

Abstract
Tumor cell invasion through the stromal extracellular matrix (ECM) is a key feature of cancer metastasis, and understanding the cellular mechanisms of invasive migration is critical to the development of effective diagnostic and therapeutic strategies. Since cancer cell migration is highly adaptable to physiochemical properties of the ECM, it is critical to define these migration mechanisms in a context-specific manner. Although extensive work has characterized cancer cell migration in two- and three-dimensional (3D) matrix environments, the migration program employed by cells to move through native and cell-derived microtracks within the stromal ECM remains unclear. We previously reported the development of an in vitro model of patterned type I collagen microtracks that enable matrix metalloproteinase-independent microtrack migration. Here we show that collagen microtracks closely resemble channel-like gaps in native mammary stroma ECM and examine the extracellular and intracellular mechanisms underlying microtrack migration. Cell-matrix mechanocoupling, while critical for migration through 3D matrix, is not necessary for microtrack migration. Instead, cytoskeletal dynamics, including actin polymerization, cortical tension, and microtubule turnover, enable persistent, polarized migration through physiological microtracks. These results indicate that tumor cells employ context-specific mechanisms to migrate and suggest that selective targeting of cytoskeletal dynamics, but not adhesion, proteolysis, or cell traction forces, may effectively inhibit cancer cell migration through preformed matrix microtracks within the tumor stroma.
Keywords: tumor invasion, migration mechanisms, microtracks
as one of the earliest steps of metastasis, local tissue invasion represents a critical transition from local deregulated cell growth to potentially lethal disease. Tissue invasion is a multistep biophysical process during which carcinoma cells bypass the epithelial basement membrane and migrate through the underlying interstitial stroma (8, 37). Classically, stromal invasion has been viewed as a challenging and highly selective step-wise process requiring an invading cell to actively breach the basement membrane, protrude into a collagen-rich stroma, and employ a diversity of mechanochemical migration mechanisms to overcome matrix and tissue barriers to effectively migrate (16, 38). However, recent evidence suggests that the vast cellular and microenvironmental heterogeneity within and around tumors allows significant diversity in invasion programs (20), and the context-specific mechanisms governing local tissue invasion and metastasis remain only partially understood.
Cancer cell migration through three-dimensional (3D) stromal microenvironments is adaptive and sensitive to regulation by intracellular and extracellular determinants (22, 66). While transit of tumor cells through the 3D extracellular matrix (ECM) requires coordination of a physiochemical motility program that generally involves cytoskeletal, adhesion, and contractility mechanisms (8, 35), migration strategies are context-specific, and the molecular mechanisms employed depend on biochemical and biophysical properties of the ECM. Notably, biophysical matrix parameters, including porosity, alignment, and elasticity, have emerged as key mediators of cell behavior and, together, determine the requirements for and nature of motility (68). Wolf et al. (69) recently showed that matrix porosity integrates with nuclear deformability to define the requirement for integrin- and actomyosin-mediated mechanocoupling and ECM remodeling during 3D migration. Furthermore, our group and others have demonstrated that the structural and biochemical nature of type I collagen matrix determines 3D migration efficiency and the requirement for proteolytic ECM remodeling by matrix metalloproteinase (MMP) activity (9, 10, 56, 71). In addition to imposing steric constraints, fibrillar 3D ECM can exhibit structural anisotropy, including matrix fiber alignment (6, 53, 55) and interfacial regions (5, 64), which provide topographical guidance cues to migrating cells. Nonetheless, while these and other studies have contributed greatly to our understanding of ECM-directed cell invasion strategies, conventional in vitro 3D ECM models for studying mechanisms of tumor cell behavior vastly overestimate the homogeneity of the stromal ECM, and the effects of local heterogeneity in matrix structure on cell motility remain only marginally understood (26, 67).
Cell-scaled tracklike structures in the interstitial matrix, which have been observed as conduits for trafficking tumor cells in vivo, provide physical guidance and a path of least resistance to migrating cells and, thus, have been implicated as critical determinants of tumor cell behavior (2, 11, 25, 26, 62). These structures can broadly include interfaces between tissue components, gaps and clefts between aligned collagen bundles, and organized networks of ECM pores. Recently, our group and others have shown that tumor and stromal cells can use mechanical and proteolytic matrix remodeling to generate cell-caliber microtracks through type I collagen matrix that facilitate proteinase-independent invasion of other cells (1, 10, 18, 24). Regardless of whether matrix microtracks are native to interstitial tissue (67) or a result of MMP-mediated matrix degradation by tumor/stromal cells (10, 24, 71), their presence in tissue challenges the conventional interstitial invasion model in which migrating cells must negotiate a restrictive collagen-rich ECM. Instead, cells may co-opt sufficiently wide preformed matrix tunnels to migrate in an unimpeded, MMP-independent manner (21). Notably, the presence of such migration-enabling matrix-free pathways could explain the limited ability of clinical MMP inhibition to prevent invasion and metastasis. Since these tracklike structures in the ECM provide strong proinvasive cues to tumor cells, an understanding of the mechanisms that govern cancer cell migration through cell-scale gaps in tissue will be critical to the development of therapeutic strategies to target metastasis.
We previously developed an in vitro system of patterned collagen microtracks to model tumor cell migration through cell-sized gaps in the ECM (33). The microtracks, unlike previous designs made from polydimethylsiloxane (PDMS) and polyacrylamide (3, 50), offer the advantage of more closely mimicking the mechanical and chemical properties of native ECM. We found that patterned collagen microtracks recapitulate tubelike tumor cell-derived channels within a 3D collagen matrix and enable MMP-independent migration of otherwise poorly invasive mammary epithelial cells (33). Here we use this platform alongside fibrillar 3D collagen matrix to define and compare the mechanisms that govern cancer cell migration within each environment. We show that mechanical cell-matrix interactions are needed for migration through 3D matrix, but not microtracks, where cleared matrix-free pathways provide little resistance and eliminate the need for traction generation, matrix remodeling, and cell body deformation. Conversely, migration through 3D matrix and microtracks is similarly driven by polarized protrusions and elongation at the leading edge, which are mediated by actin polymerization and reinforced by the microtubule (MT) cytoskeleton. Collectively, our findings suggest that microtracks within the interstitial stroma may provide paths of least resistance that enable tumor cell invasion by reducing the molecular machinery required for efficient migration.1
MATERIALS AND METHODS
Cell culture and reagents.
Highly metastatic MDA-MB-231 breast adenocarcinoma cells (catalog no. HTB-26, American Type Culture Collection, Manassas, VA) were maintained in minimum essential medium (Life Technologies, Grand Island, NY) supplemented with 10% (vol/vol) fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA), 100 U/ml penicillin, and 100 μg/ml streptomycin (Life Technologies). Green fluorescent protein (GFP)-expressing MDA-MB-231 cells (catalog no. AKR-201, Cell Biolabs, San Diego, CA) were maintained in Dulbecco's modified Eagle's medium (Life Technologies) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.1 mM minimum essential medium nonessential amino acids (Life Technologies). For inhibitor studies, complete medium was supplemented with 10 μM Y27632, 10 μM ML7, 50 μM blebbistatin, 10 μM nocodazole, 12 μM paclitaxel, 20 μM cytochalasin D, and 2.5 μM latrunculin A (all from Sigma-Aldrich, St. Louis, MO), 2 μg/ml CT04 (Cytoskeleton, Denver, CO), 2.5–10 μg/ml MAb 4B4/FITC-4B4 (Beckman Coulter, Brea, CA), or DMSO vehicle. All cell culture was maintained at 37°C and 5% CO2. Primary antibodies were anti-α-tubulin (catalog no. 05-829, Millipore, Billerica, MA), anti-phosphorylated myosin light chain (pMLC) 2 (catalog no. 3674S, Cell Signaling Technology, Danvers, MA), and anti-GAPDH (catalog no. MAb374, Millipore).
Murine mammary cancer model.
