

Abstract
Diseases of striated muscle linked to intermediate filament (IF) proteins are associated with defects in the organization of the contractile apparatus and its links to costameres, which connect the sarcomeres to the cell membrane. Here we study the role in skeletal muscle of synemin, a type IV IF protein, by examining mice null for synemin (synm-null). Synm-null mice have a mild skeletal muscle phenotype. Tibialis anterior (TA) muscles show a significant decrease in mean fiber diameter, a decrease in twitch and tetanic force, and an increase in susceptibility to injury caused by lengthening contractions. Organization of proteins associated with the contractile apparatus and costameres is not significantly altered in the synm-null. Elastimetry of the sarcolemma and associated contractile apparatus in extensor digitorum longus myofibers reveals a reduction in tension consistent with an increase in sarcolemmal deformability. Although fatigue after repeated isometric contractions is more marked in TA muscles of synm-null mice, the ability of the mice to run uphill on a treadmill is similar to controls. Our results suggest that synemin contributes to linkage between costameres and the contractile apparatus and that the absence of synemin results in decreased fiber size and increased sarcolemmal deformability and susceptibility to injury. Thus synemin plays a moderate but distinct role in fast twitch skeletal muscle.
Keywords: cytoskeleton, costameres, elastimetry, biomechanical properties, desmin
a large number of human diseases, ranging from skin disorders (51), to premature aging (34, 64), to skeletal and cardiac myopathies (19, 37), are linked to mutations in genes encoding intermediate filament (IF) proteins (25). Since the discovery of desmin, the major IF protein of striated muscle (49), its role and that of other IF proteins in muscle have been of keen interest to many laboratories (reviewed in 16). Our research on muscle IF proteins has addressed their role in force transmission and in organizing the sarcoplasm and stabilizing it against injury during force generation.
Force generated in muscle can be transmitted radially, from the myofibrils, across the sarcolemma to the extracellular matrix and neighboring cells (11, 67). As a result, a significant amount of force is exerted on the sarcolemma and the structures that link it to the underlying contractile apparatus and the extracellular matrix. The ability of the sarcolemma with the underlying contractile apparatus to withstand the distortions caused during isotonic contractions (2, 11, 12) depends on specialized structures, termed “costameres” (66), that mediate the radial transmission of force across the sarcolemma (11). Costameres are structures at the sarcolemma of striated muscle fibers that align circumferentially around the Z disks and the M bands of the nearest myofibrils, and longitudinally, to form a rectilinear sarcolemmal network comprised of integral membrane proteins (such as dystroglycan, the sarcoglycans, and Na-K-ATPase), proteins of the extracellular matrix (such as laminin and collagen IV), and proteins of the membrane-associated cytoskeleton (such as dystrophin, syntrophins, spectrin, ankyrin, and vinculin) (12, 23, 26, 41, 48, 66, 71, 76, 82, 87, 93, 95). Many muscular dystrophies are attributable to mutations in costameric proteins, especially to mutations of proteins of the dystrophin-glycoprotein complex (2, 6, 10, 12, 15, 27, 42, 63, 74, 95).
Costameres are linked to the superficial myofibrils of the contractile apparatus by microfilaments, comprised at least in part of γ-actin (26, 43, 82), and by IFs, including desmin and several forms of keratin (4, 5, 7, 10, 49, 54, 68, 73, 88, 92). Elimination of these proteins by homologous recombination leads to myopathies.
IFs are cytoskeletal and nucleoskeletal proteins that play a key role in determining cell shape, cell function, and nuclear architecture (33, 37), including the maintenance of the overall integrity of cytoplasm (18, 44). IFs also play a role in the modulation and propagation of signals within cells and in regulating cell motility (34, 36, 64, 65). IF proteins are encoded by more than 65 different genes (39) and fall into 6 different protein classes (25). Four of these are localized in the cytoplasm (types I–IV), one primarily in the nucleus (type V, the nuclear lamins), and one in lens and nerve cells (type VI, phakinin, nestin, and filensin). Mutations in the lamins are associated with Emery-Dreifuss and Limb-girdle muscular dystrophy type 1B (60). Nestin (type VI) plays a role in the accumulation of acetylcholine receptors at the neuromuscular junction and in the development of myogenic cells (86, 94). Muscles lacking keratin 19 (type I) or desmin (type III) are mildly myopathic and are weaker and more easily injured than wild-type (WT) (1, 54, 88). The role in muscle of synemin, a type IV IF protein that can associate with IFs formed by desmin or keratins (4, 56, 59, 91), has not yet been fully explored.
Like other IF proteins, synemin consists of NH2- and COOH-terminal domains flanking a central α-helical rod domain (3, 50), but it requires other IF proteins, such as desmin, to form heterofilaments (91). Unlike desmin and keratins, which are small IF proteins (40–70 kDa), synemin is large, due to its long COOH-terminal domain, and it can be expressed in either α (∼210 kDa)- or β (∼180 kDa)-isoforms (3, 7, 8), generated by alternative splicing. In addition to desmin, synemin can associate with keratin IFs and with the costameric proteins α-dystrobrevin, dystrophin, utrophin, vinculin, and talin, as well as with α-actinin at Z disks (4, 17, 35, 40, 59, 90, 91). Synemin therefore has the potential to link costameres to the contractile apparatus at Z disks. In cardiomyocytes, α-synemin stabilizes junctional complexes, whereas β-synemin appears to mediate the association of desmin with Z disks (56). Synemin is also an A-kinase anchoring protein (AKAP) that is involved in regulating the phosphorylation of proteins at the sarcolemma and Z disks via protein kinase A (80, 83). Thus synemin may have both structural and signaling functions in muscle.
We have studied mice lacking synemin to assess the consequences of its absence on the structure and function of skeletal muscle. Our results suggest that, although synemin's role in maintaining the organization of the contractile apparatus and of costameres is minimal, fast twitch skeletal muscle lacking synemin (synm-null) is myopathic. In particular, synm-null skeletal muscle is smaller than control and is more susceptible to injury. Furthermore, costameres are more weakly associated with the underlying contractile apparatus than in controls, and the sarcolemma, once isolated from contractile structures, is more easily damaged by suction pressure. We conclude that synemin plays a significant role in the physiology and biomechanics of skeletal muscle.
MATERIALS AND METHODS
Animals.
C57Bl/6 mice null for synemin were generated by homologous recombination [Synmtm1.1(KOMP)Vlcg; ID: MGI:2661187; The International Mouse Phenotyping Consortium]. Mice homozygous were bred and genotyped from tail snips (38) using the DNeasy (Qiagen, Valencia, CA) kit. The PCR reaction used the Accuprime PFX kit (Invitrogen, Carlsbad, CA). Specific forward and reverse primers were used to screen the DNA of each sample for the β-galactosidase insertion and the synemin sequence, with the following primers: for synemin forward: 5′-AAACACTCCCCCATAGATCC-3′; reverse: 5′-GCACCCCTCTTTCTTTTACAC-3′; and for β-galactosidase forward: 5′-GGTAAACTGGCTCGGATTAGGG-3′; reverse: 5′-TTGACTGTAGCGGCTGATGTTG-3′. PCR products for control and synm-null yielded bands of 405 and 210 bp, respectively. We used male age-matched C57Bl/6 control (WT) and synm-null mice from 12 to 16 wk of age for all studies reported here, unless otherwise stated. Animals were anesthetized with 2.5% isofluorane inhalation (VetEquip, Pleasanton, CA) with oxygen at 0.8 l/min at 14.7 psi (21°C) and were euthanized by cervical dislocation. All of our protocols were approved by the Institutional Animal Care Committee of the University of Maryland School of Medicine.
RT-PCR.
mRNA was prepared with TRIzol (Invitrogen) and used in the SuperScript First Strand Synthesis System (Invitrogen). PCR was used to probe the reverse-transcribed mRNAs for synemin sequences (GenBank), using the following primers: for α-synemin, forward: 5′-ATGGTTATGGTGGAAGGTTC-3′; reverse: 5′-CGGGTCCAGCTGCGGATCGC-3′; and for β-synemin, forward: 5′-GTGGACGTAAAGAAAGTCCA-3′; reverse: 5′-GGAGGCCCCAGAGCCAGTGC-3′.
Cell culture and transfection.
COS7 cells (Sigma-Aldrich, St. Louis, MO) were cultured in F-12:DMEM medium (American Type Culture Collection, Manassas, VA) supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 1% penicillin/streptomycin (Thermo-Fischer Scientific, Rockville, MD). We used 12 μg of either α- or β-synemin cDNA and 24 μl of Lipofectamine 2000 reagent (Invitrogen) for transfecting cells at ∼90% confluency. Cells were harvested at 48 h posttransfection and stored at −80°C for further analysis.
Antibody generation.
The anti-synemin antibody was made against GST fused to the COOH-terminal 108 amino acid residues of Mus musculus synemin (21st Century Biochemicals, Marlborough, MA). The antibody was purified on Sepharose beads activated with CNBr (GE Healthcare, Laurel, MD) and coupled covalently to a fusion protein of MBP and the last 108 amino acids of synemin. This antibody recognizes two proteins at 210 and 180 kDa from transfected COS7 cells, which correspond to α- and β-synemin, respectively.
Western blotting.
