

Abstract
Uremic cardiomyopathy (UCM) is characterized by metabolic remodelling, compromised energetics, and loss of insulin-mediated cardioprotection, which result in unsustainable adaptations and heart failure. However, the role of mitochondria and the susceptibility of mitochondrial permeability transition pore (mPTP) formation in ischemia-reperfusion injury (IRI) in UCM are unknown. Using a rat model of chronic uremia, we investigated the oxidative capacity of mitochondria in UCM and their sensitivity to ischemia-reperfusion mimetic oxidant and calcium stressors to assess the susceptibility to mPTP formation. Uremic animals exhibited a 45% reduction in creatinine clearance (P < 0.01), and cardiac mitochondria demonstrated uncoupling with increased state 4 respiration. Following IRI, uremic mitochondria exhibited a 58% increase in state 4 respiration (P < 0.05), with an overall reduction in respiratory control ratio (P < 0.01). Cardiomyocytes from uremic animals displayed a 30% greater vulnerability to oxidant-induced cell death determined by FAD autofluorescence (P < 0.05) and reduced mitochondrial redox state on exposure to 200 μM H2O2 (P < 0.01). The susceptibility to calcium-induced permeability transition showed that maximum rates of depolarization were enhanced in uremia by 79%. These results demonstrate that mitochondrial respiration in the uremic heart is chronically uncoupled. Cardiomyocytes in UCM are characterized by a more oxidized mitochondrial network, with greater susceptibility to oxidant-induced cell death and enhanced vulnerability to calcium-induced mPTP formation. Collectively, these findings indicate that mitochondrial function is compromised in UCM with increased vulnerability to calcium and oxidant-induced stressors, which may underpin the enhanced predisposition to IRI in the uremic heart.
Keywords: uremic cardiomyopathy, mitochondrial dysfunction, mitochondrial permeability transition pore, chronic kidney disease
cardiovascular mortality accounts for approximately half of all deaths in chronic kidney disease (CKD) patients, and the risk of cardiovascular death in patients diagnosed with CKD far outweighs the risk of disease progression (16, 40). The uremic milieu contains a number of cardiac risk factors that give rise to a distinct cardiac pathology termed uremic cardiomyopathy (UCM) (59), of which left ventricular hypertrophy (LVH) is the most prevalent (19). A characteristic feature of LVH in the uremic heart is metabolic remodelling, a partial recapitulation of the fetal gene program that manifests as a shift in myocardial substrate preference from fatty acids toward glucose utilization (19, 62). Frequently observed in LVH of different etiologies, these adaptations, although initially beneficial, are unsustainable, and decompensation ultimately ensues, resulting in heart failure (39).
Mitochondrial dysfunction is a central feature of heart failure, where diminished respiratory function underlies impaired energetic flux, leading to compromised contractile function (55). Our previous experimental studies on UCM have demonstrated a reduction in cardiac phosphocreatine content (52), suggesting a compromised energetic flux. However, mitochondrial function has yet to be evaluated in the uremic heart. In addition to a decreased metabolic capacity, dysfunctional mitochondria can compromise cardiac function via formation of the mitochondrial permeability transition pore (mPTP) (25). This occurs in response to increased matrix calcium and is sensitized by reactive oxygen species (ROS) and depletion of adenine nucleotides (23). Because these conditions occur in the reperfused myocardium, mPTP formation is considered a critical mediator of ischemia-reperfusion injury (IRI). Its formation results in the collapse of the mitochondrial membrane potential, cessation of oxidative phosphorylation, and mitochondrial swelling. Severely compromised ATP production and release of proapoptotic proteins from the intermembrane space can trigger necrotic and apoptotic cell death, respectively, and thus the sensitivity of mitochondria to pore formation can determine the ultimate fate of the cell (23).
Patients with a background of CKD have poorer outcomes after acute myocardial infarction (AMI) compared with those with normal renal function. Indeed, observational data show a hospital mortality of 23% in patients with advanced CKD vs. 12.6% in non-CKD patients following AMI (61). Experimentally, the uremic heart has an increased susceptibility to IRI; however, the mechanisms of enhanced injury are unclear (12). In vitro models of IRI have revealed an increase in tissue damage, reduction in phosphocreatine content, and impaired postischemic contractile function in uremia (52, 60). Uremic rats have also exhibited progressive impairment of left ventricular function following myocardial infarction (13).
There is indirect evidence that enhanced susceptibility to IRI in uremia may be associated with altered calcium handling. Decreased activities of sarcoplasmic reticulum Ca2+ ATPase 2a and Na+/K+-ATPase have been reported in experimental models of CKD in association with prolongation of diastolic calcium transients (32, 42). Impaired calcium extrusion upon reperfusion may promote tissue injury through hypercontracture as well as through enhanced mitochondrial calcium uptake and subsequent mPTP formation. Impaired calcium handling may be sufficient to explain the enhanced susceptibility to IRI in UCM. However, recent evidence has identified alterations in the cardioprotective signaling pathways in the uremic heart, further implicating a potential role of mitochondrial dysfunction (60).
Recent data have indicated a loss of insulin-mediated cardioprotection in UCM in the absence of insulin resistance (60). Stimulation of the insulin-signaling cascade leads to the activation of a number of downstream effectors, including Akt, mammalian target of rapamycin, and protein kinase C, which in turn confer protection via mechanisms that include enhanced glucose uptake and lipolysis (47). One key downstream kinase, glycogen synthase kinase-3β (GSK-3β), can modify the sensitivity of mPTP, conferring cardioprotection (31). Therefore, the loss of insulin-mediated protection in uremia may result in enhanced opening of mPTP. However, despite its centrality in mediating IRI, the susceptibility to mPTP formation has not yet been characterized in the uremic heart.
This study tested the hypothesis that enhanced susceptibility to IRI in UCM is mediated by an increased sensitivity to mPTP formation. Using the surgical remnant kidney model of chronic uremia, we characterized the oxidative capacity of mitochondria in UCM. The sensitivity of mitochondria to ischemia-reperfusion mimetic oxidant and calcium stressors has subsequently been assessed in isolated cardiomyocytes and isolated mitochondria to determine the sensitivity to mPTP formation in uremia.