Mice were maintained under barrier conditions according to Cornell University Animal Care guidelines, and the methods were approved by the Cornell Institutional Animal Care and Use Committee (protocol 2009-0101; R. M. Williams). The murine mammary cancer model was established as previously described (14). Briefly, 3- to 4-wk-old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (Jackson Laboratory, Bar Harbor, ME) were placed in an induction chamber and anesthetized with 3% isoflurane (Butler Animal Health Supply, Dublin, OH) for 2–5 min and subsequently moved to a custom-designed surgical platform. Developing glandular tissue was removed from the fourth and seventh inguinal mammary glands, and 105-106 GFP-expressing MDA-MB-231 cells were injected into each cleared fat pad. Surgical incisions were repaired using wound clips (Fisher Scientific, Atlanta, GA), and mice were resuscitated and monitored for 3 wk prior to in vivo and ex vivo imaging.
In vivo multiphoton imaging.
After 3 wk of orthotopic tumor growth, in vivo imaging was performed. Mice were anesthetized as described for surgery and immediately moved to a custom-designed imaging stage. Isoflurane concentration was adjusted to 2% to maintain respiration at ∼60 breaths/min (19). Mice were vivisected to expose MDA-MB-231-derived mammary tumors, and multiphoton (MP) and second harmonic generation imaging were performed as previously described (65, 76). An 880-nm beam from a mode-locked Ti:sapphire laser illuminated MDA-MB-231/GFP-derived tumors and the surrounding mammary stroma in situ on a modified Olympus AX70 upright microscope equipped with an Olympus water-immersion ×20/0.95 numerical aperture Olympus objective. The emission signal was separated into two channels to collect the second harmonic generation signal from collagen and MP fluorescence from GFP-labeled MDA-MB-231 cells. After in situ MP imaging, MDA-MB-231-derived tumors and the surrounding mammary stromal tissue were harvested for ex situ imaging; tissues were either fixed for 30 min in 3.7% (vol/vol) formaldehyde in PBS or embedded within collagen matrix (see below) for time-lapse confocal imaging.
Preparation of collagen matrices and microtracks.
Collagen matrices and microtracks were prepared as previously described using acid-extracted type I collagen from rat tail tendons (10). Collagen stock solution (10 mg/ml) was diluted to the desired concentration (1.5–5 mg/ml) using ice-cold culture medium and neutralized with sodium hydroxide. For 3D matrix experiments, dissociated MDA-MB-231 cells were incorporated sparsely into neutralized collagen solution prior to polymerization for 30 min at 37°C. For ex situ time-lapse imaging, excised tumors were similarly embedded within collagen matrix for stabilization. Collagen microtracks with dimensions of 10 × 1,100 × 20 μm (width × length × depth) were prepared as previously described (33). Briefly, micropatterned PDMS stamps were rendered nonadhesive with bovine serum albumin, washed with neutralized collagen solution, and inverted over a drop of neutralized collagen solution. Collagen was polymerized for 90 min at 37°C, PDMS stamps were removed, and MDA-MB-231 cells were seeded onto patterned collagen matrices at low density to minimize cell-cell interactions. Finally, patterned collagen matrices were covered with a collagen lid to form 3D microtracks. Except where indicated in Fig. 2, all in vitro studies were performed in 1.5 mg/ml collagen matrix (3D) and 3.0 mg/ml collagen microtracks.
Fig. 2.
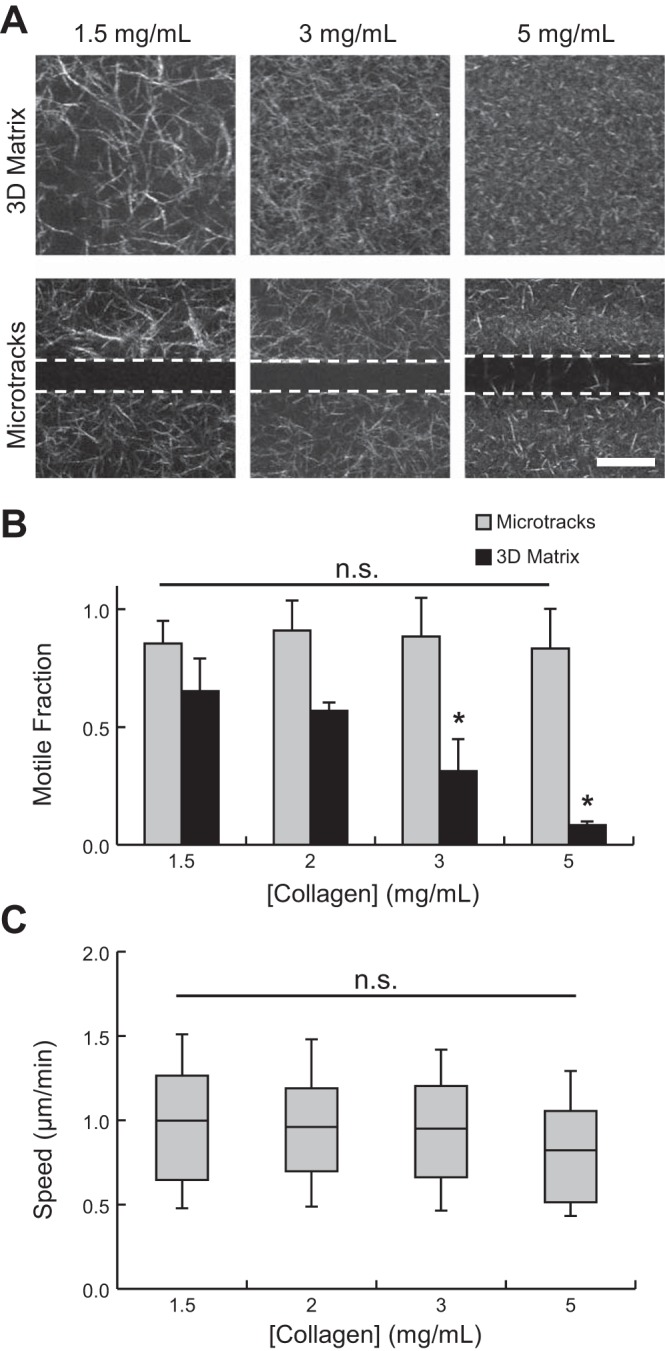
Density-dependent cell migration through 3-dimensional (3D) collagen matrix and microtracks. A: confocal reflectance images of collagen matrix structure in 3D matrix (top) and microtracks (bottom). Dashed lines indicate microtrack edges. Scale bar = 25 μm. B: fraction of motile cells within 3D matrix and collagen microtracks as a function of matrix density. C: single-cell speeds of motile cells within collagen microtracks. *P < 0.05; n.s., not significant.
Confocal and time-lapse imaging.
Confocal fluorescence and reflectance images were acquired as previously described (10) using a Zeiss LSM700 confocal microscope on a Zeiss Axio Observer Z1 inverted stand equipped with a long-working-distance water-immersion C-Apochromat ×40/1.1 numerical aperture Zeiss objective. Fluorescent labeling and imaging of actin and MTs (α-tubulin) were performed as previously described (32). The ImageJ (version 1.49b, National Institutes of Health, Bethesda, MD) plugin OrientationJ was used to quantify and colorize actin organization from confocal fluorescence images as previously described (10). Briefly, gray-scale images were analyzed using a 0.6-μm Gaussian window, and angular distributions of pixel orientation were normalized to microtrack angle. The mean and standard deviation of distributions were quantified and compared for 8–10 cells per condition. Phase-contrast images were acquired using a Zeiss Axio Observer Z1 inverted phase-contrast microscope equipped with a Hamamatsu ORCA-ER camera. Time-lapse phase-contrast and confocal imaging were performed in custom temperature-, humidity-, and CO2-controlled microscope incubation chambers.
Cell migration studies and analysis.
