

Abstract
This paper summarizes the advances made by the DiFrancesco and Noble (DFN) model of cardiac cellular electrophysiology, which was published in Philosophical Transactions B in 1985. This model was developed at a time when the introduction of new techniques and provision of experimental data had resulted in an explosion of knowledge about the cellular and biophysical properties of the heart. It advanced the cardiac modelling field from a period when computer models considered only the voltage-dependent channels in the surface membrane. In particular, it included a consideration of changes of both intra- and extracellular ionic concentrations. In this paper, we summarize the most important contributions of the DiFrancesco and Noble paper. We also describe how computer modelling has developed subsequently with the extension from the single cell to the whole heart as well as its use in understanding disease and predicting the effects of pharmaceutical interventions. This commentary was written to celebrate the 350th anniversary of the journal Philosophical Transactions of the Royal Society.
Keywords: heart, computer model, pacemaker, calcium
1. Introduction
It is almost exactly 30 years since Dario DiFrancesco and Denis Noble published their seminal paper modelling the electrical activity of cardiac muscle [1]. This was by no means the first paper to model such electrical activity. Indeed one of the authors (Noble) had much earlier, in 1962, produced the first mathematical model to reproduce the basic electrical properties of cardiac tissue [2]. This had been followed by various other models [3,4]. A PubMed search for ‘mathematical model heart action potential’ shows that in the first five years following the Noble 1962 model there were an average of two papers a year. This had increased to 178 per year in the period from 2009 to 2013. An obvious question is: what is special about the DiFrancesco and Noble paper to pay attention to it now? Our view is that it marks the transition from simply modelling the behaviour of surface membrane channels to considering the whole biology of the cell. The majority of previous models had considered the behaviour of surface membrane channels. They had, however, paid less attention to changes of intracellular ionic concentrations. The time leading up to the publication of the model had been a fertile one leading to many experimental observations which were ripe for embedding in a model. In the remainder of this paper, we consider some of the major ones and, in addition, how the field has moved on from the stimulus of this paper.
2. The pacemaker current
In the normal healthy heart, spontaneous beating is only observed in the sinoatrial node. Other sites, such as the atrioventricular node and His-Purkinje system, have the capacity to produce spontaneous activity but their intrinsic rate is slower than that of the sinoatrial node. Spontaneous beating results from a slow depolarization of the resting potential (pacemaker potential) leading to generation of an action potential. At the time of the paper, the underlying pacemaker current had recently undergone a reinterpretation. It had been thought that the pacemaker depolarization resulted from the decrease of a potassium conductance known as iK2 [5]. DiFrancesco showed, by contrast [6], that it was caused by an increase of an inward current, carried largely by sodium ions. Unlike most known currents which activate on depolarization, this channel activated on hyperpolarization. This funny (peculiar rather than ha-ha) nature of this current led to it being termed if. An interesting question is that of how a sodium current could have masqueraded as a potassium one? A major factor that had led to this conclusion was the voltage dependence of the current. As the membrane potential was initially stepped more negative, following the initial jump, the current trace became more inward/less outward with time. As the magnitude of the hyperpolarization was increased, the time-dependent change of current reversed. These results were consistent with a decreasing potassium current which would be expected to reverse at a potential given by the Nernst potential of K+. However, as shown by DiFrancesco et al. [6,7] and modelled in fig. 17 of [1], a similar apparent reversal potential can result from the combination of (i) an increasing inward current and (ii) potassium entering the cell leading to a decrease in extracellular potassium concentration which would increase the outward current.
Subsequent work has led to the realization that pacemaker activity in the heart is even more complicated. It is now known that calcium is released from the sarcoplasmic reticulum (SR) during the pacemaker depolarization [8,9] and that this activates the electrogenic Na–Ca exchange (NCX) contributing to the depolarization. This calcium release component of pacemaker activity has been termed the ‘calcium clock’ and has been extensively modelled [10]. There is still considerable discussion about the relative contributions of this and the funny current to pacemaker activity [11]. The subject is of clinical interest as it underlies the development of drugs such as ivabradine which are designed to inhibit if and thereby slow the heart [12].
3. Extracellular potassium
Not long before publication of the paper [1], movement of potassium across the surface membrane had been shown to result in appreciable changes of potassium concentration in the restricted extracellular spaces of isolated cardiac tissue [13]. Considerable attention had been paid to the effect that these concentration changes would have on membrane currents [14–16]. As mentioned above, this problem had resulted in confusion of the identity of the pacemaker current. The fact that, at least in isolated tissues, extracellular potassium concentration can be different from that in the bulk bathing solution also affects the analysis of activation of the Na–K pump by external potassium ions [1,17]. The paper explicitly models changes of extracellular potassium. What is still unclear, however, is the extent to which such changes of extracellular K+ concentration occur in the normal, blood-perfused heart.