Tibialis anterior (TA) muscles from synm-null and control mice were dissected, snap frozen, and stored at −80° C. Muscles were homogenized in a TissueLyser (Qiagen, Hilden, Germany) in buffer placed on ice [Mammalian Protein Extraction Reagent (Pierce, Rockford, IL), 300 mM NaCl, 40 mM EDTA, 0.6 M KCl, 10 mM PBS, 6 M urea, 1% Triton X-100, and 10 μg/ml protease inhibitor cocktail (Sigma-Aldrich)]. Lysates were subjected to centrifugation at 4°C at 10,000 g for 15 min. Aliquots containing 20 μg of supernatant fraction were separated by electrophoresis on 4–15% gradient acrylamide gels (Bio-Rad, Hercules, CA), and proteins were transferred at 4°C to a nitrocellulose membrane (Invitrogen) in transfer buffer with 20% methanol at a voltage of 15 V overnight. The membrane was incubated with “blocking buffer” (0.1% Tween-20, 5% BSA in TBS pH 7.4) for 30 min and then incubated with anti-synemin (1:25) or anti-GAPDH (1:20,000; Ambion, Austin, TX) antibody, diluted in blocking buffer, overnight at 4°C. The membrane was washed in blocking buffer for 30 min at room temperature and then incubated for 1 h in either horseradish peroxidase (HRP)-conjugated anti-rabbit IgG, for synemin, or HRP-conjugated anti-mouse IgG, for GAPDH (1:20,000; KPL, Gaithersburg, MD). After being washed with TBS/0.1% Tween-20 for 30 min, the bands were visualized by chemiluminiscence (Super Signal Chemiluminiscent Substrate; Thermo Scientific Waltham, MA).
Immunofluorescence labeling.
TA, extensor digitorum longus (EDL), quadriceps, and soleus muscles were perfusion fixed in situ with 4% paraformaldehyde and were then dissected, snap frozen, cryosectioned, and labeled by immunofluorescence, as described previously (62). We used chicken antibodies against βI-spectrin (92) to label costameres. Rabbit antibodies were to synemin (diluted 1:25) and dystrophin (Thermo-Scientific; diluted 1:200); mouse antibodies were to dystrophin (Vector Laboratories, Burlingame, CA; diluted 1:50), obscurin (46), α-actinin (Sigma-Aldrich; diluted 1:50), and slow myosin heavy chain (MHC; Sigma-Aldrich; diluted 1:1,000); and goat antibodies were to desmin (Santa Cruz Biotechnology; diluted 1:200). Frozen muscle tissue was sectioned at 16 μm in thickness and mounted with DAPI (Vector Laboratories) to label nuclei. Immunostaining was visualized under confocal optics (LSM 510; Carl Zeiss, Oberkochen, Germany) with a ×40/NA 1.4 or ×63/NA 1.4 objective with the pinhole set from 75 to 101 and the Airy unit <1. The relative sizes of myofibers were determined from images of frozen cross sections labeled with dystrophin and DAPI. The minimal Feret's diameter was measured (13) with a digital caliper from nine randomly selected visual fields in cross sections of at least three TA, EDL, quadriceps, and soleus muscles per group (62).
Hematoxylin and eosin staining.
Cross sections 12–16 μm in thickness from the midbelly of unfixed, snap frozen TA muscles were fixed with cold acetone, air dried, immersed in Harris hematoxylin (Sigma-Aldrich) for 3 min, and then immersed in Scotts Bluing reagent (Thermo Fisher Scientific) for 1 min, followed by three rapid dips in Wright eosin staining and finally in 95% ethanol, with rinsing steps with room temperature tap water between solution changes. Coverslips were mounted with Permount mounting medium (Thermo Fisher Scientific). The sections were observed under light microscopy (Zeiss Axioscope, ×20 objective and ×10 eyepiece; Carl Zeiss), and digital images of nine randomly selected visual fields across the sections of five TA muscles per group were obtained for analysis of necrotic fibers. Necrotic fibers were identified based on their pale appearance and fragmented myoplasm, with or without invasion by mononuclear cells (77). The number of centrally nucleated fibers (CNFs) was obtained by counting myofibers with central nuclei in frozen sections stained with hematoxylin and eosin or double labeled with DAPI and antibodies to dystrophin in TA, EDL, quadriceps, and soleus. The counts of necrotic fibers and CNFs are presented as means ± SE. Similar methods were applied to EDL, quadriceps, and soleus muscles in some experiments.
Ultrastructure.
Perfusion-fixed TA muscles were removed and incubated overnight in 2% glutaraldehyde, 2 mg/ml tannic acid, and 0.2 M cacodylate pH 7.2, dehydrated, postfixed in 1% osmium tetroxide in 0.5 M acetate buffer, dehydrated, embedded in Spurr's resin, sectioned for electron microscopy at 90 nm, and viewed with a FEI Tecnai T12 (FEI, Hillsboro, OR). Images were taken on a CCD Camera AMT (Woburn, MA) at 80 kV. Magnification was ×4,400.
Biomechanical properties.
The tension and pressure at which the cell membrane separated from nearby myofibrils in single isolated fibers were measured as described in detail previously (29, 30). The EDL muscles from synm-null (n = 18) and control mice (n = 20) were dissected in Kreb's solution (in mM: 135 NaCl, 5 KCl, 1 MgCl2, 15 NaHCO3, 11 glucose, 1 Na2HPO4, and 2.5 CaCl2, equilibrated with 95% O2-5% CO2 to a pH of 7.0) and placed in relaxing solution [in mM: 185 K (C2H5COO), 2.5 Mg (CH3COO)2 4H2O, 10 imidazole propionate, 2.5 Na2ATP, and 5 EGTA pH 7.1]. A single fiber attached to its tendons was mechanically isolated from each muscle, clamped by its tendons, and stretched to the desired average sarcomere length (SL) and measured first by eye through a calibrated micrometer positioned inside a ×25 eyepiece and then with ImageJ software (National Institutes of Health, Bethesda, MD). A suction pipette was placed on the surface of the fiber, and suction pressure was applied. The pipette was attached to an open, U-shaped manometer system filled with mercury. The manometer was connected at one end to the pipette and at the other to a syringe piston. We followed a model (75) that analyses the elastic behavior of the distortion and tension lines formed by myofibrils and the sarcolemma in response to suction pressure, P, applied over a small area. P applied to the outer surface of the myofiber was calculated from P = ρghman, where ρ = 13.5 g/cm3, g = 981 cm/s2, and hman is the difference of levels in the manometer relative to P = 0. By applying P through the pipette, we induced formation of a bleb of variable height (h), from which we calculated a set of mechanical properties, including displacement of the bleb as a function of pressure, pressure at which the cell membrane separated from nearby myofibrils (Psep), and the pressure at which the sarcolemma burst (Pbursting). For each applied P, the height of the bleb (h) formed inside the suction pipette was measured in micrometers. Photomicrographs of the bleb were taken with a 10.2 Mpix digital camera (Sony α-330, Tokyo, Japan) through a ×10 eyepiece and ×40, N.A. 0.75, water immersion objective, and analyzed with ImageJ. Experiments were done at 8°C, controlled by a thermistor connected to a temperature controller (Yellow Springs Instrument, Ohio, OH), which maintained the temperature through a two Peltier modulus system (Midland Ross, Cambridge, MA). Tension of the whole system, γtotal, the maximal tension sustained by the costameres, γcostameres, and the maximal sarcolemmal tension γsarcolemma (all measured in dyn/cm), were estimated using the Young-Laplace equation as previously described (29, 31, 75). The SLs studied were longer than optimal length to eliminate the contribution of invaginations and caveolae at SLs <3 μm (22).
Muscle force measurements.
Contractile function of isolated synm-null and control TA (n = 5 per group) muscle was measured as described previously (54). While the mice were under anesthesia, the tendon of the TA was released and attached to a load cell. Single twitches (rectangular pulse, 1 ms) were applied at different muscle lengths to determine the optimal length (Lo). The muscle at Lo was subjected to a series of three isometric twitches (1 ms) and a tetanic contraction (200 ms, 100 Hz at a constant current of 5 mA). These were elicited to establish contractile responses before any additional experimental procedures were performed. With muscles set at Lo, we gradually increased the stimulation frequency to establish a force-frequency relationship (10, 30, 40, 60, 80, and 100 Hz). We used the value that gave 100% maximal stimulation intensity to induce maximal force (Po). With muscles set at Lo, fatigue was induced through tetanic contractions (100 Hz for 100 ms) delivered every 2 s for 5 min. Maximal tetanic force was measured during continuous stimulation and expressed as a percentage of Po to provide an index of fatigue. We used the term specific force (mN/mg) as the maximal force (mN) normalized by muscle weight (mg) (53).
Muscle injury.
Lengthening contractions were performed to produce injury, as described previously (53, 78). Briefly, anesthetized mice (n = 4 per group) were placed in a supine position, with the hindlimb stabilized and the foot secured onto a plate linked to a stepper motor (NMB Technologies, Chatsworth, CA) and torque sensor. The anterior muscles of the lower hindlimb ankle (dorsiflexors) were tetanically stimulated via the peroneal nerve, and 200 ms later during continued contraction the foot was forced into plantarflexion through a 90° arc of motion (−10–80°, with the foot orthogonal to the tibia considered as 0°) at an angular velocity of 900°/s. The stimulation and forced plantarflexion were repeated 15 times over a period of 15 min. This procedure induced a reproducible injury, which was quantified as a loss of torque.
Treadmill stress test.