After cell seeding, 3D matrices and microtracks were overlaid with complete culture medium and incubated for 6–8 h to allow cell adhesion and spreading prior to time-lapse imaging. To study the molecular mechanisms underlying cell migration through 3D matrix and microtracks, inhibitors of cell-matrix adhesion, contractility, and cytoskeletal dynamics were applied immediately prior to imaging or after 4–5 h of control imaging. For phase-contrast time-lapse imaging, images were acquired at 5-min intervals for 16 h. Cells that divided or interacted with other cells during this time were excluded from analysis, and ImageJ was used to measure cells' morphologies and track the positions of cell centroids over time. To account for heterogeneity of cell migration behavior, two migration parameters were measured: motile fraction and migration speed. A cell was considered motile if its centroid moved more than one cell diameter during the observation period, and motile fraction was determined by dividing the number of motile cells by the total number of cells in each frame of view. Cell migration speed within microtracks was quantified for motile cells as previously reported (33). Motile fraction and migration speed were quantified posttreatment for >40 cells per condition from two to three independent experiments. To quantify cell morphodynamics during microtrack migration, cells were classified as amoeboid (rounded; aspect ratio <4) or mesenchymal (elongated; aspect ratio >4) as indicated in Fig. 3F. Cell aspect ratio during microtrack migration was tracked for ∼35 cells per condition to measure the rate at which cells underwent amoeboid → mesenchymal and mesenchymal → amoeboid transitions.
Fig. 3.

Effect of β1-integrin function blocking on microtrack migration. A: flow cytometry data showing fluorescence signal from cells treated with 0–30 μg/ml FITC-4B4. MFI, mean fluorescence intensity; AU, arbitrary units. B: fraction of motile cells within 3D matrix and collagen microtracks treated with 10 μg/ml β1-integrin function-blocking antibody 4B4. C: single-cell speeds of motile cells within microtracks under increasing concentrations of 4B4. D and E: time-lapse image series of control and 4B4-treated (10 μg/ml) cells migrating in 3D matrix and microtracks. Scale bars = 25 μm. F: aspect ratios of cells in E. Aspect ratios <4 correspond to rounded, amoeboid morphologies (dark gray region); aspect ratios >4 correspond to elongated, mesenchymal morphologies (light gray region). G: frequency of amoeboid → mesenchymal (A → M) and mesenchymal → amoeboid (M → A) transitions during microtrack migration. Each data point represents an individual cell. Horizontal lines indicate means. *P < 0.05
Polyacrylamide gel synthesis and traction force microscopy.
Polyacrylamide substrates with Young's moduli of 5 kPa were synthesized, functionalized with N-6-[(acryloyl)amido]hexanoic acid, coated with 100 μg/ml type I rat tail collagen (Becton Dickinson, Franklin Lakes, NJ), and used for traction force microscopy as previously described (13, 31, 32). Cells were seeded on polyacrylamide substrates and allowed to adhere and spread for 6–8 h before treatments were initiated. After 10 h of treatment, traction force microscopy experiments were performed, and the traction field was derived from bead displacements using the LIBTRC analysis library developed by Dr. Micah Dembo (Boston University) (13). Data are presented as means ± SE of total force magnitude, |F|, which is the integral of the traction field over the area of the cell, for 20–60 cells per condition.
Western blotting.
After cell seeding in 3D collagen matrices (1.5 mg/ml), samples were incubated for 24 h and then treated with inhibitors of cell contractility for 4 h. Total protein was extracted from snap-frozen gels with preheated (95°C) 2× Laemmli sample buffer, and 20 μl of protein extract were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Protein was electrotransferred onto a polyvinylidene difluoride membrane, and the blots were incubated with primary antibody overnight at 4°C and then with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. Peroxidase signal was revealed with the SuperSignal West Pico or West Femto kit (Thermo Scientific, Rockford, IL). For quantification, pMLC signal was normalized to GAPDH before comparison with control (no treatment). Data are presented as means ± SE for two independent experiments.
Flow cytometry.
Cell adhesion to the ECM via β1-integrin was blocked with the mouse monoclonal function-blocking antibody 4B4. Surface expression of β1-integrin was determined using a 4B4-FITC conjugate and fluorescence-activated cell sorting analysis with a FACSAria high-speed flow cytometer (BD Biosciences, Franklin Lakes, NJ). In separate experiments, MDA-MB-231 cells were transfected with GFP-Lifeact (a kind gift from Jan Lammerding, Cornell University), isolated using a FACSAria high-speed flow cytometer, and seeded into collagen microtracks for confocal imaging.
Statistical analysis.
Values are shown as means ± SE or box-and-whisker plots, where boxes indicate medians and 25th/75th percentiles and bars represent 5th/95th percentiles. Means were compared using analysis of variance, with a post hoc Tukey's honestly significant difference test or Dunnett's test where appropriate, using JMP software (version 10, SAS Institute, Cary, NC). Microtrack migration speeds were compared using a nonparametric Wilcoxon test. Amoeboid → mesenchymal and mesenchymal → amoeboid transition frequencies were compared by two-tailed Student's t-test. Actin alignment distributions were compared using Levene's test for equal variances. Statistical significance was considered with P < 0.05.
RESULTS
Cell-sized gaps in native stromal ECM and microfabricated collagen tracks support malignant cell invasion.
Previously we showed that microfabricated collagen tracks closely mimic the tubelike proteolytic tracks created by metastatic cancer cells migrating in 3D collagen matrix (33). Using this system as a model for “follower cell” migration, we found that microtracks provide 3D space through collagen matrix that enables MMP-independent migration of highly metastatic MDA-MB-231 cells, as well as migration of noninvasive MCF-10A mammary epithelial cells. Here we used an orthotopic murine mammary cancer model to observe interactions between breast cancer cells and the native stromal ECM during tumor invasion. At 3 wk after implantation of GFP-expressing MDA-MB-231 cells into the cleared mammary fat pad, palpable tumors had grown and cancer cells had begun to expand into and invade through the stromal ECM. Ex situ confocal (Fig. 1A) and in situ MP (Fig. 1B) imaging revealed channel-like gaps in the collagenous stromal ECM adjacent to mammary tumors, as indicated by the absence of reflected light and second harmonic signals, respectively (Fig. 1, A and B, double arrowheads). Since confocal reflectance microscopy depends on interfacial light scattering due to refractive index mismatch, lack of reflectance signal indicates a lack of light-scattering material within the aqueous interstitial space (23). Notably, cells at the tumor periphery were observed squeezing into and through these spaces (Fig. 1A, right, asterisk). Confocal reflectance microscopy of patterned collagen microtracks showed that collagen matrix structure around in vitro microtracks resembled ECM structure around in vivo microtracks, with aligned ECM fibers bounding an ∼10- to 15-μm-wide track on all sides (Fig. 1, A, C, and D). To monitor cell migration through the stromal ECM, tumors were excised and observed ex vivo using time-lapse confocal imaging (Fig. 1E), which revealed cells migrating through gaps in the ECM (Fig. 1E, double arrowheads). Similarly, MDA-MB-231 cells readily migrated through patterned collagen microtracks in vitro (Fig. 1F).
Fig. 1.

Native stromal extracellular matrix (ECM) contains channel-like gaps that are mimicked by microfabricated collagen tracks. A: ex situ confocal images of green fluorescent protein (GFP)-expressing MDA-MB-231 cells (green) migrating through the stromal ECM (purple) in the murine mammary fat pad following 3 wk of orthotopic tumor growth (×3-magnified image of cells and matrix at the indicated tumor/stroma interface). Asterisk and dashed outlines indicate tumor cells extending into open matrix pathway. B: in situ two-photon image of mammary fat pad showing tumor cells and stromal ECM. C: confocal reflectance images of microtrack patterned in 3 mg/ml collagen. Asterisk indicates cell migrating within channel. D: orthogonal views of a patterned collagen microtrack. E: time-lapse ex situ confocal reflectance and fluorescence images of cells migrating through mammary stroma (left) and RGB overlay of cell position over time (right). F: time-lapse phase-contrast image series of MDA-MB-231 cell migrating through collagen microtrack. Dashed lines indicate microtrack edges. Arrowheads in A–E denote cell-scale gaps within the ECM. Scale bars = 25 μm.
Migration through collagen microtracks is independent of collagen matrix density.
