

Abstract

A series of pyrido[3,4-d]azepines that are potent and selective 5-HT2C receptor agonists is disclosed. Compound 7 (PF-04781340) is identified as a suitable lead owing to good 5-HT2C potency, selectivity over 5-HT2B agonism, and in vitro ADME properties commensurate with an orally available and CNS penetrant profile. The synthesis of a novel bicyclic tetrasubstituted pyridine core template is outlined, including rationale to account for the unexpected formation of aminopyridine 13 resulting from an ammonia cascade cyclization.
Keywords: Tetrasubstituted pyridines; pyrido[3,4-d]azepine; 5-HT2C receptor agonist; CNS penetration
Serotonin (5-hydroxytryptamine, 5-HT, 1) acts as a neurotransmitter agonist of at least 14 different receptors classified into seven major families, 5-HT1–7. The 5-HT2 class of GPCR receptors comprises three members 5-HT2A, 5-HT2B, and 5-HT2C. Agonism of 5-HT2C in the CNS has been recognized to have potential for the treatment of obesity, urinary incontinence, psychiatric disorders, and sexual dysfunction.1 However, it has been established that selectivity over agonism of structurally related receptors 5-HT2A and 5-HT2B is required. Poorly selective agonists have been linked to clinical adverse events in humans. These include hallucinations and cardiovascular effects due to 5-HT2A agonism2,3 and chronic cardiac valvulopathy and pulmonary hypertension caused by 5-HT2B agonism.4 Notably the antiobesity treatment Fen-Phen was withdrawn in 1997 for causing irreversible valvulopathy, which has been attributed to chronic 5-HT2B agonism.
The resulting search for selective 5-HT2C agonists identified vabicaserin (2) (SCA-136) as a potential therapy for schizophrenia and lorcaserin (3) (APD-356), which was approved in 2012 as Belviq for treatment of obesity (Figure 1).5 Numerous other preclinical 5-HT2C agonists have also been reported.6−8
Figure 1.

Selected 5-HT2C agonists.
Previously Pfizer disclosed several 5-HT2C agonist series,9−14 including a pyrimidine-fused azepine template that led to the discovery of PF-03246799 (4), which offered good levels of in vitro and in vivo potency.14,15 However, compound 4, despite offering excellent selectivity for 5-HT2C over 5-HT2A, still showed weak but measurable agonism of 5-HT2B at 10 μM in both recombinant cell systems and native human tissue.14 It was later discovered that 4-methylamino substitution 5 could offer an enhancement to 5-HT2C agonist potency and simultaneously offer superior selectivity over 5-HT2B.13 However, these structural changes rendered amino-substituted pyrimidine compound 5 a substrate for multidrug resistance P-glycoprotein (P-gp), identified by a large efflux ratio (ER = 10) as measured using an in vitro transfected MDCK cell line (Figure 2).16 A previous correlation analysis of all compounds tested in this MDCK-MDR1 assay concluded that compounds with efflux ratios of <2.5 are unlikely to be significantly effluxed from the CNS by P-gp, whereas compounds with ratios >3.0 are at significant risk of exhibiting appreciable CNS impairment.16 In line with this result, preclinical in vivo efficacy studies of compound 5 showed prohibitive levels of CNS restriction limiting therapeutic efficacy even at high plasma concentrations.16
Figure 2.
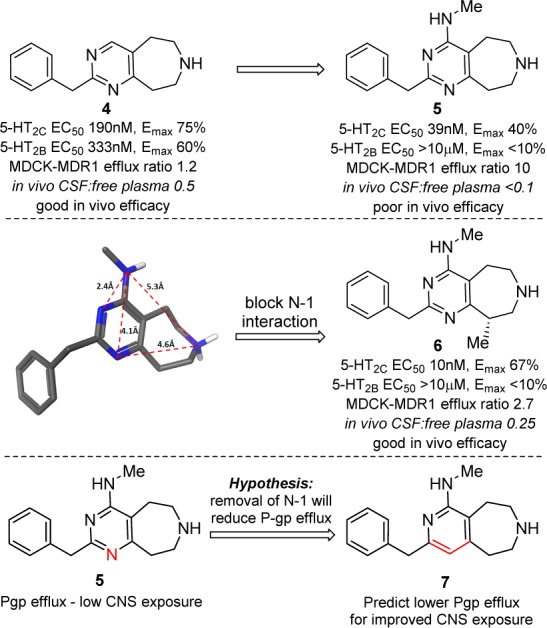
P-gp efflux and CNS exposure.
