

Abstract
The higher rate of non-synonymous over synonymous substitutions (dN/dS) of the X chromosome compared with autosomes is often interpreted as a consequence of X hemizygosity. However, other factors, such as gene expression, are also known to vary between X and autosomes. Analysing 4800 orthologues in six mammals, we found that gene expression levels, associated with GC content, fully account for the variation in dN/dS between X and autosomes with no detectable effect of hemizygosity. We also report an extensive variance in dN/dS and gene expression between autosomes.
Keywords: X chromosome, non-synonymous substitutions, synonymous substitutions, fast-X, hemizygosity, gene expression
1. Introduction
In mammals and other groups, the X-linked protein-coding genes accumulate non-synonymous substitutions at a faster rate than autosomes [1,2]. The most popular explanation for this pattern invokes hemizygosity. Because the X chromosome in males is haploid, recessive beneficial mutations on the X are exposed to selection when heterozygous, which increases their fixation probability [3]. The lack of an X copy in males also results in a decreased (by a factor of ¾ under even sex ratio) effective population size for X-linked genes compared with autosomal genes. This might also contribute to the fast-X effect by promoting the fixation of slightly deleterious, codominant mutations through genetic drift. The combination of these two factors is widely believed to explain the higher rate of non-synonymous over synonymous substitutions (dN/dS) observed on the X chromosome.
Recently, however, mammalian X-linked genes were found to experience a lower expression level than autosomal genes, on average [4]. Because a negative relationship exists between expression level, expression breadth and dN/dS [5,6], we hypothesize that gene expression might play a role in the higher dN/dS of X-linked genes. Here, we test this hypothesis in placental mammals. We report that levels of gene expression, associated with GC content, fully account for the variation in dN/dS between X and autosomes.
2. Material and methods
(a). Expression level
We used the ‘constitutive aligned exons' dataset of Brawand et al. [7], which provides normalized estimation of transcription outputs in six species of placental mammals: human, chimpanzee, gorilla, orangutan, macaque and mouse. For each gene, we computed the median of expression level between samples (sexes and tissues), expressed in reads per kilobase, per million sequenced reads (RPKM).
We measured the expression specificity (τ) of each gene as:
where N(t) is the number of tissues, RPKMi is the expression level in tissue i and RPKMmax is the maximum expression level among all tissues. τ ranges from 0 (same level of expression across all tissues) to 1 (only expressed in a single tissue). Genes with no detected expression level were excluded from the analyses.
(b). Sequence data and substitution rate
We extracted coding alignments of genes orthologous between the six species from OrthoMam v. 6. The current version (v. 8 [8]) yielded an unexpectedly high dN/dS for gorilla (electronic supplementary material, figure S1). This problem was created by misalignment of very small exons in gorilla genome assembly gorGor3.1 (EnsEMBL v59) that were not present in earlier annotations (e.g. EnsEMBL v56). Alignments were cleaned using Gblocks [9] (options: −t = c −b2 = 0.5 −b4 = 5 −b5 = All). We estimated branch-specific dN/dS using the mapping method proposed by Romiguier et al. [10] and implemented in the mapNH software (http://biopp.univ-montp2.fr/forge/testnh). The estimated dN/dS values, based on substitution counts, were divided by 3 to make them comparable with classical ‘codeml’ dN/dS, thus assuming that 25% of mutations in coding sequences are synonymous and 75% non-synonymous. We used the same topology for all the analyses: ((((human, chimpanzee), gorilla), orangutan), macaque, mouse).
For each terminal branch, genes with less than one non-synonymous substitution, less than two synonymous substitutions, or divergence (dS or dN) above the mean plus four times the standard deviation of the complete distribution were excluded.
(c). Polymorphism
Human polymorphism data were obtained from Frazer et al. [11]. We computed average heterozygosity as 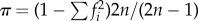 , where f is the frequency of allele i and n is the sample size. Synonymous and non-synonymous average heterozygosity were computed only for genes present in the orthologue dataset. Using these data, we computed the direction of selection (DoS) statistics [12], a modified version of the neutrality index: DoS = dN/(dN + dS) – pN/(pN + pS) with pN and pS being the number of non-synonymous and synonymous single-nucleotide polymorphisms.
