

Abstract
The progression in nonalcoholic fatty liver disease (NAFLD) to nonalcoholic steatohepatitis is a serious health concern, but the underlying mechanisms remain unclear. We hypothesized that chronic inhibition of nitric oxide (NO) synthase (NOS) via Nω-nitro-l-arginine methyl ester (l-NAME) would intensify liver injury in a rat model of obesity, insulin resistance, and NAFLD. Obese Otsuka Long-Evans Tokushima fatty (OLETF) and lean Long-Evans Tokushima Otsuka (LETO) rats received control or l-NAME (65–70 mg·kg−1·day−1)-containing drinking water for 4 wk. l-NAME treatment significantly (P < 0.05) reduced serum NO metabolites and food intake in both groups. Remarkably, despite no increase in body weight, l-NAME treatment increased hepatic triacylglycerol content (+40%, P < 0.05) vs. control OLETF rats. This increase was associated with impaired (P < 0.05) hepatic mitochondrial state 3 respiration. Interestingly, the opposite effect was found in LETO rats, where l-NAME increased (P < 0.05) hepatic mitochondrial state 3 respiration. In addition, l-NAME induced a shift toward proinflammatory M1 macrophage polarity, as indicated by elevated hepatic CD11c (P < 0.05) and IL-1β (P = 0.07) mRNA in OLETF rats and reduced expression of the anti-inflammatory M2 markers CD163 and CD206 (P < 0.05) in LETO rats. Markers of total macrophage content (CD68 and F4/80) mRNA were unaffected by l-NAME in either group. In conclusion, systemic NOS inhibition in the obese OLETF rats reduced hepatic mitochondrial respiration, increased hepatic triacylglycerol accumulation, and increased hepatic inflammation. These findings suggest an important role for proper NO metabolism in the hepatic adaptation to obesity.
Keywords: Nω-nitro-l-arginine methyl ester, obese Otsuka Long-Evans Tokushima fatty rat, nitric oxide, liver
nonalcoholic fatty liver disease (NAFLD) comprises a spectrum of liver pathologies from steatosis to nonalcoholic steatohepatitis (NASH) and cirrhosis (31). In the United States, NAFLD prevalence doubled from 1988 to 2008 and currently accounts for ∼75% of the national chronic liver disease burden (41). While hepatic steatosis is relatively benign, progression of the disease to NASH and fibrosis is associated with substantially increased health risk. In a large cohort of NAFLD patients, those with biopsy-confirmed NASH had a 16% liver-related mortality rate compared with 3% in the non-NASH patients during a 150-mo follow-up (36). Thus efforts to understand the mechanisms that contribute to advancing NAFLD severity could provide valuable insight into the development of therapeutic options.
Nitric oxide (NO) is ubiquitously produced via two constitutive NO synthase (NOS) isoforms, neuronal (nNOS) and endothelial (eNOS), and an inducible isoform (iNOS). The important physiological role of constitutive NO is well appreciated in the cardiovascular and nervous systems, where it is exerts potent vasodilatory and anti-inflammatory effects. Alternatively, iNOS is upregulated in inflammatory conditions and can produce excess NO, which can cause cellular damage. Limited evidence indicates that constitutive NO production can confer hepatoprotection by preventing lipopolysaccharide-induced necrosis (21) and oxidative damage (11) and maintaining appropriate hepatic perfusion (33). In addition, NO can influence key processes involved in NAFLD progression, including mitochondrial function (3) and biogenesis (19), fatty acid oxidation (37), Kupffer cell polarization (38), and hepatic stellate cell fibrogenesis (40). However, there is a paucity of data regarding the role of hepatic NO as it relates to NAFLD severity that warrants attention.
The spontaneously hyperphagic Otsuka Long-Evans Tokushima fatty (OLETF) rat is a well-characterized model of obesity that develops NAFLD in a pattern similar to humans (30). Hepatic mitochondrial dysfunction, a key feature of NAFLD in this model, is present through all stages of the disease progression (30). Importantly, NO influences mitochondrial biogenesis (7) and electron transport chain (ETC) function (3). We recently reported an association between reduced hepatic eNOS activation status and NASH onset in OLETF rats (34). This finding led us to hypothesize in the current investigation that impaired constitutive NO production may contribute to NAFLD severity. If true, such a concept could have an important and rapid clinical impact, given the wide array of NO-based therapies (10).
To assess the role of constitutive NO production in NAFLD, we administered Nω-nitro-l-arginine methyl ester (l-NAME) for 4 wk in OLETF rats at an age when they have significant hepatic steatosis but lack features of NASH, including inflammation (and, therefore, increased iNOS expression). In addition, lean counterparts of the OLETF [Long-Evans Tokushima Otsuka (LETO)] rats were studied to identify specific obesity-dependent processes in the liver that are affected by NOS inhibition. We hypothesized that l-NAME-treated OLETF rats would demonstrate features of more advanced NAFLD/NASH, including mitochondrial dysfunction, hepatic inflammation, and fibrosis, compared with control OLETF rats.
METHODS
Animal protocol.
OLETF and LETO rats were housed in a climate-controlled room with a 12:12-h light-dark cycle from 4 to 20 wk of age with ad libitum access to water and standard rat chow (Formulab Diet 5008, LabDiet, St. Louis, MO). At 16 wk of age, OLETF and LETO rats were randomly selected to receive control drinking water or drinking water containing l-NAME (60–70 mg·kg−1·day−1) for 4 wk. Body weight and food consumption were measured weekly. On the day they were euthanized, the animals were anesthetized at 0800 with pentobarbital sodium (1 ml/kg) after an overnight fast. Body composition was determined via dual-energy X-ray absorptiometry. Blood was collected via cardiac puncture and stored at −80°C for later processing, and animals were euthanized via cardiacectomy. The liver was rapidly removed, and portions consistently taken from the same anatomic location were placed in mitochondrial isolation buffer (see below), fixed in formalin, or snap-frozen in liquid nitrogen. All animal protocols were approved by the University of Missouri Animal Care and Use Committee.