To retain the high 5-HT2C potency and selectivity of compound 5 but with improved CNS penetration, compounds were sought to provide reduced P-gp efflux. Literature pharmacophore models for P-gp have highlighted the role of aromatic hydrophobic interactions and intramolecular hydrogen bond Acc–Acc distances of ∼2.5 and ∼4.6 Å as P-gp recognition features.17 As illustrated in Figure 2, compound 5 has Acc–Acc distances of 2.4, 4.1, and 4.6 Å suggesting close similarity to this P-gp pharmacophore pattern of hydrogen bonds.18,19
This pointed to N-1 in compound 5 being potentially instrumental to P-gp recognition when combined with a 4-amino substituent. Furthermore, SAR from related templates suggested that the N-1 interaction would not be required for 5-HT2C activity. To test this hypothesis, several compounds were designed to reduce the propensity for N-1 to interact with P-gp. This led to compounds such as chiral methyl azepine compound 6 that retained good 5-HT2C potency, selectivity, and reduced P-gp efflux (ER = 2.7) that translated to improved in vivo efficacy.13,15 It was further proposed that removing N-1 altogether, to give fused aminopyridine azepine 7, would offer good 5-HT2C agonist potency without significant P-gp efflux liability.
The controlled syntheses of tri- and tetrasubstituted pyridines, despite their favorable characteristics and popularity within medicinal chemistry, present formidable challenges. Preferred synthetic methods typically comprise the selective functionalization of a pre-existing pyridine ring or de novo ring synthesis. However, in this instance, the need for a fused bicyclic tetrasubstituted pyridine meant that most known methods were not compatible owing to either not supporting fused ring construction or providing the wrong substitution pattern.20 As a result, it was necessary to develop suitable chemistry to access amino-pyridine fused azepine template 7. A route was proposed based on limited precedent for biaryl ring synthesis via ammonia cyclization of an alkyne 8 to give isoquinolone 9 (Scheme 1).21,22
Scheme 1.

Reagents and conditions: (a) Tf2O, NaOtBu, CH2Cl2, 23 °C, 0.5 h, then Tf2O, 23 °C, 2 h; (b) BnC≡CH, DIPEA, CuI, Pd(PPh3)2Cl2, DMF, 23 °C, 2 h; (c) NH3, MeOH, 80 °C, 15 h.
Carboxybenzyl protected azepine β-ketoester 10 was converted to corresponding vinyl triflate 11 in 81% yield by treatment with triflic anhydride under basic conditions (Scheme 1). Sonogashira coupling with benzylacetylene then provided the desired yne-ene-ester 12 in preparation for the key cascade cyclization to the corresponding pyridinone. Treatment of 12 with excess ammonia in methanol at 80 °C led to conversion of starting material to a single product. Rather than being the anticipated pyridinone, the product was instead determined to be aminopyridine 13.
This unexpected result was repeated to provide gram quantities of aminopyridine 13, and a sample was crystallized from CD3OD, enabling an X-ray structure to be obtained to further confirm structure assignment (CCDC 1024393 and Supporting Information).
In order to discount a metal-mediated reaction,23 the ammonia cyclization was also carried out using yne-ene-ester 12 that had been pretreated overnight with various metal scavenger resins (QPTU, QMTU, QSMP; 1 g of resin per 0.25 mmol of 12). However, these pretreatments did not alter yield or product distribution of the cyclization.
To investigate the mechanism of the cyclization cascade and further establish the general applicability of this reaction, several related alkyne systems were tested under the same reaction conditions (Table 1).