It has been established that native mammary stoma exhibits significant heterogeneity of biophysical and biochemical ECM properties at the micro- and macroscales (26). Therefore, to investigate the effects of matrix density and tissue structure on metastatic cancer cell migration, cells were seeded in 3D collagen matrix or collagen microtracks of varying collagen concentration. As shown by confocal reflectance microscopy, all matrices were composed of interconnected networks of collagen fibers and pores, the structure and organization of which were concentration-dependent: matrices of increasing density were more tightly packed and contained smaller fibers and pores (Fig. 2A). Two motility metrics were used to quantify cell movement throughout the study: 1) motile fraction, which describes the portion of the total cell population that is migratory, and 2) cell speed. As previously shown, increasing collagen density significantly decreased motile fraction within 3D collagen matrices (9, 69), and <10% of cells were migratory within 5 mg/ml collagen matrices (Fig. 2B). In contrast, collagen density did not significantly affect motile fraction (Fig. 2B) or single-cell migration speed (Fig. 2C) of cells within microtracks. For all collagen concentrations tested, 80–90% of cells were motile in microtracks, and migration speeds consistently ranged from ∼0.5 to 1.5 μm/min. While collagen density exerts a modest effect on patterned microtrack dimensions (33), these changes did not significantly impact microtrack migration. Together, these findings indicate that cell migration through microtracks is independent of the density of the surrounding matrix.
β1-Integrin adhesion mediates elongated microtrack migration.
The independence of microtrack migration on matrix density suggests that cell-matrix interactions play a unique role in microtracks compared with two-dimensional (2D) and 3D environments. Therefore, the β1-integrin function-blocking MAb 4B4 was used to probe the role of cell-matrix adhesion in cancer cell migration through 3D matrix and collagen microtracks. Flow cytometry was used to verify that maximal β1-integrin inhibition was achieved using 10 μg/ml 4B4 (Fig. 3A). Treatment with 4B4 did not significantly affect motile fraction in microtracks or 3D matrix, where ∼85% and ∼60% of cells were motile, respectively (Fig. 3B). As previously shown (69, 75), inhibition of β1-integrin decreased cell elongation and migration speed in 3D matrix (Fig. 3D; see Supplemental Movie S1 in Supplemental Material for this article, available online at the Journal website). Interestingly, β1-integrin blocking did not affect single-cell migration speed in microtracks (Fig. 3C), but treatment with 4B4 did alter the morphology of migrating cells (Fig. 3E; see Supplemental Movie S1). Whereas control cells exhibited consistently elongated, mesenchymal-like (high aspect ratio) morphologies during microtrack migration, β1-integrin-inhibited cells showed a saltatory pattern of morphology, oscillating between elongated, mesenchymal-like (high aspect ratio) and rounded, amoeboid-like (low aspect ratio) morphologies during migration (Fig. 3F). Since the majority of control cells retained an elongated, mesenchymal morphology during microtrack migration, morphology switching was infrequent (0.04 ± 0.02 transitions/h) compared with 4B4-treated cells, which underwent morphology transitions ∼10 times more frequently (0.45 ± 0.08 transitions/h). Thus, while β1-integrin adhesion is not required to maintain migration speed within microtracks, it does facilitate elongated mesenchymal morphology during migration.
Actomyosin contractility, but not traction generation, is required for efficient microtrack migration.
Actomyosin-mediated contractility underlies several biomechanical functions central to invasive cell migration, including cell body/nuclear deformation and traction force generation (8). Myosin light chain (MLC) phosphorylation is the enabling molecular step in cell contractility and is controlled by several upstream regulators, including the Rho GTPase/Rho-associated protein kinase (ROCK) pathway and MLC kinase (MLCK) (61). To elucidate the role of cell contractility in microenvironment-dependent migration, pharmacological inhibitors targeting several players within the actomyosin contractility network were applied to cells in 3D matrix and microtracks. Inhibition of Rho (CT04), ROCK [Y27632 (Y27)], MLCK (ML7), or myosin II (blebbistatin) significantly reduced motile fraction within 3D matrix but had no effect on motile fraction within microtracks (Fig. 4A), indicating a requirement for cell contractility during migration through restrictive 3D matrix environments. As demonstrated by time-lapse imaging, contractility-deficient cells in 3D matrix exhibited a protrusive, nonmotile phenotype characterized by 1) extension of anuclear, branching pseudopodia and 2) failure to translocate the cell nucleus and body (Fig. 4B; see Supplemental Movie S1). Interestingly, while inhibition of Rho, ROCK, and MLCK alone significantly reduced cell traction forces (Fig. 4C), these treatments had no effect on cell migration speed within microtracks (Fig. 4D), indicating that traction forces are not explicitly required for microtrack migration. However, when both MLCK and ROCK were inhibited (ML7 + Y27) or myosin II activity was inhibited directly (blebbistatin), microtrack migration speed was significantly reduced (Fig. 4D), indicating that myosin activity contributes to maintenance of cell speed within microtracks. In addition to a significant reduction in traction force magnitude with blebbistatin treatment (Fig. 4C), analysis of cell morphology prior to and following treatment indicated that the blebbistatin-induced decrease in microtrack migration speed was accompanied by impaired retraction of the trailing edge (Fig. 4E, arrowhead). This behavior was also observed using other inhibitors of contractility (see Supplemental Movie S1). These results indicate that traction generation, 3D matrix migration, and microtrack migration can be pharmacologically decoupled and suggest that these behaviors may operate under the control of different mechanistic elements of the cell contractility pathway.
Fig. 4.

Role of cell contractility and traction forces in microtrack migration. A: fraction of motile cells within 3D matrix and collagen microtracks treated with Y27632 (Y27; a Rho-associated protein kinase inhibitor), ML7 [a myosin light chain (MLC) kinase inhibitor], CT04 (a Rho inhibitor), ML7 + Y27, and blebbistatin (Bleb; a myosin II inhibitor). B: representative frames from time-lapse series of cells migrating in 3D matrix under control and blebbistatin treatment. C: total traction force magnitude, |F|, of cells treated as described in A. D: single-cell speeds of motile cells within collagen microtracks under conditions described in A. E: time-lapse image series of cell migrating in microtrack treated with blebbistatin at minute 20. Arrowheads indicate lagging cytoplasmic extensions after treatment. Scale bars = 25 μm. F: Western blot for phosphorylated MLC (pMLC) in 3D matrix after 4 h of treatment with inhibitors in A. G: linear correlations between pMLC and microtrack motile fraction (R2 = 0.20), 3D matrix motile fraction (R2 = 0.13), traction force magnitude (R2 = 0.86), and microtrack migration speed (R2 = 0.01). Values are means ± SE, except microtrack migration speed, which is median ± SD. *P < 0.05.
Since MLC phosphorylation enables myosin ATPase activity, which ultimately drives cell contractility, MLC phosphorylation was measured by Western blotting following 4 h of treatment with contractility inhibitors. Treatment with Y27, CT04, and ML7 + Y27 significantly reduced phosphorylated MLC, but ML7, blebbistatin, and DMSO vehicle control did not significantly change pMLC signal (Fig. 4F). Since these treatments exhibited differential effects on cell behavior (Fig. 4, A–D), relative pMLC intensity was plotted against experimentally defined cell behaviors for each treatment to understand the relationship among MLC phosphorylation, cell migration, and traction generation. Although ML7 inhibits MLCK, this treatment had no effect on MLC phosphorylation, suggesting that the effects of ML7 on 3D cell motility (Fig. 4A) and traction force generation (Fig. 4C) were due to off-target effects of the drug, rather than a reduction in MLCK-dependent MLC phosphorylation. Alternatively, compensatory MLC phosphorylation under ML7 treatment could have been achieved through signaling pathways other than MLCK (e.g., ROCK), and/or the measured cell responses could exhibit different sensitivity to ML7 dose. Because of this uncertainty and the unknown mechanism of action on the measured behaviors, ML7 data were excluded from correlation analysis. MLC phosphorylation was not correlated with motile fraction within microtracks (R2 = 0.20 for linear fit), motile fraction within 3D matrix (R2 = 0.13 for linear fit), or microtrack migration speed (R2 = 0.01 for linear fit), suggesting that MLC phosphorylation is not a unique regulator of cell migration. However, MLC phosphorylation was positively correlated with traction force generation (R2 = 0.86 for linear fit), indicating that pMLC promotes traction generation. Together, these results suggest that while myosin II activity downstream of MLCK and ROCK mediates migration through 3D matrix and retraction of the cell rear for efficient migration through microtracks, microtrack migration does not require MLC phosphorylation-dependent traction generation and is likely driven by parallel contractility mechanisms.