Liver morphology, triacylglycerol, and total nitrate + nitrite content.
Formalin-fixed tissue was mounted in paraffin, serially sliced, and stained with hematoxylin and eosin. Biochemical quantification of intrahepatic triacylglycerol (TAG) content was measured as described by our group previously (28). Content of total hepatic nitrate and nitrite (NOX), stable oxidation products of NO, was assessed in whole liver homogenate via a commercially available kit (product no. 780001, Cayman, Ann Arbor, MI).
Blood assays.
Serum insulin, glucose, TAG, free fatty acid (FFA), cholesterol, and plasma concentrations of NOX were determined as reported previously (23).
Hepatic mitochondrial isolation.
Mitochondria were isolated from freshly harvested liver as previously described (8). Total protein concentration of each isolated mitochondrial preparation was measured via bicinchoninic acid reaction and used to normalize functional values to protein loaded.
Mitochondrial respiration and fatty acid oxidation.
Mitochondrial respiration was determined using the Oxygraph 2K (Oroboros Instruments, Innsbruck, Austria) as described previously (8). State 2 respiration was stimulated by addition of 5 mM glutamate and 2 mM malate (state 2). State 3 respiration was assessed by serial addition of ADP (25–125 μM) to promote oxidative phosphorylation (state 3, complex I). State 3 respiration was further assessed by addition of 10 mM succinate to stimulate complex II activity (state 3, complexes I and II). Finally, the uncoupling agent carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (250 nmol) was added to determine maximum uncoupled respiratory capacity. Fatty acid oxidation was determined in isolated mitochondria by addition of [1-14C]palmitate as described by our group previously (28).
Western blot analysis.
Homogenates of liver and red gastrocnemius skeletal muscle were prepared for Western blot analysis. Total protein concentration of each sample was determined via a bicinchoninic acid protein kit and diluted to 3 μg/μl with 2× Laemmli buffer. Primary antibodies are as follows: eNOS (catalog no. 610297), S1177 phosphorylation-specific eNOS (p-eNOS; catalog no. 612393), and nNOS (catalog no. 610310) from BD Biosciences (San Jose, CA); iNOS (catalog no. ab3523), CD36 (catalog no. ab133625), cytochrome c oxidase (COX) subunit 1 (COX1; catalog no. ab14705), and OXPHOS mitochondrial profile (catalog no. ab110413) from Abcam (Cambridge, MA); and acetyl-CoA carboxylase (ACC; catalog no. 3662), S79 phosphorylation-specific ACC (catalog no. 3661), fatty acid synthase (FAS; catalog no. 3189), microsomal triglyceride transfer protein (MTTP; catalog no. ab186446), cytochrome c (catalog no. 4272), α-AMP-activated protein kinase (AMPK; catalog no. 2603), T172 phosphorylation-specific AMPK (p-AMPK; catalog no. 2531), Akt (catalog no. 9272), and S473 phosphorylation-specific Akt (catalog no. 9271) from Cell Signaling (Danvers, MA). Blots were analyzed via densitometric analysis (Image Lab Beta 3, Bio-Rad Laboratories, Burlingame, CA). Total protein was assessed with amido black (0.1%; Sigma) to control for differences in protein loading and transfer as previously described by our group (28).
Gene expression.
RNA was extracted from homogenized liver tissue by a commercially available RNeasy Mini Kit (product no. 74104, Qiagen, Valencia, CA), and reverse transcriptase was used to synthesize cDNA. RNA and cDNA quality was determined by a spectrophotometer (Nanodrop 2000c, Thermo Scientific, Waltham, MA). Quantitative real-time PCR was completed with the ABI 7500 Fast Sequence Detection System (Applied Biosystems, Carlsbad, CA) using Fast SYBR Green Master Mix (Applied Biosystems). Primer pairs were obtained from Sigma (St. Louis, MO) and are listed in Table 1. Dissociation melt curves were analyzed to verify primer specificity. Transcript abundance of β-actin was not different among groups and was used as the reference gene to calculate expression levels of genes of interest using the comparative threshold (2−ΔΔCT) method. All data are normalized to expression levels of the LETO control group.
Table 1.
SYBR Green PCR primer pairs
| Primers (5′ → 3′) |
||
|---|---|---|
| Gene | Forward | Reverse |
| α-SMA | TGAAGAGGAAGACAGCCACAGC | CCAACCATCACTCCCTGGTG |
| β-Actin | CAGAGCAAGAGAGGCATCCTC | GTCCAGACGCAGGATGGCATG |
| CD11c | CTGTCATCAGCAGCCACGA | ACTGTCCACACCGTTTCTCC |
| CD68 | TCACAAAAAGGCTGCCACTCTT | TCGTAGGGCTTGCTGTGCTT |
| CD163 | TGTAGTTCATCATCTTCGGTCC | CACCTACCAAGCGGAGTTGAC |
| CD206 | GAGGACTGCGTGGTGATGAA | CAGCGAACGTTGAAAGGGTG |
| Col-1α | GATGGACTCAACGGTCTCCC | CGGCCACCATCTTGAGACTT |
| F4/80 | GGAGGACCAATGTTCCAGGG | TGGGCAAGAACAGCTGTAGG |
| HIF-1α | TCAGCTCAGGACACTGATTTAGA | ATTCATCAGTGGTGGCAGTTG |
| ICAM-1 | CACAAGGGCTGTCACTGTTCA | CCCTAGTCGGAAGATCGAAAGTC |
| IL-1β | CCTATGTCTTGCCCGTGGAG | CACACACTAGCAGGTCGTCA |
| MCP-1 | CTGTCTCAGCCAGATGCAGTTAA | AGCCGACTCATTGGGATCAT |
| PGC-1α | TTGACTGGCGTCATTCAGGA | GGCAGCACACTCTATGTCACT |
| SOD1 | CTGAAGGCGAGCATGGGTT | GCTGGACCGCCATGTTTCTT |
| SOD2 | CACCGAGGAGAAGTACCACG | TGGGTTCTCCACCACCCTTA |
| TFAM | GAATGTGGGGCGTGCTAAGA | CAGATAAGGCTGACAGGCGA |
| TGF-β | CTGCTGACCCCCACTGATAC | AGCCCTGTATTCCGTCTCCT |
α-SMA, α-smooth muscle actin; Col-1α, collagen type 1 subunit α; HIF-1α, hypoxia-inducible factor 1α; MCP-1, monocyte chemotactic protein 1; PGC-1α, peroxisome proliferator-activated receptor-γ coactivator 1α; TFAM, transcription factor A mitochondrial; TGF-β, transforming growth factor-β.