METHODS
Experimental model of uremia.
This investigation was carried out in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health under assurance no. A5634-01. All procedures and perioperative management conformed to the UK Animals (Scientific Procedures) Act of 1986 (project licence 60/3862) and were approved by the University of Hull Ethical Review Process. Uremia was induced in male Sprague-Dawley rats (∼250 g; Charles River Laboratories, Sussex, UK) via a one-stage subtotal nephrectomy, as described previously (62). Animals were maintained for 12 wk post-induction of uremia, housed individually, and pair-fed along with control animals. Cardiac hypertrophy was assessed at the time of euthanasia by determining wet heart weight/tibia length (HW/TL).
Serum and urine metabolite analysis.
Urine samples were collected over 24 h from animals housed in metabolic cages and analyzed for creatinine and total protein using a RX Monza analyzer (Randox, Antrim, UK). Serum samples collected upon euthanasia were analyzed for urea, creatinine, and albumin as described above.
Isolation of mitochondria.
Cardiac mitochondria were isolated using a modified method of Boehm et al. (3). Briefly, excised ventricles were minced in isolation medium containing (in mM) sucrose (300), HEPES (10), and EGTA (2), pH 7.2, followed by trypsin digestion (0.125 mg/ml) for 15 min at 4°C. At 5-min intervals during the digestion, the tissue was gently homogenized with a single pass of a loose-fitting Teflon glass homogenizer for a total of three passes. The digestion was stopped with the addition of isolation buffer containing 1 mg/ml BSA and 1 mg/ml trypsin inhibitor (from Glycine max; Sigma-Aldrich). The resultant tissue was homogenized with a single pass of a tight-fitting Teflon glass homogenizer and centrifuged at 600 g for 10 min at 4°C. The supernatant was decanted and centrifuged at 8,000 g for 15 min at 4°C. The resulting pellet was resuspended in isolation buffer containing 1 mg/ml BSA and centrifuged at 8,000 g for 15 min at 4°C. Finally, the mitochondrial pellet was resuspended in isolation medium and stored on ice. Mitochondrial protein concentration was determined using the Bio-Rad protein assay.
Mitochondrial respiration.
Mitochondrial oxygen consumption was determined at 25°C using a Clark-style oxygen electrode (Rank Brothers, Cambridge, UK) in buffer: 125 mM KCl, 10 mM Tris, 20 mM MOPS, 0.5 mM EGTA, 2.5 mM KH2PO4, and 2.5 mM MgCl2 (pH 7.2). Mitochondrial preparations (0.4 mg/ml) were incubated in the presence of 5 mM glutamate and 1 mM malate, 5 mM succinate and 1 μM rotenone, or 40 μM palmitoyl carnitine and 5 mM malate. ADP (0.5 mM) was added to initiate state 3 respiration. Once a steady state had been reached, state 4 respiration was initiated by the addition of 1 μg/ml oligomycin (3).
Isolated heart perfusion.
Hearts were rapidly excised and perfused ex vivo in the isovolumic Langendorff mode, as previously described (62).
Induction of IRI.
After 20 min of normoxic perfusion, hearts were immersed in buffer at 37°C, and perfusion ceased for 25 min. On reperfusion, the ventricular balloon was deflated for 5 min and reinflated to produce an end-diastolic pressure of 5–7 mmHg (20). Indices of cardiac function were measured for 25 min.
Isolation of cardiomyocytes.
Ventricular cardiomyocytes were isolated using a modified method of Smolenski et al. (63). Cells were resuspended in fresh oxygenated incubation medium at a density of 2.5 × 105 cells/ml. Viability was determined following incubation with 0.25% trypan blue, with typical values of 50–70%.
H2O2-induced stress in isolated cardiomyocytes.
Freshly isolated cardiomyocytes were incubated with 200 μM H2O2 for 60 min at room temperature. Cell viability was determined as before using 1% trypan blue (2). FAD autofluorescence was assessed before and following 60-min exposure to H2O2. Only rod-shaped myocytes were analyzed, and fluorescence intensity was expressed relative to control cells at baseline.
Mitochondrial membrane potential assay.
In a separate study, rates of mitochondrial membrane depolarization were monitored spectrofluorometrically in isolated mitochondria using rhodamine 123, using a modification of Lecoeur et al. (37). Samples were loaded into 96-well opaque-walled, clear-bottomed microplates (Grenier) and incubated for 5 min at 37°C. After the addition of 20 or 40 μM CaCl2, samples were excited at 485 nm, and fluorescence emission at 520 nm was monitored every 30 s for 15 min using a Fluostar Optima fluorescence plate reader (BMG Labtech). The increase in fluorescence (as a result of dequenching due to mitochondrial depolarization) is linearly related to Δψm (17). Fluorescence intensities were calculated relative to control incubated with 1 μM of the potassium ionophore valinomycin. Maximal pseudolinear rates of dequenching were then calculated as an indication of mitochondrial depolarization.
Statistical analysis.
Results are expressed as means ± SE. Statistical significance was determined using an unpaired t-test (for single mean comparisons) or one-way ANOVA where appropriate (using Tukey's post hoc test). Pearson's analysis was used for the significance of bivariate correlations. Statistical analysis was performed using GraphPad Prism version 6.0 (GraphPad Software, San Diego, CA). A P value of <0.05 was considered statistically significant.
RESULTS
Characterization of the experimental model.
Both serum creatinine and urea concentrations were significantly elevated in uremic animals, as observed previously (Table 1) (60, 62). Further characterization of renal function revealed a 45% reduction in creatinine clearance as an estimate of GFR, whereas a more than fourfold increase in urinary protein excretion rate indicated the presence of a marked proteinuria (P < 0.01). Uremic animals exhibited cardiac hypertrophy, as evidenced by significant increases in wet heart weight and HW/TL (P < 0.01), consistent with previous findings (62). Diastolic, systolic, and mean arterial pressures are already markedly elevated by 3 wk post-induction of uremia in this model (53).
Table 1.