4. Intracellular sodium and the sodium pump
The first continuous measurements of intracellular sodium concentration (using sodium-sensitive microelectrodes) were published only seven years before DiFrancesco and Noble's paper [18]. Three years later, the activity of the sodium pump was directly measured from the electrogenic pump current it produces [17,19,20]. This experimental work was incorporated into the DFN model making it possible to consider the effects of changes of sodium pump activity. At least two aspects of sodium regulation were not apparent at the time this paper was written and could not therefore be included in the model. (i) As well as the voltage-dependent Na current and NCX, there is a significant contribution to Na+ entry from sodium hydrogen exchange [21,22]. (ii) There is also good evidence that the cardiac cell expresses more than one isoform of the sodium pump and the different isoforms are expressed in different cellular compartments (for review, see [23]). (iii) More recent work has also shown that the properties of the inward sodium current are different from what was known at the time of the paper. In particular, following the inactivation of the current, there is a persistent, non-inactivating component. DiFrancesco and Noble modelled one component of this current, the so-called ‘window’ current which results from the fact that over a narrow voltage range, the activation and inactivation curves overlap [24]. It is now known, however, that there is a further persistent Na+ current which is increased by hypoxia [25] and is a target for therapeutic intervention [26].
5. Intracellular calcium
As in many of its other aspects, mentioned above, the paper was a turning point in the incorporation of Ca signalling into electrophysiological models. The first measurements of changes of intracellular Ca during the heart beat had been obtained in frog cardiac muscle in 1978 [27]. Measurements in mammalian cardiac muscle appeared two years later [28]. The available data provided some estimate of the levels of systolic Ca but were not sensitive enough to record the lower, diastolic levels. Calcium release from the SR through the ryanodine receptor (RyR) was modelled in terms of calcium-induced calcium release [29], where Ca entering the cell triggered the release of much more from the SR. DiFrancesco and Noble were careful to point out, ‘We should emphasise that this part of the modelling is not thought to be too secure. There are too many arbitrary factors … ’ (p. 372). Unknown at the time, a major revolution in understanding of calcium release from the SR would take place only a few years later. A major concern with models of calcium-induced calcium release was how the release could be graded as opposed to being ‘all or none’. The problem was that release is initiated by calcium entering the cell via the L-type Ca channels. However, the calcium that is released from the SR will also increase intracellular Ca concentration ([Ca2+]i) and trigger further release. One would expect that such a process would result in the complete emptying of the SR. By contrast, it was already known that contraction and presumably therefore [Ca2+]i was a continuous function of Ca entry [30]. Related to this, under normal conditions a local rise of [Ca2+]i does not propagate along the cell. Only under conditions of elevated cell and SR Ca is such propagation observed [31]. Again, this suggested that something limits the strength of calcium-induced calcium release. This paradox was eventually resolved by Stern who demonstrated theoretically that graded release could result if the SR was controlled only by Ca channels in its immediate vicinity [32] (so-called ‘local control theory’). The experimental verification of this hypothesis was provided a year later with the observation of the Ca ‘spark’ representing release from a small cluster of SR Ca release channels [33]. Subsequent work showed that a major means for regulating the size of the Ca transient was by recruiting more and more of these release sites [34]. Models which incorporate this local release are much more complicated as the stochastic behaviour of every ion channel has to be allowed for.
Another, interesting aspect of modelling Ca regulation is the question of the various mechanisms by which Ca2+ enters the cell. At a normal resting potential, essentially all the L-type Ca channels are closed. Ca2+ is still being pumped out of the cell, however, by NCX. This raises the question of the route by which Ca2+ enters to balance this efflux. DiFrancesco and Noble assigned a background, leak influx in their model. A later, experimental paper found that when the L-type channel and NCX are inhibited, hyperpolarization indeed increases [Ca2+]i in a manner expected for such a leak [35]. An unanswered question is the molecular basis of this leak. Whatever its origin, it may well not be important in the beating heart where the regular influx of Ca into the cell on the L-type current will presumably dominate over this entry.
6. Sodium–calcium exchange
At the time that the paper appeared, a revolution had recently occurred in understanding the importance of NCX. NCX uses the energy provided by Na+ ions entering the cell down their electrochemical gradient to pump Ca2+ out of the cell. For several years after its identification in the heart and squid axon [36,37], it was unclear how many Na+ exchanged for each Ca2+. It was realized that an electroneutral NCX (2 Na+ per Ca2+) could not provide enough energy to account for the low resting [Ca2+]i. A value of 3 or more Na+ would explain the resting [Ca2+]i but would mean that the NCX generated an electrical current. This idea was championed by Lorin Mullins, who also suggested that the resulting current could account for aspects of electrical behaviour of the heart [38,39], which was then put into a cardiac model [40]. There was, however, a lack of direct evidence showing this experimentally [41]. Indeed the direct demonstration of an electrogenic NCX current in the heart appeared only after DiFrancesco and Noble's paper was published [42,43]. DiFrancesco and Noble assumed a value of 3 Na+ which correctly anticipated the subsequent experimental finding.
7. Other currents
Several membrane currents which contribute to cardiac electrophysiology were unknown at the time of writing the paper. These include the ATP-sensitive K current [44]. The division of the Ca current into L- and T-type [45] was also unknown. The DFN model includes the transient outward current which underlies the initial phase of repolarization. This was modelled as a Ca-activated potassium current. More recent work has shown the complexity of this transient outward current. It comprises a voltage-activated K current [46] and, in some species, there is a contribution from a Ca-activated chloride current [47]. There is evidence for the existence of Ca-activated K currents, particularly in heart failure [48,49], but their precise functional role still remains somewhat unclear. It may seem surprising that the model does such a good job of reproducing the properties of the action potential. This highlights a challenge for computer modelling as the ability to reproduce the action potential is no guarantee that the correct mechanisms are included.