Male mice at 3 (n = 5) and 8 (n = 5) mo of age were acclimated to the treadmill (Columbus Instruments 1055M-Exer 6M, Columbus, OH) before running in three steps: 1) placing them on the stationary treadmill belt for 10 min/day for 3 days, 2) gentle running on the belt at v = 2 m/min for 10 min, and 3) running at v = 5 m/min on the following 3 days. For testing, mice ran on the treadmill at v = 10 m/min with an inclination of 7°; v was increased by 1.5 m/min every 2 min until mice were unable to continue running and instead remained on the electrical shock grid (54).
Statistics.
All values are reported as SD. Statistical significance was accessed with the paired Student's t-test, one-way ANOVA. For fiber diameter, we used Fischer's exact test and χ2-determinants; for subsarcolemmal organization, we used Fisher's exact test. We also report the statistical population variance (V) of pressure measurements, which indicates the degree of dispersion or scattering of the experimental data. *P < 0.05 is considered statistically significant.
RESULTS
Synm-null mice were generated by inserting cDNA encoding β-galactosidase into the synemin gene by homologous recombination. Mice homozygous for the synm-null genotype were viable and fertile. We studied fast-twitch muscles from synm-null mice to assess the role of this IF protein in muscle structure and function.
PCR and Western blot.
Genetic characterization of control and synm-null mice, performed by PCR of DNA extracted from tail snips, confirmed the disruption of the Synm gene in the knockout (Fig. 1A). Assays using reverse transcriptase coupled with PCR (RT-PCR) confirmed the presence of mRNA encoding synemin in control but not in synm-null muscle (Fig. 1B). To test if the synemin protein was absent in muscles from synm-null mice, Western blot analysis from TA muscles was performed using a rabbit polyclonal antibody (R238; Ref. 54) that specifically recognizes α-synemin. Control muscles showed a clear band of immunolabeling for α-synemin at ∼210 kDa, as expected, that was absent in the synm-null sample (Fig. 1C).
Fig. 1.
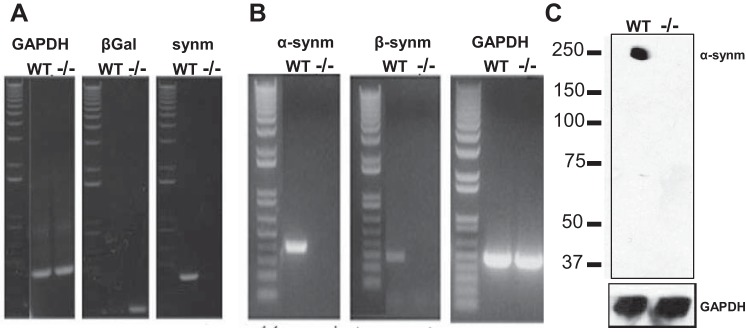
Analysis of mutant mice for synemin. A: PCR of DNA isolated from tail snips of mice null for synemin (synm-null) and control [wild-type (WT)] mice show the disruption of the synemin gene in the homozygote mouse. The band for synemin at ∼405 bp in WT is replaced by a band for β-galactosidase (LacZ) at ∼210 bp in the knockout. B: RT-PCR analysis of mRNA from skeletal muscle of synm-null and WT mice shows that mRNA encoding synemin is absent from the synm-null but present in WT. C: Western blot analysis with an antibody specific for α-synemin performed on protein supernatants prepared from extracts of tibialis anterior (TA) muscles from synm-null and WT mice shows that α-synemin is present in WT but not in the knockout. GAPDH DNA, mRNA, and protein were used as positive controls.
An antibody was generated against the COOH-terminal 108 amino acids shared by both α- and β-synemin, and it recognizes both proteins (Fig. 2, A and B). We confirmed that synemin was absent from the myofibers of TA muscles from synm-null mice by immunofluorescence labeling of cross sections (Fig. 2, C–H). Synemin in WT fibers was present in a reticular structure that surrounds the myofibrils within the interior of each fiber (e.g., Fig. 2, D and E, inset) and was frequently also concentrated at the sarcolemma, which appeared in cross sections to be almost uniformly labeled by antibodies to dystrophin (Fig. 2E). No labeling for synemin was apparent in the capillaries, and little additional labeling was apparent in the connective tissue surrounding the fibers. Labeling for synemin both within and surrounding the myofibers was completely absent in cross sections of synm-null TA muscles (Fig. 2G).
Fig. 2.

Specificity of the new anti-synemin antibody tested in COS7 cells (A and B) and in cross sections of WT myofibers (C–E) and its absence in synemin-null fibers (F–H). A: immunoblot of COS7 cells transfected with α-synemin shows 2 bands, corresponding to α (∼210 kDa)- and β (∼180 kDa)-synemin. B: transfected cells with β-synemin show a band at ∼180 kDa as expected. TA muscles from control and synm-null mice were snap frozen, cryosectioned, and fluorescently immunolabeled with antibodies against dystrophin to visualize the sarcolemma of the myofibers (shown in green) and synemin (shown in red). Structures that contain both labels are shown in yellow. Synemin in WT fibers is present at the sarcolemma (D, arrow) and in a reticular pattern in the fiber interior (D, arrowhead; E, inset). It is absent in synm-null muscle (G and H). Scale bar = 5 μm.
These results indicate that the gene encoding synemin is disrupted and that consequently synemin mRNA and protein are missing in skeletal muscle of the synm-null mouse.
Morphometric analysis.
We examined the morphology of TA muscles to learn if the absence of synemin affects muscle structure or stability. We found that the mean minimal Feret's diameter was significantly smaller in synm-null TA and quadriceps muscles (13%; P < 0.05 in both cases; Fig. 3A and Table 1) compared with controls. Interestingly minimal Feret's diameter was not affected in EDL or soleus muscles. We also quantitated the number of CNFs, a marker of ongoing degeneration and regeneration in adult skeletal muscle (28) and found that the percentage of CNFs was higher in synm-null TA (2.1%; Fig. 3, B and C) and EDL (1.4%) muscles (Table 1), compared with controls (1.0 and 0.9% respectively; P < 0.05 paired Student's t-test; Fig. 3, B and C, and Table 1). We did not observe a significant increase in the number of muscle fibers that stained for MHCs of slow-twitch muscle, suggesting that the absence of synemin did not result in the conversion of muscle fibers from fast-twitch to slow-twitch, even in soleus (Table 1). The difference in muscle mass between synm-null and control TA muscles was significant (Table 2; P < 0.05; paired Student's t-test).
Fig. 3.
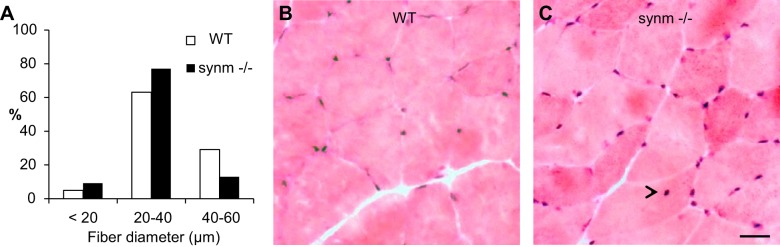
Minimal Feret's diameter and centrally nucleated myofibers (CNFs) in TA muscle. A: minimal Feret's diameter in control and synm-null TA fibers. Bar histograms represent the size distribution of synm-null and WT muscle fibers classified in 3 groups. Cryosections of TA muscles from WT (B) and synm-null (C) mice were stained with hematoxylin and eosin. Arrowhead indicates 1 example of a CNF. Scale bar = 10 μm.
Table 1.
Characterization of muscle fibers of synemin null and normal mice
| Slow MHC, % |
Minimal Feret's Diameter, μm |
CNFs, % |
||||
|---|---|---|---|---|---|---|
| C57Bl6 | Synm−/− | C57Bl6 | Synm−/− | C57Bl6 | Synm−/− | |
| TA | 7.2 ± 1.9 | 6.8 ± 1.7 | 34.0 ± 9.9 | 29.6 ± 8.6* | 1.0 ± 0.1 | 2.1 ± 0.1* |
| EDL | 8.6 ± 1.7 | 5.7 ± 2.1 | 40.1 ± 6.6 | 40.2 ± 6.8 | 0.9 ± 0.3 | 1.4 ± 0.1* |
| Quadriceps | 7.3 ± 1.9 | 5.0 ± 0.7* | 46.0 ± 9.7 | 39.9 ± 10.3* | 0 ± 0 | 0.8 ± 0.2 |
| Soleus | 32.6 ± 1.8 | 33.3 ± 5.1 | 40.2 ± 7.5 | 38.3 ± 9.6 | 0.7 ± 0.3 | 1.0 ± 0.4 |
Values of Feret's diameter and centrally nucleated fibers (CNFs) are means ± SD. Data for fibers with slow myosin heavy chain (MHC) are means ± SE. TA, tibialis anterior; EDL, extensor digitorum longus; Synm−/−, synemin null mice.
P < 0.05.
Table 2.
Morphological properties of synemin null and normal mice
| C57Bl6 | Synm−/− | |
|---|---|---|
| Animal body weight, g | 28.5 ± 2.5 | 24.2 ± 1.8* |
| TA weight, mg | 44.3 ± 2.6 | 41.5 ± 1.9* |
| Specific twitch force, mN/mg | 7.5 ± 0.6 | 6.0 ± 0.5 |
| Specific tetanic force, mN/mg | 24.4 ± 0.9 | 23.8 ± 1.4 |
Values are means ± SD.