Morphological features and renal function
| Control | Uremic | |
|---|---|---|
| Body weight, g | 570 ± 8.4 (n = 25) | 563 ± 9.0 (n = 25) |
| Wet heart weight, g | 1.83 ± 0.06 (n = 25) | 2.07 ± 0.06 (n = 25)* |
| Heart weight/tibia length, g/cm | 0.41 ± 0.01 (n = 24) | 0.46 ± 0.01 (n = 25)* |
| Serum creatinine, μM | 47.4 ± 1.5 (n = 21) | 89.7 ± 6.0 (n = 20)* |
| Serum urea, mM | 6.2 ± 0.4 (n = 11) | 14.8 ± 1.9 (n = 10)* |
| Serum albumin, g/dl | 3.85 ± 0.07 (n = 11) | 3.53 ± 0.06 (n = 10)* |
| Creatinine clearance, ml·min−1·kg body wt−1 | 6.07 ± 0.48 (n = 9) | 3.37 ± 0.38 (n = 7)* |
| Urine protein excretion rate, mg/24 h | 20.2 ± 1.1 (n = 10) | 89.8 ± 17.4 (n = 10)* |
Values are means ± SE.
P < 0.01.
Mitochondrial respiration in normoxia.
Maximal (state 3) respiration rates were unaltered in uremia and were similar for all substrates tested (Fig. 1A) Uremic mitochondria exhibited a degree of uncoupling, as evidenced by moderate elevations in state 4 respiration (44 and 23% increases, respiring on complex I- and complex II-linked substrates, respectively; Fig. 1B). Consequently, respiratory control ratios were depressed in uremia in the presence of glutamate and malate (P < 0.01) and palmitoyl carnitine and malate (P < 0.05) as substrates (Fig. 1C).
Fig. 1.

Respiration in mitochondria isolated from normoxic hearts. A: state 3 respiration. B: state 4 respiration. C: respiratory control ratio (RCR). Respiratory substrates: 5 mM glutamate + 1 mM malate (GM; n = 9), 5 mM succinate + 1 μM rotenone (SR; n = 9), and 40 μM palmitoyl carnitine + 5 mM malate (PC; control n = 5, uremic n = 4). †P < 0.01 vs. control; *P < 0.05 vs. control.
Cardiac function and mitochondrial respiration post-IRI.
Normoxic functional parameters were unaltered in uremia [left ventricular-developed pressure (mmHg): control 120 ± 18 vs. uremic 116 ± 3]. Following IRI, functional recovery was markedly depressed in uremic hearts, consistent with previous findings (60) (Fig. 2A). At the end of IRI, uremic cardiac function recovered to 9.3% of preischemic levels compared with 42.9% in controls (P < 0.01).
Fig. 2.

Mitochondrial respiration post-ischemia-reperfusion. A: Post-ischemic cardiac function expressed as %normoxic rate pressure product (RPP). B: state 3 respiration. C: state 4 respiration. D: RCR. Respiratory substrates: GM and SR (n = 4). †P < 0.01 vs. respective control; *P < 0.05 vs. respective control.
Rates of respiration were assessed in mitochondria isolated from hearts following the induction of IRI (Fig. 2, B–D). Complex I-linked respiratory parameters were similar between both experimental groups. In the presence of complex II-linked substrates, state 3 respiration did not differ in uremia (Fig. 2B); however, uremic mitochondria exhibited a 58% increase in state 4 respiration (P < 0.05; Fig. 2C), giving rise to a reduction in respiratory control ratio (P < 0.01; Fig. 2D).
Oxidant-induced stress in isolated cardiomyocytes.
The effect of oxidant stress on mitochondrial redox state was assessed in H2O2-treated isolated cardiomyocytes. Initial cell viabilities at baseline were similar between experimental groups (cell viability 50.7 ± 3.5 vs. 55.0 ± 0.3% in controls). Cardiomyocytes from uremic rats displayed an enhanced vulnerability to oxidant-induced cell death, as evidenced by a 30% reduction in cell viability following exposure to 200 μM H2O2 compared with controls (cell viability 35.0 ± 2.6 vs. 50.0 ± 1.1% in controls, P < 0.05).
Baseline FAD autofluorescence was similar between groups, characterized by a diffuse pattern of fluorescence (Fig. 3, A and B). Following a 60-min incubation with 200 μM H2O2, cells were characterized by the appearance of foci of enhanced fluorescence (arrows in Fig. 3, C and D). Uremic cardiomyocytes displayed a reduction in mitochondrial redox state following incubation with 200 μM H2O2, exhibiting a 38% increase in fluorescence intensity (P < 0.01; Fig. 3E).
Fig. 3.

FAD autofluorescence in isolated cardiomyocytes following 60-min incubation with 200 μM H2O2. Representative images are shown. A and B: baseline autofluorescence in control and uremic cardiomyocytes. C and D: autofluorescence following incubation with 200 μM H2O2. E: fluorescence intensity determined as an average of 20 cells/preparation expressed relative to baseline controls. *P < 0.01 vs. control baseline; n = 3.
Ca2+-induced permeability transition in isolated mitochondria.
The susceptibility to calcium-induced permeability transition was assessed in isolated mitochondria via rhodamine 123 fluorescence. Representative time courses of fluorescence dequenching in the presence of 0 and 20 μM Ca2+ are shown in Fig. 4, A and B, respectively. Maximum rates of depolarization were enhanced in uremia by 79 and 43% for 0 and 20 μM CaCl2, respectively (Fig. 4, C and D). Preincubation with cyclosporine inhibited depolarization to a similar extent in both groups.
Fig. 4.

Calcium-induced mitochondrial membrane depolarization. A: basal depolarization trace. B: 20 μM Ca2+ depolarization trace. C: maximum rate of mitochondrial depolarization derived from pseudolinear gradients at baseline. #P < 0.05 vs. control; *P < 0.05 vs. control + 1 μM cyclosporine and uremic + 1 μM cyclosporine; n = 8. D: maximum rate of mitochondrial depolarization derived from pseudolinear gradients in the presence of 20 μM CaCl2. #P < 0.05 vs. control; *P < 0.05 vs. control + 1 μM cyclosporine; $P < 0.05 vs. uremic + 1 μM cyclosporine; n = 3.
DISCUSSION
Cardiac mitochondria are uncoupled in UCM.
