

Abstract
Acute pancreatitis is an inflammatory process of the pancreatic gland that eventually may lead to a severe systemic inflammatory response. A key event in pancreatic damage is the intracellular activation of NF-κB and zymogens, involving also calcium, cathepsins, pH disorders, autophagy, and cell death, particularly necrosis. This review focuses on the new role of redox signaling in acute pancreatitis. Oxidative stress and redox status are involved in the onset of acute pancreatitis and also in the development of the systemic inflammatory response, being glutathione depletion, xanthine oxidase activation, and thiol oxidation in proteins critical features of the disease in the pancreas. On the other hand, the release of extracellular hemoglobin into the circulation from the ascitic fluid in severe necrotizing pancreatitis enhances lipid peroxidation in plasma and the inflammatory infiltrate into the lung and up-regulates the HIF–VEGF pathway, contributing to the systemic inflammatory response. Therefore, redox signaling and oxidative stress contribute to the local and systemic inflammatory response during acute pancreatitis.
Keywords: Acute pancreatitis, Oxidative stress, Glutathione, Acute inflammation
Abbreviations: AP, acute pancreatitis; AP-1, activator protein-1; ASC, apoptosis-associated speck-like protein containing a carboxy-terminal CARD; ASK1, apoptosis signal-regulating kinase-1; CARS, compensatory anti-inflammatory response syndrome; CBP, CREB-binding protein; CCK, cholecystokinin; DAMPs, damage-associated molecular pattern molecules; DTT, dithiothreitol; ERCP, endoscopic retrograde cholangiopancreatography; ERK, extracellular signal-regulated kinases; ETC, Electron transport chain; FAEE, fatty acid ethyl esters; GCL, glutamate cysteine ligase; GSH, reduced glutathione; GSSG, oxidized glutathione; HATs, histone acetyltransferases; HDACs, histone deacetylases; HIF, hypoxia inducible factor; HMGB1, high-mobility group Box 1 protein; IKK, IκB kinase; IL, interleukin; IκB, inhibitor of kappa B; IP3R, inositol 1,4,5-trisphosphate receptor type 3; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MDA, malondialdehyde; MKPs, MAPK phosphatases; mRNA, messenger ribonucleic acid; NAC, N-acetyl cysteine; NADH, nicotinamide adenine dinucleotide; NADPH, nicotinamide adenine dinucleotide phosphate; NF-κB, nuclear factor kappa B; NLRs, nucleotide-binding oligomerization domain (NOD) like receptors; NO, nitric oxide; NOS, nitric oxide synthase; pHe, extracellular pH; PTPs, protein tyrosine phosphatases; RNS, reactive nitrogen species; ROS, reactive oxygen species; RyR, endoplasmic reticulum membrane ryanodine receptors; SAP, severe acute pancreatitis; SHP, small heterodimer partner; STAT3, signal transducer and activator of transcription 3; STIM1, stromal interaction molecule 1; TLRs, toll-like receptors; TNFR, tumor necrosis factor receptor; TNF-α, tumor necrosis factor alpha; TRADD, tumor necrosis factor receptor type 1-associated DEATH domain protein; TRAFs, TNF receptor associated factors; TRPC3, transient receptor potential channel 3; VCAM-1, Vascular Cell adhesion protein 1; VEGF, vascular endothelial growth factor; XDH, xanthine dehydrogenase; XO, xanthine oxidase
Introduction
Acute pancreatitis (AP) is a localized inflammation of the pancreatic gland that often leads to local and systemic complications, such as lung injury and renal failure. The mortality rate in patients with AP is approximately 5%, but this percentage rises to 17–20% in patients with necrotizing pancreatitis. Mortality in AP is ascribed to multiple organ failure that may be triggered by the associated systemic inflammatory response [1]. AP should be considered, together with sepsis, trauma, burns, and surgery, as a pathologic condition that may eventually lead to the systemic inflammatory response syndrome together with multiple organ failure [2].
Although the etiology of disease is diverse, alcohol abuse and duct obstruction by gallstones are the major causes of AP [3]. Obesity is a prognostic factor for severity in the course of AP, as local and systemic complications are more frequent in obese than in non-obese patients [3,4]. AP is characterized by interstitial edema, cell vacuole accumulation and inflammatory infiltrate of macrophages and neutrophils together with necrosis of the pancreatic tissue [1]. Infection and pancreatic necrosis seem to determine severity and complications in AP [5,6]. Most patients exhibit mild interstitial edematous pancreatitis, which is self-limiting and responds rapidly to conservative management. However, around 20% of patients suffer severe acute pancreatitis (SAP), which can progress to a significant mortality [7]. Pulmonary, renal, cardiovascular, central nervous system, and coagulation are most commonly affected in acute pancreatitis [8].
Clinically, SAP occurs in two stages. In the first phase, a systemic inflammatory response syndrome might arise leading to multiple organ failure within the first few days [8,9]. If this process is not restrained or stopped, the second phase will appear, developing local complications and a subsequent compensatory anti-inflammatory response syndrome (CARS). The function of the immune system is diminished or impaired during CARS, which facilitates nosocomial infections, including infected pancreatic necrosis, one of the most feared complications of the disease [10]. The initiation and development of these stages of SAP is mediated by diverse pathophysiological mechanisms involved in the systemic inflammatory response. This review focuses on the mechanisms related to oxidative stress and redox signaling involved in the initiation and progression of the disease.
Early events in acute pancreatitis
The mechanisms involved in the pathogenesis of AP and its associated pancreatic injury are not completely elucidated, but they are believed to begin in the acinar cell [11]. Since excessive alcohol consumption and gallstones are the main causes of acute pancreatitis, both etiological factors play pivotal roles in triggering acute pancreatitis [3].
Alcohol's toxicity is mediated mainly by its oxidative and non-oxidative metabolism. Alcohol metabolism leads to an increase in fatty acid ethyl ester (FAEE) levels through its non-oxidative metabolism, and to the accumulation of acetaldehyde, acetate, and NADH through its oxidative metabolism [12]. FAEEs exert several deleterious effects, such as disruption of calcium homeostasis in acinar cells [13], pathologic zymogen activation [14], and activation of transcription factors (NF-κB and AP-1) [15]. The latter effect is enhanced by acetaldehyde leading to increased production of pro-inflammatory cytokines. In addition, alcohol modifies the intracellular redox state of the cell by diminishing the [NAD]/[NADH] ratio and increasing the [lactate]/[pyruvate] ratio, leading to metabolic alterations and acinar injury [16].
Duct obstruction by gallstones is associated with an increase in duct pressure and acinar cell exposure to bile acids. This obstruction may block acinar exocytosis leading to co-localization of zymogen and lysosomal granules and early activation of pancreatic enzymes [17]. Thus, the intrapancreatic activation of trypsinogen and other zymogen enzymes; inhibition of secretion; alterations in calcium homeostasis; and activation of cell death pathways are early events in AP [1,18] leading to inflammation of the pancreatic tissue. In those early events of AP, oxidative stress plays a pivotal role since reactive oxygen species (ROS) cause direct oxidative damage to lipids and proteins and modulate redox sensitive transcription factors and redox-sensitive signal transduction pathways [19].
Early events in acute pancreatitis will lead to the activation of several pathophysiological mechanisms that could produce local and systemic complications and organ failure, which is responsible for the initial mortality of the disease [8,9]. The main mechanisms responsible for this systemic progression are pro-inflammatory cytokines, chemokines, ROS, Ca2+, platelet activating factor, and adenosine, as well as neuronal and vascular responses [1,20–22]. Furthermore, acinar cells can behave as inflammatory cells synthesizing and releasing cytokines, chemokines and adhesion molecules [9,23,24]. Hence, acinar cells act jointly with leukocytes triggering the inflammatory response after the local damage of the pancreas.
Zymogen activation
The premature intracellular zymogen activation is one of the earliest events of AP. The physiological conversion of inactive trypsinogen to trypsin is catalyzed by the intestinal enzyme enterokinase in the gut. Trypsinogen is the most important zymogen in the initiation of pancreatic injury [25,26]. A recent study demonstrates that expression of trypsin in acinar cells is enough to induce cell death and inflammation in pancreatic tissue, using a conditional trypsinogen construct activated by post-translational modification [27]. Several mechanisms are involved in zymogen activation such as calcium influx, co-localization of lysosomes, pH modifications and autophagy.
Role of Ca2+
Hypercalcemia is a risk factor for pancreatitis, consequently the role of cytosolic Ca2+ has been described as a key modulator of the initiation and development of AP. The continuous increase of cytosolic Ca2+ in acinar cells [28] and the blockade of calcium influx lead to increase of trypsinogen activation [29].
In this context, the study of the primary sources of Ca2+ is crucial in the development of the disease. Orabi et al. [30] examined the role of endoplasmic reticulum membrane ryanodine receptors (RyR) using a pharmacologic antagonist, dantrolene. This study suggests that RyR plays an important role in mediating early acinar cell events during in vivo pancreatitis and contributes to the severity of the disease. Inhibition of transient receptor potential channel 3 (TRPC3), a channel involved in the membrane Ca2+ influx, caused reduced receptor-stimulated Ca2+ influx and a lower sustained Ca2+ increase, demonstrating that TRPC3 deficiency prevents the intra-acinar zymogen activation and pancreatitis severity in vivo [31]. Moreover, ethanol and bile acid metabolites cause a sustained pathologic Ca2+ increase due to ATP depletion [32].
Another relevant focus is the downstream target of Ca2+, such as calcineurin, which is an Ca2+-activated protein phosphatase. Mice pretreated with tacrolimus, a calcineurin inhibitor, showed reduced protease activation and decreased severity of the pancreatitis episode after cerulein hyperstimulation [33].
Colocalization of lysosomes
During decades, the major hypothesis to explain zymogen activation within acinar cells has been the co-localization of lysosomes with pancreatic digestive enzymes [34,35]. This hypothesis suggests that during the early stages of AP, pancreatic-derived digestive zymogens become co-localized with lysosomal hydrolases in cytoplasmic vacuoles in acinar cell resulting in trypsinogen activation produced by lysosomal hydrolases, such as cathepsin B [36].
Nevertheless, subcellular redistribution of cathepsin B induces neither spontaneous trypsinogen activation nor pancreatitis [37]. This finding suggests that co-localization needs another condition, most likely low pH. In vitro studies showed that a low pH increases the catalytic capacity of cathepsin B [35]. Thus, stimulation of vacuolar ATPase activity appears to be required for zymogen activation in the acinar cell [38].
pH effects
Low extracellular pH (pHe) promotes the development of pancreatitis. Reducing pHe from 7.4 to 7.0 sensitizes acinar cells to pancreatitis-like responses both in vitro and in vivo in a cerulein model of pancreatitis [39].
The injurious effects of low pHe on the acinar cell are likely mediated through changes in Ca2+ signaling. RyR inhibitors significantly reduce the sensitizing effects of low pHe on zymogen activation and cellular injury. This finding suggests that enhanced RyR-mediated Ca2+ signaling in the basolateral region of the acinar cell is responsible for the effects of low pHe in the exocrine pancreas [40].
In addition, Behrendorff et al. [41] demonstrated that exocytosis of zymogen granules from acinar cells leads to luminal acidification, and this process could contribute to tissue injury in cases of AP.
Autophagy in acute pancreatitis
The autophagic response is a complex process involving lysosomal-dependent recycling of intracellular components. Autophagy occurs at a basal rate in most cells, where it acts as a quality control mechanism to eliminate protein aggregates and damaged or unneeded organelles. Basal autophagy rate is higher in mouse exocrine pancreas than in liver, kidney, heart, or endocrine pancreas [42], since the exocrine pancreas is characterized by a high rate of protein synthesis.
Another interesting feature of AP is the accumulation of autophagic vacuoles in the acinar cell [43], but the role of autophagy in AP is still under debate. On the one hand, autophagy exerts damage effects in acinar cells during the onset of AP by activating trypsinogen to trypsin through delivering trypsinogen to the lysosome [44]. On the other hand, Marerinova et al. [45] showed that retarded autophagy is associated with an imbalance between cathepsin L, which degrades trypsinogen and trypsin, and cathepsin B, which converts trypsinogen into trypsin, resulting in intra-acinar accumulation of active trypsin in pancreatitis. Thus, a deficient lysosomal degradation may be a dominant mechanism for increased intra-acinar trypsin in pancreatitis.
Recently, another group described autophagy as a selective process, called zymophagy, that modulates pancreatitis-induced intracellular zymogen activation. This process seems to prevent cell death in early pancreatitis [46].
Inflammatory response in acute pancreatitis
Role of NF-κB
It has been described that activation of NF-κB occurs early in AP simultaneously with intracellular trypsinogen activation [27]. By contrast, expression of active trypsin in vitro failed to activate NF-κB, suggesting that these two events are independents [47].
NF-κB is a transcription factor that plays a pivotal role in regulating the inflammatory response in mammals [48]. The NF-κB family includes five members: p50, p52, RelA/p65, c-Rel, and RelB [48]. NF-κB develops a crucial role in the pathogenesis of AP [49]. It is activated early in AP, not only in leukocytes but also within pancreatic acinar cells [50]. Adenoviral-mediated overexpression of the active RelA/p65 NF-κB subunit induced severe pancreatitis in mice, characterized by NF-κB activation, up-regulation of NF-κB target genes, neutrophil infiltration, and widespread damage to pancreatic acinar cells [51]. Furthermore, constitutive overexpression of active IKK2 – a key mediator of the canonical NF-κB pathway – in pancreatic acinar cells was sufficient to induce AP [49]. Indeed, it caused edema, necrosis, leukocyte infiltration, as well as increased serum lipase activity and up-regulation of NF-κB target genes in the pancreas [49]. Histological damage and TNF-α expression were reduced in cerulein-induced pancreatitis in mice deficient in NF-κB [52].
Role of inflammatory cytokines in acute pancreatitis
Since cytokines exhibit a cross-talk with oxidative stress and are key players in the systemic response in acute pancreatitis [53], we have also reviewed their role. Cytokines are low molecular weight soluble proteins produced during stress or injury in numerous cell types as means of cell-to-cell communication [54,55]. Activated leukocytes are the main source of cytokines, which are consequently essential components of the inflammatory cascade. The primary members of the cytokine inflammatory family, particularly interleukin 1β (IL-1β) and tumor necrosis factor alpha (TNF-α), induce their own expression as well as other cytokines expression by positive feedback mechanisms that lead to amplification of the inflammatory response and are critically involved in the systemic inflammatory response syndrome [55,56]. Since dexamethasone treatment does not affect trypsinogen activation, it seems that cytokine up-regulation and inflammation occur immediately after this zymogen activation [57].
Leukocyte recruitment within the inflamed pancreas begins as early as 3 hours after AP induction with rolling and adhesion of the circulating leukocytes to the endothelium [58]. Activated macrophages release pro-inflammatory cytokines, such as IL-1β, IL-6 and TNF-α, in response to the local damage of the pancreas [9]. As indicated above, local cells contribute to increase serum levels of IL-1β, IL-6 and TNF-α in experimental AP [23]. These levels correlate with the degree of pancreatic inflammation [59–61]. Thus, peripheral blood monocyte and neutrophil counts correlate with plasma inflammatory cytokines and TNF-α soluble receptors in AP [62].
TNF-α and interleukin-1β (IL-1β) are considered primary cytokines initiating and propagating most of the consequences of the systemic inflammatory response in AP [21,22]. They amplify the inflammatory cascade by activating mitogen-activated protein kinases (MAPKs) and nuclear factor-κB (NF-κB), which induce the release of chemokines and other cytokines, and a positive feedback loop, which up-regulates their own expression [63]. Accordingly, the systemic inflammatory response in severe necrotizing AP was restrained by inhibiting tissue injury, pro-inflammatory cytokine production, acinar cell death, and cytokine production via inhibition of NF-κB and ERK1/2 activation [64].