Table 1. Ammonia-Mediated Cyclizations.
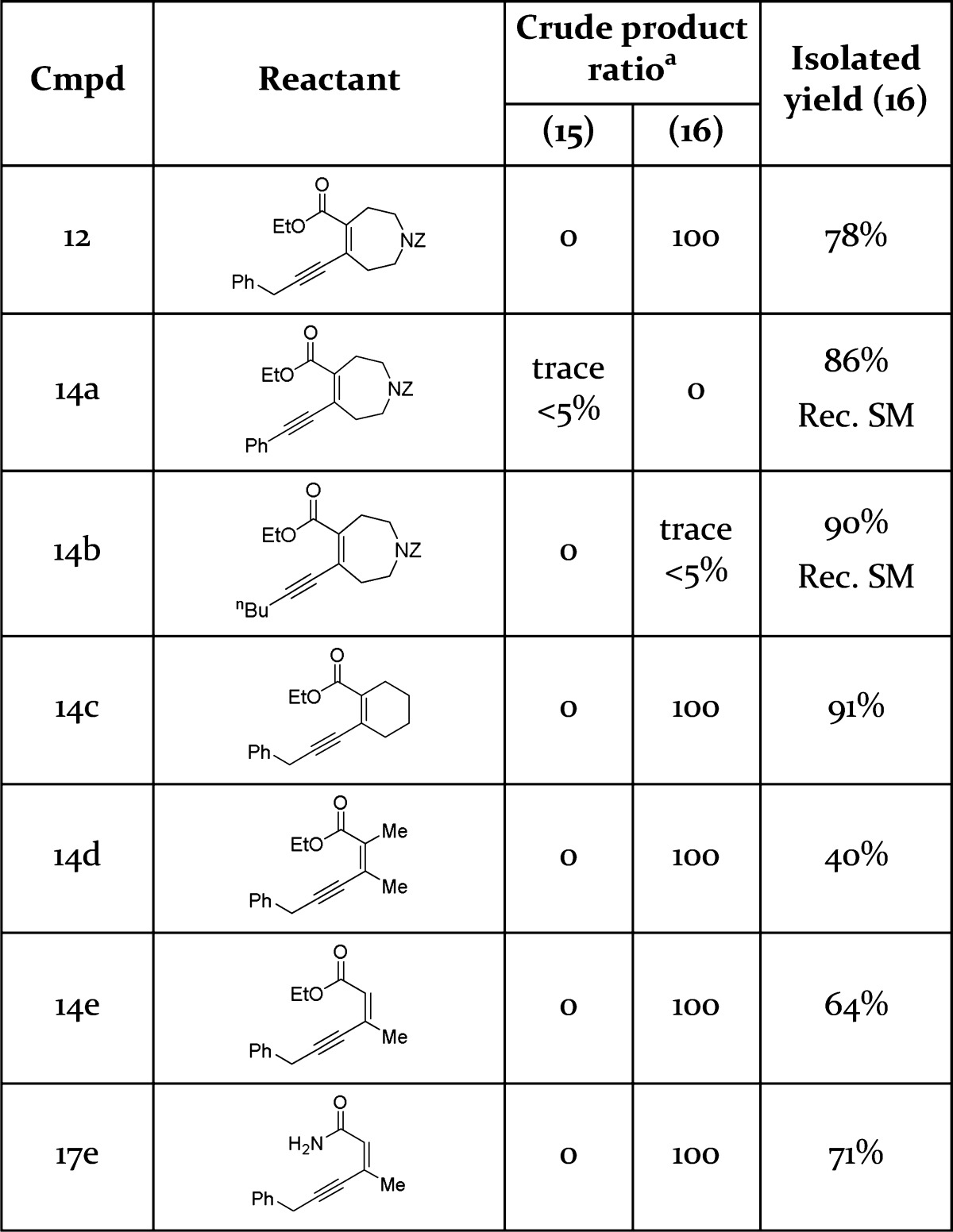
Product ratio determined by crude 1H NMR integration. Rec. SM dentoes yield of recovered starting material
Interestingly if R1 = Bn was replaced by R1 = Ph 14a or R1 = nBu 14b, then the reaction did not proceed, instead returning mostly unreacted starting material. However, when the cyclization reactant contained a benzylic R1 and aliphatic R2 and R3 (12, 14c-e), then cyclization proceeded to consistently give the corresponding aminopyridines 13 and 16c–e in good yields. To rationalize these results it is proposed that systems where R1 = Bn 14ii undergo rapid rearrangement to allenes on treatment with ammonia, driven by extended conjugative stabilization of the allene with the Bn aromatic ring (Scheme 2). The allene system likely reacts with excess ammonia to form primary amide 18, either directly or via transient cyclization of the ester carbonyl to form an activated electrophilic oxonium. Amide 18 then cyclizes onto the allene via a 6-exo-dig ring closure preferentially through oxygen due to superior orbital overlap versus the nitrogen with the exoallene π* orbital to form a reactive hemiaminal. An ammonia mediated ring opening to form keto-amidine 19 is then followed by a 6-exo-trig closure to provide the product aminopyridine 16ii. Further support for this mechanism comes from the reaction of preformed primary amide 17e with ammonia to successfully provide aminopyridine 16e, suggesting amide 17e to be an intermediate on the reaction cascade.
Scheme 2.

In contrast, aromatic alkyne-ester 14f, under identical reaction conditions, provided pyridinone 15f exclusively, with no evidence for formation of the aminopyridine 16f. However, if preformed primary amide 17f was exposed to the reaction conditions, the anticipated pyridinone product did not form, resulting in a mixture favoring aminopyridine 16f. This suggests that an alternative mechanistic pathway predominates for substrate 14f (Scheme 2).