The present study demonstrates that mitochondrial respiratory function is compromised in the uremic heart. State 3 respiration rates did not differ in uremia and were similar for all substrates tested. In contrast, state 4 respiration was enhanced in the presence of complex I- and complex II-linked substrates. While a nonsignificant increase was observed in the presence of palmitoyl carnitine and malate, this trend was consistent with the other substrates, suggesting a non-substrate-specific uncoupled phenotype in uremic mitochondria. Compromised mitochondrial respiration is a consistent finding in the failing heart (7, 56). Alterations in state 3 respiration have been shown to become evident only at the onset of systolic dysfunction, suggesting that mitochondrial dysfunction is a late development in the transition to heart failure (15). In contrast, mitochondrial respiration is considered to be unaltered in compensated hypertrophy (1, 41). Indeed, the observed increase in state 4 respiration in the presence of normal state 3 respiration is a less common finding. Such a respiratory profile has been reported in the hyperthyroid heart, where uncoupling is associated with an upregulation of uncoupling protein (UCP)2 and UCP3 (3, 28). UCPs mediate a partial dissipation of the proton gradient independent of ADP phosphorylation (18), although whether uncoupling in the uremic heart is associated with altered UCP expression remains unclear.
Stimulation of mitochondrial uncoupling through the induction of uncoupling proteins or via chemical uncouplers has been shown to be beneficial in the acute setting following IRI (57). In contrast, the consequences of a chronically uncoupled phenotype on cardiac function are less well known. Uncoupling reduces the efficiency of respiration, which may manifest as a decreased metabolic reserve. Although the mitochondrial network may be able to support contractile function at basal cardiac activity, uncoupled mitochondria may be less capable of providing bioenergetic support at increased workloads. Indeed, the reserve respiratory capacity is lost in failing hearts (22). Certainly in the present study oxidative capacity appears sufficient to support basal contraction, since cardiac functional parameters were unaltered in uremia. This is consistent with previous observations of both in vitro and in vivo cardiac function in this model (60, 62). However, it is possible that, under increased workloads, mitochondrial uncoupling may impair ATP production. Indeed, this has been demonstrated in an experimental model of obesity where, despite normal baseline cardiac function, enhanced workloads led to an impairment of contractile function (5). A recent editorial recommended monitoring cardiac reserve capacity in CKD patients to detect early dysfunction (9). Since a decrease in mitochondrial reserve capacity precedes cardiac dysfunction, the present data reinforce that argument. Furthermore, with the continued development of positron emission tomography radionuclides for in vivo monitoring of mitochondrial function, noninvasive imaging of cardiac mitochondrial respiratory activity may enable earlier identification of cardiac abnormalities in patients with CKD (67).
Post-ischemia-reperfusion mitochondrial respiration.
Cardiac function was significantly compromised in uremia following ischemia-reperfusion, consistent with previous findings (14, 60). Impaired post-ischemia-reperfusion cardiac function in the uremic heart is associated with increased cardiac injury, as shown previously through increased ischemic inosine release (52) and more recently in an in vivo model of ischemia-reperfusion by increased infarct size and caspase 3 activation (64). Maximal rates of mitochondrial respiration post-ischemia-reperfusion were similar in both groups; however, state 4 respiration was enhanced in uremia in the presence of complex II-linked substrates. From these data, it is unclear whether the mitochondrial uncoupling in uremia contributes to this functional impairment or is reflective of a more significant mitochondrial injury. The time-dependent changes in mitochondrial respiration have been characterized previously in control hearts throughout IRI (4). Mitochondria isolated at the end of ischemia were shown to exhibit an increase in state 4 respiration, which peaked by 2-min reperfusion before falling to preischemic levels by 40-min reperfusion. These increases in state 4 respiration were paralleled by those in mitochondrial swelling, which similarly normalized by the end of the reperfusion period. These authors suggested the uncoupling was attributable to enhanced proton leak, consistent with mPTP opening. The recovery of respiration and normalization of mitochondrial volume indicate that mPTP opening was transient. Indeed, transient pore opening has been proposed as a protective mechanism at the onset of reperfusion to flush accumulated calcium from the matrix (36). Persistence of elevated state 4 respiration at the end of reperfusion, as has been observed in the present study, may reflect a sustained opening of mPTP in uremic hearts following IRI (Fig. 5).
Fig. 5.
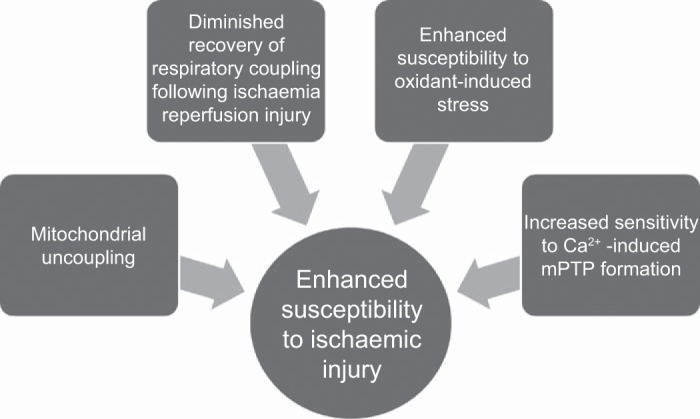
Perturbations in mitochondrial function combine to predispose the uremic heart to ischemia-reperfusion injury (IRI). Persistent mitochondrial uncoupling may compromise energetic reserve capacity, rendering the uremic heart more vulnerable to metabolic stress. Diminished recovery of respiratory coupling following an IRI insult indicates greater susceptibility of uremic mitochondria to ischemic stress. Increased sensitivity to IRI-associated oxidant and calcium stressors enhances the propensity to mitochondrial permeability transition pore (mPTP) formation and subsequent catastrophic cellular injury.
Oxidant-induced stress in isolated cardiomyocytes.