Enzyme assays.
Citrate synthase and β-hydroxyacyl-CoA dehydrogenase activities were determined in freeze-fractured isolated hepatic mitochondria using the methods of Srere (35) and Bass et al. (2), respectively, and as described previously by our group (28). Superoxide dismutase (SOD) activity was determined in whole liver homogenate via a commercially available kit (product no. 706002, Cayman). Hepatic NOS activity was assessed in whole liver homogenate via a commercially available kit (product no. 781001, Cayman) according to the manufacturer's instructions using [1-14C]arginine. Briefly, this assay assesses the catalytic conversion of [1-14C]arginine to [1-14C]citrulline in the presence of essential NOS cofactors. Snap-frozen liver samples were homogenized and incubated in reaction medium at 37°C for 15 min. To assess the relative contribution of Ca2+-dependent NOS (eNOS and nNOS), Ca2+ was omitted from the reaction medium and 5 mM EDTA was added to chelate residual Ca2+. Background counts were subtracted from each reading and were assessed by incubation of each sample without Ca2+ plus addition of 1 mM N-nitro-l-arginine (l-NNA), a nonspecific NOS inhibitor. By inference, the Ca2+-independent (i.e., iNOS) contribution to total hepatic NOS activity was considered to be the difference in counts between Ca2+-depleted and l-NNA-inhibited counts.
Statistical analysis.
Statistical significance (P < 0.05) was assessed via one-way ANOVA for each measure using the open-access statistical software R (version 2.15.1). When a significant main effect was observed, Fisher's least significant difference test was used for post hoc comparisons. Values are means ± SE of 8–10 observations per group.
RESULTS
Food intake, body composition, and blood characteristics for the animals are reported elsewhere (23) and summarized in Table 2. Four weeks of l-NAME treatment significantly reduced plasma NOX in LETO and OLETF rats. OLETF rats consumed more food, gained more weight, and had a higher percent body fat than LETO rats. l-NAME treatment in OLETF rats caused a significant reduction in weight gain, food intake, and percent body fat relative to the OLETF control group during the 4-wk treatment period. In addition, OLETF rats had elevated concentrations of plasma glucose and insulin, and l-NAME treatment in OLETF rats significantly reduced plasma glucose compared with the OLETF control group. Remarkably, despite lower percent body fat and circulating insulin, l-NAME-treated OLETF rats had a marked increase in histological hepatic steatosis (Fig. 1A) compared with the OLETF control group, which was quantitatively confirmed by biochemical assessment of hepatic TAG content (Fig. 1B).
Table 2.
Animal and metabolic characteristics
| LETO |
OLETF |
|||
|---|---|---|---|---|
| Control | + l-NAME | Control | + l-NAME | |
| Serum NOX, μM | 0.33 ± 0.05a | 0.19 ± 0.04b,c | 0.25 ± 0.04a,b | 0.13 ± 0.02c |
| Body weight at euthanization, g | 419.2 ± 9.8a | 405.3 ± 6.2a | 588.4 ± 12.6b | 557.4 ± 13.6b |
| Food consumption during treatment | ||||
| Absolute, g/wk | 159.4 ± 10.9a | 145.8 ± 9.2a | 235.3 ± 9.6b | 186.0 ± 12.2c |
| Relative, g·wk−1·g body wt−1 | 0.38 ± 0.03a | 0.36 ± 0.02a,b | 0.40 ± 0.02a | 0.33 ± 0.02b |
| Weight gain during treatment, g | 18.8 ± 4.0a | 5.8 ± 3.2b | 34.7 ± 4.0c | −7.3 ± 8.5b |
| DEXA body fat, % | 13.5 ± 0.7a | 14.0 ± 0.5a | 27.2 ± 0.8b | 24.6 ± 0.9c |
| Epididymal + retroperitoneal fat pad mass, g | 6.8 ± 0.3a | 7.4 ± 0.5a | 17.6 ± 2.0b | 17.1 ± 2.2b |
| Plasma glucose, mg/dl | 186.0 ± 13.7a | 178.1 ± 5.7a | 309.8 ± 23.5b | 312.1 ± 19.7b |
| Plasma insulin, ng/ml | 8.1 ± 1.2a | 10.1 ± 1.6a | 32.0 ± 3.8b | 22.4 ± 3.1c |
| Plasma FFA, mM | 0.31 ± 0.03a | 0.32 ± 0.04a | 0.61 ± 0.04b | 0.61 ± 0.03b |
| Plasma TAG, mg/dl | 40.1 ± 2.4a | 52.9 ± 2.6a | 142.8 ± 10.0b | 162.5 ± 9.0b |
Values are means ± SE (n = 10/group).
LETO, Long-Evans Tokushima Otsuka; OLETF, Otsuka Long-Evans Tokushima fatty; l-NAME, Nω-nitro-l-arginine methyl ester; NOx, nitrite + nitrate; DEXA, dual-energy X-ray absorptiometry; FFA, free fatty acid; TAG, triacylglycerol.
Different letter superscripts denote significant differences (P < 0.05). Data have been previously reported (23).
Fig. 1.