TNF-α is released from different tissues in the course of AP. There is an induction of its mRNA and protein in pancreas [65]. Although acinar cells may produce TNF-α, leukocytes from the inflammatory infiltrate within the pancreatic tissue are considered as the predominant source [55]. Fig. 1 shows the presence of TNF-α in rat pancreatic tissue after induction of acute pancreatitis with 3.5% of sodium taurocholate (3 h). Necrotic tissue and inflammatory infiltrate are associated with the presence of TNF-α [66].
Fig. 1.
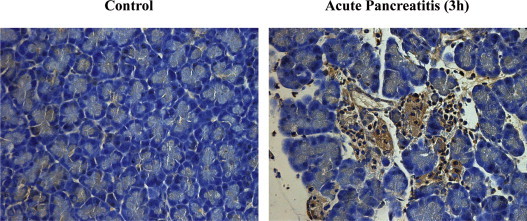
Immunohistochemical detection of TNF-α production by acinar cells in rat pancreas 3 h after AP induction with 3.5% of sodium taurocholate.
TNF-α triggers cell death through divergent mechanisms mediated by protein kinase C and causes NF-κB activation leading to pro-inflammatory up-regulation [67]. We found that inhibition of TNF-α production by pentoxifylline markedly diminished leukocyte infiltrate, edema and glutathione depletion in pancreas as well as reduced serum lipase activity after cerulein-induced pancreatitis in rats [68].
The production of IL-1β in AP may be also pancreatic and extrapancreatic. Similarly to TNF-α, few hours after pancreatic IL-1β levels increase, both its mRNA and protein are induced in lungs and liver [65]. Leukocytes from the inflammatory infiltrate within the pancreatic tissue appear to be the predominant source of IL-1β [55]. IL-1 production is closely associated with induction of other genes within the same gene-family, such as that encoding for interleukin 1β-converting enzyme (ICE), which is necessary for cleavage of pro-IL-1 protein into its active form [55].
IL-1 exhibits similar actions to TNF-α. Thus, it induces the release of other cytokines, such as IL-2 by T-helper lymphocytes and cellular adhesion molecules, which extend the inflammatory response [9]. IL-1β seems to be a pivotal inflammatory mediator in cell death associated with sterile inflammation [69], which is an important event in AP.
Finally, the inflammatory response in AP is modulated by a balance between pro-inflammatory and anti-inflammatory mediators. IL-6 and IL-8 are markers of severity in AP. IL-6 mediates the acute-phase response, whereas IL-8 participates in neutrophil chemotaxis, activation, and degranulation [70]. Nevertheless, the elevation of IL-6 levels should not be considered necessarily pathogenic, because it also induces the expression of suppressor of cytokine signals as a compensatory response mediated by STAT3 that limits the extent of inflammation [71]. IL-10 and pancreatitis-associated protein-1 (PAP-1) are major anti-inflammatory mediators in AP. IL-10 diminishes pancreatic damage [72] and exhibits anti-inflammatory actions mainly through inhibition of IL-1β and TNF-α [73]. Pap-1 knockout mice exhibited enhanced pancreatic inflammation in mild cerulein-induced pancreatitis [74]. PAP-1 reduced the inflammatory response by blocking NF-κB activation and decreasing the expression of pro-inflammatory cytokines and adhesion molecules [75,76].
Role of DAMPs and inflammasome in acute pancreatitis
Cell necrosis releases damage associated molecular patterns (DAMPs) to the extracellular space, which stimulate the inflammatory response through DAMP-receptors, mainly TOLL and purinergic receptors [77]. DAMPs contribute to the pathogenesis of acute pancreatitis as blockade of the DAMP HMBG1 reduces the inflammatory response and cell necrosis in severe acute pancreatitis [78].
DAMPs trigger activation of the large cytoplasmic multiprotein complex inflammasome that leads to pro-IL1β cleavage and IL-1β secretion [79]. Caspase-1, ASC, and NLRP3 are central components of the inflammasome. Hoque et al. have demonstrated that the inflammasome is markedly activated during pancreatitis through DAMP-receptors TLR9 and P2X7 [80]. Furthermore, caspase 1, NLRP3 and ASC are required for NF-κB activation and maximal pancreatitis. Accordingly, antagonists of TLR9 and P2X7 reduced markedly the inflammatory response in acute pancreatitis [80].
Interestingly, mitochondrial ROS act as signal-transducing molecules that up-regulate pro-inflammatory cytokine production via inflammasome-dependent pathways [81–83]. Hence, further studies are needed to clarify the role of mitochondrial reactive oxygen species (ROS) in the inflammatory response in pancreatitis.
Oxidative stress in acute pancreatitis
During last decades numerous studies have highlighted the role of oxidative stress in the acute inflammatory response, particularly in the pancreatic injury associated with AP [84–86]. In the eighties, Sanfey and co-workers reported the beneficial effects of pre-treatments with antioxidants, such as superoxide dismutase (SOD) and catalase (CAT), providing an indirect proof for the involvement of oxidative stress in AP [87]. They also emphasized the role of the pro-oxidant enzyme xanthine oxidase (XO) in AP. Accordingly they suggested that allopurinol, an inhibitor of XO, could be a potential therapy for the disease [87].
Later several groups including ours studied in depth oxidative stress, XO, and other pro-oxidants and antioxidant agents in AP [88,89]. As a result, oxidative stress is presently considered as a key mediator not only of the early local events associated with AP, but also of the associated systemic inflammatory response syndrome [89].
The in situ demonstration of oxygen free radical formation during pancreatitis in rats and humans was performed by Telek and co-workers using cerium capture of oxygen free radicals [90,91]. The source of ROS may differ depending on the experimental model of AP. In mild AP caused by overstimulation with cerulein, free radical generation would be mainly associated with infiltration of activated neutrophils, whereas in necrotic AP induced by taurocholate retrograde perfusion it seems to be mainly due to the conversion of XO dehydrogenase (XDH) to XO [92]. Other pro-oxidant enzymes that contribute to pancreatic inflammation are cytochrome P450 [93,94] and specially NADPH oxidase [95].
Experimental AP is characterized by marked depletion of reduced glutathione in pancreas together with increased lipid peroxidation in the tissue and in plasma [96]. Therefore, oxidative stress is present in both local and systemic responses during the disease.
Moreover, clinical studies have also verified the presence of oxidative stress during AP [97]. Indeed, lipid peroxidation, myeloperoxidase activity, and protein carbonyls increase in plasma of patients with SAP [98–100]. All these parameters are generally related to severity of the disease both in clinical and experimental studies. Thus, the increase in malondialdehyde (MDA) levels correlates with tissue injury in AP [100,101]. Furthermore, superoxide radical and lipid peroxide levels also increase in blood of patients and animals with AP, and these changes correlate with the degree of AP severity [85,102]. Increased levels of MDA have also been associated with pancreatitis-associated multiple organ dysfunction [103]. Furthermore, oxidative stress markers correlate with serum phospholipase A2 and plasma polymorphonuclear elastase activities, two prognostic parameters in AP [104]. In addition, concentrations of antioxidant vitamins are diminished in AP and are inversely related to the rise in C reactive protein level in this disease [105].
Reactive nitrogen species (RNS) are also involved in the pathophysiology of AP [106]. In inflammatory processes such as AP, inducible nitric oxide synthase (iNOS) contributes to large nitric oxide (NO) formation [107,108]. It has been suggested that a limited normal amount of NO production has beneficial effects in experimental edematous AP but uncontrolled over-production of NO may be detrimental, which suggests differing roles for each NOS isoform [109,110]. Endothelial nitric oxide synthase (eNOS) reduces the severity of the initial phase of experimental AP [111]. Thus, endogenous NO protects against oxidative damage since the inhibition of NOS by N-nitro-l-arginine methyl ester increases lipid peroxidation and protein oxidation in some subcellular fractions [112]. However in mice deficient in iNOS with AP, lipid peroxidation, the expression of adhesion molecules, and tissue damage were markedly restrained [113].
NO production and NOS expression seem to be differentially regulated temporally and in magnitude in the pancreas and lungs in response to cerulein hyper-stimulation [111]. AP provoked a deleterious effect on endothelium-dependent relaxing response for acetylcholine (Ach) in vitro and other hemodynamic disturbances, which were associated with high plasma NO levels as consequence of intense inflammatory response [114].
Xanthine oxidase and acute pancreatitis
Due to a significant potential capacity to generate large amounts of free radicals when activated from xanthine dehydrogenase (XDH), this enzyme has been studied in depth in ischemia–reperfusion processes and in many other diseases. In AP, the enzyme exhibits a major role in local and systemic processes.
Two different mechanisms may account for the activation and conversion of XDH to xanthine oxidase (XO) in AP. On the one hand, it has been described that activation of chymotrypsinogen to chymotrypsin may lead to activation of XDH to XO [115], which is a typical cellular process of the disease. On the other hand, oxidation of thiol groups could also lead to XO formation [116].
XO inhibition with allopurinol or derivatives has been associated with beneficial pancreatic effects in AP [117,118]. Edema, necrosis and pancreatic inflammation were reduced by administration of XO inhibitors.
Systemic effects of XO have also been described. Closa et al. suggested that the hydrolytic activity of α-amylase that markedly increases in AP cleaves the binding of XDH to glycoproteins of endothelial cells releasing large amounts of the enzyme to systemic circulation [119,120]. Activation to XO may also be produced by proteolytic enzymes present in plasma [119,120]. Finally, lungs provide oxygen to the enzyme which is needed for free radical generation [121]. Consequently, pulmonary oxidative damage is produced by XO [121], and administration of oxypurinol, a water-soluble derivative of allopurinol, prevents pulmonary damage as it abrogates the increase in myeloperoxidase activity and leukocyte infiltration in experimental AP [122].
However clinical effects of allopurinol in preventing post-ERCP pancreatitis and in the management of chronic and AP are not clear and further well-designed large clinical studies need to be conducted in this regard [123,124]. Further review of antioxidant therapies is discussed below.
Glutathione and acute pancreatitis
Reduced glutathione (GSH) is the major non-protein thiol in mammalian cells and plays a central role as antioxidant. It is in equilibrium with oxidized glutathione (GSSG) and the ratio between GSSG and GSH is a reliable indicator of oxidative stress because it reflects the balance between antioxidant status and pro-oxidant reactions in cells [125,126]. Thus, the glutathione redox status – characterized by high abundance of the reduced form – maintains the intracellular thiol/disulfide redox status. GSH concentration in the pancreas is one of the largest in the body and this tissue exhibits active transsulfuration pathway and GSH synthesis despite the relatively low activity of glutamate cysteine ligase (GCL) [106].
GSH depletion in the pancreatic tissue is a hallmark during the initial phase of AP [127,128]. Pre-treatments with glutathione monoethyl ester exhibited beneficial effects in AP by increasing pancreatic GSH levels [128], whereas inhibition of GSH synthesis with L-buthionine-(S,R)-sulfoximine (BSO) led to more pancreatic necrosis and reduced survival in rats with AP [129]. Furthermore, an association between certain genetic polymorphisms of glutathione S-transferase and severe AP has been reported [130]. Consequently, GSH depletion may contribute to the progression from mild to severe AP [131].
The biological reason for early GSH depletion in AP is not well-established. It was suggested that GSH depletion could allow a premature activation of digestive enzymes inside acinar cells triggering the inflammatory process [132]. However, glutathione depletion itself does not produce AP [133,134].
Our group found early glutathione depletion but not glutathione oxidation in pancreas in the early stage of experimental AP [68,135]. Therefore, ROS detoxification associated with the inflammatory process does not appear to be the major cause for the early glutathione depletion. Alternatively, Meister suggested that depletion of pancreatic glutathione may be due, at least in part, to the activation of pro-enzymes, since activated proteases such as carboxypeptidase may cleave GSH [136]. In fact, trypsinogen activation is accompanied by glutathione depletion in experimental AP [137].
In addition, we also showed that early up-regulation of GCL expression in pancreas occurs only in experimental mild edematous AP but not in severe necrotic AP [131]. Accordingly, rapid recovery of GSH levels occurs only in mild AP, while in the necrotic model the failure in up-regulation of GCL avoids the rapid increase in GSH levels. The marked increase in cytosolic pancreatic ribonuclease that occurs only during severe necrotic pancreatitis might be responsible for GCL mRNA degradation specifically in the severe form of the disease [131].
Pancreatic glutathione levels may be also associated with the increased severity of AP in obesity since pancreatic basal GSH levels and GSH levels after AP are decreased in obese animals in comparison with lean animals [138].
Extracellular hemoglobin and acute pancreatitis
Recently our group has demonstrated that severe acute pancreatitis is associated with marked increase in extracellular hemoglobin in plasma that comes from the ascitic fluid [139]. On the one hand, extracellular hemoglobin enhances lipid peroxidation, up-regulates pro-inflammatory cytokines Il-1β and TNF-α, and lowers anti-inflammatory IL-10 levels in abdominal adipose tissue. On the other hand, extracellular hemoglobin stimulates the leukocytic infiltrate in the lung and induces the hypoxia inducible factor (HIF)–vascular endothelial growth factor (VEGF) pathway [139]. Since VEGF increases vascular permeability in the lung and it may trigger pulmonary edema [140,141], the increase in extracellular hemoglobin might critically contribute to the development of pulmonary distress syndrome as a major complication of acute pancreatitis.
In this review we focus not only in the deleterious effects of free radicals by oxidation of biomolecules in AP, but also in the modulation of cell signaling and gene expression by redox status in this disease.
Redox signaling and inflammatory response in acute pancreatitis
Recently, new and important roles of oxidative and nitrosative stress in cell signaling have been described [142,143]. In general, modulation of intracellular signaling by redox unbalance is produced by two different mechanisms. On the one hand, oxidative species such as H2O2 and α and β-unsaturated aldehydes are considered as second messengers in the inflammatory response [144]. On the other hand, redox status affects cellular signal transduction through covalent modification of redox sensors [145]. Sulfur switches of sensitive targets, which include not only cysteine (cys) but also methionine (Met) residues, allow a transient oxidation of proteins to enable transmission of a signal and subsequent enzymatic reduction to their basal oxidation state.
Redox unbalance not only causes oxidative damage but also acts as intracellular signal in inflammatory processes, particularly up-regulating pro-inflammatory genes [106]. Indeed, ROS act as inflammatory mediators through the activation, migration, and adhesion of leukocytes, as well as by enhancing the expression of other mediators, such as cytokines, chemokines, and adhesion molecules [146,147]. In pancreatic acinar cells ROS induce activation of NF-κB as described above [148]. Furthermore, ROS generated by XO during AP induce up-regulation of lung P-selectin, an important mediator of neutrophil infiltration [146].
H2O2 is a second messenger of NADPH oxidases, which appear to be the major source of ROS in inflammation [149]. The endosomal production of superoxide by NADPH oxidase promotes the redox-dependent recruitment of TRAFs to the TNFR1/TRADD complex and subsequent NF-κB activation [150]. NF-κB may be activated by H2O2 and other pro-oxidants in many cell types, and its activation can be inhibited by antioxidants, as mentioned before [151]. However, the ability of oxidative stress to activate NF-κB depends on the cell type, the ROS generating system, and the antioxidant levels [152]. Moreover, H2O2 also modulates activation of MAPK kinase apoptosis signal-regulating kinase-1 (ASK1), an upstream kinase that activates the JNK and p38 MAPK pathways [153,154]. In addition, ROS are involved in NADPH oxidase-dependent signaling platforms associated with caveolae, lipid rafts, and nucleus [149].
