxia, we next microinjected the general NOS antagonist L-NAME into the NTS of CON and CSH rats and reexamined ventilatory responses to normoxia and acute hypoxia. L-NAME did not affect any ventilatory parameter in either of the CON or CSH treatment groups. Following L-NAME microinjection, V̇i in CSH rats was significantly greater than in CON rats post-L-NAME (744 vs. 495 ml·min−1·kg−1; n = 10 for each; Fig. 2A), and this change was not different from pre-L-NAME controls. Also similar to pre-L-NAME controls, the difference in V̇i between CON and CSH rats breathing normoxic gas mixtures was due to significant and similar increases in both fr and Vt in CSH rats relative to CON rats (Fig. 2, B and C). Exposure to acute hypoxia further increased V̇i in both groups to 1,014 and 819 ml·min−1·kg−1 in CSH and CON rats, respectively (Fig. 2A), and these increases were not different from changes observed in pre-L-NAME controls in both groups. Changes in V̇i in response to both acute and chronic hypoxia post-L-NAME injection were due to significant increases in both fr and Vt in CON rats and in fr in CSH rats, and these effects were nearly identical in magnitude to predrug control measurements (Figs. 2, B and C). L-NAME injection had no significant effect on the HVRs of V̇i or fr between CON and CSH groups (Fig. 2, A–C). Conversely, L-NAME injection tended to reduce the increase in Vt mediated by acute hypoxia in CSH rats. Specifically, although the absolute value of Vt was not statistically different between L-NAME and sham-treated CON rats, the slope of the HVR was reduced for Vt in L-NAME treated CSH rats relative to sham-treated CSH rats (Fig. 2C). A power analysis of the 10% O2 Vt data points indicated that >50 animals would be required to determine if there was a true statistical difference between sham- and drug-treated CSH rats. This contrasts with previous studies of various cellular signaling components that regulate the HVR and VAH, which have revealed far more robust effects of pharmacological interventions with 6–10 animals [e.g., targeting glutamatergic or dopaminergic pathways (21, 39)]. Therefore, we interpret any small effects of NOS antagonism on Vt in CSH animals breathing acute hypoxia to be physiologically minor and highly variable relative to other factors determining plasticity in the HVR and VAH.
Animals were also exposed to hypercapnia (7% CO2) as a control experiment to compare the HVR with the hypercapnic ventilatory response (HCVR). In sham-treated (ACSF injection) animals, the HCVR increased V̇i ∼2.5-fold in both CON (to 1,055 ml·min−1·kg−1) and CSH groups (to 1,787 ml·min−1·kg−1) and these increases were markedly greater than those induced by hypoxia (n = 10 for each; Fig. 2D). Elevated V̇i during hypercapnia was the product of significant increases in both fr and Vt in both groups (Fig. 2, E and F). Bilateral microinjections of L-NAME into the NTS had no effect on the HCVR in either group (Fig. 2, D–F).
Systemic nNOS antagonism did not effect changes in ventilation mediated by acute or chronic hypoxia.
Systemic nNOS antagonism attenuates VAH in mice (15); therefore, to determine whether nNOS located outside of the NTS plays a role in the HVR or VAH in rats, we next systemically injected the specific nNOS antagonist SMTC into a second group of rats before and after examining ventilation as above. Systemic ACSF injection had no effect on any ventilatory parameter examined relative to untreated preinjection controls (data not shown); therefore, we present ACSF injection data as the control condition to account for the effect of injection-related stress on breathing and examine only the effect of systemic nNOS antagonism on ventilation.
As in the NTS microinjection experiments, acute and chronic exposure to hypoxia had similar effects on ventilation in both CON and CSH rats following systemic ACSF injection (Fig. 3). Normoxic ventilation in CSH animals was ∼55% greater than in CON animals (802 vs. 522 ml·min−1·kg−1; n = 9 for each; Fig. 3A), and acute hypoxia significantly increased V̇i in CON (to 949 ml·min−1·kg−1) and CSH rats (to 980 ml·min−1·kg−1). In CON rats this increase was due to a significant increase in fr (from 108 to 183 breaths·min−1), whereas in CSH rats fr tended to increase, but this change did not reach significance (Fig. 3B). Vt tended to increase with acute hypoxia in both CON and CSH rats, but these changes did not reach significance in either group (Fig. 3C). There was no significant difference between the HVRs of V̇i, fr, or Vt between CON and CSH groups, although the V̇i HVR in CSH rats tended to be smaller than in CON rats because of a lesser fr HVR (Fig. 3, A–C).