Effects of Nω-nitro-l-arginine methyl ester (l-NAME) on hepatic steatosis (A and B) and mRNA expression of inflammatory M1 (C and D) and M2 (E and F) markers, total macrophage (Mφ) markers (G and H), and markers of cell adhesion [monocyte chemotactic protein 1 (MCP-1; I) and ICAM (J)]. OLETF, Otsuka Long-Evans Tokushima fatty; LETO, Long-Evans Tokushima Otsuka; TAG, triacylglycerol. Values are means ± SE; n = 7–10/group. Different letter superscripts denote significant differences (P < 0.05).
Transcript abundance of the M1 macrophage marker CD11c was elevated in l-NAME-treated OLETF rats compared with the other three groups (Fig. 1C). Other M1 macrophage markers, including IL-1β (Fig. 1D) and TNFα (data not shown), were not statistically different among groups, although IL-1β tended to be elevated in l-NAME-treated OLETF rats. mRNA expression of the anti-inflammatory M2 macrophage markers CD163 and CD206 was reduced in l-NAME-treated LETO rats relative to the LETO control group, whereas no effect was observed in OLETF rats (Fig. 1, E and F). No statistically significant treatment effect was observed in mRNA expression of the pan-macrophage (Mφ) markers F4/80 and CD68 (Fig. 1, G and H). Monocyte chemotactic protein 1 (MCP-1) and ICAM mRNA transcripts, surface markers that mediate monocyte recruitment and leukocyte adhesion, respectively, were elevated in the OLETF control group relative to the LETO control group, and MCP-1 expression was significantly increased by l-NAME treatment in LETO rats (Fig. 1, I and J). Markers of hepatic fibrosis (α-smooth muscle actin, collagen type 1 subunit α, and transforming growth factor-β) were not different among groups (data not shown).
We examined the effects of chronic NOS inhibition on hepatic NOS activity, NOX content, and NOS protein expression. Ca2+-dependent NOS activity was not different between groups, and Ca2+-depleted NOS activity was indistinguishable from l-NNA-inhibited (background) counts (Fig. 2B), indicating minimal, if any, hepatic iNOS activity in these animals. Despite a lack of an l-NAME treatment effect for hepatic NOS activity, hepatic NOX content, which is a better indicator of in vivo NO production, was significantly and comparably reduced with l-NAME treatment in OLETF and LETO rats (Fig. 2C). Hepatic nNOS protein content was not detected by Western blot analysis. Neither eNOS nor iNOS protein content was different among groups (Fig. 2, B and C). However, S1177 phosphorylation of eNOS (p-eNOS) at the constitutively active site was significantly elevated in l-NAME-treated OLETF rats compared with the other groups. This increase in p-eNOS was related to an increase in activated T172-phosphorylated AMPK (Fig. 2E), but not S473-phosphorylated Akt (Fig. 2F), known kinases of eNOS at S1177 (5, 9, 17). In addition, NO is known to modulate hepatic vascular resistance (33); thus NOS inhibition may affect hepatic perfusion and/or oxygenation. To address this possibility, we assessed hepatic hypoxia-inducible factor 1α mRNA content and observed no differences among groups (Fig. 2I).
Fig. 2.

Effect of l-NAME treatment on hepatic nitric oxide (NO) synthase (NOS) activity, total hepatic nitrate and nitrite (NOX), NOS isoform expression, and kinases known to regulate endothelial NOS (eNOS) activation. A: representative Western blot images. AMPK, α-AMP-activated protein kinase. B: activity of Ca2+-dependent and -independent NOS in liver homogenate. AU, arbitrary units; ND, not determined. C–I: effects of l-NAME treatment on hepatic NOX (C); protein expression of inducible NOS (iNOS; D), total eNOS (E), S1177-phosphorylated eNOS (F), T172-phosphorylated AMPK (F), and S473-phosphorylated Akt (H); and mRNA expression of hypoxia-inducible factor 1α (HIF-1α; I). Values are means ± SE; n = 10/group. Different letter superscripts denote significant differences (P < 0.05).
In an effort to understand the underlying mechanism by which l-NAME treatment caused excess hepatic TAG accumulation, we assessed the functional capacity of isolated hepatic mitochondria. Remarkably, l-NAME treatment caused a differential effect on mitochondrial respiration in OLETF vs. LETO rats (Fig. 3A). In isolated hepatic mitochondria, l-NAME treatment resulted in a significant increase in mitochondrial state 3 (glutamate + malate + ADP + succinate) respiratory rate in LETO rats but a reduction in OLETF rats (P < 0.05; Fig. 3A). A similar nonsignificant trend was observed in the other mitochondrial respiration states. In addition, isolated hepatic mitochondria from l-NAME-treated LETO rats demonstrated significantly increased complete palmitate oxidation compared with the LETO control group (Fig. 3B), and no treatment effect was observed in OLETF rats. There were no differences between groups in mitochondrial content as assessed in whole liver homogenate via OXPHOS cocktail (Fig. 3C), cytochrome c, and COX4 subunit 1 (data not shown). Furthermore, the activities of rate-limiting enzymes of the tricarboxylic acid cycle (citrate synthase) and β-oxidation (β-hydroxyacyl-CoA dehydrogenase) measured in mitochondrial isolates were not different among groups (data not shown).
Fig. 3.

Effects of l-NAME treatment on hepatic mitochondrial function (A and B), content (C), and biogenesis (D and E). PGC-1α, peroxisome proliferator-activated receptor-γ coactivator 1α; TFAM, transcription factor A mitochondrial. Values are means ± SE; n = 5–10/group. Different letter superscripts denote significant differences (P < 0.05).
l-NAME treatment in LETO rats caused a ∼50% reduction in peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) mRNA relative to the LETO control group. PGC-1α mRNA in the OLETF group was reduced 75% relative to the LETO control group, and, interestingly, no further reduction was observed in l-NAME-treated OLETF rats (Fig. 3D). In contrast, transcription factor A mitochondrial (TFAM) expression was significantly elevated in the OLETF control group relative to the other groups (Fig. 3E). l-NAME treatment caused a significant increase in hepatic SOD activity in LETO rats, whereas there was a trend (P = 0.13) for l-NAME to reduce hepatic SOD activity in OLETF rats (Fig. 4A). In addition, there was a trend for SOD1 and SOD2 mRNA to differ among groups (P = 0.09 and 0.15, respectively), with levels in l-NAME-treated OLETF rats tending to be reduced compared with those in the OLETF control group (Fig. 4, B and C).