It is postulated that in this case the ammonia undergoes nucleophilic conjugate addition to the alkyne, as opposed to facilitating allene formation, followed by 6-exo-trig ring closure to directly give pyridinone 15f. However, if primary amide 17f is preformed, this would necessitate 6-endo-dig closure to give pyridinone 15f, for which orbital overlap is suboptimal, rationalizing the observed mixture of pyridinone 15f and aminopyridine 16f products. Furthermore, when pyridinone 15f was treated with ammonia under the same reaction conditions, no reaction occurred, ruling out the formation of 16f via 15f.
Aminopyridine 13 proved to be a versatile intermediate (Scheme 3). Reductive amination with aldehydes yielded monoalkylated products 21a–f in moderate to good yields. Also, alkylation using iodomethane provided dimethylated compound 21g. Finally, the application of Sandmeyer conditions enabled conversion of aminopyridine 13 to chloropyridine 20h. The chlorine was then reduced to give trisubstituted pyridine 21h (Scheme 3). Compounds 7 and 21a–h were investigated for their ability to inhibit the binding of a Cy3B conjugated analogue of serotonin to human 5-HT2C receptor utilizing fluorescence polarization technology and cellular membrane preparations generated from recombinant Swiss 3T3 cells (Table 2, Ki values).24
Scheme 3.

Reagents and conditions: (a) aldehyde or ketone, DCE, AcOH, 23 °C, 30 min, then PS-BH3CN, 55 °C, 18–40 h; (b) Pd/C, H2, EtOH, 45 psi, 23 °C, 3–24 h; (c) MeI, K2CO3, DMF, 80 °C, 22 h; (d) CuCl2, amyl nitrite, DCE, 65 °C, 13 h; (e) Pd/C, H2, EtOH, 23 °C, 4 h.