Fig. 3.

Effects of systemic ACSF or S-methyl-l-thiocitrulline (SMTC) injections on ventilation in CON and CSH rats. Effects of acute hypoxia on V̇i (A), fr (B), and Vt (C) in CON and CSH rats before and after bilateral microinjection of 50 nl ACSF or 10 mg/kg SMTC. Effects of acute hypercapnia on V̇i (D), fr (E), and Vt (F) in CON and CSH rats before and after bilateral microinjection of 50 nl ACSF or SMTC. Data are means ± SD from n = 9 animals per group. Asterisks (*) indicate significant differences between 10% and 21% O2 values (A–C) and 0% and 7% CO2 values (D–F) (P < 0.05).
Systemic SMTC injection did not impact any ventilatory parameter in either the CON or CSH treatment groups. Following SMTC injection, baseline V̇i in CSH rats breathing acute normoxic gas mixtures was significantly greater than in CON rats (690 vs. 542 ml·min−1·kg−1; n = 9 for each; Fig. 3A), and this change was not different from pre-SMTC normoxic controls. The change in V̇i between CON and CSH rats was mediated primarily by a significant increase in fr in CSH rats relative to CON rats (Fig. 3, B and C). Baseline fr in SMTC-treated CSH rats breathing normoxia tended to be higher than in the same rats breathing normoxia preinjection; however, this difference did not reach significance. Exposure to acute hypoxia further increased V̇i in both groups to 953 and 983 ml·min−1·kg−1 in CSH and CON rats (Fig. 3A), respectively, and these increases were not different from changes observed in pre-SMTC controls in both groups. Changes in V̇i in response to both acute and chronic hypoxia following SMTC injections were due to significant increases in fr in both CON and CSH rats (Fig. 3, B and C). SMTC injection had no effect on Vt in normoxia or acute hypoxia in either group (Fig. 3C), or on the HVRs of V̇i, fr, or Vt between CON and CSH groups (Fig. 3, A–C). Lastly, systemic SMTC injection had no effect on ventilation or the HCVR in response to 0 or 7% CO2 in either group relative to control measurements made prior to injection (Fig. 3, D–F).
NOS antagonists do not affect metabolic rate.
Resting metabolic rate in chronic normoxic rats breathing normoxic gas mixtures were ∼60 ml O2·min−1·kg−1 in both drug treatment groups preinjection (Fig. 4; n = 10 rats for L-NAME, 9 rats for SMTC). CSH-acclimatized rats tended to have a slightly higher metabolic rate of ∼70 ml O2·min−1·kg−1 in both groups, but this difference was not significant relative to CON rats breathing normoxic gas. In both chronic normoxic and CSH-acclimatized rats, acute hypoxia induced a significant 35–45% reduction in metabolic rate. Systemic SMTC injection or NTS-specific L-NAME microinjection had no effect on metabolic rate in any group breathing either normoxic or hypoxic gas mixtures.
Fig. 4.

Metabolism is not changed by NTS or systemic neuronal nitric oxide synthase (nNOS) antagonism. Average metabolic rate in rats before (white bars) and after (black bars) drug injection. L-NAME was microinjected bilaterally into the NTS (A), and SMTC was injected systemically (B). Data are means ± SD from n = 10 rats per L-NAME group and 9 rats per SMTC group. Asterisks (*) indicate significant differences between 10 and 21% O2 values (P < 0.05).
Neuronal NOS expression increases in the middle and caudal NTS following CSH acclimatization.
Whole NTS changes in nNOS protein expression were analyzed with Western blot analysis in a subset of rats treated with CON or CSH (n = 3 rats per treatment). Neuronal NOS expression in the whole NTS did not change with CSH treatment (Fig. 5, A and B). To further assay the effect of CSH on regional nNOS expression, we also used immunohistochemistry to examine nNOS changes in the caudal, middle, and rostral NTS from a second subset of rats treated with CON or CSH (n = 6 rats per treatment). Relative to the total number of cells in each image (as assessed by counting DAPI-stained nuclei), the percentage of cells that stained positive for nNOS did not vary between brainstem regions or treatment groups (range = 9.0 to 13.4%; Fig. 6, A–C). Conversely, the fluorescent intensity of nNOS-positive neurons was similar between all brainstem regions in normoxic rats; however, neuronal nNOS expression increased 64 and 68% following CSH treatment in the middle and caudal NTS, respectively (Fig. 6, B and D). Neuronal NOS expression in the rostral section of the NTS was not significantly different between CON and CSH rats.