8. Spatial localization
The DFN model treated the cytoplasm as a homogeneous structure and also regarded the surface membrane as a single structure. Including cytoplasmic ion changes was a significant advance, but an enormous amount of subsequent work has shown that, for the following reasons, these were substantial simplifications. Transverse (t) tubules make up much of the surface membrane of the cell, and there is evidence that their composition of membrane proteins differs from that of the rest of the surface membrane (for review, see [50]). It is also clear that the space between the transverse tubule and the SR (the so-called ‘dyadic space’) has a very small volume. Consequently fluxes of, for example, sodium or calcium into the cell will result in much larger changes of concentration than would be the case if the ions distributed equally throughout the cytoplasm [51]. More recent models incorporate detailed ultrastructural information (e.g. [52]).
9. Computer models of subcellular signalling
The cellular events modelled by DiFrancesco and Noble do not act in isolation but are regulated by a complex array of intracellular signalling pathways. The complexity and interdependence of known cellular signalling networks is such that computer modelling can serve a useful role in (i) identifying potential mechanisms of normal function, (ii) probing how cell signalling can contribute to diseases like heart failure and (iii) identifying which pathways may be useful for drug development. For example, in recent years the β-adrenergic signalling cascade and Ca-calmodulin-dependent protein kinase II (CAMKII) cellular signalling have been implicated in disease and arrhythmogenic mechanisms. This has necessitated the development of a large number of computer models based on signalling pathways to investigate causative mechanisms. Such simulations have yielded information which can be difficult to obtain experimentally (reviewed in [53,54]). CAMKII was first incorporated into a model of canine excitation contraction coupling to understand its role in the rate dependence of Ca2+ handling [55]. This study suggested CAMKII to be important in the rate dependence of the Ca2+ transient but not the action potential duration. Modelling of the effect of CAMKII on Na+, Ca2+ and K+ currents suggests that, while individually opposing effects on action potential duration were observed (whereby effects on INa and ICa tended to prolong action potential duration but CAMKII Ito modulation shortened the action potential), combining the effects on all three channels resulted in action potential shortening, highlighting how computer modelling is able to separate individual effects of signalling molecules [56]. More recently, a computer model has been used to explore the relationship between RyR and CAMKII; this model describes how individual L-type Ca2+ channels and RyRs may respond to local CAMKII and dyadic Ca2+ [57], which would be very difficult to measure experimentally. Thus expansion of computer modelling to encompass cardiac signalling networks now aids our understanding of the underlying components of the cellular behaviours modelled by DiFrancesco and Noble.
10. Towards the whole heart
DiFrancesco and Noble modelled a single cell or Purkinje fibre. They left to future work the spread of excitation from one cell to another in a real heart consisting of billions of cells. Indeed, it would have been very difficult, if not impossible, for the computer power of the 1980s to easily allow for this. It has become increasingly clear that many important features depend on this multicellular nature, including the intrinsic electrical heterogeneity of cardiac tissue and anisotropic cell-to-cell electrical coupling arising from the arrangement of cardiac tissue fibres [58,59]. Whole heart modelling was made possible by the seminal work of Nielsen et al. [60], who reconstructed the first three-dimensional realistic computer model for the anatomic structures of cardiac tissue. Such three-dimensional anatomical models allow integration of cardiac electrophysiology with anatomical structures. However, until the early 1990s, owing to the limitation of computing power and other challenges [61], large-scale computer modelling of cardiac tissues used grossly simplified representations of cardiac electrophysiology with idealized cardiac tissue geometry [62], or biophysically detailed cardiac electrophysiology with idealized cardiac tissue geometry [63]. It then evolved into whole heart modelling with implementations of three-dimensional anatomical structures but still with simplified models of cellular electrophysiology [64]. With the rapid advance of high-performance computing and visualization techniques in the last decade, a virtual heart model that integrates both detailed electrophysiology and anatomical structures became possible [65].
Mathematically, cardiac tissue can be idealized as a spatially extended syncytium consisting of billions of electrically coupled cells that are bounded by the anatomical geometry of the heart. It should be remembered that, as well as myocytes, the heart also contains fibroblasts which are electrically coupled to the myocytes [66]. The spread of excitation can be represented by a reaction–diffusion equation, in which the electrical activities of individual cells are represented by single-cell models and the intercellular couplings are modelled via diffusive interactions of membrane potentials by a gap junctional conductance [59]. Within this mathematical frame, models of the whole heart incorporating detailed anatomical structures and electrophysiology have been developed for various species and different regions of the heart [67–71]. An example is shown in figure 1. A clear example of applications of these large-scale models of the propagation of the action potential through the heart is to investigate how the propagation can breakdown, resulting in the genesis of re-entrant excitation waves that are related to cardiac arrhythmias in normal [72] and gene mutation conditions [73].
Figure 1.