P < 0.05.
We examined longitudinal cryosections of TA muscles from control and synm-null mice to learn if the organization of the contractile apparatus (Fig. 4, A–T) and of costameres (Fig. 5, A–F) was altered in the knock-out. The organization of Z disks did not appear to be different in synm-null compared with WT when examined for desmin or α-actinin (Fig. 4, A–F and S–T). Obscurin at M bands was also unaffected (Fig. 4, M–R), while dystrophin at the sarcolemma appeared normally distributed in the synm-null muscle (Fig. 4J). Upon examination of the costameric protein, β-spectrin, we found that its organization, too, was not significantly affected by the absence of synemin (Fig. 5, A–F). These results suggest that synemin is not required for the normal architecture of the sarcomeres or costameres in murine TA muscles.
Fig. 4.

Organization of the Z disks and M bands in synm-null myofibers and at costameres at the sarcolemma. Frozen longitudinal cryosections of TA muscles from synm-null (D–F, J–L, P–R, and T) and WT (A–C, G–I, M–O, and S) mice were immunolabeled. Synm-null muscles are indistinguishable from controls. Structures that contain both labels are shown in yellow. Scale bar = 5 μm.
Fig. 5.

Organization of costameres in synm-null myofibers (A–F). Frozen longitudinal cryosections of TA muscles from synm-null (D–F) and WT (A–C) mice were immunolabeled. Scale bar = 5 μm.
We also examined synm-null TA muscles at the ultrastructural level to assess possible changes in the organization of the contractile apparatus at higher resolution. Unlike the results of our previous studies of desmin-null and K19-null muscles, which showed significant changes in the alignment of the Z disks of adjacent myofibrils (52), the Z disks of synm-null myofibers were well aligned, indistinguishable from controls (Fig. 6, A and B). Sarcomeres in synm-null appeared unchanged (Fig. 6, B and D). Approximately the same percentage of muscle fibers showed a single layer of mitochondria under the sarcolemma in both synm-null and control TA muscles (38 and 42%, respectively; e.g., Fig. 6, C and D, arrowhead), with the remainder having either no subsarcolemmal mitochondrial accumulation (45 and 53%, respectively) or multiple layers of mitochondria (not shown; 17 and 5%, respectively). These differences were not significant (P > 0.10, Fisher's exact test).
Fig. 6.

Ultrastructure of subsarcolemmal region and nearby sarcomeres of control and synm-null muscle fibers. TA muscles of control (A and C) and synm-null (B and D) mice were fixed in situ, processed for electron microscopy of longitudinal sections, and viewed near the fiber surfaces. Alignment of Z disks was similar in the two samples (A and C for WT and B and D for synm-null). Arrowhead indicates the presence of mitochondrial monolayer under the sarcolemma in control (C) and synm-null (D). Scale bar = 0.5 μm.
Changes in biomechanical properties in vitro.
We assessed the elastic properties of the sarcolemmal membrane and the strength of its connections to the underlying contractile apparatus by elastimetry. The method uses suction pressure (P) applied over a small sarcolemmal area through a suction pipette to induce the formation of a bleb of variable height, h, which is a function of both P and SL (Fig. 7, Aa-Af). At low negative pressures, the sarcolemma and the associated contractile apparatus underlying it were drawn into the pipette together (Fig. 7, Aa and Ad). Upon relaxation of P, h decreased and the bleb returned to its initial height. As the suction pressure increased, h increased, until at large negative pressures the sarcolemma separated from the contractile apparatus (Fig. 7, Ab and Ae, for WT and synm-null, respectively). Once separated, the sarcolemma remained distended even when the pressure was released (not shown). We obtained a series of photomicrographs of sarcolemmal blebs that were formed by progressively increasing suction pressures in fibers isolated from the EDL muscles of control (Fig. 7, Aa-Ac) and synm-null (Fig. 7, Ad-Af) mice. We generated vertical pressure-displacement (P-h) curves from all the fibers studied and analyzed them to determine the elastic behavior of the sarcolemma and nearby myofibrils as a function of P.
Fig. 7.

Biomechanical properties of the surface of single control and synm-null extensor digitorum longus (EDL) fibers measured by elastimetry in vitro. A: representative photomicrographs of blebs formed with progressively increasing pressures (a and d), when the sarcolemma separates from its contractile apparatus (b and e), and when the sarcolemma bursts (c and f) in control and synm-null mice, respectively. Sarcomere lengths (SLs) = 3.3 μm. Arrows indicate the burst sarcolemma. Scale bar = 5 μm. B: pressure-displacement curve in control and synm-null myofibers. Pressure was first increased in steps (points 1–5 and a-f for WT and synm-null respectively) and then decreased (points 6–8 for WT and g-j for synm-null) in WT and synm-null mice. Point S denotes Psep, the pressure at which separation of the sarcolemma from the underlying contractile structures occurred (points 4 for WT and e for synm null). Filled symbols represent data obtained before separation of the sarcolemma from the contractile elements. Open symbols represent the data obtained after separation. Bold and light continuous lines represent the best fit of the curves for the 1st (points 1u and 2u) and 3rd segment (points 1d and 2d) of the curve. SL = 3.5 μm. C: effect of SL on Psep was determined for control and synm-null myofibers. D: histogram of bursting pressure, Pbursting, at SL = 3.8 μm. P was applied to the sarcolemma of the myofibers until the surface membrane burst. E: surface tension of the total system (γtotal), of the sarcolemma (γsarcolemma), and of costameres (γcostameres) in WT and synm-null fibers. Bars indicate means ± SD; *P < 0.05.
The experimental P-h curve for control and synm-null muscles showed three distinct segments (Fig. 7B). In the first, obtained at low suction pressures (Fig. 7B, points 1–4, filled squares for WT, and a–e, filled triangles for synm-null); the sarcolemma remained closely associated with nearby myofibrils under the bleb. Morphological observations at this conditions indicated that the system formed by the contractile apparatus, costameres, and the sarcolemma remained intact. The displacement of the system, h, as a function of increasing pressure, P, was greater in the synm-null muscle fibers than in the WT at all SLs studied. This suggests that the system consisting of the sarcolemma, costameres, and nearby myofibrils is more deformable in the synm-null muscle than in controls.
In the next portion of the P-h curve (Fig. 7B, points 4 and 5 and e and f, for WT and synm-null, respectively) measured at higher suction pressures, the sarcolemma separated from the nearby myofibrils (Fig. 7B, point S), and consequently, the slope of the curve decreased with further increases in P. Our results show that the pressure at which separation occurred, Psep, was significantly lower in the synm-null fibers than in the WT at three of the four SLs studied (P < 0.05) and trended lower at the fourth (Fig. 7C). This suggests that the strength of the links between the sarcolemma and the myofibrils that occur at costameres is weaker in the synm-null fibers than in controls. We computed the statistical population variance (V) of Psep and determined that the variability of the experimental data was higher in control than in synm-null (122 ± 11 for WT and 93 ± 10 for synm-null; P < 0.05). Thus the variability of Psep is also significantly influenced by the absence of synemin.
The third segment of the curve (Fig. 7B, points 6–8, for WT, and h–j, for synm-null), obtained by reducing P after separation of the sarcolemma from underlying myofibrils, resulted in different values of h at a given value of P than before separation. Larger changes in h occurred for the same changes in P than in the rising portion of the curve. This hysteresis in the P-h curve indicates that the system is different before and after separation of the sarcolemma. The equations of the regression lines were calculated from y = mx + b, for the first part (Fig. 7B, 1u and 2u are y = 34.1x − 59.6 and y = 22.1x − 107.1 for WT and synm-null, respectively) and for the third part of the curve (Fig. 7B; 1d: y = 44.0x − 426.91 for WT and 2d: y = 30.5x − 444.2 for synm-null).
In the rising portions of the P-h curves, higher pressures were required in controls than in synm-null muscle fibers to produce the same increase in h, suggesting that the contractile elements contribute to the stiffness of the sarcolemma in the intact system. The differences between the WT and synm-null curves further indicate that synemin contributes to this stiffness in the controls. Once the sarcolemmal separation took place, the descending portions of the P-h curve showed little change, suggesting that neither the contractile elements nor synemin significantly affected the elasticity of the isolated sarcolemma.
In additional experiments, we increased the pressure in the pipet to the point that the sarcolemma burst (Pbursting: Fig. 7, Ac and Af). We found that the bursting pressures in the synm-null samples were lower than those in the controls at a SL of 3.8 μm (Fig. 7D), suggesting that synemin contributes to the stability of the isolated sarcolemma.
Using the Young-Laplace equation and the data of the P-h curves, we computed the total tension of the system, γtotal (Fig. 7Ea); the sarcolemmal tension, γsarcolemma (Fig. 7Eb); and the maximal tension sustained by the costameres and their links to the myofibrils, γcostameres (Fig. 7Ec). The γtotal, γsarcolemma, and γcostameres sustained by the system in WT fibers were larger (22%, 15%, and 35%, respectively) than for synm-null myofibers at all SLs studied (P < 0.05). Changes at SLs between 2.9 and 3.1 μm (data not shown) were not statistically significant (P > 0.46). These results are consistent with the conclusion that the deformability of the muscle cell surface, including the costameres and sarcolemma, increases in the absence of synemin.
Contractile force measurements in situ and treadmill running.