Fig. 5.

Western blot analysis of the effect of hypoxia on NTS nNOS protein expression in the whole NTS. A: summary of fold change in whole NTS nNOS protein expressions normalized to GAPDH protein expression in the same samples. B: sample Western blots of nNOS protein expression from NTS samples isolated from CON rats or rats treated with CSH for 7 days. Data are means ± SD from three separate experiments. Significance was assessed at P < 0.05.
Fig. 6.

Effect of chronic hypoxia on nNOS protein expression in the NTS. A: overview of the sections used for analysis (referenced caudal to bregma; modified from Ref. 41): caudal = 14.1 mm, middle = 13.8 mm, rostral = 13.3 mm; outlines show bilateral areas used for measurements. B: representative examples of areas from left side show increased neuronal staining for nNOS in CSH relative to CON animals. C: CSH had no effect on the percentage of nNOS-positive cells in all NTS regions. D: CSH significantly increased average nNOS expression in the middle and caudal NTS, but no difference was observed in the rostral NTS. Data are means ± SD from six separate experiments. Asterisks (*) indicate significant differences between CON and CSH values (P < 0.05).
DISCUSSION
Our results provide no evidence that neuronal nitric oxide synthase within the NTS contributes to ventilatory chemoreflex responses to acute or chronic sustained hypoxia in awake rats. Specifically, we show that systemic nNOS antagonism and NTS-specific NOS antagonism have no impact on ventilatory responses to acute or chronic hypoxia, indicating either that nNOS does not play any role in the HVR or VAH in this species or that nNOS located at multiple locations outside the NTS have equal and opposing effects on hypoxia-mediated changes in ventilation (i.e., VAH). Experiments testing the ventilatory response to CO2 also show no significant effect of systemic or NTS-specific nNOS antagonism on the HCVR in either chronic normoxic or chronic hypoxic rats. Results from rats in which nNOS was blocked via systemic injection of SMTC and from those that received NTS-specific microinjections of L-NAME are similar and consistent. Analysis of hypoxia-mediated metabolic depression demonstrates that nNOS antagonism has no effect on metabolic rate in any group examined, which is consistent with previous studies (7, 16, 24). These negative findings are an important advance over previous studies of the role of nNOS in ventilatory chemoreflex plasticity to chronic hypoxia, which are limited to systemic manipulations of nNOS in mice and plateau pika, and examine enzymatic activity and nNOS protein expression changes in whole murine medulla, as opposed to in isolated regions of the brainstem that are known to regulate ventilation (e.g., the NTS) (15, 42).
Nitric oxide formation plays a key role in the mammalian hippocampal model of long-term potentiation (LTP), wherein glutamatergic neurotransmission is enhanced by increased presynaptic glutamate release mediated by nNOS activity (32, 33). Specifically, sustained glutamate release causes receptor-mediated Ca2+ influx into postsynaptic neurons. Maintained Ca2+ influx through NMDARs leads to the dephosphorylation of nNOS in postsynaptic neurons (2, 14). When dephosphorylated, nNOSs act to increase NO formation (28, 35), which diffuses in a retrograde fashion back across the synapse and into the presynaptic neuron. Here NO positively modulates guanalyl cyclase expression and activity (6, 28), which increases the activity of cGMP-dependent protein kinase (36), and subsequently presynaptic glutamate release (37). Enhanced presynaptic glutamate release following the excitation of the presynaptic neuron then results in heightened sensitivity at the synapse by increasing the excitation of the postsynaptic neuron. Experimental evidence indicates that this glutamate receptor–NOS plasticity pathway contributes to the CNS component of short-term potentiation of ventilatory responses to acute hypoxic exposure of a few minutes in mice. In support of this, systemic nNOS inhibition or specific deletion of the nNOS gene attenuates short-term potentiation of breathing following acute hypoxia in conscious mice (24), and cGMP accumulates in the brainstem of wild-type but not mutant nNOS knockout mice following acute hypoxic exposure (26). Similar results have been reported in both anesthetized rats and cats, in which administration of NOS inhibitors prior to a hypoxic exposure reduces the acute HVR and prevents hypoxia-mediated cGMP accumulation in the brainstem following acute hypoxia (19, 38). In conscious rats, systemic nNOS antagonism reduces the acute HVR during a 30-min hypoxic stimulus but not in the first minute of hypoxic exposure (19). Similarly, NTS-specific NOS antagonism blunts the KCN-mediated increase in breathing at 5 and 10 min (but not 30 min) after treatment (17), and prevents the acute HVR but has no effect on the acute