Uremic cardiomyocytes exhibited an enhanced sensitivity to oxidant-induced cell death and mitochondrial oxidation, as determined by FAD autofluorescence. Sensitivity to mPTP opening is enhanced by the presence of ROS, a burst of which during the first minute of reperfusion is associated with mPTP opening (23, 30). Isolated cardiomyocytes were thus exposed to H2O2 to mimic this oxidant-induced stress in vitro. H2O2-treated uremic cardiomyocytes exhibited enhanced FAD fluorescence (Fig. 3E), which was indicative of a more oxidized mitochondrial matrix. Such an increase may be observed in the presence of increased rates of respiration or mitochondrial uncoupling (6). However, qualitative observations revealed the appearance of small foci of enhanced fluorescence in H2O2-treated cells (arrows in Fig. 3, C and D), contrasting with a more diffuse pattern visible in nontreated cells. Previous similar observations have attributed this pattern to individual or small clusters of oxidized mitochondria (54). Therefore, these results are indicative of an enhanced sensitivity to oxidant-induced stress in uremia, giving rise to an increased proportion of oxidized, energy-depleted mitochondria (Fig. 5). In the setting of the reperfused myocardium, this enhanced sensitivity to oxidant stress may belie a propensity of uremic mitochondria to undergo permeability transition and thus promote cellular injury.
Calcium-induced stress in isolated mitochondria.
Cyclosporine-sensitive rates of calcium-induced mitochondrial depolarization were enhanced in uremia, indicating an increased susceptibility to calcium-induced mPTP opening. Elevated matrix calcium is a primary trigger for mPTP induction, and the accumulation of cytosolic calcium is a key event in the development of ischemia-reperfusion injury (66).
Although rates of depolarization in the presence of 20 μM CaCl2 were increased in uremic mitochondria, basal rates of depolarization were also enhanced in the absence of calcium. However, these results do not indicate calcium-independent mPTP opening. Indeed, matrix calcium is essential for triggering pore opening, and other factors such as ROS, pH, and adenine nucleotides serve only to modify the pore sensitivity to calcium (23). Variation in matrix levels of these factors may promote mPTP induction in the absence of externally added calcium. Indeed, under the experimental conditions here, mitochondrial ROS production is augmented compared with state 3 respiration (29). Therefore, enhanced mitochondrial ROS production or sensitivity to ROS may promote mPTP opening in uremic mitochondria.
Importantly, these results implicate a mitochondrial defect in uremia that predisposes the heart to enhanced mPTP opening. Rather than enhanced injury occurring only as a function of impaired calcium extrusion upon reperfusion, these results support previous findings that identified alterations in the cardioprotective signaling pathways in the uremic heart (60).
An increase in mPTP sensitivity, as observed in the present data, may arise from alterations in cardioprotective signaling pathways purported to regulate the pore (44). Insulin administration prior to or at the onset of reperfusion has been shown to activate a prosurvival signaling cascade. i.e., the “reperfusion injury salvage kinase” (RISK) pathway (27). RISK activation is critically dependent upon the phosphorylation of phosphoinositide 3-kinase and Akt, although phosphorylation of extracellular signal-related kinases 1 and 2 have also been implicated (26, 68). A key downstream effector of RISK activation is the inhibition of GSK-3β, which in turn inhibits mPTP opening via a number of proposed mechanisms (43). Previously, our group reported an insensitivity to insulin-mediated cardioprotection in uremia. Critically, GSK-3β phosphorylation was unaltered in the uremic heart following IRI, suggesting an alternative mechanism of impaired signaling (60). Interestingly, however, phosphorylated Akt was significantly enhanced in the uremic heart prior to ischemia, whereas phosphorylated Akt2, the isoform purported to mediate RISK signaling (10), was reduced post-IRI. Chronic Akt activation may prevent the signaling apparatus from mounting a response to IRI stimuli. Indeed, an experimental model of chronic Akt activation has shown greater susceptibility to IRI (46).
Although GSK-3β is unaltered in uremia, defective Akt signaling may modulate mPTP sensitivity unfavorably through endothelial nitric oxide synthase (eNOS), which is activated in response to insulin-mediated Akt activation (21). Nitric oxide has been shown to inhibit mPTP opening in cardiac mitochondria (69), and the mechanism of protection is thought to involve the formation of membrane protein S-nitrothiols (38). Cardiac eNOS expression has been reported as unchanged in uremia (35); however, insulin-mediated activation of the Akt/eNOS pathway has not been investigated in UCM.
An increase in sensitivity to mPTP opening may also arise as a result of modified inner mitochondrial membrane lipids, more specifically cardiolipin (48). Cardiolipin is a phospholipid found almost exclusively in the inner mitochondrial membrane, where it plays a critical role in the function of respiratory complexes as well as the adenine nucleotide translocase, a possible regulatory component of mPTP (34, 65). There is an age-dependent decrease in cardiac cardiolipin content, with a concomitant rise in mitochondrial ROS production and oxidized cardiolipin levels (50). The aged heart is also characterized by an enhanced susceptibility to mPTP opening (51). Selective peroxidation of cardiolipin has been shown to sensitize mitochondria to calcium-induced mPTP opening (49). Cardiolipin biosynthesis and remodeling enzymes are altered during the development of heart failure (58), whereas loss of cardiolipin has been shown to correlate with enhanced state 4 respiration in the failing heart (45). Further study is necessary to investigate whether mitochondrial cardiolipin composition is altered in the uremic heart.
Two separate studies have recently demonstrated that the uremic heart is sensitive to cardioprotection mediated by ischemic conditioning (8, 33). Furthermore, both uremic and control hearts exhibited a similar degree of RISK pathway activation. These results contrast with our previous findings that the uremic heart is insensitive to insulin-mediated cardioprotection, which challenge Dikow and Hardt's (11) observation that the intracellular signaling mechanisms may be intact in uremic cardiomyocytes. Collectively, these observations suggest a differential response of the uremic heart to cardioprotective strategies that is dependent upon the mode of cardioprotection. Taken in concert with the results presented here, modulation of mPTP sensitivity in the uremic heart may represent a major therapeutic target.
Conclusions.