In experimental AP, NADPH oxidase expression and activity are increased in pancreas [155]. Interestingly, mice deficient in NADPH oxidase exhibited attenuation of the cerulein-induced trypsin activation in the pancreas [156]. Free radical generation through NADPH oxidase would be related not only to infiltration of activated neutrophils [92], but also to acinar cell apoptosis [157]. Indeed, NADPH oxidase up-regulates IL-6 and mediates ROS-induced apoptosis in pancreatic AR42J acinar cells stimulated with the cholecystokinin analog cerulein [157].
In inflammatory processes, other oxidized molecules such as isoprostanes may trigger signaling through G-protein coupled receptors [158], whereas 4-hydroxy-2-nonenal causes signaling through JNK, protein kinase C, Keap1 and protein tyrosine phosphatase SHP1 [159–161]. In addition, oxidized phospholipids and cholesterol esters are “damage-associated molecular patterns” (DAMPs) that activate “pattern recognition receptors”, such as toll-like receptor 4 and CD36, triggering an immune response with up-regulation of pro-inflammatory genes as previously mentioned in [162]. Pancreatic-associated fat necrosis and ascites increased the concentrations of isoprostanes, lipoperoxides, lipid chlorohydrins, and free fatty acid which markedly enhance the activation of endothelial cells and inflammation [163].
On the other hand, it should be taken into account that although ROS are associated with SIRS, the formation of ROS in acinar cells might be protective by triggering apoptotic cell death instead of necrosis [164].
Role of ROS in NF-κB activation in acute pancreatitis
NF-κB activation may be subjected to redox regulation through upstream protein kinases and phosphatases prone to oxidation or thiolation [165,166]. Thus, ROS-dependent tyrosine phosphorylation of IκB may trigger its dissociation from NF-κB dimers and subsequently NF-κB activation [165]. Accordingly, NF-κB may be activated by hydrogen peroxide (H2O2) and other pro-oxidants in many cell types, and its activation can be abrogated by antioxidants [151]. However, as mentioned before the ability of oxidative stress to activate NF-κB depends on the cell type and the ROS-generating system [106,152,167]. Indeed, NADPH oxidase was required for ROS production and NF-κB activation induced by IL-1β in monocytes, whereas 5-lipoxygenase was the source of ROS in IL-1β-stimulated lymphoid cells and it was required for NF-κB activation in these cells [71]. Furthermore, certain cell types such as mouse alveolar epithelial cells did not trigger NF-κB activation upon H2O2 exposure, and even H2O2 may diminish TNF-α-induced NF-κB nuclear translocation [168]. Reduced NF-κB activation by H2O2 was mediated by inhibition of IKK activity due to oxidation of cysteine residues present in the IKK complex [168]. In addition, NF-κB, particularly p50, is a direct target for oxidation that can decrease its ability to bind to DNA [169]. Cys-62 of p50 is oxidized in the cytoplasm and needs to be reduced in the nucleus for the DNA binding activity of NF-κB [165,169].
ROS seem to promote NF-κB activation in the early course of pancreatitis [106]. Pretreatment with N-acetylcysteine (NAC) abrogated NF-κB activation in various experimental models of AP [170–175]. Pancreatic acinar cells respond to oxidants through NF-κB activation. Thus, H2O2 induced NF-κB activation in vitro in pancreatic acinar cells [176] and NAC inhibited NF-κB activation in acinar cells in response to cerulein hyper-stimulation [177].
ROS and calcium signaling
Disruption of Ca2+ signaling in pancreatic acinar cells seems to play a pivotal role in pathogenesis of AP [28,29,178]. Thus, stimulation of acinar cells with CCK causes a sustained increase in cytosolic Ca2+, intracellular digestive enzyme activation, and necrosis [179]. Conversely, blockade of Ca2+ channels reduces the severity of experimental acute pancreatitis [180].
It is well known that Ca2+ homeostasis is sensitive to cellular redox status [181], and hence, their interplay affects the onset and development of acute pancreatitis. IP3R and RyR receptors exhibit redox-sensitive cysteine residues that may explain the regulatory role of ROS in Ca2+ channels [45,182]. Importantly, thiol oxidation in these residues increases the activity of endoplasmic reticulum Ca2+ channels, and consequently rise cytosolic Ca2+ levels leading to premature intracellular activation of trypsinogen [183,184].
Recently, a novel mechanism of Ca2+ entry in acinar cells related to formation of a STIM1-Orai complex has been reported [185]. It is worth noting that orai1 is a redox-sensitive protein involved in Ca2+ signaling [186], and thus it may contribute to the redox regulation of Ca2+ levels in acinar cells.
Another protein involved in the control of Ca2+ homeostasis in the pancreas is the plasma membrane Ca2+-ATPase pump. Bruce et al. reported that high H2O2 concentrations inhibit the pump activity and diminished the Ca2+ clearance through the plasma membrane [187]. They also found that this pump failure was due to mitochondrial depolarization, which produced a decrease in the ATP production [188].
Cell death by apoptosis or necrosis depends on the duration and severity of the disturbance of cytosolic calcium levels in pancreatic acinar cells. For example, a transient elevation of cytosolic Ca2+ levels induced by bile acids increases Ca2+ uptake into mitochondria, generates mitochondrial ROS, and causes caspase activation and apoptosis [189]. In addition, ROS promotes cytochrome c release and apoptosis in acinar cells [190]. The administration of NAC, ETC inhibitors (rotenone and antimycin-A) or Ca2+ chelators (BAPTA-AM) decreases mitochondrial ROS production and prevents bile acid-induce apoptosis [189,191]. However, Ca2+ exerted two opposite effects via ROS on cytochrome c release in acinar cells. Cytochrome c release, caspase activation, and apoptosis are stimulated by Ca2+ and ROS, but inhibited by the Ca2+-induced loss of mitochondrial membrane potential (ΔΨm) leading in this case to necrosis [190]. Thus, it seems that pancreatic mitochondria are more sensitive to Ca2+ than other mitochondria, such as liver ones [190].
On the other hand, bile acids, CCK, or non-oxidative ethanol metabolites may trigger sustained elevations of cytosolic Ca2+ causing necrosis in acinar cells [179,192]. The massive influx of Ca2+ into the mitochondrial matrix may be explained by the opening of the mitochondrial permeability transition pore (MPTP) channel that is regulated by the intracellular Ca2+ and ROS levels [193].
Disulfide stress in acute pancreatitis
We have recently shown that in acute pancreatitis glutathione depletion in pancreas is not associated with glutathione oxidation, but it is rather triggered by glutathione breakdown that yields to a marked increase in cysteine and gamma-glutamyl cysteine [135]. Furthermore, cysteine is oxidized to cystine and gamma-glutamyl cysteine to bis-gamma glutamyl cystine promoting protein cysteinylation and gamma-glutamyl cysteinylation without significant increases in protein glutathionylation [135]. This specific oxidation of thiols in proteins upon acute inflammation would be a model of disulfide stress that leads to inactivation of protein phosphatases, such as serine protein phosphatase 2A and tyrosine phosphatase SHP1, which would favor MAPK activation and amplification of the inflammatory cascade. Other relevant redox-signaling thiols targets of disulfide stress include Keap1, thioredoxin 1, disulfide isomerase, peroxiredoxin, and endonuclease APE1/Ref1 [135]. Their oxidation should certainly contribute to acinar cell damage and death in acute pancreatitis. In addition, redox buffers such as albumin and ribonuclease inhibitor are also oxidized in acute pancreatitis and their contribution to cell damage should be further explored.
As mentioned above, oxidative stress and redox status act as a cell signaling modulator by covalent modification of sulfur residues of proteins within amino acids such as cysteine or methionine. Protein phosphatases are an important example of redox signaling as their activity is modulated by oxidative stress. They are key sensors of the cellular redox state since reversible oxidation of thiols (–SH) in protein phosphatases to form intramolecular disulfide bridges (–S–S–) or sulfenyl-amide bonds leads to their transient inactivation [83,194]. Protein phosphatases are classified into two major groups: serine/threonine phosphatases (PPs) – PP1, PP2A, PP2B (calcineurin) and the metallo-dependent phosphatase PP2C – and protein tyrosine phosphatases (PTPs) [195]. This latter group includes membrane protein phosphatases such as CD45, cytosolic phosphatases such as SHP1 and SHP2, and dual-specificity phosphatases also called MAPK phosphatases (MKPs).
Redox unbalance promotes activation of phosphorylation pathways such as MAPKs through inactivation of protein phosphatases. The activation of MAPKs by ROS seems to be mediated by inhibition of MKPs via different signaling pathways [196]. MKPs belong to a large group of protein tyrosine phosphatases, which are critical molecular targets of ROS since their catalytic cysteine is much more sensitive to reversible oxidation than other cysteines due to its low pKa [197,198]. Mild oxidative stress causes reversible oxidation of the catalytic cysteine to sulfenic acid (–S–OH), while intense oxidative stress may trigger the irreversible formation of sulfinic (–S–O2H) and sulfonic acids (–S–O3H) [199]. Nevertheless, members of the PTP superfamily exhibit large variation in their sensitivity towards oxidation. Some PTPs are highly sensitive to oxidation – such as PTEN or Sac1 –, whereas others – such as myotubularin lipid phosphatase – are resistant [200,201].
Serine/threonine phosphatase PP2A activity decreases when cysteines from the active site of the catalytic subunit PP2Ac form intramolecular bonds with vicinal thiols or intermolecular disulfide bonds with regulatory subunits [202,203]. Accordingly, PP2A activity is inhibited by GSSG or H2O2, being this effect prevented by DTT [204]. Calcineurin (PP2B) may also be inactivated by H2O2 [205,206]. Indeed, the formation of disulfide bonds in PP2B decreases both its affinity towards Ca2+ and transactivation of target genes [207]. Methionine oxidation may diminish calcineurin activity too [208]. We have shown that disulfide stress causes oxidation of PP2A through formation of intramolecular disulfides in experimental AP, and moreover the loss of pancreatic PP2A activity that occurs in AP may be rescued by treatment with N-acetyl cysteine [135].
Consequently, ROS and particularly those formed by NADPH oxidases may govern the balance between MAPKs and some phosphatases controlling the inflammatory response by redox signaling [209]. Activation of these phosphorylation pathways, NF-κB, and other activators of transcription leads to changes in chromatin structure with a special role of histone acetylation in order to trigger the expression of inflammatory genes [144].
Histone acetylation and redox signaling in acute pancreatitis
During last decades, it has been reported that CBP/p300 histone acetyltransferase (HAT) complex regulates the expression of pro-inflammatory cytokines through NF-κB and STAT pathways [210,211]. Indeed, CBP and p300 HATs are coactivators of RelA/p65 and are required for transcription of numerous targets of NF-κB, such as IL-6, IL-8, iNOS, E-selectin, and VCAM-1 [212]. Furthermore, transcription factor STAT3, which decisively participates in the acute-phase inflammatory response, is regulated by acetylation and phosphorylation [213]. Indeed, STAT3 acetylation is triggered by p300 and is critical to form STAT3 dimers that are required for transcriptional activation of cytokine IL-6 [213].
Redox signaling may occur in inflammation through inactivation of histone deacetylases (HDACs). In fact, glutathione depletion and ROS generation in macrophages may lower HDACs activity leading to increased IL-8 expression [214]. Regarding antioxidant defense, Nrf-2 deficiency leads to decreased HDAC2 levels in the lung and hence to enhanced inflammatory response [215].
In acute pancreatitis, modulation of NF-κB, ERK, and histone acetylation in inflammatory processes may be regulated by redox-sensitive serine/threonine protein phosphatase PP2A. We found that PP2A activity markedly decreases during acute pancreatitis as well as serine/threonine protein phosphatase PP2B and PP2C activities [216]. Administration of pentoxifylline, a phosphodiesterase inhibitor, prevented the loss of PP2A activity without affecting the other serine/threonine protein phosphatases, and abrogated the recruitment of HATs CBP and P/CAF to the promoters of pro-inflammatory genes, such as TNF-α and iNOS [216]. In addition to this effect on HATs, it also reduced the recruitment of transcriptions factors regulated by phosphorylation, such as NF-κB and C/EBPβ [216].
We also studied the epigenetic regulation of pro-inflammatory genes by kinetic analysis of the recruitment of transcriptions factors to their promoters [66]. Our results showed that up-regulation of early (EGR-1) and late (TNF-α) genes occurred by sequential recruitment of HATs and transcriptions factors in the course of acute pancreatitis [66]. Importantly, the recovery of PP2A activity by pentoxifylline was also associated with lowered IL-6 and ICAM-1 expression [216].
Therefore, phosphorylation and chromatin remodeling by HATs may be key processes in the regulation of pro-inflammatory genes in acute pancreatitis. HATs recruitment and HDACs inactivation are complex mechanisms that play a key role in the regulation of inflammatory genes through phosphorylation and oxidative processes, being HDACs more sensitive to cellular redox status [200].
Antioxidant supplementation
The beneficial effects found with antioxidants, such as allopurinol, glutathione monoethyl ester, or N-acetyl cysteine [128,172–176], in rodents using different models of acute pancreatitis encouraged clinical trials to study the benefits of antioxidants in the disease. Unfortunately, although a large body of experimental evidence has shown that free radicals play a key role in the pathogenesis of acute pancreatitis, the clinical benefit of antioxidants remains far to be clear [164,165].
Often the beneficial effect in experimental models was observed when antioxidants were administered before the induction of acute pancreatitis, and hence it may explain its lack of effectiveness once the disorder is well established as it occurs in the clinical management.
Some studies were designed to evaluate the potential benefit of different antioxidants in preventing acute pancreatitis after ERCP, since it is well known that pancreatitis is the most common complication after this clinical procedure. Although some studies showed partial beneficial effects of allopurinol [217] or beta carotene [218], NAC failed in preventing post-ERCP pancreatitis [219,220]. Furthermore, a meta-analysis including eleven different studies from 1999 to 2011 stated that antioxidant supplementation with selenium, allopurinol, beta carotene, NAC, and pentoxifylline is not justified in the treatment of post-ERCP pancreatitis [221].
Regarding the clinical benefits of antioxidants in spontaneous acute pancreatitis, the results are even more confusing and yielded mixed results [12]. Different antioxidants, alone or in combination, have been tested in the disease following treatments administrated once acute pancreatitis was established. Initially, studies performed in Germany showed benefits such as reduced mortality and decreased complications using selenium in acute pancreatitis [222,223]. In agreement with these findings, 20 patients with recurrent attacks and/or constant pancreatic pain were treated with an antioxidant cocktail containing selenium, vitamin C, beta carotene, vitamin E, and methionine, and these antioxidants were able to reduce the number of attacks in the treated group vs. placebo [224]. Moreover, treatment with high dose of vitamin C resulted in lower complications and shorter in-ward days compared with control group [225]. In contrast, other studies showed statistically non-significant reduction of hospital stay and a similar complication rate between a group receiving vitamin C, NAC and antoxyl forte, or placebo [226]. Finally, other trials observed no effect using the different anti-oxidant combinations of selenium, NAC and ascorbic acid [227–229].
Conclusions and future research directions
In conclusion, different molecular mechanisms are involved in local and systemic effects of AP triggering inflammatory gene expression and stimulating the production of pro-inflammatory cytokines. Fig. 2 shows briefly some of these mechanisms and their relationship with oxidative stress. The latter is emerging as a crucial modulator of cell signaling and a key promoter of the inflammatory cascade, but its role is more complex than expected and it is still beginning to be elucidated. Despite recent advancements in the understanding of the pathophysiology of the disease, the interaction of antioxidants and redox status with gene expression is far to be completely clarified. More studies should be conducted in order to elucidate the complex interactions between oxidative stress and the early events in AP, in particular focused on the role of oxidative stress and redox status in the epigenetic modulation of pro-inflammatory genes in acute pancreatitis.