Actin and MT dynamics are required for microtrack migration.
Cytoskeletal dynamics downstream of the ubiquitous Rho family of GTPases are largely responsible for determination of cell morphology and motility (40, 48). Since coordination of and cross talk between the actin and MT cytoskeletons are particularly important for the initiation and development of cell protrusions (15, 59), the roles of actin and MT cytoskeletal dynamics in microtrack migration were assessed. Cells migrating within collagen microtracks contained primarily cortical F-actin and a polarized MT network aligned with the microtrack (Fig. 5A). Inhibition of MT polymerization with nocodazole and MT depolymerization with paclitaxel were verified by α-tubulin immunofluorescence (Fig. 5A, right). Both treatments significantly decreased motile fraction in 3D matrix and microtracks (Fig. 5B), reduced microtrack migration speed by ∼50% (Fig. 5C), and promoted more rounded cell morphology within microtracks (Fig. 5A, right). Inhibition of F-actin polymerization with cytochalasin D or latrunculin A prevented cell motility in 3D matrix and microtracks (Fig. 5D). To more closely examine the role of actin polymerization in microtrack migration, Lifeact-transfected cells in microtracks were imaged with time-lapse confocal microscopy before and after treatment with cytochalasin D. Migrating cells exhibited distinct front-back polarity of the actin cytoskeleton, with dynamic F-actin-rich protrusive structures at the leading edge (Fig. 5E; see Supplemental Movie S2). Upon treatment with cytochalasin D, cell migration was immediately arrested, and the aligned cortical F-actin network dissociated into small, randomly oriented actin bundles throughout the cell body (Fig. 5E, arrowhead; see Supplemental Movie S2). OrientationJ was used to pseudocolor actin as a function of feature orientation relative to the microtrack (Fig. 5F). To quantify alignment, the mean and standard deviation were extracted from actin orientation distributions (Fig. 5G, vertical solid and dashed lines, respectively). While control and cytochalasin D-treated cells showed alignments centered around 0°, cytochalasin D treatment resulted in a significant increase in distribution variance as determined by Levene's test for equal variances, indicating that treatment significantly reduced actin alignment (Fig. 5H). These results indicate that migration through physiological collagen microtracks 1) is associated with a well-organized and aligned cortical actin cytoskeleton, 2) requires actin polymerization-driven protrusion, and 3) is enhanced by MT dynamics that support directional protrusion and elongation.
Fig. 5.

Requirement for cytoskeletal dynamics during migration through physiological ECM. A: confocal fluorescence images of actin and microtubule (MT) organization in control and nocodazole (Noco)- and paclitaxel (Ptx)-treated microtrack-migrating cells. B: fraction of motile cells within 3D matrix and collagen microtracks treated with nocodazole (a MT polymerization inhibitor) and paclitaxel (a MT stabilizer). C: single-cell speeds of motile cells within collagen microtracks under conditions described in A. Box-and-whisker plots show medians, 25th/75th, and 5th/95th percentiles. D: fraction of motile cells within 3D matrix and collagen microtracks treated with actin polymerization inhibitors cytochalasin D (Cyto D) and latrunculin A (Lat A). †No motile cells detected. E: confocal fluorescence time-lapse image series of Lifeact-transfected cell migrating through microtrack (from Supplemental Movie S2), with Cyto D added as shown. F: selected panels from E pseudocolored with OrientationJ to indicate actin alignment relative to vertical microtrack (0°). Dashed yellow lines indicate microtrack edges. G: actin alignment distributions of control and cytochalasin D-treated cells in F. μ, Distribution means (solid vertical lines); σ, distribution standard deviations (dashed vertical lines). H: actin alignment distributions and descriptive statistics from multiple cells. Scale bars = 25 μm. *P < 0.05.
DISCUSSION
Local tissue invasion is an inherently biophysical process that requires cells to coordinate adhesive, cytoskeletal, contractile, and proteolytic cellular machinery to negotiate matrix and tissue barriers in the tumor microenvironment. Tumor cell migration is highly adaptive, and the specific molecular mechanisms employed by cells to invade are determined largely by physiochemical properties of the stromal ECM, including composition, architecture, and mechanics (62, 66). Here we used an in vitro collagen microtrack platform (33) to define the biochemical and biophysical mechanisms that mediate migration through physiologically relevant cell-scale tracks within interstitial tissue. We demonstrate that mechanisms of migration within collagen microtracks are distinct from those required for migration through 3D collagen matrix. Unlike mesenchymal tumor cell migration in 3D matrix, microtrack migration is matrix density-independent and does not require β1-integrin adhesion. We show that migration through collagen microtracks, but not 3D matrix, can proceed in the absence of pMLC-dependent traction forces and that actin and MT cytoskeletal dynamics regulate cell migration through collagen microtracks and 3D matrix. Together, these findings represent the first description of cell migration mechanisms within physiological collagen microtracks and suggest that once cells reach preformed paths of least resistance within the interstitial stroma, the molecular machinery required for efficient migration may be considerably reduced, effectively lowering the mechanistic threshold for local tissue invasion.
Modeling migration through the stromal ECM.
The stromal ECM surrounding tumors primarily consists of type I collagen organized into interconnected fibrillar networks, the physiochemical properties of which, including stiffness (36, 49, 75), composition (41, 44), and architecture, regulate 3D invasive behavior. While recent work has highlighted the roles of matrix porosity (9, 69), alignment (6, 55), and interfacial features (5) in modulating 3D migration, in vitro models for the study of migration through collagenous ECM generally overestimate matrix uniformity (67), and the effect of local heterogeneity in ECM structure remains poorly understood. Notably, cell-scale gaps in the stromal ECM, including clefts between ECM fibers, interfaces between tissue components, organized networks of ECM pores, and cell-derived proteolytic microtracks, have been identified as conduits for tumor cell migration in vitro (10, 18, 24, 27, 29) and in vivo (2, 11, 26, 62). Here we show that microfabricated collagen tracks closely resemble the channel-like gaps found in the native mammary gland stromal ECM, providing a path of least resistance and highly aligned features that promote rapid, MMP-independent cancer cell migration (33).
Mechanisms of microtrack migration.
During cancer progression, homeostatic ECM maintenance is lost, leading to deregulation of biophysical matrix properties (7, 12, 39). Thus, invasive carcinoma stroma exhibits a desmoplastic response (34) that can include increased collagen I density and cross-linking (36, 54). While migration through 3D matrix is negatively regulated by matrix density due to increased steric resistance, we show here that microtracks overcome this restriction by providing cell-caliber space, enabling matrix-density-independent migration (9, 29, 69). Since changes in matrix density can alter adhesion ligand availability, which has been shown to modulate cell migration in 2D (46) and 3D (74, 75) environments, these results suggest that microtrack migration is ligand-density-independent. Furthermore, while β1-integrin inhibition in 3D matrix results in inefficient, rounded migration, microtrack migration speed is not affected by β1-integrin blocking. Together, these results are consistent with recent findings by Wolf et al. (69) that sufficiently large pores in 3D collagen matrix reduce the need for matrix remodeling and integrin-mediated mechanocoupling during mesenchymal migration. Interestingly, cells treated with the MAb 4B4 oscillate between mesenchymal and amoeboid morphologies during migration, indicating that persistent cell elongation within microtracks requires β1-integrin activity. Thus, while β1-integrin ligation is not required for microtrack migration per se, β1-integrin may function as part of integrin signaling and trafficking pathways that integrate with Rho GTPases, including Rac1 (4), the activity of which promotes actin-dependent membrane protrusion and maintains cell polarity (73). Our findings complement previous work in 3D matrix illustrating that integrin blocking in mesenchymal cells restricts cell elongation and reduces migration speed (69, 75) and indicate that cell spreading and migration in microtracks involve cell-matrix interaction programs that are unique to those employed in 2D and 3D microenvironments.