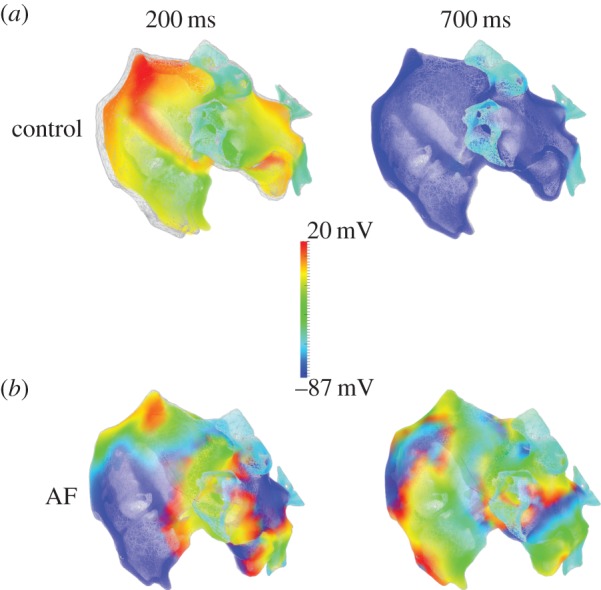
Simulated electrical and mechanical activities of the human atria during control and chronic atrial fibrillation conditions. Atrial electrical excitation waves are presented by colour coded cellular action potentials (see colour key), and mechanical contraction is represented by superimposition of the atrial mesh on its original geometry (grey; i.e. the geometry before electrical activation). (a) Snapshots of atrial electromechanical activity at 200 ms (during contraction) and 700 ms (after repolarization and tissue relaxation). (b) During atrial fibrillation (AF), showing negligible contraction and multiple re-entrant wavelets which are maintained and persist at 700 ms. (Figure was produced by Dr Ismail Adeniran and H.Z.)
Pumping blood around the body requires a sequence of rhythmic mechanical contractions from the heart; these are triggered by the propagation of excitation waves. The conduction of excitation waves in the heart generates an electrical field in the surface of the body, which can be measured as the body surface ECGs. Current development of computing power also allows development of multi-scale electromechanical models of the heart, which integrate coupling among cardiac electrophysiology, cellular contraction mechanisms and mechanic deformation [74,75]. The whole heart model can be embedded into the torso model, enabling simulations of the body surface potential and ECGs in normal and pathological conditions [76].
11. Broader perspectives
In the future, it is hoped that computer models will not only help us to understand mechanisms of cardiac function in health and disease, but actually guide therapy to aid in the treatment of disease. The vision for the Virtual Physiological Human initiative is to generate a customized computer model of a patient's condition across multiple organ systems by creating an infrastructure to link models at different biological levels to allow prediction of personalized medication [77]. Current work, however, has already made important steps using computer models to facilitate patient treatment. At the organ level, compared to other tissues, the computational model of the heart is one of the most highly advanced [78]. This has been facilitated by the long history of cardiac modelling and seminal contributions such as that of DiFrancesco and Noble. Models of the whole heart are being used to investigate how resynchronization therapy may be improved in dyssyncronous heart failure [79]. Advanced imaging methods make the construction of a patient-specific model possible. Such a personalized model has been used to simulate ventricular tachycardia (VT), opening up the possibility of using computer modelling to assess the risk of VT in a given patient and also plan potential ablation strategies for treatment [80]. The ability of computer models to screen for drugs which may be arrhythmogenic has also been demonstrated. While reduction of INa in the Cardiac Arrhythmia Suppression Trial (CAST) [81] was expected to decrease arrhythmias, sudden cardiac death actually increased, an effect which was not predicted by conventional pharmacological studies [82]. A computer model of single cells showed that both lidocaine and flecainide lowered excitability but cells were still able to generate action potentials. When the model was scaled up to incorporate coupled groups of cells it showed that flecainide caused serious conduction block at high heart rates which was confirmed experimentally in rabbit heart. The model was also able to successfully predict concentrations of both drugs at which adverse effects were seen, which was again validated experimentally in the rabbit heart [83]. This type of model may therefore be able to highlight drugs which exacerbate arrhythmias in the future. As with all computer models, it is important to stress that data should be validated using experiments which are separate from those used to define assumptions within the model itself. Computer models will never completely replace experiments on tissues and animals.
12. Summary
Looking back 30 years, the DiFrancesco and Noble paper still appears as a superb mathematical model of cardiac electrophysiology. Equally importantly, it can be seen as a vital bridge in the development of mathematical models of the heart which took the field from simple biophysics to systems biology. In particular, its consideration of changes of ionic concentrations can be seen as the forerunner of countless subsequent models.
Author profiles

Katharine Dibb received her PhD from Leeds University and is currently a lecturer at the University of Manchester.
Andrew Trafford is a British Heart Foundation Senior Fellow and Professor of Cardiac Pathophysiology at the University of Manchester. He qualified as a veterinary surgeon and obtained his PhD at the University of Liverpool.
Henggui Zhang received his PhD from Leeds University and is currently Professor of Biological Physics at the University of Manchester.

David Eisner is currently British Heart Foundation (BHF) Professor of Cardiac Physiology at the University of Manchester. From 1976 to 1979, he studied for his D. Phil in Denis Noble's laboratory in Oxford at the same time that Dario DiFrancesco was a visiting scientist. He can still remember the excitement when Dario first revolutionized cardiac pacemaker activity.