Fig. 4.
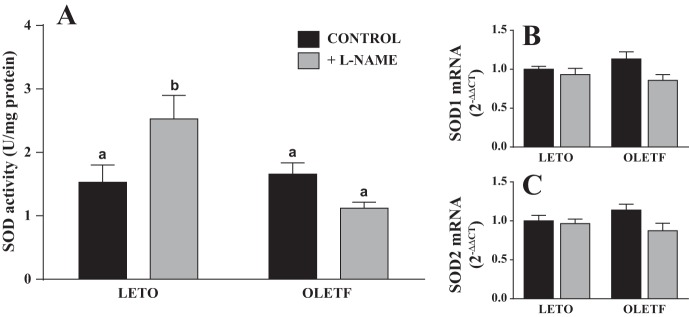
Effects of l-NAME treatment on total hepatic SOD activity (A) and mRNA expression of SOD1 (B) and SOD2 (C). Values are means ± SE; n = 10/group. Different letter superscripts denote significant differences (P < 0.05).
In an attempt to identify other processes that could explain the increase in liver TAGs in l-NAME-treated OLETF rats, we assessed markers of hepatic de novo lipogenesis (ACC and FAS), lipid export (MTTP), fatty acid import (CD36), and skeletal muscle insulin sensitivity (Akt activation). Hepatic MTTP content did not differ among groups (Fig. 5B). CD36 protein content was significantly reduced in l-NAME-treated OLETF rats relative to the OLETF control group (Fig. 5C). Total ACC and FAS protein was elevated in OLETF relative to LETO rats, with no effect of l-NAME treatment (Fig. 5, D–F). Phosphorylated (inactive) ACC relative to total ACC was reduced in the OLETF control group relative to the LETO control group, and this reduction was partially prevented with l-NAME treatment in OLETF rats (Fig. 5E). Finally, l-NAME treatment significantly increased the S473 phosphorylation of Akt in red gastrocnemius (RG) skeletal muscle from LETO rats, and a similar trend was observed in OLETF rats (Fig. 5G).
Fig. 5.

Increased hepatic steatosis in l-NAME-treated OLETF rats is independent of markers of lipid export [microsomal triglyceride transfer protein (MTTP; B)], fatty acid uptake (C), de novo lipogenesis [acetyl-CoA carboxylase (ACC; D), S79-phosphorylated ACC (p-ACC; E), and fatty acid synthase (FAS; F)], or red gastrocnemius (RG) skeletal muscle S473 phosphorylation-specific Akt (p-Akt; G). Values are means ± SE; n = 10/group. Different letter superscripts denote significant differences (P < 0.05).
DISCUSSION
The progression in NAFLD severity to NASH is strongly linked to liver-related and all-cause mortality; thus efforts to elucidate the mechanisms underlying this progression are critical. We previously reported an association between reduced hepatic eNOS activation and NASH development in 40-wk-old diabetic OLETF rats that was not apparent at younger ages (34). This prompted us to test the hypothesis that chronic inhibition of NOS in the OLETF rat earlier in the disease progression would induce a NASH-like phenotype. Consistent with this hypothesis, 4 wk of l-NAME treatment in insulin-resistant OLETF rats increased hepatic TAG accumulation, impaired hepatic mitochondrial respiration, and induced a shift toward M1 macrophage polarity. Remarkably, we report a different effect in lean LETO rats, where l-NAME treatment reduced expression of markers of anti-inflammatory M2 macrophage polarity, increased hepatic mitochondrial respiration, and increased hepatic SOD activity. These results demonstrate a novel role for hepatic NO in the adaptation to obesity in the liver. Efforts to exploit hepatic NO production and signaling may have important clinical implications in the treatment of NAFLD.
Our group (32) and others (16, 20, 24) have demonstrated that caloric restriction in OLETF rats rapidly and effectively improves systemic metabolic parameters and resolves hepatic steatosis. Similarly, in the current investigation, l-NAME treatment reduced food intake in OLETF rats and was associated with reduced adiposity and insulinemia (Table 1) (23). Conversely, these rats accumulated 40% more hepatic TAG content than control OLETF rats. In contrast, in a previous report, 12 wk of l-NAME treatment (100 mg·kg−1·day−1) in mice fed a high-fat diet (80% saturated fat) caused a reduction in liver TAG content compared with controls fed a high-fat diet (39). However, the composition and duration of the diet likely induced significant hepatic inflammation, and the resulting impact of l-NAME on hepatic TAG content may be attributed to the inhibition of iNOS in the previous report. In fact, in rats fed a high-fat diet, treatment with the iNOS-specific inhibitor 1400W induced a nearly threefold reduction in liver TAG (25). In this regard, the current report is unique, as OLETF rats were studied prior to the onset of robust hepatic inflammation to gain insight into the role of constitutive, rather than inducible, NO production in NAFLD. Indeed, the vast majority of hepatic NOS activity was observed to be constitutive (Ca2+-dependent, primarily eNOS), rather than inducible, across all groups (Fig. 2B). Thus our results point to a novel protective role for hepatic constitutive NOS in NAFLD.