The application of cytoskeletal tension to the ECM results in traction forces that enable cell and matrix deformation and promote migration through restrictive 3D microenvironments (69, 72). Here we show that inhibition of actomyosin contractility or its upstream regulators MLCK and Rho/ROCK significantly reduces cell traction force and abolishes 3D matrix migration. Excessive cell elongation and indiscriminate generation of branching cytoplasmic projections under contractility inhibition are consistent with release of cortical tension (17) and failure to deform the cell body and/or matrix to squeeze through pores (69). Interestingly, inhibition of single upstream mediators of myosin II activity does not affect microtrack migration, but migration is hindered when myosin II is directly inhibited with blebbistatin or both MLCK and ROCK are inhibited with ML7 and Y27632. These results suggest that the MLCK and Rho/ROCK pathways may provide compensatory activation of myosin II to maintain rapid microtrack migration. Indeed, MLCK and Rho/ROCK have been shown to play distinct spatial and temporal roles during adhesion and migration (43, 61), and while we do not observe such specificity in microtracks here, it remains that these upstream regulators may act on myosin II activity through parallel and compensatory pathways. In support of this hypothesis, the contractility inhibitors used here elicit a variety of effects on MLC phosphorylation, which we show is a poor indicator of migration behavior. Conversely, pMLC is correlated with traction force generation, confounding the link between pMLC-dependent traction generation and migration through 3D matrix and microtracks. While we previously observed traction generation and ECM displacement during microtrack migration (33), we demonstrate here that this behavior is not required for migration, since inhibition of Rho with CT04 reduces traction forces and pMLC by ∼90% but has no effect on microtrack migration. Instead, as indicated by lagging cytoplasmic extensions in migration-deficient cells, actomyosin contractility is likely utilized in microtracks to retract the cell rear, which is a characteristic requirement of mesenchymal-type migration strategies (52, 63). Notably, our group and others have shown that cell contractility is required for matrix reorganization (6, 55) and microtrack generation (10, 24), but not perception and response to matrix structure. The present results are consistent with this conclusion and, together, suggest that preformed space in the ECM alleviates the need for actomyosin-based matrix remodeling and pMLC-dependent traction generation in cancer cell motility, which is likely under the control of a number of parallel cell contractility pathways.
Our results indicate that cell-matrix mechanocoupling, which is critical for cancer cell migration in both planar and 3D settings (42, 69), is largely dispensable for migration within collagen microtracks. Alternatively, we show that actin and MT cytoskeletal dynamics mediate protrusive, polarized microtrack migration. Cells within microtracks exhibit a cortical actin network and polarized MT cytoskeleton, which are disrupted by inhibitors of actin polymerization and MT dynamics, respectively. Inhibition of MT polymerization (nocodazole) or depolymerization (paclitaxel) limits cell spreading and reduces motility in 3D matrix and microtracks, which is consistent with previous work indicating that MT dynamics enable 3D cell spreading (32) and promote elongated 3D cell protrusions (30, 59). As previously suggested, MTs may provide means for intracellular delivery of molecular cargo needed for elongation and stabilization of actin-based protrusions (59). In support of this hypothesis, slower, more rounded microtrack migration can continue with inhibition of MT dynamics, but migration is immediately arrested with inhibition of actin polymerization. Treatment with cytochalasin D terminates growth of actin-rich protrusions at the leading edge and results in the loss of cortical actin localization and alignment, suggesting that actin dynamics contribute to both protrusion and maintenance of cortical actomyosin tension, which drive microtrack migration (17, 45). Together, these results indicate that pseudopodial protrusions at the leading edge, which are initiated by actin polymerization and supported by MTs, provide the primary impetus for physiological collagen microtrack migration.
The ability to readily adapt cell phenotype (i.e., phenotypic plasticity) is a characteristic feature of many cancer cells (20, 51, 58). It is presumed that such plasticity is advantageous to cancer cells, as it enables cells to continue their objective (to invade) under a variety of microenvironmental conditions. Here we directly demonstrate that MDA-MB-231 cells adapt their mode of migration upon confronting different structural microenvironments (3D matrix vs. microtracks) and when challenged pharmacologically. This well-documented switch of migration strategies by tumor cells can be induced by both cellular and extracellular influences (22, 66) and is accompanied by a shift in molecular mechanisms underlying migration (47, 57). Briefly, amoeboid migration is generally observed in low-density ECM, where rounded, blebbing cells use low cell-ECM adhesion and strong propulsive forces and cytoskeletal contraction to negotiate matrix. Conversely, mesenchymal migration occurs in low- to high-density ECM, where elongated cells utilize step-wise and cyclical protrusion, adhesion, traction generation, and tail retraction to move through matrix (70). Here we show that microtrack migration can exhibit elements of both mesenchymal and amoeboid migration and further demonstrate that microtracks provide a microenvironmental context that enables cells to adapt their migration mode and, thus, potentially evade inhibitors of cell migration (e.g., integrin-blocking and cell contractility inhibitors). The oscillatory migration strategy exhibited by 4B4-treated cells within microtracks provides direct evidence of this dynamic switching and contrasts with the stable mesenchymal → amoeboid transition exhibited by cells treated with integrin and MMP inhibitors in 3D matrix (69, 70, 75). Interestingly, the metric of motile fraction emerges as an indicator of migration plasticity, since it describes the subset of the cell population that is able to move under a given experimental or microenvironmental condition. Thus, as motile fraction approaches 0 (e.g., high 3D matrix density, inhibition of cell contractility in 3D matrix, inhibition of actin polymerization), cells have exhausted options for adaptation and cease migrating. Critically, our findings that inhibition of MT dynamics or actin polymerization arrest cell migration are consistent with literature suggesting that Rho GTPases and cytoskeletal dynamics are required to coordinate migration mode plasticity (47, 58).
Ultimately, it will be necessary to transition findings back to an in vivo tumor/stroma model, but, in light of results of our present study and those reported by others (62) highlighting the critical role of the microenvironment in regulating mechanisms of cell motility, this transition will require an increasingly thorough understanding of the in vivo stromal context. Since the composition and structure of in vivo stroma remain only marginally understood (2), rationally designed in vitro models of the tumor microenvironment represent an effective approach to defining the mechanisms of cell-matrix interactions during tumor invasion. Importantly, our collagen microtrack migration model differs from microfluidic models of confined migration fabricated from rigid, nonporous PDMS (60) and polyacrylamide (50), in that migrating cells are bounded by physiological, fibrillar collagen matrix on all sides within our system (33). Recently, several independent studies showed that confinement within narrow microfluidic PDMS or polyacrylamide channels eliminates the need for cell contractility and actin polymerization in migration (3, 50, 60). While these findings are in contrast to our results, they are consistent with a computational model of confined dendritic cell migration wherein a pressure buildup in the cell that is caused by the confinement induced by rigid channel walls is sufficient to produce forward movement (28). This model shows that migration under confinement relies on increasing friction caused by the pressure buildup and is independent of substrate adhesion and cell contractility. An alternative model of confined cell migration suggests that water permeation across cell membranes leads to polarized changes in cell volume and migration in the absence of actin polymerization (60). In the context of these experimental and theoretical models, our results suggest that collagen microtracks, being inherently compliant and porous substrates, may not be able to induce mechanical and osmotic pressures within the cell and, therefore, require a cell to utilize actin polymerization for migration. This difference highlights the need to carefully consider all aspects of the microenvironment through which cells migrate when designing experimental models and interpreting results.