References
- 1.DiFrancesco D, Noble D. 1985. A model of cardiac electrical activity incorporating ionic pumps and concentration changes. Phil. Trans. R. Soc. Lond. B 307, 353–398. ( 10.1098/rstb.1985.0001) [DOI] [PubMed] [Google Scholar]
- 2.Noble D. 1962. A modification of the Hodgkin–Huxley equations applicable to Purkinje fibre action and pace-maker potentials. J. Physiol. (Lond.) 160, 317–352. ( 10.1113/jphysiol.1962.sp006849) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McAllister RE, Noble D, Tsien RW. 1975. Reconstruction of the electrical activity of cardiac Purkinje fibres. J. Physiol. (Lond.) 251, 1–59. ( 10.1113/jphysiol.1975.sp011080) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beeler GW, Reuter H. 1977. Reconstruction of the action potential of ventricular myocardial fibers. J. Physiol. (Lond.) 268, 177–210. ( 10.1113/jphysiol.1977.sp011853) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noble D, Tsien RW. 1968. The kinetics and rectifier properties of the slow potassium current in cardiac Purkinje fibres. J. Physiol. (Lond.) 195, 185–214. ( 10.1113/jphysiol.1968.sp008454) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DiFrancesco D. 1981. A new interpretation of the pace-maker current in calf Purkinje fibres. J. Physiol. (Lond.) 314, 359–376. ( 10.1113/jphysiol.1981.sp013713) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DiFrancesco D, Ohba M, Ojeda C. 1979. Measurement and significance of the reversal potential for the pace-maker current (ik2) in sheep Purkinje fibres. J. Physiol. (Lond.) 297, 135–162. ( 10.1113/jphysiol.1979.sp013032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rigg L, Terrar DA. 1996. Possible role of calcium release from the sarcoplasmic reticulum in pacemaking in guinea-pig sino-atrial node. Exp. Physiol. 81, 877–880. ( 10.1113/expphysiol.1996.sp003983) [DOI] [PubMed] [Google Scholar]
- 9.Huser J, Blatter LA, Lipsius SL. 2000. Intracellular Ca2+ release contributes to automaticity in cat atrial pacemaker cells. J. Physiol. (Lond.) 524, 415–422. ( 10.1111/j.1469-7793.2000.00415.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maltsev VA, Lakatta EG. 2009. Synergism of coupled subsarcolemmal Ca2+ clocks and sarcolemmal voltage clocks confers robust and flexible pacemaker function in a novel pacemaker cell model. Am. J. Physiol. 296, H594–H615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DiFrancesco D, Lakatta EG. 2009. What keeps us ticking, a funny current, a calcium clock, or both? J. Mol. Cell Cardiol. 47, 157–170. ( 10.1016/j.yjmcc.2009.03.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dobre D, et al. 2014. Heart rate: a prognostic factor and therapeutic target in chronic heart failure. The distinct roles of drugs with heart rate-lowering properties. Eur. J. Heart Fail. 16, 76–85. ( 10.1093/eurjhf/hft129) [DOI] [PubMed] [Google Scholar]
- 13.Kunze DL. 1977. Rate-dependent changes in extracellular potassium in the rabbit atrium. Circ. Res. 41, 122–127. ( 10.1161/01.RES.41.1.122) [DOI] [PubMed] [Google Scholar]
- 14.Baumgarten CM, Isenberg G. 1977. Depletion and accumulation of potassium in the extracellular clefts of cardiac Purkinje fibers during voltage clamp hyperpolarization and depolarization. Pflügers Archiv. 386, 19–31. ( 10.1007/BF01063450) [DOI] [PubMed] [Google Scholar]
- 15.Attwell D, Cohen I, Eisner DA. 1979. Membrane potential and ion concentration stability conditions for a cell with a restricted extracellular space. Proc. R. Soc. Lond. B 206, 145–161. ( 10.1098/rspb.1979.0098) [DOI] [PubMed] [Google Scholar]
- 16.Attwell D, Eisner DA, Cohen I. 1979. Voltage clamp and tracer flux data: effects of a restricted extra-cellular space. Q. Rev. Biophys. 12, 213–261. ( 10.1017/S0033583500005448) [DOI] [PubMed] [Google Scholar]
- 17.Eisner DA, Lederer WJ. 1980. Characterization of the electrogenic sodium pump in cardiac Purkinje fibres. J. Physiol. Lond 303, 441–474. ( 10.1113/jphysiol.1980.sp013298) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ellis D. 1977. The effects of external cations and ouabain on the intracellular sodium activity of sheep heart Purkinje fibres. J. Physiol. (Lond.) 273, 211–240. ( 10.1113/jphysiol.1977.sp012090) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eisner DA, Lederer WJ. 1980. The relationship between sodium pump activity and twitch tension in cardiac Purkinje fibres. J. Physiol. (Lond.) 303, 475–494. ( 10.1113/jphysiol.1980.sp013299) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gadsby DC. 1980. Activation of electrogenic Na+/K+ exchange by extracellular K+ in canine cardiac Purkinje fibers. Proc. Natl Acad. Sci. USA 76, 1783–1787. ( 10.1073/pnas.76.4.1783) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bers DM, Barry WH, Despa S. 2003. Intracellular Na+ regulation in cardiac myocytes. Cardiovasc. Res. 57, 897–912. ( 10.1016/S0008-6363(02)00656-9) [DOI] [PubMed] [Google Scholar]