We assessed the contribution of synemin to the function of muscle tissue by measuring maximal isometric contractile force produced by the TA muscles in anesthetized animals. The tendon of the TA was released and attached to a load cell. Resting length (the length at which the muscle generates maximal force, Lo) did not differ significantly between controls and muscles lacking synemin. Figure 8A shows twitch (single action potential stimulation) and maximal tetanic (100 Hz stimulation) force responses from control and synm-null mice. Stimulation at 100 Hz was used to elicit peak tetanic force. Mutant mice showed a significant 25% decrease in twitch specific force (Po) generated from a single action potential, compared with controls. The differences were not as great with tetanic contraction generated between the groups (9%). Po vs. frequency of stimulation relationship is depicted in Fig. 8B. At lower stimulation frequencies (30, 40, and 60 Hz), there was no significant difference between control and synm-null mice. This result suggests that force production in fast-twitch muscle decreased a small but significant extent in synm-null mice. When normalized to muscle mass weight, the values for maximal twitch and maximal tetanic force, termed specific force, did not differ significantly from controls (Table 2). Thus the weakness demonstrated by synm-null muscle is likely due to the decrease in the average weight of the synm-null muscle (Table 2), rather than to changes in actin-myosin interactions. We also tested the rate of muscle fatigue in situ (57) and observed a faster decline after 4 min of stimulation in synm-null mice (Fig. 8, C and D). At earlier time points in the fatigue protocol (1–3 min), both control and synm-null muscles showed similar levels of fatigue. At later time points, however, the synm-null TA muscles showed a small but significant increase in fatigue (P < 0.05), as indicated by the lower values in maximal isometric force measured at 4 and 5 min of stimulation (Fig. 8D).
Fig. 8.

Measurements of contractile force and fatigability. A: single twitch and maximal tetanic force of TA muscles were measured in situ in WT and synm-null mice. The absence of synemin yields a decrease in twitch and tetanic force. B: force-frequency relationship for control and synm-null TA muscles. Difference between synm-null and control muscles is significant at 80 and 100 Hz. C: representative trace recordings of fatigue in WT and synm-null TA muscles. D: rate of muscle fatigue in situ at maximal isometric force. Synm-null muscles showed a higher amount of fatigue after 4 min. E: resistance of synm-null and WT to treadmill running. Synm-null mice fatigue, on average, the same as controls. All data are presented as means ± SD. * P < 0.05
We also tested the endurance of the mice while running on a treadmill. After acclimatizing the mice to the treadmill, we increased the speed (v) of the treadmill by 1.5 m/min every 2 min until the mice, running uphill at an angle of 7°, were exhausted and no longer avoided the shock grid. Control and synm-null mice were evaluated at 3 and 8 mo of age. Both WT and synm-null mice at these ages were able to run for average times of 19 and 11 min, respectively (Fig. 8E). There were no significant differences between them. Thus the absence of synemin does not affect the ability of mice to run uphill on a treadmill at 3 and 8 mo of age.
In vivo susceptibility to injury.
We measured maximal isometric torque and its loss upon injury, using a previously described model in which injury results from 15 forced lengthening contractions (54, 88). Maximal isometric torque of the ankle dorsiflexors, of which the TA is the dominant muscle (57), was significantly lower in the synm-null mouse, consistent with our measurements of absolute isometric force. (Fig. 9, A and B). Five minutes after a series of lengthening contractions, both the WT and the synm-null mice showed a significant loss in torque (Fig. 9C), but the overall loss relative to the initial values was greater in the synm-null than in the WT (Fig. 9C). This indicates that synm-null muscle is more susceptible to loss of function than controls following injury caused by lengthening contractions. We examined the injured muscles histologically at 3 days after injury, when necrotic fibers can be more easily detected (78). The number of necrotic fibers was significantly increased in the synm-null muscles compared with controls (Fig. 10, C and D, arrowheads, and E; P < 0.05). Although we did not quantitate them, inflammatory infiltrates were also more frequent in the injured synm-null muscles (e.g., Fig. 10D, arrowheads). Together with our torque data, these results indicate that synm-null muscles are significantly more susceptible than controls to injurious lengthening contractions.
Fig. 9.
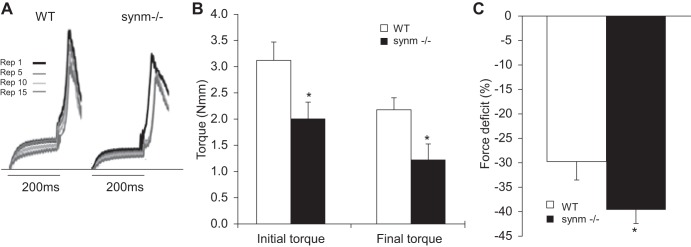
Assessment of injury. A: representative trace recordings of torque from control and synm-null mice show the progression of injury in the synm-null mouse. “Rep,” repetitions. B: initial maximal isometric torque and final torque following lengthening contractions in control and synm-null mice. C: loss in maximal torque due to injury is summarized. Torque after injury decreases 24% more in the synm-null than in WT. *P < 0.05.
Fig. 10.

Necrotic fibers increase at 3 days after injury. A–D: necrotic fibers were rare in hematoxylin and eosin-stained cross sections of uninjured TA muscle from control TA muscles, but they were more common in synm-null TA muscles. Their frequency increased at 3 days postinjury in both samples, but necrotic fibers remained more common in the synm-null samples. E: quantitation of necrotic fibers before and 3 days after injury. Synm-null muscle had low levels in before injury, although at ∼3% this was significantly greater than controls. Necrotic fibers in both WT and synm-null muscles tripled in number at 3 days after injury. Synm-null muscle showed significantly more necrotic fibers than WT (*P < 0.05 for both conditions). Arrowheads indicate necrotic fibers. Scale bar = 20 μm.
DISCUSSION
IFs, composed of desmin, synemin, and keratins (16, 86), are considered to be mediators of mechanical stress and force transduction in striated muscle (36, 47). The major IF protein is desmin, which surrounds the contractile apparatus at Z disks and serves as an important linkage to costameres at the sarcolemma (16). Synemin, which copolymerizes with desmin (59, 91), is also present in striated muscle, although at lower levels (40, 88). Keratin 19 is present in striated muscle (54, 88) as well. The networks of intermediate filaments formed by these proteins organize around the contractile apparatus at Z disks and link the contractile machinery to the sarcolemma through interactions with the dystrophin-associated protein complex (DAPC). In previous studies, we examined the effects on skeletal muscle of null mutants of desmin and keratin 19 (54, 88). Here we examine the effects of eliminating synemin. Our results suggest that the phenotype of fast twitch skeletal muscle in synm-null mice is milder than that of muscle lacking either desmin or K19. Nevertheless, our results indicate that synemin plays a significant role in determining muscle fiber size and susceptibility to injury, as well as influencing the elasticity of the muscle cell surface and the stability of the sarcolemma. As synemin is also involved in regulating signaling (96), it is likely to play a key role in the health of muscle and in influencing the effects of mutations lead to neuromuscular disease.
Despite the mild phenotype we found in synm-null skeletal muscle, the association of synemin with disease is high, primarily in neuropathologies. Pathologic conditions can induce synemin expression in diverse cell types, including reactive astrocytes, liver fibrotic cells (55, 82, 84), and glioblastomas (45, 70). Synemin expression is also altered in myoepithelial cells of breast carcinomas due to changes in the methylation of the synemin gene (61). It is abundant in glioblastoma tumors (45) and in reactive astrocytes after neurotrauma and in Alexander disease (69). Other reports indicate high levels of synemin in Parkinson's and Alzheimer's diseases (50).
Although synemin can in principle play a structural role in skeletal muscle together with desmin, with which it concentrates in a reticulum surrounding the Z disks, our results suggest that its absence does not significantly alter the organization of the sarcoplasm or of costameres at the sarcolemma. α-Actinin at Z disks, and desmin and obscurin surrounding Z disks and M bands, respectively, retain their normal architecture. Ultrastructural studies of the contractile apparatus indicate that the organization of A, I, and M bands and of Z disks in synm-null muscle is indistinguishable from controls. Notably, unlike the absence of desmin or of keratin 19, both of which result in displacement of Z disks in neighboring myofibrils and an increase in subsarcolemmal mitochondria (16, 54, 62, 68, 85), the absence of synemin has no significant effect on alignment of Z disks across myofibers nor does it lead to a significant accumulation of mitochondria under the sarcolemma.
As synemin interacts with proteins at the sarcolemma, its absence is likely to cause substantial changes in the biomechanical properties of the muscle cell surface. We measured these properties using novel methods developed in our laboratory designed to assess the elasticity of the sarcolemma and nearby cytoplasm in the intact muscle cell, the strength of the attachment of the sarcolemma to the underlying contractile apparatus, and the stability of the sarcolemma once it has separated from nearby myofibrils. Our results indicate that the myofiber surface is more elastic in synm-null muscle and that the sarcolemma separates from the contractile structures at lower suction pressures. Both these observations are consistent with a possible role for synemin in stabilizing costameres and promoting their function at the sarcolemma.
Our observation that weaker costameres are associated with the absence of synemin, as they are in the absence of dystrophin (29), is consistent with synemin's identification as a ligand of both dystrophin and dystrobrevin (7, 59), a component of the dystrophin-dystroglycan complex, as well as with the changes that occur in synm-null cardiomyocytes (56). Thus its absence could also lead to the reduced stability of the sarcolemma that we observe in synm-null muscle when we increased the pressure in the pipet to the point that the sarcolemma burst, which is also seen in dystrophin-null muscle and is linked to a loss in sarcolemmal stability (30).