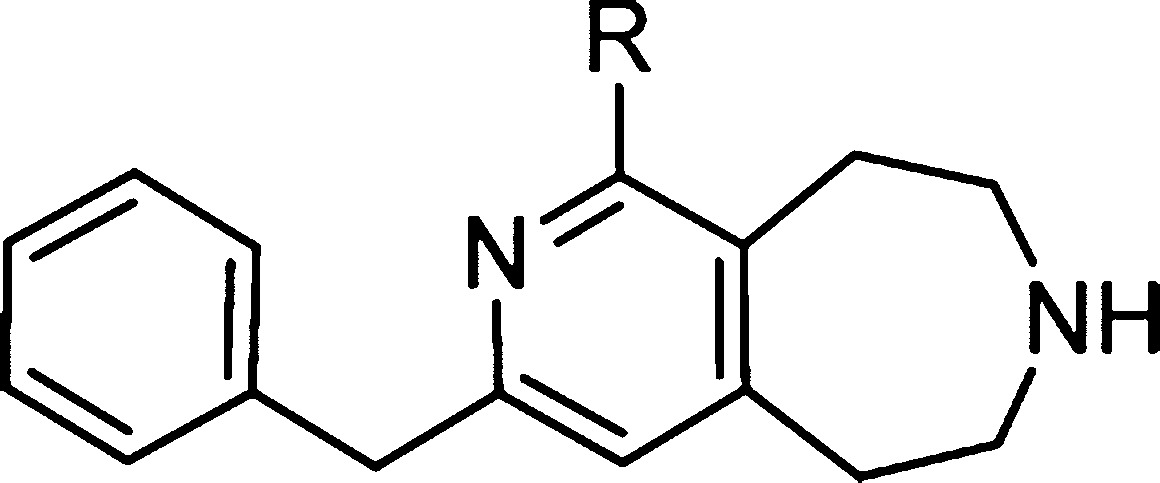
Table 2. 5-HT2C & 5-HT2B Activity, Microsomal Stability and in vitro Permeability Data for Compounds.
| 5-HT2C |
5-HT2B |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | R | logD | EC50 (nM)a | Emaxa,b | Ki (nM)a | EC50 (nM)a | Emaxa,c | Kb (nM)a | HLM Clint (mL min–1 mg–1) | RRCK (×10–6 cm/s) | MDCK-MDR1 AB Papp (×10–6 cm/s) | MDCK-MDR1 ER (BA/AB) |
| 7 | NHMe | 0.4 | 9 | 99% | 3 | 1484 | 69% | 19 | 8 | 2.5 | 2.8 | |
| 21a | NHEt | 0.6 | 11 | 79% | 3 | 18 | 28% | 13 | 7 | 1.8 | 2.3 | |
| 21b | NHCH2iPr | 1.7 | 36 | 95% | 12 | nt | nt | nt | nt | nt | nt | |
| 21c | NHCH2cPr | 1.2 | 21 | 100% | 0.5 | 22 | 51% | 27 | 2 | 0.8 | 6.1 | |
| 21d | NHnPr | 1.0 | nt | nt | 4 | 27 | 38% | 11 | 4 | 1.5 | 3.5 | |
| 21e | NHiPr | 1.0 | nt | nt | 13 | 33 | 32% | nt | 12 | nt | nt | |
| 21f | NHBn | 1.7 | 158 | 37% | 22 | 121 | 35 | 2 | 0.6 | 3.6 | ||
| 21g | NMe2 | 0.6 | nt | nt | 12 | 30 | 38% | 43 | 8 | 2.1 | 2.2 | |
| 21h | H | 0.5 | nt | nt | 35 | 27 | 53% | <8 | 18 | 3.9 | 1.5 | |
Values are geometric means of up to five experiments. Differences of <2-fold should not be considered significant.
Percent activation by maximum asymptote at 10 μM relative to 5-HT.
Percent activation by maximum asymptote at 30 μM relative to 5-HT; nt denotes not tested.
The 5-HT2C and 5-HT2B functional agonist activities of selected compounds were evaluated relative to 5-HT (1) by measuring ability to induce G-protein activation via recruitment of GTPγS and mobilization of intracellular calcium for 5-HT2C and 5-HT2B, respectively (Table 2, EC50 and Emax).13,24 Previous studies within Pfizer have shown compound Ki at the 5-HT2C receptor to be the most predictive indicator of free brain exposure required to elicit 5-HT2C related pharmacological effects in vivo(25) (see SI for cell culture and assay protocols).