- 22.Murphy E, Eisner DA. 2009. Regulation of intracellular and mitochondrial sodium in health and disease. Circ. Res. 104, 292–303. ( 10.1161/CIRCRESAHA.108.189050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aronsen JM, Swift F, Sejersted OM. 2013. Cardiac sodium transport and excitation–contraction coupling. J. Mol. Cell Cardiol. 61, 11–19. ( 10.1016/j.yjmcc.2013.06.003) [DOI] [PubMed] [Google Scholar]
- 24.Attwell D, Cohen I, Eisner DA, Ohba M, Ojeda C. 1979. The steady state TTX-sensitive (‘Window’) sodium current in cardiac Purkinje fibres. Pflügers Archiv. 379, 137–142. ( 10.1007/BF00586939) [DOI] [PubMed] [Google Scholar]
- 25.Ju YK, Saint DA, Gage PW. 1996. Hypoxia increases persistent sodium current in rat ventricular myocytes. J. Physiol. (Lond.) 497, 337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zaza A, Belardinelli L, Shryock JC. 2008. Pathophysiology and pharmacology of the cardiac ‘late sodium current’. Pharmacol. Ther. 119, 326–339. ( 10.1016/j.pharmthera.2008.06.001) [DOI] [PubMed] [Google Scholar]
- 27.Allen DG, Blinks JR. 1978. Calcium transients in aequorin-injected frog cardiac muscle. Nature 273, 509–513. ( 10.1038/273509a0) [DOI] [PubMed] [Google Scholar]
- 28.Allen DG, Kurihara S. 1980. Calcium transients in mammalian ventricular muscle. Eur. Heart J. 1, 5–15. ( 10.1093/eurheartj/1.suppl_1.5) [DOI] [PubMed] [Google Scholar]
- 29.Fabiato A. 1983. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 245, C1–C14. [DOI] [PubMed] [Google Scholar]
- 30.Beeler GW, Reuter H. 1970. The relation between membrane potential, membrane currents and activation of contraction in ventricular myocardial fibres. J. Physiol. (Lond.) 207, 211–229. ( 10.1113/jphysiol.1970.sp009057) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trafford AW, O'Neill SC, Eisner DA. 1993. Factors affecting the propagation of locally activated systolic Ca transients in rat ventricular myocytes. Pflügers Archiv. 425, 181–183. ( 10.1007/BF00374521) [DOI] [PubMed] [Google Scholar]
- 32.Stern MD. 1992. Theory of excitation–contraction coupling in cardiac muscle. Biophys. J. 63, 497–517. ( 10.1016/S0006-3495(92)81615-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng H, Lederer WJ, Cannell MB. 1993. Calcium sparks: elementary events underlying excitation–contraction coupling in heart muscle. Science 262, 740–744. ( 10.1126/science.8235594) [DOI] [PubMed] [Google Scholar]
- 34.Cannell MB, Cheng H, Lederer WJ. 1995. The control of calcium release in heart muscle. Science 268, 1045–1049. ( 10.1126/science.7754384) [DOI] [PubMed] [Google Scholar]
- 35.Kupittayanant P, Trafford AW, Diaz ME, Eisner DA. 2006. A mechanism distinct from the L-type Ca current or Na–Ca exchange contributes to Ca entry in rat ventricular myocytes. Cell Calcium 39, 417–423. ( 10.1016/j.ceca.2006.01.011) [DOI] [PubMed] [Google Scholar]
- 36.Reuter H, Seitz N. 1968. The dependence of calcium efflux from cardiac muscle on temperature and external ion composition. J. Physiol. (Lond.) 195, 451–470. ( 10.1113/jphysiol.1968.sp008467) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baker PF, Blaustein MP, Hodgkin AL, Steinhardt RA. 1969. The influence of calcium on sodium efflux in squid giant axons. J. Physiol. (Lond.) 200, 431–458. ( 10.1113/jphysiol.1969.sp008702) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mullins LJ. 1979. The generation of electric currents in cardiac fibers by Na/Ca exchange. Am. J. Physiol. 236, C103–C110. [DOI] [PubMed] [Google Scholar]
- 39.Mullins LJ. 1981. Ion transport in the heart. New York, NY: Raven Press. [Google Scholar]
- 40.Fischmeister R, Vassort G. 1981. The electrogenic Na–Ca exchange and the cardiac electrical activity. I. Stimulation on Purkinje fibre action potential. J. Physiol. 77, 705–709. [PubMed] [Google Scholar]
- 41.Eisner DA, Lederer WJ. 1985. Na–Ca exchange: stoichiometry and electrogenicity. Am. J. Physiol. 248, C189–C202. [DOI] [PubMed] [Google Scholar]
- 42.Kimura J, Noma A, Irisawa H. 1986. Na–Ca exchange current in mammalian heart cells. Nature 319, 596–597. ( 10.1038/319596a0) [DOI] [PubMed] [Google Scholar]
- 43.Mechmann S, Pott L. 1986. Identification of Na–Ca exchange current in single cardiac myocytes. Nature 319, 597–599. ( 10.1038/319597a0) [DOI] [PubMed] [Google Scholar]
- 44.Noma A. 1983. ATP-regulated K+ channels in cardiac muscle. Nature 305, 147–148. ( 10.1038/305147a0) [DOI] [PubMed] [Google Scholar]
- 45.Nilius B, Hess P, Lansman JB, Tsien RW. 1985. A novel type of cardiac calcium channel in ventricular cells. Nature 316, 443–446. ( 10.1038/316443a0) [DOI] [PubMed] [Google Scholar]