In addition to hepatic NOS activity, we assessed the effects of l-NAME treatment on hepatic expression of NOS isoforms. Neither eNOS nor iNOS protein content was different among groups. nNOS protein was not detected by Western blotting, suggesting that constitutive hepatic NOS activity is primarily due to eNOS. However, the phosphorylation and activation status of eNOS was enhanced in l-NAME-treated OLETF rats. This increase was associated with increased activated AMPK, a kinase of eNOS at S1177 (5), which can promote glycolytic pathway activity to maintain energy homeostasis in mitochondrial failure (1). In light of the current finding that l-NAME impaired mitochondrial function in OLETF rats (see below), these data suggest that proper hepatic NO production is essential for the hepatic response to metabolic excess.
The increase in liver TAG content in l-NAME-treated OLETF rats led us to hypothesize that NO is essential for normal hepatic lipid handling. Thus we focused on identifying mechanisms whereby NO could influence liver fat accumulation. Common indexes of hepatic de novo lipogenesis (ACC and FAS), fatty acid uptake (CD36), and TAG export (MTTP) do not explain the current data (Fig. 5). In addition, l-NAME treatment actually reduced fasting insulin (Table 2), reduced homeostatic model assessment of insulin resistance (HOMA-IR) (23), and enhanced Akt phosphorylation in skeletal muscle (Fig. 5G). Furthermore, it is unlikely that l-NAME treatment increased hepatic exposure to and uptake of FFAs due to elevated systemic lipolysis, because serum FFAs did not differ with l-NAME treatment and reduced hepatic CD36 protein expression likely indicates suppressed hepatic FFA uptake (1). Finally, we previously demonstrated in these animals an increased proinflammatory gene expression profile in the aorta, but not in various adipose tissue depots, following l-NAME administration (23), which is in contrast to our finding in the present study that l-NAME increased hepatic proinflammatory CD11c expression (Fig. 1) and suggests a novel effect of l-NAME that is somewhat unique to the liver. Instead, l-NAME treatment in OLETF rats reduced respiratory function in isolated hepatic mitochondria, which was independent of changes in mitochondrial content, β-oxidation, or tricarboxylic acid cycle function. Importantly, mitochondrial dysfunction is a key feature underlying NAFLD development and progression (30) and is present in patients with NASH (27). Thus, although the specific mechanism remains unclear, available data suggest that NOS inhibition caused a reduction of hepatic mitochondrial respiratory capacity, leading to the accumulation of intrahepatic TAG in the hyperphagic OLETF rat.
An unexpected, but intriguing, observation of the current investigation is that, in contrast to the OLETF rats, l-NAME-treated LETO rats exhibited an increase in hepatic mitochondrial respiration. Physiological NO concentrations can modify ETC flux through competitive reversible inhibition of COX (3, 14). Speculatively, we propose a model in which 1) excess nutrient availability in obesity increases driving pressure (i.e., reducing equivalents) through the ETC and 2) NOS inhibition increases ETC conductance through removal of the tonic inhibitory influence of NO on COX. As a result, the combined effect of increased driving pressure and increased electron conductance (l-NAME-treated OLETF rats) is greater than that of increased driving pressure (control OLETF rats) or electron conductance (l-NAME-treated LETO rats) alone. Increased respiration without altered ATP demand will increase the mitochondrial membrane potential, which can cause superoxide generation and ETC dysfunction (12, 13). Thus one interpretation of the current findings is that NOS inhibition leads to increases in mitochondrial respiration in lean normophagic LETO rats that compensated by increasing hepatic SOD activity to prevent mitochondrial reactive oxygen species (ROS)-induced damage. Alternatively, the obese hyperphagic OLETF rats were unable to adapt in the same manner, resulting in hepatic mitochondrial dysfunction, inflammation, and TAG accumulation. These differential findings suggest that the metabolically unhealthy OLETF rats were more susceptible to the negative effects of the systemic NOS inhibition, but whether that was related to the environment of overnutrition or obesity or both needs more thorough examination. Future work should also include short-term (e.g., 1, 3, and 5 days) and long-term studies of NOS inhibition in OLETF and LETO rats with assessment of in vivo ROS production, mitochondrial membrane potential, and mitochondrial respiration to determine if OLETF rats respond in the short-term similar to the LETO rats and whether longer-term NOS inhibition in LETO animals would eventually cause hepatic mitochondrial ROS-induced damage and suppression of mitochondrial function. To support this concept involving eNOS and the nutritional environment, our preliminary data show that hepatic mitochondrial respiration is higher in eNOS knockout mice fed a low-fat diet but lower in those fed a Western (high-fat, high-sucrose, and high-cholesterol) diet than in wild-type controls (R. S. Rector, unpublished observations).
One cellular response to counter mitochondrial damage/dysfunction is an increase in mitochondrial turnover. NO stimulates mitochondrial biogenesis via a cGMP/PGC-1α-dependent mechanism (19). Indeed, we observed a 50% reduction in PGC-1α mRNA in l-NAME-treated LETO rats compared with controls but no effect of l-NAME on PGC-1α mRNA in OLETF rats. Interestingly, TFAM mRNA was elevated in control, but not l-NAME-treated, OLETF rats. Given that indexes of mitochondrial content were not different between groups, these data suggest that increased mitochondrial turnover may be an adaptive mechanism to increase the amount of new, healthy mitochondria entering the mitochondrial pool. Thus it appears that l-NAME-treated OLETF rats have both an increased propensity for mitochondrial dysfunction and a relative inability to replace damaged mitochondria. This possibility warrants future study.
Another differential effect of l-NAME in lean vs. obese rats was the impact on hepatic macrophage polarization. l-NAME treatment in OLETF rats significantly increased the mRNA expression of a key M1 marker, CD11c, which is strongly associated with insulin resistance and systemic inflammation (26). In LETO rats, l-NAME treatment caused a significant reduction of M2 markers (CD163 and CD206) but had no effect on M1 markers. Along these lines, the increase in MCP-1 expression in l-NAME-treated LETO rats may represent a compensatory response to replenish the M2 pool. These findings encourage further evaluation of hepatic macrophage polarization as it relates to NAFLD and NO bioavailability.