Fig. 2.
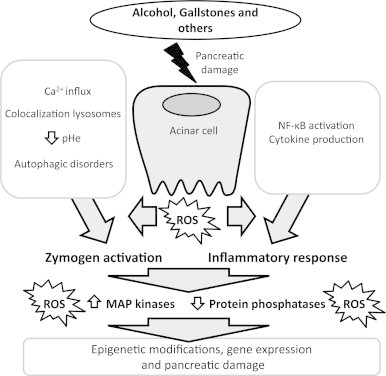
Illustration showing the main mechanisms triggering acute pancreatitis.
So far, clinical trials testing antioxidants in acute pancreatitis have been insufficient and conflicting, in some cases showing limited effectiveness. It seems that targeting only oxidative stress is not be powerful enough to stop the fast progression of acute pancreatitis, a disease that starts up quickly and simultaneously different pathogenic mechanisms. Therefore, future studies should be also conducted in order to design more efficient therapeutic strategies based on antioxidants or redox modulators together with direct anti-inflammatory therapies. In this regard, we tested some years ago a co-treatment with oxypurinol – as water-soluble xanthine oxidase inhibitor, and pentoxifylline – as inhibitor of TNF-alpha production – administered intravenously in rats with AP, which was able to abrogate the activation of ERK, JNK, and p38α simultaneously in the pancreas leading to a marked reduction in the local and systemic inflammatory response [122]. Perhaps, this co-treatment could serve as a basis for a future efficient combined therapy.
Acknowledgments
This work was supported by Grant SAF2012-39694 with FEDER funds to J.S., from the Spanish Ministry of Economy and Competitiveness.
References
- 1.Pandol S.J., Saluja A.K., Imrie C.W., Banks P.A. Acute pancreatitis: bench to the bedside. Gastroenterology. 2007;132(3):1127–1151. doi: 10.1053/j.gastro.2007.01.055. 17383433 [DOI] [PubMed] [Google Scholar]
- 2.Nystrom P.O. The systemic inflammatory response syndrome: definitions and aetiology. Journal of Antimicrobial Chemotherapy. 1998;41(Suppl. 1):1–7. doi: 10.1093/jac/41.suppl_1.1. 9511080 [DOI] [PubMed] [Google Scholar]
- 3.Jha R.K., Ma Q., Sha H., Palikhe M. Acute pancreatitis: a literature review. Medical Science Monitor. 2009;15(7):RA147–RA156. 19564840 [PubMed] [Google Scholar]
- 4.Martínez J., Johnson C.D., Sánchez-Payá J., de Madaria E., Robles-Díaz G., Pérez-Mateo M. Obesity is a definitive risk factor of severity and mortality in acute pancreatitis: an updated meta-analysis. Pancreatology. 2006;6(3):206–209. doi: 10.1159/000092104. 16549939 [DOI] [PubMed] [Google Scholar]
- 5.Kaiser A.M., Saluja A.K., Sengupta A., Saluja M., Steer M.L. Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. American Journal of Physiology. 1995;269(5 1):C1295–C1304. doi: 10.1152/ajpcell.1995.269.5.C1295. 7491921 [DOI] [PubMed] [Google Scholar]
- 6.Xue P., Deng L.H., Zhang Z.D., Yang X.N., Wan M.H., Song B., Xia Q. Infectious complications in patients with severe acute pancreatitis. Digestive Diseases and Sciences. 2009;54(12):2748–2753. doi: 10.1007/s10620-008-0668-1. 19104931 [DOI] [PubMed] [Google Scholar]
- 7.Whitcomb D.C. Clinical practice. Acute pancreatitis. New England Journal of Medicine. 2006;354(20):2142–2150. doi: 10.1056/NEJMcp054958. 16707751 [DOI] [PubMed] [Google Scholar]
- 8.Kusske A.M., Rongione A.J., Reber H.A. Cytokines and acute pancreatitis. Gastroenterology. 1996;110(2):639–642. doi: 10.1053/gast.1996.v110.agast960639. 8566616 [DOI] [PubMed] [Google Scholar]
- 9.Neoptolemos J.P., Raraty M., Finch M., Sutton R. Acute pancreatitis: the substantial human and financial costs. Gut. 1998;42(6):886–891. doi: 10.1136/gut.42.6.886. 9691932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayerle J., Dummer A., Sendler M., Malla S.R., van den Brandt C., Teller S., Aghdassi A., Nitsche C., Lerch M.M. Differential roles of inflammatory cells in pancreatitis. Journal of Gastroenterology and Hepatology. 2012;27(Suppl. 2):47–51. doi: 10.1111/j.1440-1746.2011.07011.x. 22320916 [DOI] [PubMed] [Google Scholar]
- 11.Steer M.L. Early events in acute pancreatitis. Baillieres Best Practice & Research Clinical Gastroenterology. 1999;13(2):213–225. doi: 10.1053/bega.1999.0020. 11030602 [DOI] [PubMed] [Google Scholar]
- 12.Robles L., Vaziri N.D., Ichii H. Role of oxidative stress in the pathogenesis of pancreatitis: effect of antioxidant therapy. Pancreatic Disorders & Therapy. 2013;3(1):112. doi: 10.4172/2165-7092.1000112. 24808987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Criddle D.N., Murphy J., Fistetto G., Barrow S., Tepikin A.V., Neoptolemos J.P., Sutton R., Petersen O.H. Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of ATP synthesis. Gastroenterology. 2006;130(3):781–793. doi: 10.1053/j.gastro.2005.12.031. 16530519 [DOI] [PubMed] [Google Scholar]
- 14.Gerasimenko J.V., Lur G., Sherwood M.W., Ebisui E., Tepikin A.V., Mikoshiba K., Gerasimenko O.V., Petersen O.H. Pancreatic protease activation by alcohol metabolite depends on Ca2+ release via acid store IP3 receptors. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(26):10758–10763. doi: 10.1073/pnas.0904818106. 19528657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gukovskaya A.S., Mouria M., Gukovsky I., Reyes C.N., Kasho V.N., Faller L.D., Pandol S.J. Ethanol metabolism and transcription factor activation in pancreatic acinar cells in rats. Gastroenterology. 2002;122(1):106–118. doi: 10.1053/gast.2002.30302. 11781286 [DOI] [PubMed] [Google Scholar]
- 16.Lieber C.S. Metabolism of etanol. In: Lieber C.S., editor. Medical and Nutritional Complicatios of Alcoholism: Mechanism and Management. Plenum Publishing Corporation; New York: 1992. pp. 1–35. [Google Scholar]
- 17.Saluja A., Saluja M., Villa A., Leli U., Rutledge P., Meldolesi J., Steer M. Pancreatic duct obstruction in rabbits causes digestive zymogen and lysosomal enzyme colocalization. Journal of Clinical Investigation. 1989;84(4):1260–1266. doi: 10.1172/JCI114293. 2477393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cosen-Binker L.I., Gaisano H.Y. Recent insights into the cellular mechanisms of acute pancreatitis. Canadian Journal of Gastroenterology. 2007;21(1):19–24. doi: 10.1155/2007/930424. 17225878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.F. Soriano, E.C.S. Rios, Molecular biology of acute pancreatitis, in: L. Rodrigo (Ed.), Acute Pancreatitis, 2012.
- 20.Grady T., Liang P., Ernst S.A., Logsdon C.D. Chemokine gene expression in rat pancreatic acinar cells is an early event associated with acute pancreatitis. Gastroenterology. 1997;113(6):1966–1975. doi: 10.1016/s0016-5085(97)70017-9. 9394737 [DOI] [PubMed] [Google Scholar]
- 21.Pereda J., Sabater L., Aparisi L., Escobar J., Sandoval J., Viña J., López-Rodas G., Sastre J. Interaction between cytokines and oxidative stress in acute pancreatitis. Current Medicinal Chemistry. 2006;13(23):2775–2787. doi: 10.2174/092986706778522011. 17073628 [DOI] [PubMed] [Google Scholar]
- 22.Escobar J., Pereda J., Arduini A., Sandoval J., Sabater L., Aparisi L., López-Rodas G., Sastre J. Cross-talk between oxidative stress and pro-inflammatory cytokines in acute pancreatitis: a key role for protein phosphatases. Current Pharmaceutical Design. 2009;15(26):3027–3042. doi: 10.2174/138161209789058075. 19754377 [DOI] [PubMed] [Google Scholar]
- 23.Gukovskaya A.S., Gukovsky I., Zaninovic V., Song M., Sandoval D., Gukovsky S., Pandol S.J. Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-alpha. Role in regulating cell death and pancreatitis. Journal of Clinical Investigation. 1997;100(7):1853–1862. doi: 10.1172/JCI119714. 9312187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Dios I., Ramudo L., Alonso J.R., Recio J.S., García-Montero A.C., Manso M.A. CD45 expression on rat acinar cells: involvement in pro-inflammatory cytokine production. FEBS Letters. 2005;579(28):6355–6360. doi: 10.1016/j.febslet.2005.10.017. 16263122 [DOI] [PubMed] [Google Scholar]
- 25.Hofbauer B., Saluja A.K., Lerch M.M., Bhagat L., Bhatia M., Lee H.S., Frossard J.L., Adler G., Steer M.L. Intra-acinar cell activation of trypsinogen during caerulein-induced pancreatitis in rats. American Journal of Physiology. 1998;275(2 1):G352–G362. doi: 10.1152/ajpgi.1998.275.2.G352. 9688663 [DOI] [PubMed] [Google Scholar]
- 26.Halangk W., Lerch M.M. Early events in acute pancreatitis. Clinics in Laboratory Medicine. 2005;25(1):1–15. doi: 10.1016/j.cll.2004.12.006. 15749229 [DOI] [PubMed] [Google Scholar]
- 27.Gaiser S., Daniluk J., Liu Y., Tsou L., Chu J., Lee W., Longnecker D.S., Logsdon C.D., Ji B. Intracellular activation of trypsinogen in transgenic mice induces acute but not chronic pancreatitis. Gut. 2011;60(10):1379–1388. doi: 10.1136/gut.2010.226175. 21471572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Criddle D.N., Gerasimenko J.V., Baumgartner H.K., Jaffar M., Voronina S., Sutton R., Petersen O.H., Gerasimenko O.V. Calcium signalling and pancreatic cell death: apoptosis or necrosis? Cell Death & Differentiation. 2007;14(7):1285–1294. doi: 10.1038/sj.cdd.4402150. 17431416 [DOI] [PubMed] [Google Scholar]
- 29.Saluja A.K., Bhagat L., Lee H.S., Bhatia M., Frossard J.L., Steer M.L. Secretagogue-induced digestive enzyme activation and cell injury in rat pancreatic acini. American Journal of Physiology. 1999;276(4 1):G835–G842. doi: 10.1152/ajpgi.1999.276.4.G835. 10198325 [DOI] [PubMed] [Google Scholar]
- 30.Orabi A.I., Shah A.U., Ahmad M.U., Choo-Wing R., Parness J., Jain D., Bhandari V., Husain S.Z. Dantrolene mitigates caerulein-induced pancreatitis in vivo in mice. American Journal of Physiology - Gastrointestinal and Liver Physiology. 2010;299(1):G196–G204. doi: 10.1152/ajpgi.00498.2009. 20448143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim M.S., Lee K.P., Yang D., Shin D.M., Abramowitz J., Kiyonaka S., Birnbaumer L., Mori Y., Muallem S. Genetic and pharmacologic inhibition of the Ca2+ influx channel TRPC3 protects secretory epithelia from Ca2+-dependent toxicity. Gastroenterology. 2011;140(7):2107–2115. doi: 10.1053/j.gastro.2011.02.052. 21354153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Voronina S.G., Barrow S.L., Simpson A.W., Gerasimenko O.V., da Silva Xavier G., Rutter G.A., Petersen O.H., Tepikin A.V. Dynamic changes in cytosolic and mitochondrial ATP levels in pancreatic acinar cells. Gastroenterology. 2010;138(5):1976–1987. doi: 10.1053/j.gastro.2010.01.037. 20102715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shah A.U., Sarwar A., Orabi A.I., Gautam S., Grant W.M., Park A.J., Shah A.U., Liu J., Mistry P.K., Jain D., Husain S.Z. Protease activation during in vivo pancreatitis is dependent on calcineurin activation. American Journal of Physiology – Gastrointestinal and Liver Physiology. 2009;297(5):G967–G973. doi: 10.1152/ajpgi.00181.2009. 20501444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watanabe O., Baccino F.M., Steer M.L., Meldolesi J. Supramaximal caerulein stimulation and ultrastructure of rat pancreatic acinar cell: early morphological changes during development of experimental pancreatitis. American Journal of Physiology. 1984;246(4 1):G457–G467. doi: 10.1152/ajpgi.1984.246.4.G457. 6720895 [DOI] [PubMed] [Google Scholar]
- 35.Lerch M.M., Saluja A.K., Dawra R., Saluja M., Steer M.L. The effect of chloroquine administration on two experimental models of acute pancreatitis. Gastroenterology. 1993;104(6):1768–1779. doi: 10.1016/0016-5085(93)90658-y. 8500736 [DOI] [PubMed] [Google Scholar]
- 36.Saluja A.K., Donovan E.A., Yamanaka K., Yamaguchi Y., Hofbauer B., Steer M.L. Cerulein-induced in vitro activation of trypsinogen in rat pancreatic acini is mediated by cathepsin B. Gastroenterology. 1997;113(1):304–310. doi: 10.1016/s0016-5085(97)70108-2. 9207291 [DOI] [PubMed] [Google Scholar]
- 37.Meister T., Niehues R., Hahn D., Domschke W., Sendler M., Lerch M.M., Schnekenburger J. Missorting of cathepsin B into the secretory compartment of CI-MPR/IGFII-deficient mice does not induce spontaneous trypsinogen activation but leads to enhanced trypsin activity during experimental pancreatitis − without affecting disease severity. Journal of Physiology and Pharmacology. 2010;61(5):565–575. 21081800 [PubMed] [Google Scholar]
- 38.Waterford S.D., Kolodecik T.R., Thrower E.C., Gorelick F.S. Vacuolar ATPase regulates zymogen activation in pancreatic acini. Journal of Biological Chemistry. 2005;280(7):5430–5434. doi: 10.1074/jbc.M413513200. 15582989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhoomagoud M., Jung T., Atladottir J., Kolodecik T.R., Shugrue C., Chaudhuri A., Thrower E.C., Gorelick F.S. Reducing extracellular pH sensitizes the acinar cell to secretagogue-induced pancreatitis responses in rats. Gastroenterology. 2009;137(3):1083–1092. doi: 10.1053/j.gastro.2009.05.041. 19454288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reed A.M., Husain S.Z., Thrower E., Alexandre M., Shah A., Gorelick F.S., Nathanson M.H. Low extracellular pH induces damage in the pancreatic acinar cell by enhancing calcium signaling. Journal of Biological Chemistry. 2011;286(3):1919–1926. doi: 10.1074/jbc.M110.158329. 21084290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Behrendorff N., Floetenmeyer M., Schwiening C., Thorn P. Protons released during pancreatic acinar cell secretion acidify the lumen and contribute to pancreatitis in mice. Gastroenterology. 2010;139(5):1711–1720. doi: 10.1053/j.gastro.2010.07.051. 20691184 [DOI] [PubMed] [Google Scholar]
- 42.Mizushima N., Yamamoto A., Matsui M., Yoshimori T., Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Molecular Biology of the Cell. 2004;15(3):1101–1111. doi: 10.1091/mbc.E03-09-0704. 14699058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fortunato F., Bürgers H., Bergmann F., Rieger P., Büchler M.W., Kroemer G., Werner J. Impaired autolysosome formation correlates with lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology. 2009;137(1):350–360. doi: 10.1053/j.gastro.2009.04.003. 19362087 [DOI] [PubMed] [Google Scholar]