These altered biomechanical properties of the sarcolemma and its links to the underlying contractile apparatus are likely to contribute to the increased susceptibility of synm-null skeletal muscle to injury (31), a feature that it also shares with muscle lacking desmin or K19 (54) and that is a general feature of dystrophic muscles (52, 53, 72). Weaker costameres and a more unstable sarcolemma are expected to lead more readily to damage muscle fibers, which, in the absence of efficient repair mechanisms, can become necrotic. Even without injuries caused by lengthening contractions, synm-null muscles show an approximately twofold more necrotic fibers than controls. Following injury, the number of necrotic fibers triples, and loss of contractile function relative to control muscles, injured similarly, is significantly enhanced.
Contractile force, torque, and fatigability of synm-null muscle are also compromised. Although the absence of synemin influences contractile function during the later stages of fatigue, much of its effect is due to the fact that synm-null muscle fibers are smaller in diameter than WT and as a result the muscles are smaller. Corrected for size, however, synm-null muscles show the same specific force of contraction as controls. Thus, with the exception of its effects on fatigue, synemin does not seem to regulate the contractile properties of fast twitch skeletal muscle, consistent with synm-null muscles maintaining a normal ultrastructure and sarcomeric organization.
Recent work published by Zhenlin et al. (96) report a mild phenotype of skeletal muscle of synm-null mice and specifically no change in the size of TA muscle. They did, however, report that the sarcolemma had a wrinkled appearance and that sarcomeres were moderately disorganized. Our observations differ in some respects, as we find that the sarcolemma and sarcomeres of synm-null TA muscles are normal in appearance but that the TA muscle is reduced in size. The differences may be due to our use of different methods to generate the null mice or to technical differences. For example, sarcolemmal wrinkles can appear at SLs <2.0 μm (22, 87, 89), the rather short SLs studied by Zhenlin et al. (96). We limited our observations to TA muscle fibers in which the average SLs were 2.1 μm (Fig. 6). Despite these minor differences, however, both we and Zhenlin et al. (96) find that synm-null muscle showed compromised membrane integrity, a higher number of CNFs, and Z disks and M bands that were normal in appearance. Zhenlin et al. also report a decrease in contractile force measurements in fast muscle, as we have observed.
It seems unlikely that the smaller size of synm-null myofibers is due to synemin's structural role; rather, it may be due to synemin's role in signaling, which Zhenlin et al. (96) have linked to the regulation of atrophy and hypertrophy. Although several factors play significant roles (80), our work and that of Zhenlin et al. (96) indicate that synemin also contributes to regulate fiber size and stability. Synemin is a phosphoprotein (83) that modulates Akt signaling (96), and, as an A-kinase anchoring protein (AKAP), it may be involved in regulating the phosphorylation of proteins at the sarcolemma and Z disks via protein kinase A (80). Both protein kinase A and Akt mediate the plasticity of growth of striated muscle (80, 81, 96). Notably, the absence of synemin does not affect fast and slow muscles in the same way. The pathways regulating Akt also vary with fiber type and can result in pronounced atrophy in fast muscle but minimal atrophy in slow fibers (9, 14), consistent with our observations. Furthermore, as AKAPs play an important role in the process (21, 58, 79), synemin's AKAP activity could contribute to PKA's ability to concentrate adjacent to the Z disks of each sarcomere. Loss of synemin's function as an AKAP would likely alter the relationship between contractile activity and the signaling that regulates muscle size, including the capacity to atrophy, when synemin is absent. Conversely, when synemin is present and active, it and the enzymes to which it binds should modulate signaling cascades associated with hypertrophy associated with mechanical overload, including changes in phosphorylation of key proteins, such as Akt, CREB, rS6, and PKA-RIIa, which have been reported by Zhenlin et al. (96). Synemin's role as an AKAP and as a regulator of signal transduction via PKA and Akt may also be linked to the greater fatigability of synm-null muscles after several minutes of continuous activity, as fatigability, too, is regulated in part by PKA (24).
As noted above, synemin also associates with proteins of the dystrophin-dystroglycan complex, in particular with α-dystrobrevin and with dystrophin itself (7, 59). Interestingly, muscle fibers of patients with desmin myopathy contain large numbers of inclusions (32) enriched not only in desmin and other IFs but also in the proteins of the dystrophin-glycoprotein complex (20). In control muscle, synemin, like dystrophin and dystrophin-associated proteins, is present in costameres, where, in addition to stabilizing the connections between the contractile apparatus and the sarcolemma, it could regulate the properties of the membrane-cytoskeletal complexes responsible for sarcolemmal organization and stability. We speculate that the changes we observed in Pbursting of synm-null sarcolemma may be due in part to the absence of synemin's AKAP activity. The role of synemin as a sarcolemmal AKAP and its possible contribution to the biomechanical properties and the regulation of growth of muscle remain to be determined.
GRANTS
Our research has been supported in part by a Physiological Genomics Fellowship and a CONACyT Fellowship (to K. P. García-Pelagio) and by National Institutes of Health Grants R01-AR-059179 (to R. M. Lovering), R01-HL-79134 and R01-AG-16613 (to M. Bond), and R01-AR-055928 (to R. J. Bloch).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: K.P.G.-P. conception and design of research; K.P.G.-P., J.M., A.O., P.F.D., R.M.L., L.L., and M.B. performed experiments; K.P.G.-P. analyzed data; K.P.G.-P. interpreted results of experiments; K.P.G.-P. prepared figures; K.P.G.-P. drafted manuscript; K.P.G.-P., J.M., A.O., P.F.D., R.M.L., L.L., M.B., and R.J.B. approved final version of manuscript; R.J.B. edited and revised manuscript.
ACKNOWLEDGMENTS
We express our appreciation to our late colleague Hugo Gonzalez-Serratos, who developed the biomechanical methods we used here and whose love of nature spurred his enduring enthusiasm for science. We thank Alyssa Collier and Stephen Pratt for technical assistance; Daniel Weinreich, Adair Oesterle, and Jaclyn P. Kerr for useful advice; and John Strong and the Imaging Core Facility at the University of Maryland, Baltimore, for providing the ultrastructural results.
REFERENCES
- 1.Agbulut O, Li Z, Perie S. Lack of desmin results in abortive muscle regeneration and modifications in synaptic structure. Cell Motil Cytoskeleton 49: 51–66, 2001. [DOI] [PubMed] [Google Scholar]
- 2.Anastasi G, Cutroneo G, Santoro G, Arco A, Rizzo G, Bramanti P, Rinaldi C, Sidoti A, Amato A, Favaloro A. Costameric proteins in human skeletal muscle during muscular inactivity. J Anat 213: 284–295, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becker B, Bellin RM, Sernett SW, Huiatt TW, Robson RM. Synemin contains the rod domain of intermediate filaments. Biochem Biophys Res Commun 213: 796–802, 1995. [DOI] [PubMed] [Google Scholar]
- 4.Bellin R, Huiatt W, Critchley R, Robson M. Synemin may function to directly link muscle cell intermediate filaments to both myofibrillar Z-lines and costameres. J Biol Chem 276: 32330–32337, 2001. [DOI] [PubMed] [Google Scholar]
- 5.Bellin RM, Sernett SW, Becker B, Ip W, Huiatt TW, Robson RM. Molecular characteristics and interactions of the intermediate filament protein synemin. Interactions with alpha-actinin may anchor synemin-containing heterofilaments. J Biol Chem 274: 29493–29499, 1999. [DOI] [PubMed] [Google Scholar]
- 6.Bhat HF, Adams ME, Khandayc FA. Syntrophin proteins as Santa Claus: role(s) in cell signal transduction. Cell Mol Life Sci 70: 2533–2554, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhosle RC, Michele D, Campbell KP, Li Z, Robson RM. Interactions of intermediate filament protein synemin with dystrophin and utrophin. Biochem Biophys Res Commun 346: 768–777, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Bilak SR, Sernett SW, Bilak MM, Bellin RM, Stromer MH, Huiatt TW, Robson RM. Properties of the novel intermediate filament protein synemin and its identification in mammalian muscle. Arch Biochem Biophys 355: 63–76, 1998. [DOI] [PubMed] [Google Scholar]
- 9.Blaauw B, Schiaffino S, Reggiani C. Mechanisms modulating skeletal muscle phenotype. Compr Physiol 13: 1645–1687, 2013. [DOI] [PubMed] [Google Scholar]
- 10.Blake DJ, Martin-Rendon E. Intermediate filaments and the function of the dystrophin-protein complex. Trends Cardiovasc Med 12: 224–228, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Bloch RJ, Capetanaki Y, O'Neill A, Reed P, Williams M, Resneck W, Porter N, Ursitti J. Costameres: repeating structures at the sarcolemma of skeletal muscle. Clin Orthop Relat Res 403: S203–S210, 2002. [DOI] [PubMed] [Google Scholar]