HCVR after 1 min (38). Conversely, in our experiments we do not observe any effect of nNOS antagonism on the acute HVR within the first 30 min of hypoxic exposure; however, in the case of systemic SMTC injection experiments, the time required to return the animal to the chamber postinjection (∼2 to 3 min) limits our ability to detect changes in this very early postinjection period. It is therefore possible that the detection of a response is prevented at this time point. However, studies from other laboratories suggest that any sensitivity to NOS antagonism should be maintained for at least 5–20 min following injection. We are unable therefore to explain the discrepancy between our NTS-specific study and previous similar studies of the effect of NOS antagonism on the acute HVR, although it is notable that some previous studies have reported conflicting results of the effects of hypoxic stimuli on this putative connection. For example, studies in awake nNOS knockout mice clearly support a role for nNOS in the acute HVR (24, 26); however, systemic blockade of nNOS with SMTC has no effect of the acute HVR in awake mice (15).
In addition to examining the physiological impact of nNOS manipulation on the HVR and VAH in rats, we also examined the effect of CSH exposure on the expression of nNOS in the rat NTS for the first time by using two approaches. First, Western blot analysis indicated that nNOS expression does not change significantly in the whole NTS with CSH. However, more fine-scale analysis of nNOS expression by IHC revealed increases of 64–68% in nNOS expression with CSH in nNOS-positive neurons in the middle and caudal NTS, where the majority of carotid body afferents are thought to synapse (20). The discrepancy between our IHC and Western blot results is most likely explained by a higher signal-to-noise ratio in the whole NTS biopsies used for Western blots. Consider that nNOS expression changes with chronic hypoxia in only two thirds of the NTS regions examined. Therefore, our failure to detect changes in nNOS expression in the whole NTS with Western blots may be due to the lack of change in the rostral region, which has the largest surface area of the NTS regions studied (see Fig. 6A). This likely diluted the impact of the changes observed in the rostral and middle NTS sections on whole NTS nNOS expression, resulting in no significant change with CSH across the whole NTS (Fig. 5). Nonetheless, regardless of the degree to which nNOS expression changes with CSH in the NTS, the lack of a physiological relevance of nNOS in VAH is clearly demonstrated by the in vivo antagonist experiments.
Indeed, the change in nNOS protein expression presently reported in rat middle and caudal NTS is considerably smaller and more localized than in the brainstem of mice exposed to 2 wk of CSH, in which whole medulla nNOS protein expression increases approximately threefold, whereas nNOS mRNA expression doubles (15). This difference in the magnitude of CSH-mediated changes to NTS nNOS expression between mice and rats may underlie the apparently divergent role for nNOS in VAH in these two species. More specifically, mice exhibit a robust and medulla-wide increase in nNOS protein and mRNA expression and enzymatic activity following CSH; blocking nNOS prevents VAH in this species. Conversely, in rats the impact of CSH acclimatization on the expression of nNOS proteins in the NTS is smaller and localized to a small region of the medulla (i.e., the middle and caudal NTS); nNOS antagonism has no observable effect on VAH. It is important to note, however, that our IHC analysis examines the whole NTS region, which includes neurons that regulate a variety of physiological responses in addition to ventilation. Therefore, changes in nNOS expression in the caudal NTS with CSH may be localized to nonventilatory neurons and thus may not be indicative in changes in nNOS activity specific to the synaptic connections between ventilatory neurons.
Although our study aimed to test the hypothesis that nNOS contributes to VAH in CSH-acclimatized rats, we found no evidence to support this hypothesis. However, our ability to definitively conclude that nNOS does not contribute to VAH is limited by our experimental approach and design. A key limitation of our study is that we utilized systemic SMTC injections to test the involvement of nNOS in VAH. Although this approach undoubtedly blocks nNOS in the NTS that may contribute to VAH, nonspecific blockade of systemic nNOS signaling likely effects other pathways and systems that may impact the control of breathing. Nonetheless, this approach has successfully revealed a key role for nNOS signaling in VAH in mice and pika (15, 42). Furthermore, in these species the impact of systemic SMTC treatment on VAH is robust and easily detectable. The lack of even a small effect of systemic SMTC administration on VAH in rats is thus all the more striking.