- 44.Hashimoto D., Ohmuraya M., Hirota M., Yamamoto A., Suyama K., Ida S., Okumura Y., Takahashi E., Kido H., Araki K., Baba H., Mizushima N., Yamamura K. Involvement of autophagy in trypsinogen activation within the pancreatic acinar cells. Journal of Cell Biology. 2008;181(7):1065–1072. doi: 10.1083/jcb.200712156. 18591426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mareninova O.A., Hermann K., French S.W., O'Konski M.S., Pandol S.J., Webster P., Erickson A.H., Katunuma N., Gorelick F.S., Gukovsky I., Gukovskaya A.S. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. Journal of Clinical Investigation. 2009;119(11):3340–3355. doi: 10.1172/JCI38674. 19805911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grasso D., Ropolo A., Lo Ré A., Boggio V., Molejón M.I., Iovanna J.L., Gonzalez C.D., Urrutia R., Vaccaro M.I. Zymophagy, a novel selective autophagy pathway mediated by VMP1-USP9x-p62, prevents pancreatic cell death. Journal of Biological Chemistry. 2011;286:8308–8324. doi: 10.1074/jbc.M110.197301. 21173155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ji B., Gaiser S., Chen X., Ernst S.A., Logsdon C.D. Intracellular trypsin induces pancreatic acinar cell death but not NF-kappaB activation. Journal of Biological Chemistry. 2009;284(26):17488–17498. doi: 10.1074/jbc.M109.005520. 19383608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ghosh S., May M.J., Kopp E.B. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annual Review of Immunology. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. 9597130 [DOI] [PubMed] [Google Scholar]
- 49.Baumann B., Wagner M., Aleksic T., von Wichert G., Weber C.K., Adler G., Wirth T. Constitutive IKK2 activation in acinar cells is sufficient to induce pancreatitis in vivo. Journal of Clinical Investigation. 2007;117(6):1502–1513. doi: 10.1172/JCI30876. 17525799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vaquero E., Gukovsky I., Zaninovic V., Gukovskaya A.S., Pandol S.J. Localized pancreatic NF-kappaB activation and inflammatory response in taurocholate-induced pancreatitis. American Journal of Physiology – Gastrointestinal and Liver Physiology. 2001;280(6):G1197–G1208. doi: 10.1152/ajpgi.2001.280.6.G1197. 11352813 [DOI] [PubMed] [Google Scholar]
- 51.Chen X., Ji B., Han B., Ernst S.A., Simeone D., Logsdon C.D. NF-kappaB activation in pancreas induces pancreatic and systemic inflammatory response. Gastroenterology. 2002;122(2):448–457. doi: 10.1053/gast.2002.31060. 11832459 [DOI] [PubMed] [Google Scholar]
- 52.Altavilla D., Famulari C., Passaniti M., Galeano M., Macrì A., Seminara P., Minutoli L., Marini H., Calò M., Venuti F.S., Esposito M., Squadrito F. Attenuated cerulein-induced pancreatitis in nuclear factor-kappaB-deficient mice. Laboratory Investigation. 2003;83(12):1723–1732. doi: 10.1097/01.lab.0000101734.82054.be. 14691290 [DOI] [PubMed] [Google Scholar]
- 53.Gómez-Cambronero L.G., Sabater L., Pereda J., Cassinello N., Camps B., Viña J., Sastre J. Role of cytokines and oxidative stress in the pathophysiology of acute pancreatitis: therapeutical implications. Current Drug Targets – Inflammation & Allergy. 2002;1(4):393–403. doi: 10.2174/1568010023344544. 14561185 [DOI] [PubMed] [Google Scholar]
- 54.Dinarello C.A. The interleukin-1 family: 10 years of discovery. FASEB Journal. 1994;8(15):1314–1325. 8001745 [PubMed] [Google Scholar]
- 55.Norman J. The role of cytokines in the pathogenesis of acute pancreatitis. American Journal of Surgery. 1998;175(1):76–83. doi: 10.1016/s0002-9610(97)00240-7. 9445247 [DOI] [PubMed] [Google Scholar]
- 56.Dinarello C.A., Gelfand J.A., Wolff S.M. Anticytokine strategies in the treatment of the systemic inflammatory response syndrome. Journal of the American Medical Association. 1993;269(14):1829–1835. 8459516 [PubMed] [Google Scholar]
- 57.Muller C.A., Belyaev O., Appelros S., Buchler M., Uhl W., Borgstrom A. Dexamethasone affects inflammation but not trypsinogen activation in experimental acute pancreatitis. European Surgical Research. 2008;40(4):317–324. doi: 10.1159/000118027. 18303267 [DOI] [PubMed] [Google Scholar]
- 58.Vonlaufen A., Apte M.V., Imhof B.A., Frossard J.L. The role of inflammatory and parenchymal cells in acute pancreatitis. Journal of Pathology. 2007;213(3):239–248. doi: 10.1002/path.2231. 17893879 [DOI] [PubMed] [Google Scholar]
- 59.Grewal H.P., Mohey el Din A., Gaber L., Kotb M., Gaber A.O. Amelioration of the physiologic and biochemical changes of acute pancreatitis using an anti-TNF-alpha polyclonal antibody. American Journal of Surgery. 1994;167(1):214–219. doi: 10.1016/0002-9610(94)90076-0. 8311136 [DOI] [PubMed] [Google Scholar]
- 60.Norman J.G., Fink G.W., Franz M.G. Acute pancreatitis induces intrapancreatic tumor necrosis factor gene expression. Archives of Surgery. 1995;130(9):966–970. doi: 10.1001/archsurg.1995.01430090052018. 7661681 [DOI] [PubMed] [Google Scholar]
- 61.Mayer J., Rau B., Gansauge F., Beger H.G. Inflammatory mediators in human acute pancreatitis: clinical and pathophysiological implications. Gut. 2000;47(4):546–552. doi: 10.1136/gut.47.4.546. 10986216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Naskalski J.W., Kusnierz-Cabala B., Kedra B., Dumnicka P., Panek J., Maziarz B. Correlation of peripheral blood monocyte and neutrophil direct counts with plasma inflammatory cytokines and TNF-alpha soluble receptors in the initial phase of acute pancreatitis. Advances in Medical Sciences. 2007;52:129–134. 18217404 [PubMed] [Google Scholar]
- 63.Escobar J., Pereda J., Arduini A., Sandoval J., Sabater L., Aparisi L., Vento M., López-Rodas G., Sastre J. Role of redox signaling, protein phosphatases and histone acetylation in the inflammatory cascade in acute pancreatitis. Therapeutic implications. Inflammation & Allergy – Drug Targets. 2010;9(2):97–108. doi: 10.2174/187152810791292773. 20361855 [DOI] [PubMed] [Google Scholar]
- 64.Kim T.H., Bae G.S., Oh H.J., Kim M.S., Park K.C., Koo B.S., Kim B.J., Yang Y.S., Park D.E., Lee J.H., Seo S.W., Shin Y.K., Yun K.J., Sohn D.H., Kim H.J., So H.S., Park R.K., Song H.J., Park S.J. 2′,4′,6′-Tris(methoxymethoxy) chalcone (TMMC) attenuates the severity of cerulein-induced acute pancreatitis and associated lung injury. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2011;301(4):G694–G706. doi: 10.1152/ajpgi.00210.2010. 21778460 [DOI] [PubMed] [Google Scholar]
- 65.Norman J.G., Fink G.W., Denham W., Yang J., Carter G., Sexton C., Falkner J., Gower W.R., Franz M.G. Tissue-specific cytokine production during experimental acute pancreatitis. A probable mechanism for distant organ dysfunction. Digestive Diseases and Sciences. 1997;42(8):1783–1788. doi: 10.1023/a:1018886120711. 9286248 [DOI] [PubMed] [Google Scholar]
- 66.Sandoval J., Pereda J., Rodriguez J.L., Escobar J., Hidalgo J., Joosten L.A., Franco L., Sastre J., López-Rodas G. Ordered transcriptional factor recruitment and epigenetic regulation of tnf-alpha in necrotizing acute pancreatitis. Cellular and Molecular Life Sciences. 2010;67(10):1687–1697. doi: 10.1007/s00018-010-0272-3. 20130956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Satoh A., Gukovskaya A.S., Edderkaoui M., Daghighian M.S., Reeve J.R., Jr, Shimosegawa T., Pandol S.J. Tumor necrosis factor-alpha mediates pancreatitis responses in acinar cells via protein kinase C and proline-rich tyrosine kinase 2. Gastroenterology. 2005;129(2):639–651. doi: 10.1016/j.gastro.2005.05.005. 16083718 [DOI] [PubMed] [Google Scholar]
- 68.Gómez-Cambronero L., Camps B., de la Asunción J.G., Cerdá M., Pellín A., Pallardó F.V., Calvete J., Sweiry J.H., Mann G.E., Viña J., Sastre J. Pentoxifylline ameliorates cerulein-induced pancreatitis in rats: role of glutathione and nitric oxide. Journal of Pharmacology and Experimental Therapeutics. 2000;293(2):670–676. 10773043 [PubMed] [Google Scholar]
- 69.Chen C.J., Kono H., Golenbock D., Reed G., Akira S., Rock K.L. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nature Medicine. 2007;13(7):851–856. doi: 10.1038/nm1603. 17572686 [DOI] [PubMed] [Google Scholar]
- 70.Leser H.G., Gross V., Scheibenbogen C., Heinisch A., Salm R., Lausen M., Rückauer K., Andreesen R., Farthmann E.H., Schölmerich J. Elevation of serum interleukin-6 concentration precedes acute-phase response and reflects severity in acute pancreatitis. Gastroenterology. 1991;101(3):782–785. doi: 10.1016/0016-5085(91)90539-w. 1907253 [DOI] [PubMed] [Google Scholar]
- 71.Kishimoto T. Interleukin-6: from basic science to medicine—40 years in immunology. Annual Review of Immunology. 2005;23:1–21. doi: 10.1146/annurev.immunol.23.021704.115806. 15771564 [DOI] [PubMed] [Google Scholar]
- 72.Keceli M., Kucuk C., Sozuer E., Kerek M., Ince O., Arar M. The effect of interleukin-10 on acute pancreatitis induced by cerulein in a rat experimental model. Journal of Investigative Surgery. 2005;18(1):7–12. doi: 10.1080/08941930590905080. 15804946 [DOI] [PubMed] [Google Scholar]
- 73.Ogawa Y., Duru E.A., Ameredes B.T. Role of IL-10 in the resolution of airway inflammation. Current Molecular Medicine. 2008;8(5):437–445. doi: 10.2174/156652408785160907. 18691071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gironella M., Folch-Puy E., LeGoffic A., Garcia S., Christa L., Smith A., Tebar L., Hunt S.P., Bayne R., Smith A.J., Dagorn J.C., Closa D., Iovanna J.L. Experimental acute pancreatitis in PAP/HIP knock-out mice. Gut. 2007;56(8):1091–1097. doi: 10.1136/gut.2006.116087. 17409121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Folch-Puy E., Granell S., Dagorn J.C., Iovanna J.L., Closa D. Pancreatitis-associated protein I suppresses NF-kappa B activation through a JAK/STAT-mediated mechanism in epithelial cells. Journal of Immunology. 2006;176(6):3774–3779. doi: 10.4049/jimmunol.176.6.3774. 16517747 [DOI] [PubMed] [Google Scholar]
- 76.Vasseur S., Folch-Puy E., Hlouschek V., Garcia S., Fiedler F., Lerch M.M., Dagorn J.C., Closa D., Iovanna J.L. p8 improves pancreatic response to acute pancreatitis by enhancing the expression of the anti-inflammatory protein pancreatitis-associated protein I. Journal of Biological Chemistry. 2004;279(8):7199–7207. doi: 10.1074/jbc.M309152200. 14660681 [DOI] [PubMed] [Google Scholar]
- 77.Kono H., Rock K.L. How dying cells alert the immune system to danger. Nature Reviews Immunology. 2008;8(4):279–289. doi: 10.1038/nri2215. 18340345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sawa H., Ueda T., Takeyama Y., Yasuda T., Shinzeki M., Nakajima T., Kuroda Y. Blockade of high mobility group box-1 protein attenuates experimental severe acute pancreatitis. World Journal of Gastroenterology. 2006;12(47):7666–7670. doi: 10.3748/wjg.v12.i47.7666. 17171797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Becker C.E., O’Neill L.A. Inflammasomes in inflammatory disorders: the role of TLRs and their interactions with NLRs. Seminars in Immunopathology. 2007;29(3):239–248. doi: 10.1007/s00281-007-0081-4. 17805544 [DOI] [PubMed] [Google Scholar]
- 80.Hoque R., Sohail M., Malik A., Sarwar S., Luo Y., Shah A., Barrat F., Flavell R., Gorelick F., Husain S., Mehal W. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology. 2011;141(1):358–369. doi: 10.1053/j.gastro.2011.03.041. 21439959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nakahira K., Haspel J.A., Rathinam V.A., Lee S.J., Dolinay T., Lam H.C., Englert J.A., Rabinovitch M., Cernadas M., Kim H.P., Fitzgerald K.A., Ryter S.W., Choi A.M. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature Immunology. 2011;12(3):222–230. doi: 10.1038/ni.1980. 21151103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi: 10.1038/nature09663. 21124315 [DOI] [PubMed] [Google Scholar]
- 83.Naik E., Dixit V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. Journal of Experimental Medicine. 2011;208(3):417–420. doi: 10.1084/jem.20110367. 21357740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sweiry J.H., Mann G.E. Role of oxidative stress in the pathogenesis of acute pancreatitis. Scandinavian Journal of Gastroenterology. 1996;Suppl. 219:10–15. doi: 10.3109/00365529609104992. 8865464 [DOI] [PubMed] [Google Scholar]
- 85.Tsai K., Wang S.S., Chen T.S., Kong C.W., Chang F.Y., Lee S.D., Lu F.J. Oxidative stress: an important phenomenon with pathogenetic significance in the progression of acute pancreatitis. Gut. 1998;42(6):850–855. doi: 10.1136/gut.42.6.850. 9691925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sevillano S., de la Mano A.M., Manso M.A., Orfao A., de Dios I. N-acetylcysteine prevents intra-acinar oxygen free radical production in pancreatic duct obstruction-induced acute pancreatitis. Biochimica et Biophysica Acta. 2003;1639(3):177–184. doi: 10.1016/j.bbadis.2003.09.003. 14636949 [DOI] [PubMed] [Google Scholar]
- 87.Sanfey H., Bulkley G.B., Cameron J.L. The role of oxygen-derived free radicals in the pathogenesis of acute pancreatitis. Annals of Surgery. 1984;200(4):405–413. doi: 10.1097/00000658-198410000-00003. 6207783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Escobar J., Pereda J., Arduini A., Sandoval J., Moreno M.L., Pérez S., Sabater L., Aparisi L., Cassinello N., Hidalgo J., Joosten L.A., Vento M., López-Rodas G., Sastre J. Oxidative and nitrosative stress in acute pancreatitis. Modulation by pentoxifylline and oxypurinol. Biochemical Pharmacology. 2012;83(1):122–130. doi: 10.1016/j.bcp.2011.09.028. 22000995 [DOI] [PubMed] [Google Scholar]