- 46.Dixon JE, Shi W, Wang HS, McDonald C, Yu H, Wymore RS, Cohen IS, McKinnon D. 1996. Role of the Kv4.3 K+ channel in ventricular muscle. A molecular correlate for the transient outward current. Circ. Res. 79, 659–668. ( 10.1161/01.RES.79.4.659) [DOI] [PubMed] [Google Scholar]
- 47.Sipido KR, Callewaert G, Carmeliet E. 1993. [Ca2+]i transients and [Ca2+]i dependent chloride current in single purkinje cells from rabbit hearts. J Physiol. (Lond.) 468, 641–667. ( 10.1113/jphysiol.1993.sp019793) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang XD, et al. 2014. Critical roles of a small conductance Ca2+-activated K+ channel (SK3) in the repolarization process of atrial myocytes. Cardiovasc. Res. 101, 317–325. ( 10.1093/cvr/cvt262) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Terentyev D, Rochira JA, Terentyeva R, Roder K, Koren G, Li W. 2014. Sarcoplasmic reticulum Ca2+ release is both necessary and sufficient for SK channel activation in ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 306, H738–H746. ( 10.1152/ajpheart.00621.2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brette F, Orchard C. 2003. T-tubule function in mammalian cardiac myocytes. Circ. Res. 92, 1182–1192. ( 10.1161/01.RES.0000074908.17214.FD) [DOI] [PubMed] [Google Scholar]
- 51.Lederer WJ, Niggli E, Hadley RJ. 1990. Sodium–calcium exchange in excitable cells: fuzzy space. Science 248, 283 ( 10.1126/science.2326638) [DOI] [PubMed] [Google Scholar]
- 52.Cannell MB, Kong CHT, Imtiaz MS, Laver DR. 2013. Control of sarcoplasmic reticulum Ca2+ release by stochastic RyR gating within a 3D model of the cardiac dyad and importance of induction decay for CICR termination. Biophys. J. 104, 2149–2159. ( 10.1016/j.bpj.2013.03.058) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang JH, Saucerman JJ. 2011. Computational models reduce complexity and accelerate insight into cardiac signaling networks. Circ. Res. 108, 85–97. ( 10.1161/CIRCRESAHA.110.223602) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roberts BN, Yang PC, Behrens SB, Moreno JD, Clancy CE. 2012. Computational approaches to understand cardiac electrophysiology and arrhythmias. Am. J. Physiol. Heart Circ. Physiol. 303, H766–H783. ( 10.1152/ajpheart.01081.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hund TJ, Rudy Y. 2004. Rate dependence and regulation of action potential and calcium transient in a canine cardiac ventricular cell model. Circulation 110, 3168–3174. ( 10.1161/01.CIR.0000147231.69595.D3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grandi E, Puglisi JL, Wagner S, Maier LS, Severi S, Bers DM. 2007. Simulation of Ca-calmodulin-dependent protein kinase II on rabbit ventricular myocyte ion currents and action potentials. Biophys. J. 93, 3835–3847. ( 10.1529/biophysj.107.114868) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hashambhoy YL, Greenstein JL, Winslow RL. 2010. Role of CaMKII in RyR leak, EC coupling and action potential duration: a computational model. J. Mol. Cell Cardiol. 49, 617–624. ( 10.1016/j.yjmcc.2010.07.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fink M, et al. 2011. Cardiac cell modelling: observations from the heart of the cardiac physiome project. Progr. Biophys. Mol. Biol. 104, 2–21. ( 10.1016/j.pbiomolbio.2010.03.002) [DOI] [PubMed] [Google Scholar]
- 59.Clayton RH, et al. 2011. Models of cardiac tissue electrophysiology: progress, challenges and open questions. Progr. Biophys. Mol. Biol. 104, 22–48. ( 10.1016/j.pbiomolbio.2010.05.008) [DOI] [PubMed] [Google Scholar]
- 60.Nielsen PM, Le Grice IJ, Smaill BH, Hunter PJ. 1991. Mathematical model of geometry and fibrous structure of the heart. Am. J. Physiol. Heart Circ. Physiol. 260, H1365–H1378. [DOI] [PubMed] [Google Scholar]
- 61.Noble D, Bett G. 1993. Reconstructing the heart: a challenge for integrative physiology. Cardiovasc. Res. 27, 1701–1712. ( 10.1093/cvr/27.10.1701) [DOI] [PubMed] [Google Scholar]
- 62.Holden AV, Zhang H. 1993. Modelling propagation and re-entry in anisotropic and smoothly heterogeneous cardiac tissue. J. Chem. Soc. 89, 2833–2837. [Google Scholar]
- 63.Winslow RL, Kimball AL, Varghese A, Noble D. 1993. Simulating cardiac sinus and atrial network dynamics on the Connection Machine. Physica D 64, 281–298. ( 10.1016/0167-2789(93)90260-8) [DOI] [Google Scholar]
- 64.Aliev RR, Panfilov AV. 1996. Modeling of heart excitation patterns caused by a local inhomogeneity. J. Theor. Biol. 181, 33–40. ( 10.1006/jtbi.1996.0112) [DOI] [PubMed] [Google Scholar]