In addition, we confirmed our systemic results with NTS-specific microinjections of a general NOS antagonist. This second experimental approach is also limited, however, by the fact that localized injections to the NTS likely spill over and effect nonventilatory neurons. For example, pulmonary stretch receptor afferent neurons that prevent lung overinflation also synapse in the NTS and rely on glutamatergic neurotransmission (4). Spillover onto these neurons may explain the nonsignificant reduction in Vt we observe in L-NAME-treated CSH rats breathing acute hypoxia by limiting lung expansion and thus Vt (Fig. 2C and 3C). Furthermore, in addition to nNOS, L-NAME inhibits endothelial and inducible NOS (eNOS and iNOS, respectively), and there is evidence that eNOS knockout mice have an abnormal HVR (27). Taken together, these nonspecific effects due to the regional application of drugs and the pharmacological actions of NTS L-NAME microinjections may impact a variety of systemic response to hypoxia, including cardiac responses, local vascular tone, baroreflexes, and immune signaling, all of which may impact the HVR or VAH (11, 17, 43). It is notable, however, that the confounding nonspecific effects of regional drug injection on the synaptic control of cardiac and ventilatory responses to hypoxia will also occur if SMTC is injected directly into the NTS in place of L-NAME. Similarly, genetic approaches targeting nNOS with adenoviruses microinjected into the NTS would also not be specific to only the neurons that regulate ventilation or to ventilatory neurons in the NTS alone (30). Despite these limitations, NTS microinjection of a nonspecific NOS inhibitor has been used previously in awake rats to evaluate the role of nNOS in the HVR (38). However, to definitively determine whether or not nNOS regulate VAH will likely require an optigenetic approach specifically targeted to the population of NTS neurons that are identified as active in the control of breathing. This was beyond the scope of our present study.
To our knowledge, only a few studies have examined the role of NOS in ventilatory responses to chronic hypoxia. In mice, systemic nNOS antagonism has no effect on the magnitude of the HVR but decreases VAH (15). Similarly, we also report that nNOS antagonism has no effect on the magnitude of the HVR; however, converse to in mice, our study does not support a role for nNOS in VAH in rats. Therefore, other mechanisms of plasticity must be at play in this response in this species. For example, recent electrophysiological studies of NTS second-order neurons in ex vivo brainstem slices show that in neurons from nonacclimatized rats, hypoxia activates an inhibitory ATP-sensitive K+ (KATP) channel-mediated conductance that opposes neuronal excitation (53). In neurons from CSH rats, this current is markedly reduced, likely because of a significant reduction of KATP channel subunit expression in the NTS. This mechanism would render NTS second-order neurons considerably more excitable to stimulation following acclimatization to CSH and may underlie plasticity in the CNS component of VAH after chronic hypoxia. Changes in glutamate receptor expression or phosphorylation may also contribute to CSH-mediated plasticity, and we have recently reported changes in glutamate receptor phosphorylation state in the NTS of CSH rats (39). Alternatively, alterations of the immune system may underlie the central component of VAH. Recent studies show that CSH induces the expression of proinflammatory cytokines and chemokines in the brainstem, and concurrent treatment with ibuprofen during CSH acclimatization blocks both the CNS gain of the VAH and also CSH-mediated increases in inflammatory cytokines in the brainstem (43). Further studies are required to determine the plasticity mechanisms in the CNS component of this pathway.
GRANTS
Support for this study was provided by NHLBI Grant 1R01HL081823 (to F. Powell).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
M.E.P., Z.F., and F.L.P. conception and design of research; M.E.P., A.G., and Z.F. performed experiments; M.E.P., A.G., Z.F., and F.L.P. analyzed data; M.E.P. and F.L.P. interpreted results of experiments; M.E.P. prepared figures; M.E.P. drafted manuscript; M.E.P. and F.L.P. edited and revised manuscript; M.E.P. and F.L.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We graciously thank Ms. Isabelle Marfone and Mr. Shashank Gupta for technical assistance.