In summary, this study is the first to systematically analyze the mechanisms guiding cell migration within physiological collagen microtracks. Using a micropatterned collagen microtrack platform that recapitulates matrix-free space within the stromal ECM, we demonstrate that adhesion and contractility mechanisms uniquely regulate migration through 3D collagen matrix and collagen microtracks. Microtrack migration is insensitive to matrix density and is independent of cell-matrix mechanocoupling, both of which are significant regulators of migration within 3D matrix. Actin and MT cytoskeletal dynamics at the leading edge and actomyosin contractility at the cell rear promote morphological polarity, which enables rapid, MMP-independent migration through microtracks. Collectively, our data provide insight into the unique collection of mechanisms required by tumor cells to migrate through preexisting spaces in the stromal ECM and indicate that path-following migration during leader-follower collective invasion and transit along preformed paths of least resistance are less mechanistically demanding strategies for local tissue invasion.
GRANTS
This work was supported by the Cornell Center on the Microenvironment and Metastasis through National Cancer Institute Grant U54 CA-143876 and National Science Foundation-National Institutes of Health Physical and Engineering Sciences in Oncology Grant 1233827 to C. A. Reinhart-King and also by National Science Foundation Graduate Research Fellowships to S. P. Carey, A. Rahman, and C. M. Kraning-Rush.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.P.C., R.M.W., and C.A.R.-K. developed the concept and designed the research; S.P.C., A.R., C.M.K.-R., B.R., S.S., and O.M.T. performed the experiments; S.P.C., A.R., C.M.K.-R., S.S., and O.M.T. analyzed the data; S.P.C. and C.A.R.-K. interpreted the results of the experiments; S.P.C. prepared the figures; S.P.C. drafted the manuscript; S.P.C. and C.A.R.-K. edited and revised the manuscript; S.P.C., A.R., C.M.K.-R., B.R., S.S., O.M.T., R.M.W., and C.A.R.-K. approved the final version of the manuscript.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the use of equipment and resources at the Cornell NanoScale Science and Technology Facility.
Footnotes
This article is the topic of an Editorial Focus by Dominika A. Rudzka and Michael F. Olson (55a).
REFERENCES
- 1.Alcoser TA, Bordeleau F, Carey SP, Lampi MC, Kowal DR, Somasegar S, Varma S, Shin SJ, Reinhart-King CA. Probing the biophysical properties of primary breast tumor-derived fibroblasts. Cell Mol Bioeng. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander S, Weigelin B, Winkler F, Friedl P. Preclinical intravital microscopy of the tumour-stroma interface: invasion, metastasis, and therapy response. Curr Opin Cell Biol 25: 659–671, 2013. [DOI] [PubMed] [Google Scholar]
- 3.Balzer EM, Tong Z, Paul CD, Hung WC, Stroka KM, Boggs AE, Martin SS, Konstantopoulos K. Physical confinement alters tumor cell adhesion and migration phenotypes. FASEB J 26: 4045–4056, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berrier AL. The integrin beta tail is required and sufficient to regulate adhesion signaling to Rac1. J Cell Sci 115: 4285–4291, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Bordeleau F, Tang LN, Reinhart-King CA. Topographical guidance of 3D tumor cell migration at an interface of collagen densities. Phys Biol 10: 065004, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brownfield DG, Venugopalan G, Lo A, Mori H, Tanner K, Fletcher DA, Bissell MJ. Patterned collagen fibers orient branching mammary epithelium through distinct signaling modules. Curr Biol 23: 703–709, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carey SP, Charest JM, Reinhart-King CA. Forces during cell adhesion and spreading: implications for cellular homeostasis. In: Cellular and Biomolecular Mechanics and Mechanobiology, edited by Gefen A. Berlin: Springer-Verlag, 2011, p. 29–69. [Google Scholar]
- 8.Carey SP, D'Alfonso TM, Shin SJ, Reinhart-King CA. Mechanobiology of tumor invasion: engineering meets oncology. Crit Rev Oncol Hematol 83: 170–183, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carey SP, Kraning-Rush CM, Williams RM, Reinhart-King CA. Biophysical control of invasive tumor cell behavior by extracellular matrix microarchitecture. Biomaterials 33: 4157–4165, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carey SP, Starchenko A, McGregor AL, Reinhart-King CA. Leading malignant cells initiate collective epithelial cell invasion in a three-dimensional heterotypic tumor spheroid model. Clin Exp Metastasis 30: 615–630, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Condeelis J, Segall JE. Intravital imaging of cell movement in tumours. Nat Rev Cancer 3: 921–930, 2003. [DOI] [PubMed] [Google Scholar]
- 12.Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech 4: 165–178, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dembo M, Wang YL. Stresses at the cell-to-substrate interface during locomotion of fibroblasts. Biophys J 76: 2307–2316, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deome KB, Faulkin LJ, Bern HA, Blair PB. Development of mammary tumors from hyperplastic alveolar nodules transplanted into gland-free mammary fat pads of female C3H mice. Cancer Res 19: 515–520, 1959. [PubMed] [Google Scholar]
- 15.Etienne-Manneville S. Actin and microtubules in cell motility: which one is in control? Traffic 5: 470–477, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Fidler IJ. Tumor heterogeneity and the biology of cancer invasion and metastasis. Cancer Res 38: 2651–2660, 1978. [PubMed] [Google Scholar]
- 17.Fischer RS, Gardel M, Ma X, Adelstein RS, Waterman CM. Local cortical tension by myosin II guides 3D endothelial cell branching. Curr Biol 19: 260–265, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fisher KE, Sacharidou A, Stratman AN, Mayo AM, Fisher SB, Mahan RD, Davis MJ, Davis GE. MT1-MMP- and Cdc42-dependent signaling co-regulate cell invasion and tunnel formation in 3D collagen matrices. J Cell Sci 122: 4558–4569, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flesken-Nikitin A, Williams RM, Zipfel WR, Webb WW, Nikitin AY. Use of multiphoton imaging for studying cell migration in the mouse. Methods Mol Biol 294: 335–345, 2005. [DOI] [PubMed] [Google Scholar]
- 20.Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147: 992–1009, 2011. [DOI] [PubMed] [Google Scholar]
- 21.Friedl P, Wolf K. Tube travel: the role of proteases in individual and collective cancer cell invasion. Cancer Res 68: 7247–7249, 2008. [DOI] [PubMed] [Google Scholar]
- 22.Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol 188: 11–19, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friedl P. Dynamic imaging of cellular interactions with extracellular matrix. Histochem Cell Biol 122: 183–190, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol 9: 1392–1400, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Goetz JG, Minguet S, Navarro-Lérida I, Lazcano JJ, Samaniego R, Calvo E, Tello M, Osteso-Ibáñez T, Pellinen T, Echarri A, Cerezo A, Klein-Szanto AJ, Garcia R, Keely PJ, Sánchez-Mateos P, Cukierman E, Del Pozo MA. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 146: 148–163, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gritsenko PG, Ilina O, Friedl P. Interstitial guidance of cancer invasion. J Pathol 226: 185–199, 2012. [DOI] [PubMed] [Google Scholar]
- 27.Guiet R, Van Goethem E, Cougoule C, Balor S, Valette A, Al Saati T, Lowell CA, Le Cabec V, Maridonneau-Parini I. The process of macrophage migration promotes matrix metalloproteinase-independent invasion by tumor cells. J Immunol 187: 3806–3814, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hawkins RJ, Piel M, Joanny JF, Prost J. Pushing off the walls: a mechanism of cell motility in confinement. Phys Rev Lett 102: 058103, 2009. [DOI] [PubMed] [Google Scholar]
- 29.Ilina O, Bakker GJ, Vasaturo A, Hoffman RM, Friedl P. Two-photon laser-generated microtracks in 3D collagen lattices: principles of MMP-dependent and -independent collective cancer cell invasion. Phys Biol 8: 015010, 2011. [DOI] [PubMed] [Google Scholar]
- 30.Kikuchi K, Takahashi K. WAVE2- and microtubule-dependent formation of long protrusions and invasion of cancer cells cultured on three-dimensional extracellular matrices. Cancer Sci 99: 2252–2259, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kraning-Rush CM, Carey SP, Califano JP, Reinhart-King CA. Quantifying traction stresses in adherent cells. In: Methods in Cell Biology, edited by Asthagiri AR, Arkin A. New York: Elsevier, p. 139–178. [DOI] [PubMed] [Google Scholar]