, where f is the frequency of allele i and n is the sample size. Synonymous and non-synonymous average heterozygosity were computed only for genes present in the orthologue dataset. Using these data, we computed the direction of selection (DoS) statistics [12], a modified version of the neutrality index: DoS = dN/(dN + dS) – pN/(pN + pS) with pN and pS being the number of non-synonymous and synonymous single-nucleotide polymorphisms.
(d). Statistical analyses
Most of the statistical analyses were carried out under the multivariate linear model in R (v. 3.1.1). We used type II ANOVA (from the ‘car’ package) and the model selection procedure implemented in the package ‘glmulti’ [13] using default parameters. Models are presented in the Results section following the ‘model formulae’ syntax of R where the ‘ +’ operator is for additive effects and ‘:’ represents interaction between variables.
3. Results and discussion
Our curated dataset was composed of 4789 orthologues (electronic supplementary material, table S1). Both a lower expression level and a higher dN/dS on the X chromosome were well recovered by our dataset (not shown). We found significant differences between species, with human and chimpanzee having a higher dN/dS than orangutan and macaque, which in turn have a higher dN/dS than mouse (Tukey's test, p < 0.001).
(a). Expression pattern and GC content contribute to the fast-X effect
To evaluate how much of the fast-X effect could be explained by variation in gene expression level variation, we used the linear model ‘log(dN/dS) ∼ Chromosome Types + log(RPKM) + τ + GC3 + Species', where GC3 is the GC content computed at third codon positions. GC content is a relevant variable because it covaries with many features of the mammalian genome such as recombination rate (through GC-biased gene conversion) and gene density [14] that both might influence dN/dS. We would expect Chromosome Type (i.e. X versus autosomes) to have a significant effect on dN/dS if hemizygosity was influencing the rate of non-synonymous substitution. This is not what we found. Chromosome Type has no significant effect on the dN/dS in this multivariate analysis (table 1, p = 0.55), whereas expression level and GC3 have a strong negative effect, and τ has a positive effect (table 1, p < 0.001). Excluding GC3 or variables linked to expression pattern from the model leads to a moderate recovery of the significance of Chromosome Type (p = 0.05 and p = 0.01 for GC3 and expression, respectively). The lack of influence of Chromosome Type on dN/dS is confirmed by the model selection procedure evaluating the effect of the all the variables and their pairwise interactions. The best-selected model is ‘log(dN/dS) ∼ Species + log(RPKM) + t + GC3 + τ : log(RPKM) + GC3 : log(RPKM) + GC3 : τ + Species : log(RPKM) + Species : τ + Species : GC3'. This model has an R2 of 18% and Chromosome Type is not retained as an informative variable either alone or in interaction.
Table 1.
Result of the linear model log(dN/dS) ∼ Chromosome Types + Species + log(RPKM) + τ + GC3. Estimates and standard error of the estimates are provided only for continuous variables. F statistics are computed using the type II ANOVA.
| variables | estimates | s.e. | F | p-value |
|---|---|---|---|---|
| Chromosome Types | 0.346 | 0.56 | ||
| Species | 445.08 | <2.2 × 10−16 | ||
| log(RPKM) | −0.049 | 0.004 | 151.04 | <2.2 × 10−16 |
| expression specificity (τ) | 0.246 | 0.030 | 66.42 | 3.885 × 10−16 |
| GC3 | −1.016 | 0.040 | 642.58 | <2.2 × 10−16 |
(b). Extensive variation in expression levels and dN/dS between autosomes
In all the analysed species, we detected a significant variation in mean expression level between autosomes (ANOVA; p < 0.001 in all the species). The X chromosome always had the lowest median expression levels, but the variation between autosomes was extensive and of similar magnitude to the variation between X and autosomes. In humans, for example, chromosome 19 has a median expression level that is 2.5 times higher than the median of the other autosomes (Mann–Whitney test, p = 0.002), whereas X-linked genes have a median expression that is 2.5 times lower. Moreover, chromosome-wide dN/dS (computed as the sum of dN over the sum of dS across genes) is negatively correlated with median expression level in all the species. Considering the 136 individual chromosomes from the six species as statistical units, we found that a model combining expression level and species effect explained 79% of the variation in dN/dS between chromosomes, with a strong effect of both expression (F = 43.58, p < 0.001) and species (F = 65.03, p < 0.001) but no interaction between the two (p = 0.60, figure 1). As for the analysis at the gene level, there was no statistically significant difference between autosomes and X once expression level was taken into account (model: log(ΣdN/ΣdS) ∼ Species + log(RPKM) + Chromosome Type, Chromosome effect F = 0.35, p = 0.55, figure 1). This result is confirmed by the fact that excluding X chromosomes from the analyses did not alter the explanatory power of the model (R2 = 0.80), with expression level keeping a highly significant effect (F = 31.04, p < 0.001).