REFERENCES
- 1.Aaron EA, Powell FL. Effect of chronic hypoxia on hypoxic ventilatory response in awake rats. J Appl Physiol 74: 1635–1640, 1993. [DOI] [PubMed] [Google Scholar]
- 2.Baader SL, Schilling K. Glutamate receptors mediate dynamic regulation of nitric oxide synthase expression in cerebellar granule cells. J Neurosci 16: 1440–1449, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bisgard GE, Neubauer MS. Peripheral and central effects of hypoxia. In: Regulation of Breathing, edited by Dempsey JA and Pack AIJ. New York: Marcel Dekker, 1995, p. 617–618. [Google Scholar]
- 4.Bonham AC, Coles SK, McCrimmon DR. Pulmonary stretch receptor afferents activate excitatory amino acid receptors in the nucleus tractus solitarii in rats. J Physiol 464: 725–745, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braga VA, Soriano RN, Braccialli AL, de Paula PM, Bonagamba LG, Paton JF, Machado BH. Involvement of L-glutamate and ATP in the neurotransmission of the sympathoexcitatory component of the chemoreflex in the commissural nucleus tractus solitarii of awake rats and in the working heart-brainstem preparation. J Physiol 581: 1129–1145, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bredt DS, Snyder SH. Nitric oxide mediates glutamate-linked enhancement of cGMP levels in the cerebellum. Proc Natl Acad Sci U S A 86: 9030–9033, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castro-Blanco S, Encinas JM, Serrano J, Alonso D, Gomez MB, Sanchez J, Rios-Tejada F, Fernandez-Vizarra P, Fernandez AP, Martinez-Murillo R, Rodrigo J. Expression of nitrergic system and protein nitration in adult rat brains submitted to acute hypobaric hypoxia. Nitric Oxide 8: 182–201, 2003. [DOI] [PubMed] [Google Scholar]
- 8.Chugh DK, Katayama M, Mokashi A, Bebout DE, Ray DK, Lahiri S. Nitric oxide-related inhibition of carotid chemosensory nerve activity in the cat. Respir Physiol 97: 147–156, 1994. [DOI] [PubMed] [Google Scholar]
- 9.da Silva LG, Dias AC, Furlan E, Colombari E. Nitric oxide modulates the cardiovascular effects elicited by acetylcholine in the NTS of awake rats. Am J Physiol Regul Integr Comp Physiol 295: R1774–R1781, 2008. [DOI] [PubMed] [Google Scholar]
- 10.Dias AC, Colombari E. Central nitric oxide modulates hindquarter vasodilation elicited by AMPA receptor stimulation in the NTS of conscious rats. Am J Physiol Regul Integr Comp Physiol 290: R1330–R1336, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Dias AC, Vitela M, Colombari E, Mifflin SW. Nitric oxide modulation of glutamatergic, baroreflex, and cardiopulmonary transmission in the nucleus of the solitary tract. Am J Physiol Heart Circ Physiol 288: H256–H262, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Drorbaugh JE, Fenn WO. A barometric method for measuring ventilation in newborn infants. Pediatrics 16: 81–87, 1955. [PubMed] [Google Scholar]
- 13.Dwinell MR, Powell FL. Chronic hypoxia enhances the phrenic nerve response to arterial chemoreceptor stimulation in anesthetized rats. J Appl Physiol 87: 817–823, 1999. [DOI] [PubMed] [Google Scholar]
- 14.East SJ, Parry-Jones A, Brotchie JM. Ionotropic glutamate receptors and nitric oxide synthesis in the rat striatum. Neuroreport 8: 71–75, 1996. [DOI] [PubMed] [Google Scholar]
- 15.El Hasnaoui-Saadani R, Alayza RC, Launay T, Pichon A, Quidu P, Beaudry M, Leon-Velarde F, Richalet JP, Duvallet A, Favret F. Brain stem NO modulates ventilatory acclimatization to hypoxia in mice. J Appl Physiol 103: 1506–1512, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Gozal D, Torres JE, Gozal YM, Littwin SM. Effect of nitric oxide synthase inhibition on cardiorespiratory responses in the conscious rat. J Appl Physiol (1985) 81: 2068–2077, 1996. [DOI] [PubMed] [Google Scholar]
- 17.Granjeiro EM, Machado BH. NO in the caudal NTS modulates the increase in respiratory frequency in response to chemoreflex activation in awake rats. Respir Physiol Neurobiol 166: 32–40, 2009. [DOI] [PubMed] [Google Scholar]