Compounds 7 and 21a–h exhibited excellent 5-HT2C binding potency and agonist efficacy (Table 2). Varying the 2-amino substituent sampled a range of molecular weight and lipophilicity. However, despite larger and more lipophilic substituents being generally well tolerated, they appeared less ligand and lipophilic efficient, providing no appreciable improvements in 5-HT2C potency. Furthermore, although this series generally showed similar levels of 5-HT2B potency (EC50), the compounds were either weak partial agonists at 5-HT2B, characterized by low Emax values, or showed antagonism (compound 21f). Overall, all compounds also tended to exhibit good metabolic stability in human liver microsomes (HLM) and moderate to good passive permeability in RRCK cells.
Methylamino-substituted pyridine compound 7 looked the most promising on balance of physicochemistry, potency, selectivity, and metabolic stability. In accordance with the original design hypothesis, compound 7 also exhibited a low efflux ratio in the MDCK-MDR1 P-gp assay (P-gp ER = 2.8), a pronounced improvement over the equivalent pyrimidine compound 5 (P-gp ER = 10). This level of P-gp efflux (ER = 2.8) correlates well with other examples from the broader azepine series such as pyrimidine compound 6 (ER = 2.7) that previously achieved good CNS exposure and efficacy in preclinical in vivo studies.13
In summary, the rational design and synthesis of a series of pyridine-fused azepines with potent 5-HT2C agonist activity and low P-gp efflux ratios has been described to deliver lead compound 7 (PF-04781340). Chemistry was developed and rationalized to access this template, including an ammonia-mediated cascade synthesis of aminopyridine 13. These methods have also been extended to the synthesis of polysubstituted and fused bicyclic aminopyridines, illustrating potential for broader application.
Acknowledgments
We thank the Primary Pharmacology Group for screening data, Jianmin Sun for spectra, Dr. J. E. Davies for X-ray crystallography, and Asser Bassyouni for 5-HT2B selectivity data. We are grateful to the EPSRC (SVL, grant n° EP/K0099494/1 and n° EP/K039520/1) for financial support.
Glossary
Abbreviations
- CCDC
Cambridge Crystallographic Data Center
- CNS
central nervous system
- Cy3B
cyanine dye 3B
- DCE
1,2-dichloroethane
- DIPEA
N,N-diisopropylethylamine
- DMF
dimethylformamide
- HLM
human liver microsomes
- MDCK
Madin–Darby canine kidney
- MDR1
multidrug resistance gene
- P-gp
P-glycoprotein
- RRCK
Ralph Russ canine kidney cell line
- SM
starting material
- Z
carboxybenzyl
Supporting Information Available
Experimental procedures and 1H NMR and 13C NMR spectra of compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# Pfizer Neusentis, The Portway Building, Granta Park, Cambridge, CB21 6GS, U.K.
The authors declare no competing financial interest.