Figure 1.

Relationship between chromosome-wide dN/dS and median gene expression levels (RPKM) in six species of mammals. (Online version in colour.)
(c). Contrasting polymorphism and divergence in humans
A recent study in chimpanzees reported an excess of dN/dS compared with pN/pS on the X chromosome but not on autosomes [15]. This result was interpreted as the consequence of a higher rate of fixation of partial recessive beneficial alleles on the X chromosome, again a consequence of hemizygosity. To evaluate this result in humans, we computed the DoS statistic for each human chromosome. A positive value of the DoS (excess of dN/dS relative to pN/pS) is indicative of positive selection while a negative DoS reflects the influence of purifying selection. We report that the human X chromosome does indeed have a higher DoS than the autosomes (−0.134 versus −0.171 for X chromosome and median autosomes, respectively). Some autosomes have, however, a DoS very close to that of the X chromosome, for example, chromosome 21 (DoS = −0.129) and chromosome 14 (DoS = −0.139). Moreover, the confidence interval on DoS, estimated by bootstrapping genes within chromosomes (1000 replicates), was very large on the X chromosome (between −0.198 and −0.086) and overlapped with that of all the autosomes (electronic supplementary material, figure S2).
Finally, we split the dataset into 50 windows, based on gene locations, in order to obtain windows similar in gene number to the X chromosome (37 genes). This was intended to (i) control for potential variation in DoS due to sampling size and (ii) test whether there is any correlation between DoS and expression level. No relationship was observed between expression level and DoS (figure 2). More importantly, the X chromosome did not appear as a clear outlier, with seven autosomal windows having a higher average DoS than the X (figure 2).
Figure 2.

Relationship between the direction of selection (DoS) and median gene expression levels (RPKM) computed in windows of 37 genes across the human genome. (Online version in colour.)
Additional control analyses are presented as electronic supplementary material.
4. Conclusion
The X chromosome differs from the autosomes not only in ploidy level: it is an outlier with respect to several genomic features, including GC content and gene expression level, which are known to influence the rates of molecular evolution. Our results indicate that these genomic features are sufficient to explain higher dN/dS in mammals, with hemizygosity having no direct detectable effect in our analysis. The lack of any effect of hemizygosity on dN/dS is perhaps surprising based on the existing literature. It might be explained by a more efficient purging of recessive deleterious mutations on the X, which tends to reduce dN/dS, thus offsetting the increased fixation rate of recessive beneficial mutations. Positively selected mutations might well be promoted by hemizygosity but be rare enough to negligibly affect the mean dN/dS. Finally, it should be noted that positive selection affecting regulatory sequences is not considered in the present analysis [16].
We argue that comparing the X chromosome to an autosomal average, thus ignoring the variance between autosomes, can be a misleading approach. In humans, the X is no more different from the genomic average than, say, chromosome 19 or 22 with respect to dN/dS, GC content and expression level. Whether these confounding effects also apply to the other groups in which the fast-X (or fast-Z) effect is documented, such as birds, Drosophila and Lepidoptera, is an open question.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgement
We thank Sylvain Glémin for his help with the polymorphism analysis.
Data accessibility
The data underlying this study are available on Dryad: doi:10.5061/dryad.qr20n.