- 12.Bloch RJ, González-Serratos H. Lateral force transmission across costameres in skeletal muscle. Exerc Sport Sci Rev 31: 73–78, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Briguet A, Courdier-Fruh I, Foster M, Meier T, Magyar JP. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx mouse. Neuromuscul Disord 14: 675–682, 2004. [DOI] [PubMed] [Google Scholar]
- 14.Brooks NE, Myburgh KH. Skeletal muscle wasting with disuse atrophy is multi-dimensional: the response and interaction of myonuclei, satellite cells and signaling pathways. Front Physiol 5: 99, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell K, Stull T. Skeletal muscle basement membrane-sarcolemma-cytoskeleton interaction minireview series. J Biol Chem 278: 12599–12600, 2003. [DOI] [PubMed] [Google Scholar]
- 16.Capetanaki Y, Bloch RJ, Kouloumenta A, Mavroidis M, Psarras S. Muscle intermediate filaments and their links to membranes and membranous organelles. Exp Cell Res 313: 2063–2076, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Carlsson L, Li Z, Paulin D, Price M, Breckler J, Robson R, Wiche G, Thornell LE. Differences in the distribution of synemin, paranemin, and plectin in skeletal muscles of wild-type and desmin knock-out mice. Histochem Cell Biol 114: 39–47, 2000. [DOI] [PubMed] [Google Scholar]
- 18.Coulombe PA, Bousquet O, Ma L, Yamada S, Wirtz D. The “ins” and “outs” of IF organization. Trends Cell Biol 10: 420–428, 2000. [DOI] [PubMed] [Google Scholar]
- 19.Dalakas M, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb L. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med 342: 770–780, 2000. [DOI] [PubMed] [Google Scholar]
- 20.De Bleecker JL, Ertl BB, Engel AG. Patterns of abnormal protein expression in target formation and unstructured cores. Neuromuscul Disord 6: 339–349, 1996. [DOI] [PubMed] [Google Scholar]
- 21.Diviani D, Maric D, Pérez López I, Cavin S, Del Vescovo CD. A-kinase anchoring proteins: molecular regulators of the cardiac stress response. Biochim Biophys Acta 1833: 901–908, 2013. [DOI] [PubMed] [Google Scholar]
- 22.Dulhunty AF, Franzini-Armstrong C. The relative contribution of the folds and caveolae to the surface membrane of frog skeletal muscle fibers at different sarcomere length. J Physiol 250: 513–539, 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Durbeej M, Campbell KP. Muscular dystrophies involving the dystrophin- glycoprotein complex: an overview of current mouse models. Curr Opin Genet Dev 12: 349–361, 2002. [DOI] [PubMed] [Google Scholar]
- 24.Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 17: 164–184, 2013. [DOI] [PubMed] [Google Scholar]
- 25.Eriksson JE, Dechat T, Grin B, Helfand B, Mendez M, Pallari HM, Goldman R. Introducing intermediate filaments: from discovery to disease. J Clin Invest 119: 1763–1771, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ervasti J, Campbell K. Membrane organization of the dystrophin-glycoprotein complex. Cell 66: 1121–1131, 1991. [DOI] [PubMed] [Google Scholar]
- 27.Ervasti J. Costameres: the Achilles' heel of herculean muscle. J Biol Chem 278: 13591–13594, 2003. [DOI] [PubMed] [Google Scholar]
- 28.Frimel TN, Walter GA, Gibbs JD, Gaidosh GS, Vandenborne K. Noninvasive monitoring of muscle damage during reloading following limb disuse. Muscle Nerve 32: 605–612, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Pelagio K, Bloch R, Ortega A, Gonzalez-Serratos H. Biomechanics of the sarcolemma and costameres in single skeletal muscle fibers from normal and dystrophin-null mice. J Muscle Res Cell Motil 31: 323–336, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia-Pelagio K, Bloch R, Ortega A, Gonzalez-Serratos H. Elastic properties of the sarcolemma-costamere complex of muscle cells in normal mice. AIP Conf Proc 854: 51–53, 2006. [Google Scholar]
- 31.García-Pelagio K, Muriel J, O'Neill A, Desmond P, Lovering R, Lund L, Bond M, Bloch R. Characterization of skeletal muscle in the synemin knock-out mouse. AIP Conf Proc 1626: 67, 2014. [Google Scholar]
- 32.Goebel HH. Congenital myopathies with inclusion bodies: a brief review. Neuromuscul Disord 8: 162–168, 1998. [DOI] [PubMed] [Google Scholar]
- 33.Goldman R, Khuon S, Chou Y, Opal P, Steinert P. The function of intermediate filaments in cell shape and cytoskeletal integrity. J Cell Biol 134: 971–983, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goto H, Inagaki M. New insights into roles of intermediate filament phosphorylation and progeria pathogenesis. IUBMB Life 2014 Mar 23 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 35.Granger BL, Lazarides E. Synemin: a new high molecular weight protein associated with desmin and vimentin filaments in muscle. Cell 22: 727–738, 1980. [DOI] [PubMed] [Google Scholar]
- 36.Helfand B, Chang L, Goldman R. Intermediate filaments are dynamic and motile elements of cellular architecture. J Cell Sci 117: 133–141, 2004. [DOI] [PubMed] [Google Scholar]
- 37.Herrmann H, Strelkov SV, Burkhard P, Aebi U. Intermediate filaments: primary determinants of cell architecture and plasticity. J Clin Invest 119: 1772–1783, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hesse M, Franz T, Tamai Y, Taketo MM, Magin TM. Targeted deletion of keratins 18 and 19 leads to trophoglast fragility and early embryonic lethality. EMBO J 19: 5060–5070, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hesse M, Magin T, Weber K. Genes for intermediate filament proteins and the draft sequence of the human genome: novel keratin genes and a surprisingly high number of pseudogenes related to keratin genes 8 and 18. J Cell Sci 114: 2569–2575, 2001. [DOI] [PubMed] [Google Scholar]
- 40.Hijikata T, Nakamura A, Isokawa K, Imamura M, Yuasa K, Ishikawa R, Kohama K, Takeda S, Yorifuji H. Plectin 1 links intermediate filaments to costameric sarcolemma through beta-synemin, alpha-dystrobrevin and actin. J Cell Sci 121: 2062–2074, 2008. [DOI] [PubMed] [Google Scholar]
- 41.Hoffman EP, Brown R, Kunkel L. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51: 919–928, 1987. [DOI] [PubMed] [Google Scholar]
- 42.Holland A, Carberry S, Ohlendieck K. Proteomics of the dystrophin-glycoprotein complex and dystrophinopathy. Curr Protein Pept Sci 14: 680–697, 2013. [DOI] [PubMed] [Google Scholar]
- 43.Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 355: 696–702, 1992. [DOI] [PubMed] [Google Scholar]
- 44.Janmey P, Euteneuer U, Traub P, Schliwa M. Viscoelastic properties of vimentin compared with other filamentous biopolymer networks. J Cell Biol 113: 155–160, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jing R, Pizzolato G, Robson RM, Gabbiani G, Skalli O. Intermediate filament protein synemin is present in human reactive and malignant astrocytes and associates with ruffled membranes in astrocytoma cells. Glia 50: 107–120, 2005. [DOI] [PubMed] [Google Scholar]
- 46.Kontrogianni-Konstantopoulos A, Jones EM, Van Rossum DB, Bloch RJ. Obscurin is a ligand for small ankyrin 1 in skeletal muscle. Mol Biol Cell 14: 1138–1148, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kreplak L, Bär H, Leterrier JF, Hermann H, Aebi U. Exploring the mechanical behavior of single intermediate filaments. J Mol Biol 354: 569–577, 2005. [DOI] [PubMed] [Google Scholar]
- 48.Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res 94: 1023–1031, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Lazarides E, Hubbard B. Immunological characterization of the subunit of the 100 A filaments from muscle cells. Proc Natl Acad Sci USA 73: 4344–4348, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lepinoux-Chambaud C, Eyer J. Review on intermediate filaments of the nervous system and their pathological alterations. Histochem Cell Biol 140: 13–22, 2013. [DOI] [PubMed] [Google Scholar]
- 51.Li Z, Colucci-Guyon E, Pincon-Raymond M, Mericshay M, Pourin S, Paulin D, Babinet C. Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev Biol 175, 362–366, 1996. [DOI] [PubMed] [Google Scholar]
- 52.Lou J, Bi W, Zhao Y, Liu S, Zheng J, Yan C. Muscle injury induced by different types of contractions in dystrophic mdx mice. J Muscle Res Cell Motil 32: 411–419, 2012. [DOI] [PubMed] [Google Scholar]
- 53.Lovering R, De Deyne P. Contractile function, sarcolemma integrity and the loss of dystrophin after skeletal muscle eccentric contraction-induced injury. Am J Physiol Cell Physiol 286: C230–C238, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lovering R, O'Neill A, Muriel J, Prosser B, Strong J, Bloch RJ. Physiology, structure, and susceptibility to injury of skeletal muscle in mice lacking keratin 19-based and desmin-based intermediate filaments. Am J Physiol Cell Physiol 300: C803–C813, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luna G, Lewis GP, Banna CD, Skalli O, Fisher SK. Expression profiles of nestin and synemin in reactive astrocytes and Müller cells following retinal injury: a comparison with glial fibrillar acidic protein and vimentin. Mol Vis 16: 2511–2523, 2010. [PMC free article] [PubMed] [Google Scholar]