Supplementary Material
References
- Wacker D. A.; Miller K. J. Agonists of the serotonin 5-HT2C receptor: preclinical and clinical progression in multiple diseases. Curr. Opin. Drug Discovery Dev. 2008, 11, 438–445. [PubMed] [Google Scholar]
- Nichols D. Hallucinogens. Pharmacol. Ther. 2004, 101, 131–181. [DOI] [PubMed] [Google Scholar]
- Villalon C. M.; Centurion D. Cardiovascular responses produced by 5-hydroxytriptamine: a pharmacological update on the receptors/mechanisms involved and therapeutic implications. Naunyn-Schmiedeberg's Arch. Pharmacol. 2007, 376, 45–63. [DOI] [PubMed] [Google Scholar]
- Roth B. L. Drugs and valvular heart disease. N. Engl. J. Med. 2007, 356, 6–9. [DOI] [PubMed] [Google Scholar]
- Fleming J. W.; McClendon K. S.; Riche D. M. New obesity agents: lorcaserin and phentermine/topiramate. Ann. Pharmacother. 2013, 47, 1007–1016. [DOI] [PubMed] [Google Scholar]
- Monck N. J. T.; Kennett G. A. 5-HT2C ligands: recent progress. Prog. Med. Chem. 2008, 46, 281–390. [DOI] [PubMed] [Google Scholar]
- Yang H. Y.; Tae J.; Seo Y. W.; Kim Y. J.; Im H. Y.; Choi G. D.; Cho H.; Park W.-K.; Kwon O. S.; Cho Y. S.; Ko M.; Jang H.; Lee J.; Choi K.; Kim C.-H.; Lee J.; Pae A. N. Novel pyrimidoazepine analogs as serotonin 5-HT2A and 5-HT2C receptor ligands for the treatment of obesity. Eur. J. Med. Chem. 2013, 63, 558–569. [DOI] [PubMed] [Google Scholar]
- Sargent B. J.; Henderson A. J. Targeting 5-HT receptors for the treatment of obesity. Curr. Opin. Pharmacol. 2011, 11, 52–58. [DOI] [PubMed] [Google Scholar]
- Brennan P. E.; Whitlock G. A.; Ho D. K. H.; Conlon K.; McMurray G. Discovery of a novel azepine series of potent and selective 5-HT2C agonists as potential treatments for urinary incontinence. Bioorg. Med. Chem. Lett. 2009, 19, 4999–5003. [DOI] [PubMed] [Google Scholar]
- Fish P. V.; Brown A. D.; Evrard E.; Roberts L. R. 7-Sulfonamido-3-benzazepines as potent and selective 5-HT2C receptor agonists: Hit-to-lead optimization. Bioorg. Med. Chem. Lett. 2009, 19, 1871–1875. [DOI] [PubMed] [Google Scholar]
- Liu K. K. C.; Cornelius P.; Patterson T. A.; Zeng Y.; Santucci S.; Tomlinson E.; Gibbons C.; Maurer T. S.; Marala R.; Brown J.; Kong J. X.; Lee E.; Werner W.; Wenzel Z.; Vage C. Design and synthesis of orally-active and selective azaindane 5-HT2C agonist for the treatment of obesity. Bioorg. Med. Chem. Lett. 2010, 20, 266–271. [DOI] [PubMed] [Google Scholar]
- Liu K. K. C.; Lefker B. A.; Dombroski M. A.; Chiang P.; Cornelius P.; Patterson T. A.; Zeng Y.; Santucci S.; Tomlinson E.; Gibbons C. P.; Marala R.; Brown J. A.; Kong J. X.; Lee E.; Werner W.; Wenzel Z.; Giragossian C.; Chen H.; Coffey S. B. Orally active and brain permeable proline amides as highly selective 5-HT2C agonists for the treatment of obesity. Bioorg. Med. Chem. Lett. 2010, 20, 2365–2369. [DOI] [PubMed] [Google Scholar]
- Storer R. I.; Brennan P. E.; Brown A. D.; Bungay P. J.; Conlon K. M.; Corbett M. S.; DePianta R. P.; Fish P. V.; Heifetz A.; Ho D. K. H.; Jessiman A. S.; McMurray G.; de Oliveira C. A. F.; Roberts L. R.; Root J. A.; Shanmugasundaram V.; Shapiro M. J.; Sherten M.; Westbrook D.; Wheeler S.; Whitlock G. A.; Wright J. Multiparameter optimization in CNS drug discovery: design of pyrimido[4,5-d]azepines as potent 5-HT2C receptor agonists with exquisite functional selectivity over 5-HT2A and 5-HT2B receptors. J. Med. Chem. 2014, 57, 5258–5269. [DOI] [PubMed] [Google Scholar]
- Andrews M. D.; Fish P. V.; Blagg J.; Brabham T. K.; Brennan P. E.; Bridgeland A.; Brown A. D.; Bungay P. J.; Conlon K. M.; Edmunds N. J.; af Forselles K.; Gibbons C. P.; Green M. P.; Hanton G.; Holbrook M.; Jessiman A. S.; McIntosh K.; McMurray G.; Nichols C. L.; Root J. A.; Storer R. I.; Sutton M. R.; Ward R. V.; Westbrook D.; Whitlock G. A. Pyrimido[4,5-d]azepines as potent and selective 5-HT2C receptor agonists: Design, synthesis, and evaluation of PF-3246799 as a treatment for urinary incontinence. Bioorg. Med. Chem. Lett. 2011, 21, 2715–2720. [DOI] [PubMed] [Google Scholar]
- Compound 4 (PF-3246799, Catalog No. PZ0229) and compound 6 (PF-4479745, Catalog No. PZ0032) are available from Sigma Aldrich.