- 89.Que R.S., Cao L.P., Ding G.P., Hu J.A., Mao K.J., Wang G.F. Correlation of nitric oxide and other free radicals with the severity of acute pancreatitis and complicated systemic inflammatory response syndrome. Pancreas. 2010;39(4):536–540. doi: 10.1097/MPA.0b013e3181c0e199. 20084045 [DOI] [PubMed] [Google Scholar]
- 90.Telek G., Scoazec J.Y., Chariot J., Ducroc R., Feldmann G., Roz C. Cerium-based histochemical demonstration of oxidative stress in taurocholate-induced acute pancreatitis in rats. A confocal laser scanning microscopic study. Journal of Histochemistry & Cytochemistry. 1999;47(9):1201–1212. doi: 10.1177/002215549904700912. 10449541 [DOI] [PubMed] [Google Scholar]
- 91.Telek G., Regöly-Mérei J., Kovács G.C., Simon L., Nagy Z., Hamar J., Jakab F. The first histological demonstration of pancreatic oxidative stress in human acute pancreatitis. Hepato-Gastroenterology. 2001;48(41):1252–1258. 11677940 [PubMed] [Google Scholar]
- 92.Closa D., Bulbena O., Hotter G., Roselló-Catafau J., Fernández-Cruz L., Gelpí E. Xanthine oxidase activation in cerulein- and taurocholate-induced acute pancreatitis in rats. Archives Internationales de Physiologie, de Biochimie et de Biophysique. 1994;102(3):167–170. doi: 10.3109/13813459409007532. 7528065 [DOI] [PubMed] [Google Scholar]
- 93.Acheson D.W., Rose P., Houston J.B., Braganza J.M. Induction of cytochromes P-450 in pancreatic disease: consequence, coincidence or cause? Clinica Chimica Acta. 1985;153(2):73–84. doi: 10.1016/0009-8981(85)90158-5. 4064344 [DOI] [PubMed] [Google Scholar]
- 94.Weber H., Merkord J., Jonas L., Wagner A., Schröder H., Käding U., Werner A., Dummler W. Oxygen radical generation and acute pancreatitis: effects of dibutyltin dichloride/ethanol and ethanol on rat pancreas. Pancreas. 1995;11(4):382–388. doi: 10.1097/00006676-199511000-00010. 8532655 [DOI] [PubMed] [Google Scholar]
- 95.Bokoch G.M., Knaus U.G. NADPH oxidases: not just for leukocytes anymore! Trends in Biochemical Sciences. 2003;28(9):502–508. doi: 10.1016/S0968-0004(03)00194-4. 13678962 [DOI] [PubMed] [Google Scholar]
- 96.Rau B., Poch B., Gansauge F., Bauer A., Nüssler A.K., Nevalainen T., Schoenberg M.H., Beger H.G. Pathophysiologic role of oxygen free radicals in acute pancreatitis: initiating event or mediator of tissue damage? Annals of Surgery. 2000;231(3):352–360. doi: 10.1097/00000658-200003000-00008. 10714628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Salomone T., Tosi P., Di Battista N., Binetti N., Raiti C., Tomassetti P., Migliori M., Gullo L. Impaired alveolar gas exchange in acute pancreatitis. Digestive Diseases and Sciences. 2002;47(9):2025–2028. doi: 10.1023/a:1019668728058. 12353850 [DOI] [PubMed] [Google Scholar]
- 98.Park B.K., Chung J.B., Lee J.H., Suh J.H., Park S.W., Song S.Y., Kim H., Kim K.H., Kang J.K. Role of oxygen free radicals in patients with acute pancreatitis. World Journal of Gastroenterology. 2003;9(10):2266–2269. doi: 10.3748/wjg.v9.i10.2266. 14562390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Winterbourn C.C., Bonham M.J., Buss H., Abu-Zidan F.M., Windsor J.A. Elevated protein carbonyls as plasma markers of oxidative stress in acute pancreatitis. Pancreatology. 2003;3(5):375–382. doi: 10.1159/000073652. 14526146 [DOI] [PubMed] [Google Scholar]
- 100.Hernández V., Miranda M., Pascual I., Sanchiz V., Almela P., Añón R., Cuadrado E., Sanz M.I., Mínguez M., Mora F., Romero F.J., Benages A. Malondialdehyde in early phase of acute pancreatitis. Revista Española de Enfermedades Digestivas. 2011;103(11):563–569. doi: 10.4321/s1130-01082011001100002. 22149557 [DOI] [PubMed] [Google Scholar]
- 101.Czakó L., Takács T., Varga I.S., Tiszlavicz L., Hai D.Q., Hegyi P., Matkovics B., Lonovics J. Involvement of oxygen-derived free radicals in l-arginine-induced acute pancreatitis. Digestive Diseases and Sciences. 1998;43(8):1770–1777. doi: 10.1023/a:1018839821176. 9724167 [DOI] [PubMed] [Google Scholar]
- 102.Abu-Zidan F.M., Bonham M.J., Windsor J.A. Severity of acute pancreatitis: a multivariate analysis of oxidative stress markers and modified Glasgow criteria. British Journal of Surgery. 2000;87(8):1019–1023. doi: 10.1046/j.1365-2168.2000.01464.x. 10931044 [DOI] [PubMed] [Google Scholar]
- 103.Shi C., Andersson R., Zhao X., Wang X. Potential role of reactive oxygen species in pancreatitis-associated multiple organ dysfunction. Pancreatology. 2005;5(4–5):492–500. doi: 10.1159/000087063. 16020935 [DOI] [PubMed] [Google Scholar]
- 104.Wereszczyńska-Siemiatkowska A., Dabrowski A., Jedynak M., Gabryelewicz A. Oxidative stress as an early prognostic factor in acute pancreatitis (AP): its correlation with serum phospholipase A2 (PLA2) and plasma polymorphonuclear elastase (PMN-E) in different-severity forms of human AP. Pancreas. 1998;17(2):163–168. 9700948 [PubMed] [Google Scholar]
- 105.Curran F.J., Sattar N., Talwar D., Baxter J.N., Imrie C.W. Relationship of carotenoid and vitamins A and E with the acute inflammatory response in acute pancreatitis. British Journal of Surgery. 2000;87(3):301–305. doi: 10.1046/j.1365-2168.2000.01375.x. 10718798 [DOI] [PubMed] [Google Scholar]
- 106.Leung P.S., Chan Y.C. Role of oxidative stress in pancreatic inflammation. Antioxidants & Redox Signaling. 2009;11(1):135–165. doi: 10.1089/ars.2008.2109. 18837654 [DOI] [PubMed] [Google Scholar]
- 107.Sandstrom P., Brooke-Smith M.E., Thomas A.C., Grivell M.B., Saccone G.T., Toouli J., Svanvik J. Highly selective inhibition of inducible nitric oxide synthase ameliorates experimental acute pancreatitis. Pancreas. 2005;30(1):e10–e15. 15632690 [PubMed] [Google Scholar]
- 108.Tanjoh K., Tomita R., Izumi T., Kinoshita K., Kawahara Y., Moriya T., Utagawa A. The expression of the inducible nitric oxide synthase messenger RNA on monocytes in severe acute pancreatitis. Hepato-Gastroenterology. 2007;54(75):927–931. 17591094 [PubMed] [Google Scholar]
- 109.Ozturk F., Gul M., Esrefoglu M., Ates B. The contradictory effects of nitric oxide in caerulein-induced acute pancreatitis in rats. Free Radical Research. 2008;42(4):289–296. doi: 10.1080/10715760801930730. 18404527 [DOI] [PubMed] [Google Scholar]
- 110.Ang A.D., Adhikari S., Ng S.W., Bhatia M. Expression of nitric oxide synthase isoforms and nitric oxide production in acute pancreatitis and associated lung injury. Pancreatology. 2009;9(1–2):150–159. doi: 10.1159/000178886. 19077466 [DOI] [PubMed] [Google Scholar]
- 111.DiMagno M.J. Nitric oxide pathways and evidence-based perturbations in acute pancreatitis. Pancreatology. 2007;7(5–6):403–408. doi: 10.1159/000108956. 17898529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sánchez-Bernal C., García-Morales O.H., Domínguez C., Martin-Gallán P., Calvo J.J., Ferreira L., Pérez-González N. Nitric oxide protects against pancreatic subcellular damage in acute pancreatitis. Pancreas. 2004;28(1):e9–15. doi: 10.1097/00006676-200401000-00021. 14707743 [DOI] [PubMed] [Google Scholar]
- 113.Cuzzocrea S., Mazzon E., Dugo L., Serraino I., Centorrino T., Ciccolo A., Van de Loo F.A., Britti D., Caputi A.P., Thiemermann C. Inducible nitric oxide synthase-deficient mice exhibit resistance to the acute pancreatitis induced by cerulein. Shock. 2002;17(5):416–422. doi: 10.1097/00024382-200205000-00013. 12022764 [DOI] [PubMed] [Google Scholar]
- 114.Camargo E.A., Delbin M.A., Ferreira T., Landucci E.C., Antunes E., Zanesco A. Influence of acute pancreatitis on the in vitro responsiveness of rat mesenteric and pulmonary arteries. BMC Gastroenterology. 2008;8:19. doi: 10.1186/1471-230X-8-19. 18510740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Enroth C., Eger B.T., Okamoto K., Nishino T., Nishino T., Pai E.F. Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: structure-based mechanism of conversion. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(20):10723–10728. doi: 10.1073/pnas.97.20.10723. 11005854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Stirpe F., Della Corte E. The regulation of rat liver xanthine oxidase. Conversion in vitro of the enzyme activity from dehydrogenase (type D) to oxidase (type O) Journal of Biological Chemistry. 1969;244(14):3855–3863. 4308738 [PubMed] [Google Scholar]
- 117.Sanfey H., Bulkley G.B., Cameron J.L. The pathogenesis of acute pancreatitis. The source and role of oxygen-derived free radicals in three different experimental models. Annals of Surgery. 1985;201(5):633–639. doi: 10.1097/00000658-198505000-00013. 2581519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Czakó L., Takács T., Varga I.S., Tiszlavicz L., Hai D.Q., Hegyi P., Matkovics B., Lonovics J. Oxidative stress in distant organs and the effects of allopurinol during experimental acute pancreatitis. International Journal of Pancreatology. 2000;27(3):209–216. doi: 10.1385/IJGC:27:3:209. 10952403 [DOI] [PubMed] [Google Scholar]
- 119.Folch E., Closa D., Neco P., Solé S., Planas A., Gelpí E., Roselló-Catafau J. Pancreatitis induces HSP72 in the lung: role of neutrophils and xanthine oxidase. Biochemical and Biophysical Research Communications. 2000;273(3):1078–1083. doi: 10.1006/bbrc.2000.3077. 10891374 [DOI] [PubMed] [Google Scholar]
- 120.Granell S., Bulbena O., Genesca M., Sabater L., Sastre J., Gelpi E., Closa D. Mobilization of xanthine oxidase from the gastrointestinal tract in acute pancreatitis. BMC Gastroenterology. 2004;4:1. doi: 10.1186/1471-230X-4-1. 14728722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Granell S., Serrano-Mollar A., Folch-Puy E., Navajas D., Farre R., Bulbena O., Closa D. Oxygen in the alveolar air space mediates lung inflammation in acute pancreatitis. Free Radical Biology and Medicine. 2004;37(10):1640–1647. doi: 10.1016/j.freeradbiomed.2004.07.036. 15477015 [DOI] [PubMed] [Google Scholar]
- 122.Pereda J., Sabater L., Cassinello N., Gómez-Cambronero L., Closa D., Folch-Puy E., Aparisi L., Calvete J., Cerdá M., Lledó S., Viña J., Sastre J. Effect of simultaneous inhibition of TNF-alpha production and xanthine oxidase in experimental acute pancreatitis: the role of mitogen activated protein kinases. Annals of Surgery. 2004;240(1):108–116. doi: 10.1097/01.sla.0000129343.47774.89. 15213626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zheng M., Chen Y., Bai J., Xin Y., Pan X., Zhao L. Meta-analysis of prophylactic allopurinol use in post-endoscopic retrograde cholangiopancreatography pancreatitis. Pancreas. 2008;37(3):247–253. doi: 10.1097/MPA.0b013e31816857e3. 18815544 [DOI] [PubMed] [Google Scholar]
- 124.Mohseni Salehi Monfared S.S., Vahidi H., Abdolghaffari A.H., Nikfar S., Abdollahi M. Antioxidant therapy in the management of acute, chronic and post-ERCP pancreatitis: a systematic review. World Journal of Gastroenterology. 2009;15(36):4481–4490. doi: 10.3748/wjg.15.4481. 19777606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Viña J. Glutathione: Metabolism and Physiological Functions. CRC Press; Boston: 1990. [Google Scholar]
- 126.Jones D.P. Redefining oxidative stress. Antioxidants & Redox Signaling. 2006;8(9–10):1865–1879. doi: 10.1089/ars.2006.8.1865. 16987039 [DOI] [PubMed] [Google Scholar]
- 127.Schoenberg M.H., Birk D., Beger H.G. Oxidative stress in acute and chronic pancreatitis. American Journal of Clinical Nutrition. 1995;62(6 Suppl):1306S–1314S. doi: 10.1093/ajcn/62.6.1306S. 7495225 [DOI] [PubMed] [Google Scholar]
- 128.Neuschwander-Tetri B.A., Ferrell L.D., Sukhabote R.J., Grendell J.H. Glutathione monoethyl ester ameliorates caerulein-induced pancreatitis in the mouse. Journal of Clinical Investigation. 1992;89(1):109–116. doi: 10.1172/JCI115550. 1370292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Alsfasser G., Gock M., Herzog L., Gebhard M.M., Herfarth C., Klar E., Schmidt J. Glutathione depletion with l-buthionine-(S,R)-sulfoximine demonstrates deleterious effects in acute pancreatitis of the rat. Digestive Diseases and Sciences. 2002;47(8):1793–1799. doi: 10.1023/a:1016496612906. 12184532 [DOI] [PubMed] [Google Scholar]
- 130.Rahman S.H., Ibrahim K., Larvin M., Kingsnorth A., McMahon M.J. Association of antioxidant enzyme gene polymorphisms and glutathione status with severe acute pancreatitis. Gastroenterology. 2004;126(5):1312–1322. doi: 10.1053/j.gastro.2004.02.002. 15131792 [DOI] [PubMed] [Google Scholar]
- 131.Pereda J., Escobar J., Sandoval J., Rodríguez J.L., Sabater L., Pallardó F.V., Torres L., Franco L., Viña J., López-Rodas G., Sastre J. Glutamate cysteine ligase up-regulation fails in necrotizing pancreatitis. Free Radical Biology and Medicine. 2008;44(8):1599–1609. doi: 10.1016/j.freeradbiomed.2008.01.018. 18279677 [DOI] [PubMed] [Google Scholar]
- 132.Schulz H.U., Niederau C., Klonowski-Stumpe H., Halangk W., Luthen R., Lippert H. Oxidative stress in acute pancreatitis. Hepato-Gastroenterology. 1999;46(29):2736–2750. 10576339 [PubMed] [Google Scholar]
- 133.Mårtensson J., Jain A., Meister A. Glutathione is required for intestinal function. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(5):1715–1719. doi: 10.1073/pnas.87.5.1715. 2308931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lüthen R.E., Neuschwander-Tetri B.A., Niederau C., Ferrell L.D., Grendell J.H. The effect of L-buthionine-[S,R]-sulfoximine on the pancreas in mice. A model of weakening glutathione-based defense mechanisms. International Journal of Pancreatology. 1994;16(1):31–36. 7528760 [PubMed] [Google Scholar]
- 135.Moreno M.L., Escobar J., Izquierdo-Álvarez A., Gil A., Pérez S., Pereda J., Zapico I., Vento M., Sabater L., Marina A., Martínez-Ruiz A., Sastre J. Disulfide stress: a novel type of oxidative stress in acute pancreatitis. Free Radical Biology and Medicine. 2014;70:265–277. doi: 10.1016/j.freeradbiomed.2014.01.009. 24456905 [DOI] [PubMed] [Google Scholar]