- 18.Haibara AS, Bonagamba LG, Machado BH. Sympathoexcitatory neurotransmission of the chemoreflex in the NTS of awake rats. Am J Physiol Regul Integr Comp Physiol 276: R69–R80, 1999. [DOI] [PubMed] [Google Scholar]
- 19.Haxhiu MA, Chang CH, Dreshaj IA, Erokwu B, Prabhakar NR, Cherniack NS. Nitric oxide and ventilatory response to hypoxia. Respir Physiol 101: 257–266, 1995. [DOI] [PubMed] [Google Scholar]
- 20.Housley GD, Sinclair JD. Localization by kainic acid lesions of neurones transmitting the carotid chemoreceptor stimulus for respiration in rat. J Physiol 406: 99–114, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huey KA, Brown IP, Jordan MC, Powell FL. Changes in dopamine D(2)-receptor modulation of the hypoxic ventilatory response with chronic hypoxia. Respir Physiol 123: 177–187, 2000. [DOI] [PubMed] [Google Scholar]
- 22.Hupperets MD, Hopkins SR, Pronk MG, Tiemessen IJ, Garcia N, Wagner PD, Powell FL. Increased hypoxic ventilatory response during 8 weeks at 3800 m altitude. Respir Physiol Neurobiol 142: 145–152, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Jacky JP. A plethysmograph for long-term measurements of ventilation in unrestrained animals. J Appl Physiol 45: 644–647, 1978. [DOI] [PubMed] [Google Scholar]
- 24.Kline DD, Overholt JL, Prabhakar NR. Mutant mice deficient in NOS-1 exhibit attenuated long-term facilitation and short-term potentiation in breathing. J Physiol 539: 309–315, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kline DD, Prabhakar NR. Role of nitric oxide in short-term potentiation and long-term facilitation: involvement of NO in breathing stability. Adv Exp Med Biol 499: 215–219, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Kline DD, Yang T, Huang PL, Prabhakar NR. Altered respiratory responses to hypoxia in mutant mice deficient in neuronal nitric oxide synthase. J Physiol 511: 273–287, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kline DD, Yang T, Premkumar DR, Thomas AJ, Prabhakar NR. Blunted respiratory responses to hypoxia in mutant mice deficient in nitric oxide synthase-3. J Appl Physiol 88: 1496–1508, 2000. [DOI] [PubMed] [Google Scholar]
- 28.Knowles RG, Palacios M, Palmer RM, Moncada S. Formation of nitric oxide from L-arginine in the central nervous system: a transduction mechanism for stimulation of the soluble guanylate cyclase. Proc Natl Acad Sci U S A 86: 5159–5162, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar P, Prabhakar N. Peripheral chemoreceptors: function and plasticity of the carotid body. Compr Physiol 2: 141–219, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin LH, Dragon DN, Jin J, Talman WT. Targeting neurons of rat nucleus tractus solitarii with the gene transfer vector adeno-associated virus type 2 to up-regulate neuronal nitric oxide synthase. Cell Mol Neurobiol 31: 847–859, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lipski J, McAllen RM, Spyer KM. The carotid chemoreceptor input to the respiratory neurones of the nucleus of tractus solitarus. J Physiol 269: 797–810, 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron 44: 5–21, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science 285: 1870–1874, 1999. [DOI] [PubMed] [Google Scholar]
- 34.Mizusawa A, Ogawa H, Kikuchi Y, Hida W, Kurosawa H, Okabe S, Takishima T, Shirato K. In vivo release of glutamate in nucleus tractus solitarii of the rat during hypoxia. J Physiol 478: 55–66, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med 329: 2002–2012, 1993. [DOI] [PubMed] [Google Scholar]
- 36.Monfort P, Munoz MD, Kosenko E, Felipo V. Long-term potentiation in hippocampus involves sequential activation of soluble guanylate cyclase, cGMP-dependent protein kinase, and cGMP-degrading phosphodiesterase. J Neurosci 22: 10116–10122, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montague PR, Gancayco CD, Winn MJ, Marchase RB, Friedlander MJ. Role of NO production in NMDA receptor-mediated neurotransmitter release in cerebral cortex. Science 263: 973–977, 1994. [DOI] [PubMed] [Google Scholar]