- Feng B.; Mills J. B.; Davidson R. E.; Mireles R. J.; Janiszewski J. S.; Troutman M. D.; de Morais S. M. In vitro P-gp assays to predict in vivo interactions of P-gp with drugs in the central nervous system. Drug Metab. Dispos. 2008, 36, 268–275. [DOI] [PubMed] [Google Scholar]
- Seelig A. Toward understanding P-gp structure-activity relationships. Methods Princ. Med. Chem. 2009, 40, 497–519. [Google Scholar]
- Penzotti J. E.; Lamb M. L.; Evensen E.; Grootenhuis P. D. J. A computational ensemble pharmacophore model for identifying substrates of P-gp. J. Med. Chem. 2002, 45, 1737–1740. [DOI] [PubMed] [Google Scholar]
- Ferreira R. J.; dos Santos D. J. V. A.; Ferreira M.-J. U.; Guedes R. C. Toward a Better Pharmacophore Description of P-Glycoprotein Modulators, Based on Macrocyclic Diterpenes from Euphorbia Species. J. Chem. Inf. Model. 2011, 51, 1315–1324. [DOI] [PubMed] [Google Scholar]
- Donohoe T. J.; Bower J. F.; Chan L. K. M. Olefin cross-metathesis for the synthesis of heteroaromatic compounds. Org. Biomol. Chem. 2012, 10, 1322–1328. [DOI] [PubMed] [Google Scholar]
- Sakamoto T.; Kondo Y.; Yamanaka H. Studies on pyrimidine derivatives. XXVIII. Synthesis of pyridopyrimidine derivatives by cross-coupling of halopyrimidines with olefins and acetylenes. Chem. Pharm. Bull. 1982, 30, 2410–2416. [Google Scholar]
- Sakamoto T.; Kondo Y.; Yamanaka H. Condensed heteroaromatic ring systems. III. Synthesis of naphthyridine derivatives by cyclization of ethynylpyridinecarboxamides. Chem. Pharm. Bull. 1985, 33, 626–633. [Google Scholar]
- Long Y.; She Z.; Liu X.; Chen Y. Synthesis of 1-Aminoisoquinolines by Gold(III)-Mediated Domino Reactions from 2-Alkynylbenzamides and Ammonium Acetate. J. Org. Chem. 2013, 78, 2579–2588. [DOI] [PubMed] [Google Scholar]
- Cornelius P.; Lee E.; Lin W.; Wang R.; Werner W.; Brown J. A.; Stuhmeier F.; Boyd J. G.; McClure K. Design, synthesis, and pharmacology of fluorescent-labeled analogs of serotonin: application to screening of the 5-HT2C receptor. J. Biomol. Screening 2009, 14, 360–370. [DOI] [PubMed] [Google Scholar]
- Liu K. K. C.; Cornelius P.; Patterson T. A.; Zeng Y.; Santucci S.; Tomlinson E.; Gibbons C.; Maurer T. S.; Marala R.; Brown J.; Kong J. X.; Lee E.; Werner W.; Wenzel Z.; Vage C. Design and synthesis of orally-active and selective azaindane 5-HT2C agonist for the treatment of obesity. Bioorg. Med. Chem. Lett. 2010, 20, 266–271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.