A limitation of the current study is that l-NAME treatment is well known to cause hypertension in rats (18). The lack of acetylcholine-induced vasodilation in isolated aortic rings from the current cohort of l-NAME-treated rats (23) would be consistent with the notion that these animals were hypertensive. Although our data are consistent with functions attributed to NO [e.g., reduced PGC-1α expression (19) and modulation of mitochondrial respiration (3, 4)], we cannot rule out the possibility that hypertension influenced the current results. However, previous work in pigs indicates that l-NAME increases hepatic resistance and blood pressure, yielding no net change in hepatic perfusion (15), a concept that is supported by our finding of similar hypoxia-inducible factor 1α expression between groups (Fig. 2I). Nevertheless, the potential clinical importance of this report remains clear, as therapeutic interventions to improve NO availability to the liver will also affect blood pressure. Further efforts to better identify the relationship between NO, NAFLD, and hypertension are important and may provide clinical utility.
In conclusion, the results of the current study reveal an important and novel role for constitutive hepatic NOS-mediated NO production in the ability of the liver to adapt to an environment of overnutrition and obesity. Chronic inhibition of NOS in OLETF rats caused deleterious hepatic outcomes, including a marked increase in steatosis, reduced mitochondrial respiratory capacity, and evidence of increased M1 macrophage polarity. In addition, the current data reveal a novel obesity-NO interaction in the liver, as NOS inhibition caused different effects in lean LETO rats. This work may have important therapeutic implications, as it suggests that currently available NO-based therapies may exert pleiotropic effects in the livers of obese patients.
GRANTS
This work was supported by a University of Missouri Life Sciences Fellowship (R. D. Sheldon), National Heart, Lung, and Blood Institute Grants K01 HL-125503 (J. Padilla) and HL-36088 (M. H. Laughlin), and Veterans Health Administration Grant CDA2 BX001299 (R. S. Rector). This work was completed with resources and the use of facilities at the Harry S Truman Memorial Veterans Hospital.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.D.S., J.P., N.T.J., M.H.L., and R.S.R. developed the concept and designed the research; R.D.S., J.P., N.T.J., M.H.L., and R.S.R. performed the experiments; R.D.S. and R.S.R. analyzed the data; R.D.S., J.P., N.T.J., M.H.L., and R.S.R. interpreted the results of the experiments; R.D.S. prepared the figures; R.D.S. and R.S.R. drafted the manuscript; R.D.S., J.P., N.T.J., M.H.L., and R.S.R. edited and revised the manuscript; R.D.S., J.P., N.T.J., M.H.L., and R.S.R. approved the final version of the manuscript.
ACKNOWLEDGMENTS
The authors thank Grace Meers and Pam Thorne for excellent technical assistance and Drs. John P. Thyfault and E. Matthew Morris for intellectual input.
REFERENCES
- 1.Almeida A, Moncada S, Bolaños JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol 6: 45–51, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Bass A, Brdiczka D, Eyer P, Hofer S, Pette D. Metabolic differentiation of distinct muscle types at the level of enzymatic organization. Eur J Biochem 10: 198–206, 1969. [DOI] [PubMed] [Google Scholar]
- 3.Brown GC. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett 369: 136–139, 1995. [DOI] [PubMed] [Google Scholar]
- 4.Brown GC. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim Biophys Acta 1504: 46–57, 2001. [DOI] [PubMed] [Google Scholar]
- 5.Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett 443: 285–289, 1999. [DOI] [PubMed] [Google Scholar]
- 7.Clementi E, Nisoli E. Nitric oxide and mitochondrial biogenesis: a key to long-term regulation of cellular metabolism. Comp Biochem Physiol A Mol Integr Physiol 142: 102–110, 2005. [DOI] [PubMed] [Google Scholar]
- 8.Fletcher JA, Meers GM, Linden MA, Kearney ML, Morris EM, Thyfault JP, Rector RS. Impact of various exercise modalities on hepatic mitochondrial function. Med Sci Sports Exerc 46: 1089–1097, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hurt KJ, Musicki B, Palese MA, Crone JK, Becker RE, Moriarity JL, Snyder SH, Burnett AL. Akt-dependent phosphorylation of endothelial nitric-oxide synthase mediates penile erection. Proc Natl Acad Sci USA 99: 4061–4066, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ignarro LJ, Napoli C, Loscalzo J. Nitric oxide donors and cardiovascular agents modulating the bioactivity of nitric oxide: an overview. Circ Res 90: 21–28, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Kim YM, Bergonia H, Lancaster JR Jr. Nitrogen oxide-induced autoprotection in isolated rat hepatocytes. FEBS Lett 374: 228–232, 1995. [DOI] [PubMed] [Google Scholar]
- 12.Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett 416: 15–18, 1997. [DOI] [PubMed] [Google Scholar]
- 13.Lambert A, Brand M. Superoxide production by NADH: ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J 382: 511–517, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mason MG, Nicholls P, Wilson MT, Cooper CE. Nitric oxide inhibition of respiration involves both competitive (heme) and noncompetitive (copper) binding to cytochrome c oxidase. Proc Natl Acad Sci USA 103: 708–713, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McAllister RM, Newcomer SC, Pope ER, Turk JR, Laughlin MH. Effects of chronic nitric oxide synthase inhibition on responses to acute exercise in swine. J Appl Physiol 104: 186–197, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minamiyama Y, Bito Y, Takemura S, Takahashi Y, Kodai S, Mizuguchi S, Nishikawa Y, Suehiro S, Okada S. Calorie restriction improves cardiovascular risk factors via reduction of mitochondrial reactive oxygen species in type II diabetic rats. J Pharmacol Exp Ther 320: 535–543, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Montagnani M, Chen H, Barr VA, Quon MJ. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser1179. J Biol Chem 276: 30392–30398, 2001. [DOI] [PubMed] [Google Scholar]