This study has demonstrated that mitochondrial respiration in the uremic heart is chronically uncoupled but that respiratory inefficiency does not impair contractile function. Post-IRI, an increase in state 4 respiration was observed, suggesting that respiratory function is impaired in the postischemic uremic heart. Cellular studies revealed that uremic cardiomyocytes were characterized by a more oxidized mitochondrial network that is more susceptible to oxidant-induced cell death. Finally, uremic mitochondria exhibited an enhanced susceptibility to calcium-induced mPTP formation. Collectively, these findings together with previous observations indicate that mitochondrial respiratory function is compromised in the uremic heart and that an enhanced mitochondrial vulnerability to calcium and oxidant-induced stressors may underpin the increased susceptibility of the uremic heart to IRI.
GRANTS
D. Taylor was the recipient of a University of Hull Ph.D Scholarship, and the research was supported by the Hull and E. Yorkshire NHS Renal Research Fund.
DISCLOSURES
All authors declare no competing interests, financial or otherwise.
AUTHOR CONTRIBUTIONS
D.T., S.B., and A.-M.L.S. conception and design of research; D.T. performed experiments; D.T. analyzed data; D.T. and A.-M.L.S. interpreted results of experiments; D.T. prepared figures; D.T. drafted manuscript; D.T. and A.-M.L.S. edited and revised manuscript; D.T., S.B., and A.-M.L.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Jenny Foster and Laura Goodlass for their excellent technical assistance and Dr. Roger Sturmey and Henry Leese for helpful discussions.
Present Address of D. Taylor: Cedars-Sinai Heart Institute, 8700 Beverly Blvd., Advanced Health Sciences Pavilion, A9600, Los Angeles, CA 90048.
REFERENCES
- 1.Abel ED, Doenst T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc Res 90: 234–242, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschuld R, Gibb L, Ansel A, Hohl C, Kruger FA, Brierley GP. Calcium tolerance of isolated rat heart cells. J Mol Cell Cardiol 12: 1383–1395, 1980. [DOI] [PubMed] [Google Scholar]
- 3.Boehm EA, Jones BE, Radda GK, Veech RL, Clarke K. Increased uncoupling proteins and decreased efficiency in palmitate-perfused hyperthyroid rat heart. Am J Physiol Heart Circ Physiol 280: H977–H983, 2001. [DOI] [PubMed] [Google Scholar]
- 4.Bosetti F, Baracca A, Lenaz G, Solaini G. Increased state 4 mitochondrial respiration and swelling in early post-ischemic reperfusion of rat heart. FEBS Lett 563: 161–164, 2004. [DOI] [PubMed] [Google Scholar]
- 5.Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 112: 2686–2695, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Brandes R, Bers DM. Simultaneous measurements of mitochondrial NADH and Ca(2+) during increased work in intact rat heart trabeculae. Biophys J 83: 587–604, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bugger H, Schwarzer M, Chen D, Schrepper A, Amorim PA, Schoepe M, Nguyen TD, Mohr FW, Khalimonchuk O, Weimer BC, Doenst T. Proteomic remodelling of mitochondrial oxidative pathways in pressure overload-induced heart failure. Cardiovasc Res 85: 376–384, 2010. [DOI] [PubMed] [Google Scholar]
- 8.Byrne CJ, McCafferty K, Kieswich J, Harwood S, Andrikopoulos P, Raftery M, Thiemermann C, Yaqoob MM. Ischemic conditioning protects the uremic heart in a rodent model of myocardial infarction. Circulation 125: 1256–1265, 2012. [DOI] [PubMed] [Google Scholar]
- 9.Chinnappa S, Hothi SS, Tan LB. Is uremic cardiomyopathy a direct consequence of chronic kidney disease? Expert Rev Cardiovasc Ther 12: 127–130, 2014. [DOI] [PubMed] [Google Scholar]
- 10.DeBosch B, Sambandam N, Weinheimer C, Courtois M, Muslin AJ. Akt2 regulates cardiac metabolism and cardiomyocyte survival. J Biol Chem 281: 32841–32851, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dikow R, Hardt SE. The uremic myocardium and ischemic tolerance: a world of difference. Circulation 125: 1215–1216, 2012. [DOI] [PubMed] [Google Scholar]
- 12.Dikow R, Kihm LP, Zeier M, Kapitza J, Tornig J, Amann K, Tiefenbacher C, Ritz E. Increased infarct size in uremic rats: reduced ischemia tolerance? J Am Soc Nephrol 15: 1530–1536, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Dikow R, Schmidt U, Kihm LP, Schaier M, Schwenger V, Gross ML, Katus HA, Zeier M, Hardt SE. Uremia aggravates left ventricular remodeling after myocardial infarction. Am J Nephrol 32: 13–22, 2010. [DOI] [PubMed] [Google Scholar]
- 14.Dikow R, Wasserhess C, Zimmerer K, Kihm LP, Schaier M, Schwenger V, Hardt S, Tiefenbacher C, Katus H, Zeier M, Gross LM. Effect of insulin and glucose infusion on myocardial infarction size in uraemic rats. Basic Res Cardiol 104: 571–579, 2009. [DOI] [PubMed] [Google Scholar]
- 15.Doenst T, Pytel G, Schrepper A, Amorim P, Farber G, Shingu Y, Mohr FW, Schwarzer M. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc Res 86: 461–470, 2010. [DOI] [PubMed] [Google Scholar]
- 16.Drey N, Roderick P, Mullee M, Rogerson M. A population-based study of the incidence and outcomes of diagnosed chronic kidney disease. Am J Kidney Dis 42: 677–684, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Emaus RK, Grunwald R, Lemasters JJ. Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: spectral and metabolic properties. Biochim Biophys Acta 850: 436–448, 1986. [DOI] [PubMed] [Google Scholar]
- 18.Fink BD, Hong YS, Mathahs MM, Scholz TD, Dillon JS, Sivitz WI. UCP2-dependent proton leak in isolated mammalian mitochondria. J Biol Chem 277: 3918–3925, 2002. [DOI] [PubMed] [Google Scholar]