- 32.Kraning-Rush CM, Carey SP, Califano JP, Smith BN, Reinhart-King CA. The role of the cytoskeleton in cellular force generation in 2D and 3D environments. Phys Biol 8: 015009, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kraning-Rush CM, Carey SP, Lampi MC, Reinhart-King CA. Microfabricated collagen tracks facilitate single cell metastatic invasion in 3D. Integr Biol 5: 606–616, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar V, Abbas A, Fausto N, Aster J. Robbins and Contran Pathologic Basis of Disease (8th ed.). Philadelphia: Elsevier Saunders, 2010. [Google Scholar]
- 35.Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell 84: 359–369, 1996. [DOI] [PubMed] [Google Scholar]
- 36.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139: 891–906, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liotta LA, Stetler-Stevenson WG. Tumor invasion and metastasis: an imbalance of positive and negative regulation. Cancer Res 51: 5054s–5059s, 1991. [PubMed] [Google Scholar]
- 38.Liotta LA, Tryggvason K, Garbisa S, Hart I, Foltz CM, Shafie S. Metastatic potential correlates with enzymatic degradation of basement membrane collagen. Nature 284: 67–68, 1980. [DOI] [PubMed] [Google Scholar]
- 39.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol 196: 395–406, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature 461: 99–103, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maller O, Hansen KC, Lyons TR, Acerbi I, Weaver VM, Prekeris R, Tan AC, Schedin P. Collagen architecture in pregnancy-induced protection from breast cancer. J Cell Sci 126: 4108–4120, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mierke CT, Rösel D, Fabry B, Brábek J. Contractile forces in tumor cell migration. Eur J Cell Biol 87: 669–676, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morin TR, Ghassem-Zadeh SA, Lee J. Traction force microscopy in rapidly moving cells reveals separate roles for ROCK and MLCK in the mechanics of retraction. Exp Cell Res 326: 280–294, 2014. [DOI] [PubMed] [Google Scholar]
- 44.Nguyen-Ngoc KV, Cheung KJ, Brenot A, Shamir ER, Gray RS, Hines WC, Yaswen P, Werb Z, Ewald AJ. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc Natl Acad Sci USA 109: E2595–E2604, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olson MF, Sahai E. The actin cytoskeleton in cancer cell motility. Clin Exp Metastasis 26: 273–287, 2009. [DOI] [PubMed] [Google Scholar]
- 46.Palecek S, Loftus J, Ginsberg MH, Lauffenburger DA, Horwitz AF. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature 385: 537–540, 1997. [DOI] [PubMed] [Google Scholar]
- 47.Panková K, Rösel D, Novotný M, Brábek J. The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell Mol Life Sci 67: 63–71, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parri M, Chiarugi P. Rac and Rho GTPases in cancer cell motility control. Cell Commun Signal 8: 23, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Tensional homeostasis and the malignant phenotype. Cancer Cell 8: 241–254, 2005. [DOI] [PubMed] [Google Scholar]
- 50.Pathak A, Kumar S. Independent regulation of tumor cell migration by matrix stiffness and confinement. Proc Natl Acad Sci USA 109: 10334–10339, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Petersen OW, Lind Nielsen H, Gudjonsson T, Villadsen R, Rønnov-Jessen L, Bissell MJ. The plasticity of human breast carcinoma cells is more than epithelial to mesenchymal conversion. Breast Cancer Res 3: 213–217, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poincloux R, Collin O, Lizárraga F, Romao M, Debray M, Piel M, Chavrier P. Contractility of the cell rear drives invasion of breast tumor cells in 3D Matrigel. Proc Natl Acad Sci USA 108: 1943–1948, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med 4: 38, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene 28: 4326–4343, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Provenzano PP, Inman DR, Eliceiri KW, Trier SM, Keely PJ. Contact guidance mediated three-dimensional cell migration is regulated by Rho/ROCK-dependent matrix reorganization. Biophys J 95: 5374–5384, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55a.Rudzka DA, Olson MF. Microtrack migration: insights into 3D cell motility. Focus on “Comparative mechanisms of cancer cell migration through 3D matrix and physiological microtracks.” Am J Physiol Cell Physiol (January 22, 2015). doi: 10.1152/ajpcell.00016.2015. [DOI] [PubMed] [Google Scholar]
- 56.Sabeh F, Shimizu-Hirota R, Weiss SJ. Protease-dependent versus -independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. J Cell Biol 185: 11–19, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol 5: 711–719, 2003. [DOI] [PubMed] [Google Scholar]
- 58.Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ. Rac activation and inactivation control plasticity of tumor cell movement. Cell 135: 510–523, 2008. [DOI] [PubMed] [Google Scholar]
- 59.Schoumacher M, Goldman RD, Louvard D, Vignjevic DM. Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J Cell Biol 189: 541–556, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stroka KM, Jiang H, Chen SH, Tong Z, Wirtz D, Sun SX, Konstantopoulos K. Water permeation drives tumor cell migration in confined microenvironments. Cell 157: 611–623, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Totsukawa G, Yamakita Y, Yamashiro S, Hartshorne DJ, Sasaki Y, Matsumura F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J Cell Biol 150: 797–806, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tozluoǧlu M, Tournier AL, Jenkins RP, Hooper S, Bates PA, Sahai E. Matrix geometry determines optimal cancer cell migration strategy and modulates response to interventions. Nat Cell Biol 15: 751–762, 2013. [DOI] [PubMed] [Google Scholar]
- 63.Vicente-Manzanares M, Koach MA, Whitmore L, Lamers ML, Horwitz AF. Segregation and activation of myosin IIB creates a rear in migrating cells. J Cell Biol 183: 543–554, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weigelin B, Bakker GJ, Friedl P. Principles of interface guidance and microvesicle dynamics: intravital third harmonic generation microscopy of collective melanoma cell invasion. Intravital 1: 32–43, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams RM, Ellenson LH, Connolly DC, Hamilton TC, Nikitin AY, Zipfel WR, Flesken-Nikitin A. Strategies for high-resolution imaging of epithelial ovarian cancer by laparoscopic nonlinear microscopy. Transl Oncol 3: 181–194, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Willis AL, Sabeh F, Li XY, Weiss SJ. Extracellular matrix determinants and the regulation of cancer cell invasion stratagems. J Microsc 251: 250–260, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wolf K, Alexander S, Schacht V, Coussens LM, von Andrian UH, van Rheenen J, Deryugina E, Friedl P. Collagen-based cell migration models in vitro and in vivo. Semin Cell Dev Biol 20: 931–941, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wolf K, Friedl P. Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends Cell Biol 21: 736–744, 2011. [DOI] [PubMed] [Google Scholar]
- 69.Wolf K, te Lindert M, Krause M, Alexander S, te Riet J, Willis AL, Hoffman RM, Figdor CG, Weiss SJ, Friedl P. Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol 201: 1069–1084, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, Strongin AY, Bröcker EB, Friedl P. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J Cell Biol 160: 267–277, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, Stack MS, Friedl P. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol 9: 893–904, 2007. [DOI] [PubMed] [Google Scholar]
- 72.Wyckoff JB, Pinner SE, Gschmeissner S, Condeelis JS, Sahai E. ROCK- and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr Biol 16: 1515–1523, 2006. [DOI] [PubMed] [Google Scholar]
- 73.Yamazaki D, Kurisu S, Takenawa T. Involvement of Rac and Rho signaling in cancer cell motility in 3D substrates. Oncogene 28: 1570–1583, 2009. [DOI] [PubMed] [Google Scholar]
- 74.Zaman MH, Kamm RD, Matsudaira P, Lauffenburger DA. Computational model for cell migration in three-dimensional matrices. Biophys J 89: 1389–1397, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zaman MH, Trapani LM, Sieminski AL, MacKellar D, Gong H, Kamm RD, Wells A, Lauffenburger DA, Matsudaira P. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc Natl Acad Sci USA 103: 10889–10894, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zipfel WR, Williams RM, Webb WW. Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotechnol 21: 1369–1377, 2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.