- 136.Meister A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacology & Therapeutics. 1991;51(2):155–194. doi: 10.1016/0163-7258(91)90076-x. 1784629 [DOI] [PubMed] [Google Scholar]
- 137.Lüthen R., Grendell J.H., Niederau C., Häussinger D. Trypsinogen activation and glutathione content are linked to pancreatic injury in models of biliary acute pancreatitis. International Journal of Pancreatology. 1998;24(3):193–202. doi: 10.1007/BF02788422. 9873954 [DOI] [PubMed] [Google Scholar]
- 138.Pereda J., Pérez S., Escobar J., Arduini A., Asensi M., Serviddio G., Sabater L., Aparisi L., Sastre J. Obese rats exhibit high levels of fat necrosis and isoprostanes in taurocholate-induced acute pancreatitis. PLOS One. 2012;7(9):e44383. doi: 10.1371/journal.pone.0044383. 23028532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.S. Pérez, J. Pereda, L. Sabater, J. Sastre. Pancreatic ascites hemoglobin contributes to the systemic response in acute pancreatitis. Free Radic. Biol. Med. August (2014), pii: S0891-5849(14)00384-0, 10.1016/j.freeradbiomed.2014.08.008. [DOI] [PubMed]
- 140.Kaner R.J., Ladetto J.V., Singh R., Fukuda N., Matthay M.A., Crystal R.G. Lung overexpression of the vascular endothelial growth factor gene induces pulmonary edema. American Journal of Respiratory Cell and Molecular Biology. 2000;22(6):657–664. doi: 10.1165/ajrcmb.22.6.3779. 10837361 [DOI] [PubMed] [Google Scholar]
- 141.Mura M., dos Santos C.C., Stewart D., Liu M. Vascular endothelial growth factor and related molecules in acute lung injury. Journal of Applied Physiology. 2004;97(5):1605–1617. doi: 10.1152/japplphysiol.00202.2004. 15475552 [DOI] [PubMed] [Google Scholar]
- 142.D’Autréaux B., Toledano M.B. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nature Reviews Molecular Cell Biology. 2007;8(10):813–824. doi: 10.1038/nrm2256. 17848967 [DOI] [PubMed] [Google Scholar]
- 143.Lundberg J.O., Weitzberg E., Gladwin M.T. The nitrate–nitrite–nitric oxide pathway in physiology and therapeutics. Nature Reviews Drug Discovery. 2008;7(2):156–167. doi: 10.1038/nrd2466. 18167491 [DOI] [PubMed] [Google Scholar]
- 144.Escobar J., Pereda J., López-Rodas G., Sastre J. Redox signaling and histone acetylation in acute pancreatitis. Free Radical Biology and Medicine. 2012;52(5):819–837. doi: 10.1016/j.freeradbiomed.2011.11.009. 22178977 [DOI] [PubMed] [Google Scholar]
- 145.Szczesny B., Bhakat K.K., Mitra S., Boldogh I. Age-dependent modulation of DNA repair enzymes by covalent modification and subcellular distribution. Mech. Ageing Dev. 2004;125(10–11):755–765. doi: 10.1016/j.mad.2004.07.005. 15541770 [DOI] [PubMed] [Google Scholar]
- 146.Folch E., Salas A., Prats N., Panés J., Piqué J.M., Gelpí E., Roselló-Catafau J., Closa D. H(2)O(2) and PARS mediate lung p-selectin upregulation in acute pancreatitis. Free Radical Biology and Medicine. 2000;28(8):1286–1294. doi: 10.1016/s0891-5849(00)00245-8. 10889459 [DOI] [PubMed] [Google Scholar]
- 147.Chipitsyna G., Gong Q., Gray C.F., Haroon Y., Kamer E., Arafat H.A. Induction of monocyte chemoattractant protein-1 expression by angiotensin II in the pancreatic islets and beta-cells. Endocrinology. 2007;148(5):2198–2208. doi: 10.1210/en.2006-1358. 17303665 [DOI] [PubMed] [Google Scholar]
- 148.Blanchard J.A., 2nd, Barve S., Joshi-Barve S., Talwalker R., Gates L.K., Jr. Antioxidants inhibit cytokine production and suppress NF-kappaB activation in CAPAN-1 and CAPAN-2 cell lines. Digestive Diseases and Sciences. 2001;46(12):2768–2772. doi: 10.1023/a:1012795900871. 11768272 [DOI] [PubMed] [Google Scholar]
- 149.Ushio-Fukai M. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxidants & Redox Signaling. 2009;11(6):1289–1299. doi: 10.1089/ars.2008.2333. 18999986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li Q., Spencer N.Y., Oakley F.D., Buettner G.R., Engelhardt J.F. Endosomal Nox2 facilitates redox-dependent induction of NF-kappaB by TNF-alpha. Antioxidants & Redox Signaling. 2009;11(6):1249–1263. doi: 10.1089/ars.2008.2407. 19113817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Flohé L., Brigelius-Flohé R., Saliou C., Traber M.G., Packer L. Redox regulation of NF-kappa B activation. Free Radical Biology and Medicine. 1997;22(6):1115–1126. doi: 10.1016/s0891-5849(96)00501-1. 9034250 [DOI] [PubMed] [Google Scholar]
- 152.Janssen-Heininger Y.M., Poynter M.E., Baeuerle P.A. Recent advances towards understanding redox mechanisms in the activation of nuclear factor kappaB. Free Radical Biology and Medicine. 2000;28(9):1317–1327. doi: 10.1016/s0891-5849(00)00218-5. 10924851 [DOI] [PubMed] [Google Scholar]
- 153.Saitoh M., Nishitoh H., Fujii M., Takeda K., Tobiume K., Sawada Y., Kawabata M., Miyazono K., Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO Journal. 1998;17(9):2596–2606. doi: 10.1093/emboj/17.9.2596. 9564042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liu H., Zhang H., Iles K.E., Rinna A., Merrill G., Yodoi J., Torres M., Forman H.J. The ADP-stimulated NADPH oxidase activates the ASK-1/MKK4/JNK pathway in alveolar macrophages. Free Radical Research. 2006;40(8):865–874. doi: 10.1080/10715760600758514. 17015265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chan Y.C., Leung P.S. Angiotensin II type 1 receptor-dependent nuclear factor-kappaB activation-mediated proinflammatory actions in a rat model of obstructive acute pancreatitis. Journal of Pharmacology and Experimental Therapeutics. 2007;323(1):10–18. doi: 10.1124/jpet.107.124891. 17616560 [DOI] [PubMed] [Google Scholar]
- 156.Gukovskaya A.S., Vaquero E., Zaninovic V., Gorelick F.S., Lusis A.J., Brennan M.L., Holland S., Pandol S.J. Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology. 2002;122(4):974–984. doi: 10.1053/gast.2002.32409. 11910350 [DOI] [PubMed] [Google Scholar]
- 157.Yu J.H., Lim J.W., Kim K.H., Morio T., Kim H. NADPH oxidase and apoptosis in cerulein-stimulated pancreatic acinar AR42J cells. Free Radical Biology and Medicine. 2005;39(5):590–602. doi: 10.1016/j.freeradbiomed.2005.04.019. 16085178 [DOI] [PubMed] [Google Scholar]
- 158.Mohler E.R., Franklin M.T., Adam L.P. Intracellular signaling by 8-epi-prostaglandin F2 alpha is mediated by thromboxane A2/prostaglandin endoperoxide receptors in porcine carotid arteries. Biochemical and Biophysical Research Communications. 1996;225(3):915–923. doi: 10.1006/bbrc.1996.1272. 8780711 [DOI] [PubMed] [Google Scholar]
- 159.Parola M., Robino G., Marra F., Pinzani M., Bellomo G., Leonarduzzi G., Chiarugi P., Camandola S., Poli G., Waeg G., Gentilini P., Dianzani M.U. HNE interacts directly with JNK isoforms in human hepatic stellate cells. Journal of Clinical Investigation. 1998;102(11):1942–1950. doi: 10.1172/JCI1413. 9835619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Levonen A.L., Landar A., Ramachandran A., Ceaser E.K., Dickinson D.A., Zanoni G., Morrow J.D., Darley-Usmar V.M. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochemical Journal. 2004;378(2):373–382. doi: 10.1042/BJ20031049. 14616092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Castello L., Marengo B., Nitti M., Froio T., Domenicotti C., Biasi F., Leonarduzzi G., Pronzato M.A., Marinari U.M., Poli G., Chiarpotto E. 4-hydroxynonenal signalling to apoptosis in isolated rat hepatocytes: the role of PKC-delta. Biochimica et Biophysica Acta. 2005;1737(2–3):83–93. doi: 10.1016/j.bbalip.2005.10.003. 16311069 [DOI] [PubMed] [Google Scholar]
- 162.Miller Y.I., Choi S.H., Wiesner P., Fang L., Harkewicz R., Hartvigsen K., Boullier A., Gonen A., Diehl C.J., Que X., Montano E., Shaw P.X., Tsimikas S., Binder C.J., Witztum J.L. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circulation Research. 2011;108(2):235–248. doi: 10.1161/CIRCRESAHA.110.223875. 21252151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Closa D. Free radicals and acute pancreatitis: much ado about … something. Free Radical Research. 2013;47(11):934–940. doi: 10.3109/10715762.2013.829571. 23895210 [DOI] [PubMed] [Google Scholar]
- 164.Armstrong J.A., Cash N., Soares P.M., Souza M.H., Sutton R., Criddle D.N. Oxidative stress in acute pancreatitis: lost in translation? Free Radical Research. 2013;47(11):917–933. doi: 10.3109/10715762.2013.835046. 23952531 [DOI] [PubMed] [Google Scholar]
- 165.Kabe Y., Ando K., Hirao S., Yoshida M., Handa H. Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxidants & Redox Signaling. 2005;7(3–4):395–403. doi: 10.1089/ars.2005.7.395. 15706086 [DOI] [PubMed] [Google Scholar]
- 166.Pantano C., Reynaert N.L., van der Vliet A., Janssen-Heininger Y.M. Redox-sensitive kinases of the nuclear factor-kappaB signaling pathway. Antioxidants & Redox Signaling. 2006;8(9–10):1791–1806. doi: 10.1089/ars.2006.8.1791. 16987032 [DOI] [PubMed] [Google Scholar]
- 167.Bonizzi G., Piette J., Merville M.P., Bours V. Cell type-specific role for reactive oxygen species in nuclear factor-kappaB activation by interleukin-1. Biochemical Pharmacology. 2000;59(1):7–11. doi: 10.1016/s0006-2952(99)00290-7. 10605929 [DOI] [PubMed] [Google Scholar]
- 168.Korn S.H., Wouters E.F., Vos N., Janssen-Heininger Y.M. Cytokine-induced activation of nuclear factor-kappa B is inhibited by hydrogen peroxide through oxidative inactivation of IkappaB kinase. Journal of Biological Chemistry. 2001;276(38):35693–35700. doi: 10.1074/jbc.M104321200. 11479295 [DOI] [PubMed] [Google Scholar]
- 169.Nishi T., Shimizu N., Hiramoto M., Sato I., Yamaguchi Y., Hasegawa M., Aizawa S., Tanaka H., Kataoka K., Watanabe H., Handa H. Spatial redox regulation of a critical cysteine residue of NF-kappaB in vivo. Journal of Biological Chemistry. 2002;277(46):44548–44556. doi: 10.1074/jbc.M202970200. 12213807 [DOI] [PubMed] [Google Scholar]
- 170.Gukovsky I., Gukovskaya A.S., Blinman T.A., Zaninovic V., Pandol S.J. Early NF-kappaB activation is associated with hormone-induced pancreatitis. American Journal of Physiology. 1998;275(6 1):G1402–G1414. doi: 10.1152/ajpgi.1998.275.6.G1402. 9843778 [DOI] [PubMed] [Google Scholar]
- 171.Ramudo L., Manso M.A., Sevillano S., de Dios I. Kinetic study of TNF-alpha production and its regulatory mechanisms in acinar cells during acute pancreatitis induced by bile–pancreatic duct obstruction. Journal of Pathology. 2005;206(1):9–16. doi: 10.1002/path.1747. 15761843 [DOI] [PubMed] [Google Scholar]
- 172.De Dios I., Ramudo L., García-Montero A.C., Manso M.A. Redox-sensitive modulation of CD45 expression in pancreatic acinar cells during acute pancreatitis. Journal of Pathology. 2006;210(2):234–239. doi: 10.1002/path.2037. 16886168 [DOI] [PubMed] [Google Scholar]
- 173.Ramudo L., Manso M.A. N-acetylcysteine in acute pancreatitis. World Journal of Gastrointestinal Pharmacology and Therapeutics. 2010;1(1):21–26. doi: 10.4292/wjgpt.v1.i1.21. 21577291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Yubero S., Ramudo L., Manso M.A., Collía F., de Dios I. Evaluation of N-acetylcysteine treatment in acute pancreatitis-induced lung injury. Inflammation Research. 2012;61(7):699–705. doi: 10.1007/s00011-012-0462-6. 22453841 [DOI] [PubMed] [Google Scholar]
- 175.Du B.Q., Yang Y.M., Chen Y.H., Liu X.B., Mai G. N-acetylcysteine improves pancreatic microcirculation and alleviates the severity of acute necrotizing pancreatitis. Gut Liver. 2013;7(3):357–362. doi: 10.5009/gnl.2013.7.3.357. 23710319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Algül H., Tando Y., Beil M., Weber C.K., Von Weyhern C., Schneider G., Adler G., Schmid R.M. Different modes of NF-kappaB/Rel activation in pancreatic lobules. American Journal of Physiology - Gastrointestinal and Liver Physiology. 2002;283(2):G270–G281. doi: 10.1152/ajpgi.00407.2001. 12121873 [DOI] [PubMed] [Google Scholar]
- 177.Bhatia M., Brady M., Kang Y.K., Costello E., Newton D.J., Christmas S.E., Neoptolemos J.P., Slavin J. MCP-1 but not CINC synthesis is increased in rat pancreatic acini in response to cerulein hyperstimulation. American Journal of Physiology – Gastrointestinal and Liver Physiology. 2002;282(1):G77–G85. doi: 10.1152/ajpgi.00031x.2002. 11751160 [DOI] [PubMed] [Google Scholar]
- 178.Ward J.B., Petersen O.H., Jenkins S.A., Sutton R. Is an elevated concentration of acinar cytosolic free ionised calcium the trigger for acute pancreatitis? Lancet. 1995;346(8981):1016–1019. doi: 10.1016/s0140-6736(95)91695-4. 7475553 [DOI] [PubMed] [Google Scholar]
- 179.Raraty M., Ward J., Erdemli G., Vaillant C., Neoptolemos J.P., Sutton R., Petersen O.H. Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(24):13126–13131. doi: 10.1073/pnas.97.24.13126. 11087863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hughes C.B., el-Din A.B., Kotb M., Gaber L.W., Gaber A.O. Calcium channel blockade inhibits release of TNF alpha and improves survival in a rat model of acute pancreatitis. Pancreas. 1996;13(1):22–28. doi: 10.1097/00006676-199607000-00003. 8783330 [DOI] [PubMed] [Google Scholar]
- 181.Brookes P.S., Yoon Y., Robotham J.L., Anders M.W., Sheu S.S. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. American Journal of Physiology – Cell Physiology. 2004;287(4):C817–C833. doi: 10.1152/ajpcell.00139.2004. 15355853 [DOI] [PubMed] [Google Scholar]
- 182.Sun J., Xu L., Eu J.P., Stamler J.S., Meissner G. Classes of thiols that influence the activity of the skeletal muscle calcium release channel. Journal of Biological Chemistry. 2001;276(19):15625–15630. doi: 10.1074/jbc.M100083200. 11278999 [DOI] [PubMed] [Google Scholar]