- 19.Foley RN, Parfrey PS, Harnett JD, Kent GM, Martin CJ, Murray DC, Barre PE. Clinical and echocardiographic disease in patients starting end-stage renal disease therapy. Kidney Int 47: 186–192, 1995. [DOI] [PubMed] [Google Scholar]
- 20.Ganote CE, Humphrey SM. Effects of anoxic or oxygenated reperfusion in globally ischemic, isovolumic, perfused rat hearts. Am J Pathol 120: 129–145, 1985. [PMC free article] [PubMed] [Google Scholar]
- 21.Gao F, Gao E, Yue TL, Ohlstein EH, Lopez BL, Christopher TA, Ma XL. Nitric oxide mediates the antiapoptotic effect of insulin in myocardial ischemia-reperfusion: the roles of PI3-kinase, Akt, and endothelial nitric oxide synthase phosphorylation. Circulation 105: 1497–1502, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Gong G, Liu J, Liang P, Guo T, Hu Q, Ochiai K, Hou M, Ye Y, Wu X, Mansoor A, From AH, Ugurbil K, Bache RJ, Zhang J. Oxidative capacity in failing hearts. Am J Physiol Heart Circ Physiol 285: H541–H548, 2003. [DOI] [PubMed] [Google Scholar]
- 23.Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol 46: 821–831, 2009. [DOI] [PubMed] [Google Scholar]
- 25.Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta 1787: 1402–1415, 2009. [DOI] [PubMed] [Google Scholar]
- 26.Hausenloy DJ, Tsang A, Mocanu MM, Yellon DM. Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am J Physiol Heart Circ Physiol 288: H971–H976, 2005. [DOI] [PubMed] [Google Scholar]
- 27.Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc Res 61: 448–460, 2004. [DOI] [PubMed] [Google Scholar]
- 28.Heather LC, Cole MA, Atherton HJ, Coumans WA, Evans RD, Tyler DJ, Glatz JF, Luiken JJ, Clarke K. Adenosine monophosphate-activated protein kinase activation, substrate transporter translocation, and metabolism in the contracting hyperthyroid rat heart. Endocrinology 151: 422–431, 2010. [DOI] [PubMed] [Google Scholar]
- 29.Hoffman DL, Salter JD, Brookes PS. Response of mitochondrial reactive oxygen species generation to steady-state oxygen tension: implications for hypoxic cell signaling. Am J Physiol Heart Circ Physiol 292: H101–H108, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Javadov S, Karmazyn M, Escobales N. Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J Pharmacol Exp Ther 330: 670–678, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest 113: 1535–1549, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kennedy D, Omran E, Periyasamy SM, Nadoor J, Priyadarshi A, Willey JC, Malhotra D, Xie Z, Shapiro JI. Effect of chronic renal failure on cardiac contractile function, calcium cycling, and gene expression of proteins important for calcium homeostasis in the rat. J Am Soc Nephrol 14: 90–97, 2003. [DOI] [PubMed] [Google Scholar]
- 33.Kocsis GF, Sárközy M, Bencsik P, Pipicz M, Varga ZV, Pálóczi J, Csonka C, Ferdinandy P, Csont T. Preconditioning protects the heart in a prolonged uremic condition. Am J Physiol Heart Circ Physiol 303: H1229–H1236, 2012. [DOI] [PubMed] [Google Scholar]
- 34.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427: 461–465, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koleganova N, Piecha G, Ritz E, Gross ML. Calcitriol ameliorates capillary deficit and fibrosis of the heart in subtotally nephrectomized rats. Nephrol Dial Transplant 24: 778–787, 2009. [DOI] [PubMed] [Google Scholar]
- 36.Korge P, Yang L, Yang JH, Wang Y, Qu Z, Weiss JN. Protective role of transient pore openings in calcium handling by cardiac mitochondria. J Biol Chem 286: 34851–34857, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lecoeur H, Borgne-Sanchez A, Chaloin O, El-Khoury R, Brabant M, Langonne A, Porceddu M, Briere JJ, Buron N, Rebouillat D, Pechoux C, Deniaud A, Brenner C, Briand JP, Muller S, Rustin P, Jacotot E. HIV-1 Tat protein directly induces mitochondrial membrane permeabilization and inactivates cytochrome c oxidase. Cell Death Dis 3: e282, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leite AC, Oliveira HC, Utino FL, Garcia R, Alberici LC, Fernandes MP, Castilho RF, Vercesi AE. Mitochondria generated nitric oxide protects against permeability transition via formation of membrane protein S-nitrosothiols. Biochim Biophys Acta 1797: 1210–1216, 2010. [DOI] [PubMed] [Google Scholar]
- 39.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 322: 1561–1566, 1990. [DOI] [PubMed] [Google Scholar]
- 40.Marks A, Macleod C, McAteer A, Murchie P, Fluck N, Smith WC, Prescott GJ, Clark LE, Ali T, Black C. Chronic kidney disease, a useful trigger for proactive primary care? Mortality results from a large U.K. cohort. Fam Pract 30: 282–289, 2013. [DOI] [PubMed] [Google Scholar]
- 41.Matlib MA, Rembert JC, Millard RW, Ashraf M, Rouslin W, Asano G, Greenfield JC Jr, Schwartz A. Mitochondrial function in canine experimental cardiac hypertrophy. J Mol Cell Cardiol 15: 221–232, 1983. [DOI] [PubMed] [Google Scholar]
- 42.McMahon AC, Greenwald SE, Dodd SM, Hurst MJ, Raine AE. Prolonged calcium transients and myocardial remodelling in early experimental uraemia. Nephrol Dial Transplant 17: 759–764, 2002. [DOI] [PubMed] [Google Scholar]
- 43.Miura T, Tanno M. Mitochondria and GSK-3beta in cardioprotection against ischemia/reperfusion injury. Cardiovasc Drugs Ther 24: 255–263, 2010. [DOI] [PubMed] [Google Scholar]
- 44.Miura T, Tanno M. The mPTP and its regulatory proteins: final common targets of signalling pathways for protection against necrosis. Cardiovasc Res 94: 181–189, 2012. [DOI] [PubMed] [Google Scholar]