- 56.Lund M, Kerr J, Lupinetti J, Zhang Y, Russell M, Bloch R, Bond M. Synemin isoforms differentially organize cell junctions and desmin filaments in neonatal cardiomyocytes. FASEB J 1: 137–148, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Manal K, Roberts D, Buchanan T. Optimal pennation angle of the primary ankle plantar and dorsiflexors: variations with sex, contraction intensity, and limb. J Appl Biomech 22: 255–263, 2006. [DOI] [PubMed] [Google Scholar]
- 58.McConnachie G, Langeberg LK, Scott JD. AKAP signaling complexes: getting to the heart of matter. Trends Mol Med 12: 317–323, 2006. [DOI] [PubMed] [Google Scholar]
- 59.Mizuno Y, Thompson TG, Guyon JR, Lidov HG, Brosius M, Imamura M, Ozawa E, Watkins SC, Kunkel LM. Desmuslin, an intermediate filament protein that interacts with alpha-dystrobrevin and desmin. Proc Natl Acad Sci USA 98: 6156–6161, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nigro V, Savarese M. Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myol 33: 1–12, 2014. [PMC free article] [PubMed] [Google Scholar]
- 61.Noetzel E, Rose M, Sevinc E, Hilgers RD, Hartmann A, Naami A, Knüchel R, Dahl E. Intermediate filament dynamics and breast cancer: aberrant promoter methylation of the synemin gene is associated with early tumor relapse. Oncogene 29: 4814–4825, 2010. [DOI] [PubMed] [Google Scholar]
- 62.O'Neill A, Williams M, Resneck W, Milner D, Capetanaki Y, Bloch RJ. Sarcolemmal organization in skeletal muscle lacking desmin: evidence for cytokeratins associated with the membrane skeleton at costameres. Mol Biol Cell 13: 2347–2359, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ozawa E. From dystrophinopathy to sarcoglycanopathy: evolution of a concept of muscular dystrophy. Muscle Nerve 21: 421–438, 1998. [DOI] [PubMed] [Google Scholar]
- 64.Pan X, Hobbs RP, Coulombe PA. The expanding significance of keratin intermediate filaments in normal and diseased epithelia. Curr Opin Cell Biol 25: 47–56, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paramio J, Jorcano L. Beyond structure: do intermediate filaments modulate cell signaling? Bioassays 24: 836–844, 2002. [DOI] [PubMed] [Google Scholar]
- 66.Pardo JV, Siliciano J, Craig S. A vinculin-containing cortical lattice in skeletal muscle: transverse lattice elements (“costameres”) mark sites of attachment between myofibrils and sarcolemma. Proc Natl Acad Sci USA 80: 1008–1012, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Patel TJ, Lieber RL. Force transmission in skeletal muscle: from actomyosin to external tendons. Exerc Sport Sci Rev 25: 321–363, 1997. [PubMed] [Google Scholar]
- 68.Paulin D, Li Zhenlin. Desmin: a major intermediate filament protein essential for the structural integrity and function of muscle. Exp Cell Res 301: 1–7, 2004. [DOI] [PubMed] [Google Scholar]
- 69.Pekny T, Faiz M, Wilhelmsson U, Curtis MA, Matej R, Skalli O, Pekny M. Synemin is expressed in reactive astrocytes and Rosenthal fibers in Alexander disease. APMIS 122: 76–80, 2014. [DOI] [PubMed] [Google Scholar]
- 70.Pitre A, Davis N, Paul M, Orr AW, Skalli O. Synemin promotes AKT-dependent glioblastoma cell proliferation by antagonizing PP2. Mol Biol Cell 23: 1243–1253, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Porter GA, Dmytrenko GM, Winkelmann JC, Bloch RJ. Dystrophin colocalizes with beta-spectrin in distinct subsarcolemmal domains in mammalian skeletal muscle. J Cell Biol 117: 997–1005, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pratt SJ, Shah SB, Ward CW, Kerr JP, Stains JP, Lovering RM. Recovery of altered neuromuscular junction morphology and muscle function in mdx mice after injury. Cell Mol Life Sci 72: 153–164, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Price MG, Lazarides E. Expression of intermediate filament/associated proteins paranemin and synemin in chicken development. J Cell Biol 97: 1860–1874, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rando TA. The dystrophin-glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve 24: 1575–1594, 2001. [DOI] [PubMed] [Google Scholar]
- 75.Rapoport S. Mechanical properties of the sarcolemma and myoplasm in frog muscle as a function of sarcomere length. J Gen Physiol 59: 559–585, 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reed PW, Mathews KD, Mills KA, Bloch RJ. The sarcolemma in the Large(myd) mouse. Muscle Nerve 30: 585–595, 2004. [DOI] [PubMed] [Google Scholar]
- 77.Roche JA, Lovering RM, Roche R, Ru L, Reed PW, Bloch R. Extensive mononuclear infiltration and myogenesis characterize recovery of dysferlin-null skeletal muscle from contraction induced injuries. Am J Physiol Cell Physiol 298: C298–C312, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roche JA, Ru LW, Bloch RJ. Distinct effects of contraction-induced injury in vivo on four different murine models of dysferlinopathy. J Biomed Biotechnol 2012: 13403, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rudolf R, Khan MM, Lustrino D, Labeit S, Kettelhut IC, Navegantes LC. Alterations of cAMP-dependent signaling in dystrophic skeletal muscle. Front Physiol 4: 290, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rusell AP. Molecular regulation of skeletal muscle mass. Clin Exp Pharmacol Physiol 37: 378–384, 2009. [DOI] [PubMed] [Google Scholar]
- 81.Russell MA, Lund LM, Haber R, McKeegan K, Cianciola N, Bond M. The intermediate filament protein, synemin, is an AKAP in the heart. Arch Biochem Biophys 456: 204–215, 2006. [DOI] [PubMed] [Google Scholar]
- 82.Rybakova I, Patel J, Ervasti J. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J Cell Biol 150: 1209–1214, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sandoval IV, Colaco CA, Lazarides E. Purification of the intermediate filament-associated protein, synemin, from chicken smooth muscle. Studies on its physicochemical properties, interaction with desmin, and phosphorylation. J Biol Chem 258: 2568–2576, 1983. [PubMed] [Google Scholar]
- 84.Schmitt-Graeff A, Jing R, Nitschke R, Desmoulière A, Skalli O. Synemin expression is widespread in liver fibrosis and is induced in proliferating and malignant biliary epithelial cells. Hum Pathol 37: 1200–1210, 2006. [DOI] [PubMed] [Google Scholar]
- 85.Sejersen T, Lendahl U. Transient expression of the intermediate filament nestin during skeletal muscle development. J Cell Sci 106: 1291–1300, 1993. [DOI] [PubMed] [Google Scholar]
- 86.Shah SB, Davis J, Weisleder N, Kostavassili I, McCulloch AD, Raiston E, Capetanaki Y, Lieber RL. Structural and functional roles of desmin in mouse skeletal muscle during passive deformation. Biophys J 86: 2993–3008, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shear CR, Bloch RJ. Vinculin in subsarcolemmal densities in chicken skeletal muscle: localization and relationship to intracellular and extracellular structures. J Cell Biol 101: 240–256, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stone MR, O'Neill A, Lovering RM, Strong J, Resneck WG, Reed PW, Toivola DM, Ursitti JA, Omary MB, Bloch RJ. Absence of keratin 19 in mice causes skeletal myopathy with mitochondrial and sarcolemmal reorganization. J Cell Sci 120: 3999–4008, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Street SE. Lateral transmission of tension in frog myofibers: a myofibrillar network and transverse cytoskeletal connections are possible transmitters. J Cell Physiol 114: 346–364, 1983. [DOI] [PubMed] [Google Scholar]
- 90.Sun N, Critchley D, Paulin D, Li Z, Robson R. Human alpha-synemin interacts directly with vinculin and metavinculin. Biochem J 409: 657–667, 2008. [DOI] [PubMed] [Google Scholar]
- 91.Sun N, Critchley D, Paulin D, Li Z, Robson R. Identification of a repeated domain within mammalian alpha-synemin that interacts directly with talin. Exp Cell Res 314: 1839–1849, 2008. [DOI] [PubMed] [Google Scholar]
- 92.Ursitti JA, Lee PC, Resneck WG, McNally MM, Bowman AL, O'Neill A, Stone MR, Bloch RJ. Cloning and characterization of cytokeratins 8 and 19 in adult rat striated muscle. Interaction with the dystrophin glycoprotein complex. J Biol Chem 279: 41830–41838, 2004. [DOI] [PubMed] [Google Scholar]
- 93.Williams W, Bloch RJ. Differential distribution of dystrophin, and β-spectrin at the sarcolemma of fast twitch skeletal muscle fibers. J Muscle Res Cell Motil 20: 383–393, 1999. [DOI] [PubMed] [Google Scholar]
- 94.Yang J, Dominguez B, de Winter F, Gould T, Eriksson J, Lee K. Nestin negatively regulates postsynaptic differentiation of the neuromuscular synapse. Nat Neurosci 14: 324–330, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yurchenco PD, Cheng YS, Campbell K, Li S. Loss of basement membrane, receptor and cytoskeletal lattices in a laminin-deficient muscular dystrophy. J Cell Sci 117: 735–742, 2004. [DOI] [PubMed] [Google Scholar]
- 96.Zhenlin L, Parlakian A, Coletti D, Alonso-Martin S, Hourdé C, Joanne P, Gao-Li J, Blanc J, Ferry A, Paulin D, Xue Z, Agbulut O. Synemin acts as a regulator of signaling molecules in skeletal muscle hypertrophy. J Cell Sci 127: 4589–4601, 2014. [DOI] [PubMed] [Google Scholar]