Author contributions
B.N. and N.G. designed the study. P.L.N. and B.N. performed the analyses. B.N. drafted the manuscript. All authors contributed to the final form of the article.
Funding statement
This work was supported by Agence Nationale de la Recherche grants ANR-14-CE02–0002–01 ‘BirdIslandGenomic’ and ANR-10-BINF-01–02 ‘Ancestrome’. This is publication ISE-M 2015–009.
References
- 1.Lu J, Wu C-I. 2005. Weak selection revealed by the whole-genome comparison of the X chromosome and autosomes of human and chimpanzee. Proc. Natl Acad. Sci. USA 102, 4063–4067. ( 10.1073/pnas.0500436102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vicoso B, Charlesworth B. 2006. Evolution on the X chromosome: unusual patterns and processes. Nat. Rev. Genet. 7, 645–653. ( 10.1038/nrg1914) [DOI] [PubMed] [Google Scholar]
- 3.Charlesworth B, Coyne JA, Barton NH. 1987. The relative rates of evolution of sex chromosomes and autosomes. Am. Nat. 130, 113 ( 10.1086/284701) [DOI] [Google Scholar]
- 4.Julien P, et al. 2012. Mechanisms and evolutionary patterns of mammalian and avian dosage compensation. PLoS Biol. 10, e1001328 ( 10.1371/journal.pbio.1001328) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drummond DA, Wilke CO. 2008. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134, 341–352. ( 10.1016/j.cell.2008.05.042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duret L, Mouchiroud D. 2000. Determinants of substitution rates in mammalian genes: expression pattern affects selection intensity but not mutation rate. Mol. Biol. Evol. 17, 68–74. ( 10.1093/oxfordjournals.molbev.a026239) [DOI] [PubMed] [Google Scholar]
- 7.Brawand D, et al. 2011. The evolution of gene expression levels in mammalian organs. Nature 478, 343–348. ( 10.1038/nature10532) [DOI] [PubMed] [Google Scholar]
- 8.Douzery EJP, Scornavacca C, Romiguier J, Belkhir K, Galtier N, Delsuc F, Ranwez V. 2014. OrthoMaM v8: a database of orthologous exons and coding sequences for comparative genomics in mammals. Mol. Biol. Evol. 31, 1923–1928. ( 10.1093/molbev/msu132) [DOI] [PubMed] [Google Scholar]
- 9.Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. ( 10.1080/10635150701472164) [DOI] [PubMed] [Google Scholar]
- 10.Romiguier J, Figuet E, Galtier N, Douzery EJP, Boussau B, Dutheil JY, Ranwez V. 2012. Fast and robust characterization of time-heterogeneous sequence evolutionary processes using substitution mapping. PLoS ONE 7, e33852 ( 10.1371/journal.pone.0033852) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frazer KA, et al. 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851–861. ( 10.1038/nature06258) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stoletzki N, Eyre-Walker A. 2011. Estimation of the neutrality index. Mol. Biol. Evol. 28, 63–70. ( 10.1093/molbev/msq249) [DOI] [PubMed] [Google Scholar]
- 13.Calcagno V, de Mazancourt C. 2010. glmulti: an R package for easy automated model selection with (generalized) linear models. J. Stat. Softw. 34, 1–29. [Google Scholar]
- 14.Duret L, Galtier N. 2009. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu. Rev. Genomics Hum. Genet. 10, 285–311. ( 10.1146/annurev-genom-082908-150001) [DOI] [PubMed] [Google Scholar]
- 15.Hvilsom C, et al. 2012. Extensive X-linked adaptive evolution in central chimpanzees. Proc. Natl Acad. Sci. USA 109, 2054–2059. ( 10.1073/pnas.1106877109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kayserili MA, Gerrard DT, Tomancak P, Kalinka AT. 2012. An excess of gene expression divergence on the X chromosome in Drosophila embryos: implications for the faster-X hypothesis. PLoS Genet. 8, e1003200 ( 10.1371/journal.pgen.1003200) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available on Dryad: doi:10.5061/dryad.qr20n.