- 45.Mulligan CM, Sparagna GC, Le CH, De Mooy AB, Routh MA, Holmes MG, Hickson-Bick DL, Zarini S, Murphy RC, Xu FY, Hatch GM, McCune SA, Moore RL, Chicco AJ. Dietary linoleate preserves cardiolipin and attenuates mitochondrial dysfunction in the failing rat heart. Cardiovasc Res 94: 460–468, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagoshi T, Matsui T, Aoyama T, Leri A, Anversa P, Li L, Ogawa W, del Monte F, Gwathmey JK, Grazette L, Hemmings BA, Kass DA, Champion HC, Rosenzweig A. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J Clin Invest 115: 2128–2138, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ng KW, Allen ML, Desai A, Macrae D, Pathan N. Cardioprotective effects of insulin: how intensive insulin therapy may benefit cardiac surgery patients. Circulation 125: 721–728, 2012. [DOI] [PubMed] [Google Scholar]
- 48.Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Changes in the mitochondrial permeability transition pore in aging and age-associated diseases. Mech Ageing Dev 134: 1–9, 2013. [DOI] [PubMed] [Google Scholar]
- 49.Petrosillo G, Casanova G, Matera M, Ruggiero FM, Paradies G. Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: induction of permeability transition and cytochrome c release. FEBS Lett 580: 6311–6316, 2006. [DOI] [PubMed] [Google Scholar]
- 50.Petrosillo G, Matera M, Moro N, Ruggiero FM, Paradies G. Mitochondrial complex I dysfunction in rat heart with aging: critical role of reactive oxygen species and cardiolipin. Free Radic Biol Med 46: 88–94, 2009. [DOI] [PubMed] [Google Scholar]
- 51.Petrosillo G, Moro N, Paradies V, Ruggiero FM, Paradies G. Increased susceptibility to Ca(2+)-induced permeability transition and to cytochrome c release in rat heart mitochondria with aging: effect of melatonin. J Pineal Res 48: 340–346, 2010. [DOI] [PubMed] [Google Scholar]
- 52.Raine AE, Seymour AM, Roberts AF, Radda GK, Ledingham JG. Impairment of cardiac function and energetics in experimental renal failure. J Clin Invest 92: 2934–2940, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reddy V, Bhandari S, Seymour AML. Myocardial function, energy provision, and carnitine deficiency in experimental uremia. J Am Soc Nephrol 18: 84–92, 2007. [DOI] [PubMed] [Google Scholar]
- 54.Romashko DN, Marban E, O'Rourke B. Subcellular metabolic transients and mitochondrial redox waves in heart cells. Proc Natl Acad Sci USA 95: 1618–1623, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosca MG, Hoppel CL. Mitochondria in heart failure. Cardiovasc Res 88: 40–50, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, Stanley W, Sabbah HN, Hoppel CL. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res 80: 30–39, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sack MN. Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. Cardiovasc Res 72: 210–219, 2006. [DOI] [PubMed] [Google Scholar]
- 58.Saini-Chohan HK, Holmes MG, Chicco AJ, Taylor WA, Moore RL, McCune SA, Hickson-Bick DL, Hatch GM, Sparagna GC. Cardiolipin biosynthesis and remodeling enzymes are altered during development of heart failure. J Lipid Res 50: 1600–1608, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Semple D, Smith K, Bhandari S, Seymour AM. Uremic cardiomyopathy and insulin resistance: a critical role for akt? J Am Soc Nephrol 22: 207–215, 2011. [DOI] [PubMed] [Google Scholar]
- 60.Semple DJ, Bhandari S, Seymour AM. Uremic cardiomyopathy is characterized by loss of the cardioprotective effects of insulin. Am J Physiol Renal Physiol 303: F1275–F1286, 2012. [DOI] [PubMed] [Google Scholar]
- 61.Shroff GR, Frederick PD, Herzog CA. Renal failure and acute myocardial infarction: clinical characteristics in patients with advanced chronic kidney disease, on dialysis, and without chronic kidney disease. A collaborative project of the United States Renal Data System/National Institutes of Health and the National Registry of Myocardial Infarction. Am Heart J 163: 399–406, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith K, Semple D, Aksentijevic D, Bhandari S, Seymour AM. Functional and metabolic adaptation in uraemic cardiomyopathy. Front Biosci (Elite Ed) 2: 1492–1501, 2010. [DOI] [PubMed] [Google Scholar]
- 63.Smolenski RT, Schrader J, de Groot H, Deussen A. Oxygen partial pressure and free intracellular adenosine of isolated cardiomyocytes. Am J Physiol Cell Physiol 260: C708–C714, 1991. [DOI] [PubMed] [Google Scholar]
- 64.Song Y, Yu Q, Zhang J, Huang W, Liu Y, Pei H, Liu J, Sun L, Yang L, Li C, Li Y, Zhang F, Qu Y, Tao L. Increased myocardial ischemia-reperfusion injury in renal failure involves cardiac adiponectin signal deficiency. Am J Physiol Endocrinol Metab 306: E1055–E1064, 2014. [DOI] [PubMed] [Google Scholar]
- 65.Sparagna GC, Chicco AJ, Murphy RC, Bristow MR, Johnson CA, Rees ML, Maxey ML, McCune SA, Moore RL. Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J Lipid Res 48: 1559–1570, 2007. [DOI] [PubMed] [Google Scholar]
- 66.Stamm C, Friehs I, Choi YH, Zurakowski D, McGowan FX, del Nido PJ. Cytosolic calcium in the ischemic rabbit heart: assessment by pH- and temperature-adjusted rhod-2 spectrofluorometry. Cardiovasc Res 59: 695–704, 2003. [DOI] [PubMed] [Google Scholar]
- 67.Strauss HW, Schöder H. Myocardial imaging for mitochondrial membrane potential. JACC Cardiovasc Imaging 5: 293–296, 2012. [DOI] [PubMed] [Google Scholar]
- 68.Tong H, Chen W, Steenbergen C, Murphy E. Ischemic preconditioning activates phosphatidylinositol-3-kinase upstream of protein kinase C. Circ Res 87: 309–315, 2000. [DOI] [PubMed] [Google Scholar]
- 69.Wang G, Liem DA, Vondriska TM, Honda HM, Korge P, Pantaleon DM, Qiao X, Wang Y, Weiss JN, Ping P. Nitric oxide donors protect murine myocardium against infarction via modulation of mitochondrial permeability transition. Am J Physiol Heart Circ Physiol 288: H1290–H1295, 2005. [DOI] [PubMed] [Google Scholar]