- 18.Navarro J, Sanchez A, Saiz J, Ruilope L, Garcia-Estan J, Romero J, Moncada S, Lahera V. Hormonal, renal, and metabolic alterations during hypertension induced by chronic inhibition of NO in rats. Am J Physiol Regul Integr Comp Physiol 267: R1516–R1521, 1994. [DOI] [PubMed] [Google Scholar]
- 19.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299: 896–899, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Okauchi N, Mizuno A, Yoshimoto S, Zhu M, Sano T, Shima K. Is caloric restriction effective in preventing diabetes mellitus in the Otsuka Long Evans Tokushima fatty rat, a model of spontaneous non-insulin-dependent diabetes mellitus? Diabetes Res Clin Pract 27: 97–106, 1995. [DOI] [PubMed] [Google Scholar]
- 21.Ou J, Carlos TM, Watkins SC, Saavedra JE, Keefer LK, Kim YM, Harbrecht BG, Billiar TR. Differential effects of nonselective nitric oxide synthase (NOS) and selective inducible NOS inhibition on hepatic necrosis, apoptosis, ICAM-1 expression, and neutrophil accumulation during endotoxemia. Nitric Oxide Biol Chem 1: 404–416, 1997. [DOI] [PubMed] [Google Scholar]
- 23.Padilla J, Jenkins NT, Thorne PK, Lansford KA, Fleming NJ, Bayless DS, Sheldon RD, Rector RS, Laughlin MH. Differential regulation of adipose tissue and vascular inflammatory gene expression by chronic systemic inhibition of NOS in lean and obese rats. Physiol Rep 2: e00225, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park SY, Choi GH, Choi HI, Ryu J, Jung CY, Lee W. Calorie restriction improves whole-body glucose disposal and insulin resistance in association with the increased adipocyte-specific GLUT4 expression in Otsuka Long-Evans Tokushima fatty rats. Arch Biochem Biophys 436: 276–284, 2005. [DOI] [PubMed] [Google Scholar]
- 25.Pasarín M, Abraldes JG, Rodríguez-Vilarrupla A, La Mura V, García-Pagán JC, Bosch J. Insulin resistance and liver microcirculation in a rat model of early NAFLD. J Hepatol 55: 1095–1102, 2011. [DOI] [PubMed] [Google Scholar]
- 26.Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab 8: 301–309, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pérez-Carreras M, Del Hoyo P, Martín MA, Rubio JC, Martín A, Castellano G, Colina F, Arenas J, Solis-Herruzo JA. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 38: 999–1007, 2003. [DOI] [PubMed] [Google Scholar]
- 28.Rector RS, Thyfault JP, Morris RT, Laye MJ, Borengasser SJ, Booth FW, Ibdah JA. Daily exercise increases hepatic fatty acid oxidation and prevents steatosis in Otsuka Long-Evans Tokushima fatty rats. Am J Physiol Gastrointest Liver Physiol 294: G619–G626, 2008. [DOI] [PubMed] [Google Scholar]
- 30.Rector RS, Thyfault JP, Uptergrove GM, Morris EM, Naples SP, Borengasser SJ, Mikus CR, Laye MJ, Laughlin MH, Booth FW, Ibdah JA. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J Hepatol 52: 727–736, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rector RS, Thyfault JP, Wei Y, Ibdah JA. Non-alcoholic fatty liver disease and the metabolic syndrome: an update. World J Gastroenterol 14: 185–192, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rector RS, Uptergrove GM, Morris EM, Borengasser SJ, Laughlin MH, Booth FW, Thyfault JP, Ibdah JA. Daily exercise vs. caloric restriction for prevention of nonalcoholic fatty liver disease in the OLETF rat model. Am J Physiol Gastrointest Liver Physiol 300: G874–G883, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shah V, Haddad FG, Garcia-Cardena G, Frangos JA, Mennone A, Groszmann RJ, Sessa WC. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J Clin Invest 100: 2923–2930, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheldon RD, Laughlin MH, Rector RS. Reduced hepatic eNOS phosphorylation is associated with NAFLD and type 2 diabetes progression and is prevented by daily exercise in hyperphagic OLETF rats. J Appl Physiol 116: 1156–1164, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srere P. Citrate synthase. Methods Enzymol 13: 3–5, 1969. [Google Scholar]
- 36.Stepanova M, Rafiq N, Makhlouf H, Agrawal R, Kaur I, Younoszai Z, McCullough A, Goodman Z, Younossi ZM. Predictors of all-cause mortality and liver-related mortality in patients with non-alcoholic fatty liver disease (NAFLD). Dig Dis Sci 58: 3017–3023, 2013. [DOI] [PubMed] [Google Scholar]
- 37.Tateya S, Rizzo-De Leon N, Handa P, Cheng AM, Morgan-Stevenson V, Ogimoto K, Kanter JE, Bornfeldt KE, Daum G, Clowes AW. VASP increases hepatic fatty acid oxidation by activating AMPK in mice. Diabetes 62: 1913–1922, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tateya S, Rizzo NO, Handa P, Cheng AM, Morgan-Stevenson V, Daum G, Clowes AW, Morton GJ, Schwartz MW, Kim F. Endothelial NO/cGMP/VASP signaling attenuates Kupffer cell activation and hepatic insulin resistance induced by high-fat feeding. Diabetes 60: 2792–2801, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsuchiya K, Sakai H, Suzuki N, Iwashima F, Yoshimoto T, Shichiri M, Hirata Y. Chronic blockade of nitric oxide synthesis reduces adiposity and improves insulin resistance in high fat-induced obese mice. Endocrinology 148: 4548–4556, 2007. [DOI] [PubMed] [Google Scholar]
- 40.Xie G, Wang X, Wang L, Wang L, Atkinson RD, Kanel GC, Gaarde WA, DeLeve LD. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology 142: 918–927, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Younossi ZM, Stepanova M, Afendy M, Fang Y, Younossi Y, Mir H, Srishord M. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin Gastroenterol Hepatol 9: 524–530, 2011. [DOI] [PubMed] [Google Scholar]