- 183.Husain S.Z., Prasad P., Grant W.M., Kolodecik T.R., Nathanson M.H., Gorelick F.S. The ryanodine receptor mediates early zymogen activation in pancreatitis. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(40):14386–14391. doi: 10.1073/pnas.0503215102. 16186498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Suzuki Y.J., Ford G.D. Superoxide stimulates IP3-induced Ca2+ release from vascular smooth muscle sarcoplasmic reticulum. American Journal of Physiology. 1992;262(1 2):H114–H116. doi: 10.1152/ajpheart.1992.262.1.H114. 1310231 [DOI] [PubMed] [Google Scholar]
- 185.Lur G., Haynes L.P., Prior I.A., Gerasimenko O.V., Feske S., Petersen O.H., Burgoyne R.D., Tepikin A.V. Ribosome-free terminals of rough ER allow formation of STIM1 puncta and segregation of STIM1 from IP(3) receptors. Current Biology. 2009;19(19):1648–1653. doi: 10.1016/j.cub.2009.07.072. 19765991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Bogeski I., Kummerow C., Al-Ansary D., Schwarz E.C., Koehler R., Kozai D., Takahashi N., Peinelt C., Griesemer D., Bozem M., Mori Y., Hoth M., Niemeyer B.A. Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Science Signaling. 2010;3(115):24. doi: 10.1126/scisignal.2000672. 20354224 [DOI] [PubMed] [Google Scholar]
- 187.Bruce J.I., Elliott A.C. Oxidant-impaired intracellular Ca2+ signaling in pancreatic acinar cells: role of the plasma membrane Ca2+-ATPase. American Journal of Physiology – Cell Physiology. 2007;293(3):C938–C950. doi: 10.1152/ajpcell.00582.2006. 17494627 [DOI] [PubMed] [Google Scholar]
- 188.Baggaley E.M., Elliott A.C., Bruce J.I. Oxidant-induced inhibition of the plasma membrane Ca2+-ATPase in pancreatic acinar cells: role of the mitochondria. American Journal of Physiology – Cell Physiology. 2008;295(5):C1247–C1260. doi: 10.1152/ajpcell.00083.2008. 18787078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Booth D.M., Murphy J.A., Mukherjee R., Awais M., Neoptolemos J.P., Gerasimenko O.V., Tepikin A.V., Petersen O.H., Sutton R., Criddle D.N. Reactive oxygen species induced by bile acid induce apoptosis and protect against necrosis in pancreatic acinar cells. Gastroenterology. 2011;140(7):2116–2125. doi: 10.1053/j.gastro.2011.02.054. 21354148 [DOI] [PubMed] [Google Scholar]
- 190.Odinokova I.V., Sung K.F., Mareninova O.A., Hermann K., Evtodienko Y., Andreyev A., Gukovsky I., Gukovskaya A.S. Mechanisms regulating cytochrome c release in pancreatic mitochondria. Gut. 2009;58(3):431–442. doi: 10.1136/gut.2007.147207. 18596195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Criddle D.N., Gillies S., Baumgartner-Wilson H.K., Jaffar M., Chinje E.C., Passmore S., Chvanov M., Barrow S., Gerasimenko O.V., Tepikin A.V., Sutton R., Petersen O.H. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. Journal of Biological Chemistry. 2006;281(52):40485–40492. doi: 10.1074/jbc.M607704200. 17088248 [DOI] [PubMed] [Google Scholar]
- 192.Kim J.Y., Kim K.H., Lee J.A., Namkung W., Sun A.Q., Ananthanarayanan M., Suchy F.J., Shin D.M., Muallem S., Lee M.G. Transporter-mediated bile acid uptake causes Ca2+-dependent cell death in rat pancreatic acinar cells. Gastroenterology. 2002;122(7):1941–1953. doi: 10.1053/gast.2002.33617. 12055600 [DOI] [PubMed] [Google Scholar]
- 193.Petronilli V., Miotto G., Canton M., Brini M., Colonna R., Bernardi P., Di Lisa F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophysical Journal. 1999;76(2):725–734. doi: 10.1016/S0006-3495(99)77239-5. 9929477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Chiarugi P. PTPs versus PTKs: the redox side of the coin. Free Radical Research. 2005;39(4):353–364. doi: 10.1080/10715760400027987. 16028361 [DOI] [PubMed] [Google Scholar]
- 195.Den Hertog J., Groen A., van der Wijk T. Redox regulation of protein-tyrosine phosphatases. Archives of Biochemistry and Biophysics. 2005;434(1):11–15. doi: 10.1016/j.abb.2004.05.024. 15629103 [DOI] [PubMed] [Google Scholar]
- 196.Whicher J.T., Barnes M.P., Brown A., Cooper M.J., Read R., Walters G., Williamson R.C. Complement activation and complement control proteins in acute pancreatitis. Gut. 1982;23(11):944–950. doi: 10.1136/gut.23.11.944. 6922819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lee K., Esselman W.J. Inhibition of PTPs by H(2)O(2) regulates the activation of distinct MAPK pathways. Free Radical Biology and Medicine. 2002;33(8):1121–1132. doi: 10.1016/s0891-5849(02)01000-6. 12374624 [DOI] [PubMed] [Google Scholar]
- 198.Van Montfort R.L., Congreve M., Tisi D., Carr R., Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423(6941):773–777. doi: 10.1038/nature01681. 12802339 [DOI] [PubMed] [Google Scholar]
- 199.Tonks N.K. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121(5):667–670. doi: 10.1016/j.cell.2005.05.016. 15935753 [DOI] [PubMed] [Google Scholar]
- 200.Jackson M.D., Denu J.M. Molecular reactions of protein phosphatases—insights from structure and chemistry. Chemical Reviews. 2001;101(8):2313–2340. doi: 10.1021/cr000247e. 11749375 [DOI] [PubMed] [Google Scholar]
- 201.Cho S.H., Lee C.H., Ahn Y., Kim H., Kim H., Ahn C.Y., Yang K.S., Lee S.R. Redox regulation of PTEN and protein tyrosine phosphatases in H(2)O(2) mediated cell signaling. FEBS Letters. 2004;560(1–3):7–13. doi: 10.1016/s0014-5793(04)00112-7. 15017976 [DOI] [PubMed] [Google Scholar]
- 202.Ross S.H., Lindsay Y., Safrany S.T., Lorenzo O., Villa F., Toth R., Clague M.J., Downes C.P., Leslie N.R. Differential redox regulation within the PTP superfamily. Cellular Signalling. 2007;19(7):1521–1530. doi: 10.1016/j.cellsig.2007.01.026. 17346927 [DOI] [PubMed] [Google Scholar]
- 203.Foley T.D., Kintner M.E. Brain PP2A is modified by thiol-disulfide exchange and intermolecular disulfide formation. Biochemical and Biophysical Research Communications. 2005;330(4):1224–1229. doi: 10.1016/j.bbrc.2005.03.108. 15823574 [DOI] [PubMed] [Google Scholar]
- 204.Foley T.D., Petro L.A., Stredny C.M., Coppa T.M. Oxidative inhibition of protein phosphatase 2A activity: role of catalytic subunit disulfides. Neurochem. Res. 2007;32(11):1957–1964. doi: 10.1007/s11064-007-9394-x. 17562162 [DOI] [PubMed] [Google Scholar]
- 205.Foley T.D., Armstrong J.J., Kupchak B.R. Identification and H2O2 sensitivity of the major constitutive MAPK phosphatase from rat brain. Biochemical and Biophysical Research Communications. 2004;315(3):568–574. doi: 10.1016/j.bbrc.2004.01.096. 14975738 [DOI] [PubMed] [Google Scholar]
- 206.Reiter T.A., Abraham R.T., Choi M., Rusnak F. Redox regulation of calcineurin in T-lymphocytes. Journal of Biological Inorganic Chemistry. 1999;4(5):632–644. doi: 10.1007/s007750050387. 10550693 [DOI] [PubMed] [Google Scholar]
- 207.Bogumil R., Namgaladze D., Schaarschmidt D., Schmachtel T., Hellstern S., Mutzel R., Ullrich V. Inactivation of calcineurin by hydrogen peroxide and phenylarsine oxide. Evidence for a dithiol–disulfide equilibrium and implications for redox regulation. European Journal of Biochemistry. 2000;267(5):1407–1415. doi: 10.1046/j.1432-1327.2000.01133.x. 10691978 [DOI] [PubMed] [Google Scholar]
- 208.Tan R.Y., Mabuchi Y., Grabarek Z. Blocking the Ca2+-induced conformational transitions in calmodulin with disulfide bonds. Journal of Biological Chemistry. 1996;271(13):7479–7483. doi: 10.1074/jbc.271.13.7479. 8631777 [DOI] [PubMed] [Google Scholar]
- 209.Carruthers N.J., Stemmer P.M. Methionine oxidation in the calmodulin-binding domain of calcineurin disrupts calmodulin binding and calcineurin activation. Biochemistry. 2008;47(10):3085–3095. doi: 10.1021/bi702044x. 18275158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Yao H., Rahman I. Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicology and Applied Pharmacology. 2011;254(2):72–85. doi: 10.1016/j.taap.2009.10.022. 21296096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kamei Y., Xu L., Heinzel T., Torchia J., Kurokawa R., Gloss B., Lin S.C., Heyman R.A., Rose D.W., Glass C.K., Rosenfeld M.G. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85(3):403–414. doi: 10.1016/s0092-8674(00)81118-6. 8616895 [DOI] [PubMed] [Google Scholar]
- 212.Gerritsen M.E., Williams A.J., Neish A.S., Moore S., Shi Y., Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(7):2927–2932. doi: 10.1073/pnas.94.7.2927. 9096323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Yuan Z.L., Guan Y.J., Chatterjee D., Chin Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307(5707):269–273. doi: 10.1126/science.1105166. 15653507 [DOI] [PubMed] [Google Scholar]
- 214.Yang S.R., Chida A.S., Bauter M.R., Shafiq N., Seweryniak K., Maggirwar S.B., Kilty I., Rahman I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. American Journal of Physiology – Lung Cellular and Molecular Physiology. 2006;291(1):L46–L57. doi: 10.1152/ajplung.00241.2005. 16473865 [DOI] [PubMed] [Google Scholar]
- 215.Adenuga D., Caito S., Yao H., Sundar I.K., Hwang J.W., Chung S., Rahman I. Nrf2 deficiency influences susceptibility to steroid resistance via HDAC2 reduction. Biochemical and Biophysical Research Communications. 2010;403(3–4):452–456. doi: 10.1016/j.bbrc.2010.11.054. 21094147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Sandoval J., Escobar J., Pereda J., Sacilotto N., Rodriguez J.L., Sabater L., Aparisi L., Franco L., López-Rodas G., Sastre J. Pentoxifylline prevents loss of PP2A phosphatase activity and recruitment of histone acetyltransferases to proinflammatory genes in acute pancreatitis. Journal of Pharmacology and Experimental Therapeutics. 2009;331(2):609–617. doi: 10.1124/jpet.109.157537. 19671881 [DOI] [PubMed] [Google Scholar]
- 217.Martinez-Torres H., Rodriguez-Lomeli X., Davalos-Cobian C., Garcia-Correa J., Maldonado-Martinez J.M., Medrano-Muñoz F., Fuentes-Orozco C., Gonzalez-Ojeda A. Oral allopurinol to prevent hyperamylasemia and acute pancreatitis after endoscopic retrograde cholangiopancreatography. World Journal of Gastroenterology. 2009;15(13):1600–1606. doi: 10.3748/wjg.15.1600. 19340902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Lavy A., Karban A., Suissa A., Yassin K., Hermesh I., Ben-Amotz A. Natural beta-carotene for the prevention of post-ERCP pancreatitis. Pancreas. 2004;29(2):e45–e50. doi: 10.1097/00006676-200408000-00018. 15257114 [DOI] [PubMed] [Google Scholar]
- 219.Katsinelos P., Kountouras J., Paroutoglou G., Beltsis A., Mimidis K., Zavos C. Intravenous N-acetylcysteine does not prevent post-ERCP pancreatitis. Gastrointestinal Endoscopy. 2005;62(1):105–111. doi: 10.1016/s0016-5107(05)01574-9. 15990827 [DOI] [PubMed] [Google Scholar]
- 220.Milewski J., Rydzewska G., Degowska M., Kierzkiewicz M., Rydzewski A. N-acetylcysteine does not prevent post-endoscopic retrograde cholangiopancreatography hyperamylasemia and acute pancreatitis. World Journal of Gastroenterology. 2006;12(23):3751–3755. doi: 10.3748/wjg.v12.i23.3751. 16773694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Gu W.J., Wei C.Y., Yin R.X. Antioxidant supplementation for the prevention of post-endoscopic retrograde cholangiopancreatography pancreatitis: a meta-analysis of randomized controlled trials. Nutrition Journal. 2013;12:12–23. doi: 10.1186/1475-2891-12-23. 23398675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kuklinski B., Zimmermann T., Schweder R. Decreasing mortality in acute pancreatitis with sodium selenite. Clinical results of 4 years antioxidant therapy. Medizinische Klinik. 1995;90(Suppl. 1):36–41. [PubMed] [Google Scholar]
- 223.Kuklinski B., Buchner M., Müller T., Schweder R. Anti-oxidative therapy of pancreatitis--an 18-month interim evaluation. Zeitschrift für die gesamte innere Medizin und ihre Grenzgebiete. 1992;47:239–245. [PubMed] [Google Scholar]
- 224.Uden S., Bilton D., Nathan L., Hunt L.P., Main C., Braganza J.M. Antioxidant therapy for recurrent pancreatitis: placebo-controlled trial. Alimentary Pharmacology & Therapeutics. 1990;4(4):357–371. doi: 10.1111/j.1365-2036.1990.tb00482.x. 2103755 [DOI] [PubMed] [Google Scholar]
- 225.Du W.D., Yuan Z.R., Sun J., Tang J.X., Cheng A.Q., Shen D.M., Huang C.J., Song X.H., Yu X.F., Zheng S.B. Therapeutic efficacy of high-dose vitamin C on acute pancreatitis and its potential mechanisms. World Journal of Gastroenterology. 2003;9(11):2565–2569. doi: 10.3748/wjg.v9.i11.2565. 14606098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Sateesh J., Bhardwaj P., Singh N., Saraya A. Effect of antioxidant therapy on hospital stay and complications in patients with early acute pancreatitis: a randomised controlled trial. Tropical Gastroenterology. 2009;30(4):201–206. 20426279 [PubMed] [Google Scholar]
- 227.Virlos I.T., Mason J., Schofield D., McCloy R.F., Eddleston J.M., Siriwardena A.K. Intravenous N-acetylcysteine, ascorbic acid and selenium-based anti-oxidant therapy in severe acute pancreatitis. Scandinavian Journal of Gastroenterology. 2003;38(12):1262–1267. doi: 10.1080/00365520310006540. 14750647 [DOI] [PubMed] [Google Scholar]
- 228.Bansal D., Bhalla A., Bhasin D.K., Pandhi P., Sharma N., Rana S., Malhotra S. Safety and efficacy of vitamin-based antioxidant therapy in patients with severe acute pancreatitis: a randomized controlled trial. Saudi Journal of Gastroenterology. 2011;17(3):174–179. doi: 10.4103/1319-3767.80379. 21546719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Siriwardena A.K., Mason J.M., Balachandra S., Bagul A., Galloway S., Formela L., Hardman J.G., Jamdar S. Randomised, double blind, placebo controlled trial of intravenous antioxidant (N-acetylcysteine, selenium, vitamin C) therapy in severe acute pancreatitis. Gut. 2007;56(10):1439–1444. doi: 10.1136/gut.2006.115873. 17356040 [DOI] [PMC free article] [PubMed] [Google Scholar]