- 65.McCulloch A, Bassingthwaighte J, Hunter P, Noble D. 1998. Computational biology of the heart: from structure to function. Prog. Biophys. Mol. Biol. 69, 153–155. ( 10.1016/S0079-6107(98)00005-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chilton L, Giles WR, Smith GL. 2007. Evidence of intercellular coupling between co-cultured adult rabbit ventricular myocytes and myofibroblasts. J. Physiol. 583, 225–236. ( 10.1113/jphysiol.2007.135038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bishop MJ, Plank G, Burton RA, Schneider JE, Gavaghan DJ, Grau V, Kohl P. 2010. Development of an anatomically detailed MRI-derived rabbit ventricular model and assessment of its impact on simulations of electrophysiological function. Am. J. Physiol. Heart Circ. Physiol. 298, H699–H718. ( 10.1152/ajpheart.00606.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aslanidi OV, et al. 2013. Application of micro-computed tomography with iodine staining to cardiac imaging, segmentation, and computational model development. Med. Imaging IEEE Trans. 32, 8–17. ( 10.1109/TMI.2012.2209183) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Benson AP, Aslanidi OV, Zhang H, Holden AV. 2008. The canine virtual ventricular wall: a platform for dissecting pharmacological effects on propagation and arrhythmogenesis. Progr. Biophys. Mol. Biol. 96, 187–208. ( 10.1016/j.pbiomolbio.2007.08.002) [DOI] [PubMed] [Google Scholar]
- 70.Butters TD, Aslanidi OV, Zhao J, Smaill B, Zhang H. 2013. A novel computational sheep atria model for the study of atrial fibrillation. Interface Focus 3, 20120067 ( 10.1098/rsfs.2012.0067) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pravdin SF, Dierckx H, Katsnelson LB, Solovyova O, Markhasin VS, Panfilov AV. 2014. Electrical wave propagation in an anisotropic model of the left ventricle based on analytical description of cardiac architecture. PLoS ONE 9, e93617 ( 10.1371/journal.pone.0093617) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bishop MJ, Vigmond EJ, Plank G. 2013. The functional role of electrophysiological heterogeneity in the rabbit ventricle during rapid pacing and arrhythmias. Am. J. Physiol. Heart Circ. Physiol. 304, H1240–H1252. ( 10.1152/ajpheart.00894.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Adeniran I, El HA, Hancox JC, Zhang H. 2012. Proarrhythmia in KCNJ2-linked short QT syndrome: insights from modelling. Cardiovasc. Res. 94, 66–76. ( 10.1093/cvr/cvs082) [DOI] [PubMed] [Google Scholar]
- 74.Gurev V, Constantino J, Rice JJ, Trayanova NA. 2010. Distribution of electromechanical delay in the heart: insights from a three-dimensional electromechanical model. Biophys. J. 99, 745–754. ( 10.1016/j.bpj.2010.05.028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Adeniran I, Hancox J, Zhang H. 2013. In silico investigation of the short QT syndrome, using human ventricle models incorporating electromechanical coupling. Front. Physiol. 4, 166 ( 10.3389/fphys.2013.00166) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Colman MA, Aslanidi OV, Stott J, Holden AV, Zhang H. 2011. Correlation between P-wave morphology and origin of atrial focal tachycardia—insights from realistic models of the human atria and torso. IEEE Trans. Biomed. Eng. 58, 2952–2955. ( 10.1109/TBME.2011.2161305) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hunter P, et al. 2013. A vision and strategy for the virtual physiological human: 2012 update. Interface Focus 3, 20130004 ( 10.1098/rsfs.2013.0004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Winslow RL, Trayanova N, Geman D, Miller MI. 2012. Computational medicine: translating models to clinical care. Sci. Transl. Med. 4, 158rv11 ( 10.1126/scitranslmed.3003528) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Niederer SA, Shetty AK, Plank G, Bostock J, Razavi R, Smith NP, Rinaldi CA. 2012. Biophysical modeling to simulate the response to multisite left ventricular stimulation using a quadripolar pacing lead. Pacing Clin. Electrophysiol. 35, 204–214. ( 10.1111/j.1540-8159.2011.03243.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Relan J, Chinchapatnam P, Sermesant M, Rhode K, Ginks M, Delingette H, Rinaldi CA, Razavi R, Ayache N. 2011. Coupled personalization of cardiac electrophysiology models for prediction of ischaemic ventricular tachycardia. Interface Focus 1, 396–407. ( 10.1098/rsfs.2010.0041) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. 1989. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. N. Engl. J. Med. 321, 406–412. ( 10.1056/NEJM198908103210629) [DOI] [PubMed] [Google Scholar]
- 82.Rosen MR, Hoffman BF. 1973. Mechanisms of action of antiarrhythmic drugs. Circ. Res. 32, 1–8. ( 10.1161/01.RES.32.1.1) [DOI] [PubMed] [Google Scholar]
- 83.Moreno JD, et al. 2011. A computational model to predict the effects of class I anti-arrhythmic drugs on ventricular rhythms. Sci. Transl. Med. 3, 98ra83 ( 10.1126/scitranslmed.3002588) [DOI] [PMC free article] [PubMed] [Google Scholar]