- 38.Ogawa H, Mizusawa A, Kikuchi Y, Hida W, Miki H, Shirato K. Nitric oxide as a retrograde messenger in the nucleus tractus solitarii of rats during hypoxia. J Physiol 486: 495–504, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pamenter ME, Carr JA, Go A, Fu ZX, Reid SG, Powell FL. Glutamate receptors in the nucleus tractus solitarius contribute to ventilatory acclimatization to hypoxia in rat. J Physiol 592: 1839–1856, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pamenter ME, Nguyen J, Carr JA, Powell FL. The effect of combined glutamate receptor blockade in the NTS on the hypoxic ventilatory response in awake rats differs from the effect of individual glutamate receptor blockade. Physiol Rep 2: e12092, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paxinos G, Watson C. The Rat Brain Atlas in Stereotaxic Coordinates. Orlando, FL: Academic, 1997. [Google Scholar]
- 42.Pichon A, Zhenzhong B, Favret F, Jin G, Shufeng H, Marchant D, Richalet JP, Ge RL. Long-term ventilatory adaptation and ventilatory response to hypoxia in plateau pika (Ochotona curzoniae): role of nNOS and dopamine. Am J Physiol Regul Integr Comp Physiol 297: R978–R987, 2009. [DOI] [PubMed] [Google Scholar]
- 43.Popa D, Fu Z, Go A, Powell FL. Ibuprofen blocks time-dependent increases in hypoxic ventilation in rats. Respir Physiol Neurobiol 178: 381–386, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Powell FL, Milsom WK, Mitchell GS. Time domains of the hypoxic ventilatory response. Respir Physiol 112: 123–134, 1998. [DOI] [PubMed] [Google Scholar]
- 45.Reeves SR, Gozal E, Guo SZ, Sachleben LR Jr, Brittian KR, Lipton AJ, Gozal D. Effect of long-term intermittent and sustained hypoxia on hypoxic ventilatory and metabolic responses in the adult rat. J Appl Physiol 95: 1767–1774, 2003. [DOI] [PubMed] [Google Scholar]
- 46.Reid SG, Powell FL. Effects of chronic hypoxia on MK-801-induced changes in the acute hypoxic ventilatory response. J Appl Physiol 99: 2108–2114, 2005. [DOI] [PubMed] [Google Scholar]
- 47.Richter DW, Schmidt-Garcon P, Pierrefiche O, Bischoff AM, Lalley PM. Neurotransmitters and neuromodulators controlling the hypoxic respiratory response in anaesthetized cats. J Physiol 514: 567–578, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tagawa T, Imaizumi T, Harada S, Endo T, Shiramoto M, Hirooka Y, Takeshita A. Nitric oxide influences neuronal activity in the nucleus tractus solitarius of rat brainstem slices. Circ Res 75: 70–76, 1994. [DOI] [PubMed] [Google Scholar]
- 49.Vardhan A, Kachroo A, Sapru HN. Excitatory amino acid receptors in commissural nucleus of the NTS mediate carotid chemoreceptor responses. Am J Physiol Regul Integr Comp Physiol 264: R41–R50, 1993. [DOI] [PubMed] [Google Scholar]
- 50.Wilkinson KA, Fu Z, Powell FL. Ventilatory effects of substance P-saporin lesions in the nucleus tractus solitarii of chronically hypoxic rats. Am J Physiol Regul Integr Comp Physiol 301: R343–R350, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilkinson KA, Huey K, Dinger B, He L, Fidone S, Powell FL. Chronic hypoxia increases the gain of the hypoxic ventilatory response by a mechanism in the central nervous system. J Appl Physiol 109: 424–430, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Youssef F, Addae J, McRae A, Stone T. Long-term potentiation protects rat hippocampal slices from the effects of acute hypoxia. Brain Res 907: 144–150, 2001. [DOI] [PubMed] [Google Scholar]
- 53.Zhang W, Carreno FR, Cunningham JT, Mifflin SW. Chronic sustained and intermittent hypoxia reduce function of ATP-sensitive potassium channels in nucleus of the solitary tract. Am J Physiol Regul Integr Comp Physiol 295: R1555–R1562, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang W, Mifflin SW. Excitatory amino acid receptors within NTS mediate arterial chemoreceptor reflexes in rats. Am J Physiol Heart Circ Physiol 265: H770–H773, 1993. [DOI] [PubMed] [Google Scholar]