

Abstract
Farnesyl diphosphate synthase (FPPS) is an important drug target for bone resorption, cancer, and some infectious diseases. Here, we report five new structures including two having unique bound ligand geometries. The diamidine inhibitor 7 binds to human FPPS close to the homoallylic (S2) and allosteric (S3) sites and extends into a new site, here called S4. With the bisphosphonate inhibitor 8, two molecules bind to Trypanosoma brucei FPPS, one molecule in the allylic site (S1) and the other close to S2, the first observation of two bisphosphonate molecules bound to FPPS. We also report the structures of apo-FPPS from T. brucei, together with two more bisphosphonate-bound structures (2,9), for purposes of comparison. The diamidine structure is of particular interest because 7 could represent a new lead for lipophilic FPPS inhibitors, while 8 has low micromolar activity against T. brucei, the causative agent of human African trypanosomiasis.
Keywords: Farnesyl diphosphate synthase, inhibitor, T. brucei, diamidine
The bisphosphonate class of drugs1 such as alendronate (1), risedronate (2), ibandronate (3), and zoledronate (4) that are used to treat bone resorption diseases (osteoporosis, hypercalcemia due to malignancy, and Paget’s disease) function primarily by targeting the enzyme farnesyl diphosphate synthase (FPPS) in osteoclasts.2,3 They also have other interesting effects including inhibiting tumor cell invasiveness;4 activating gamma delta T-cells (containing the Vγ2 Vδ2 T-cell receptor) to kill tumor cells and bacteria;5 they switch tumor-associated macrophages from a tumor promoting (M2) to a tumor-inhibiting (M1) phenotype;6 and they have activity as antiparasitics.7,8 There is, therefore, interest in the further development of FPPS inhibitors for use against a broad range of diseases because farnesyl diphosphate (FPP) plays a key role in many different biosynthetic pathways including protein prenylation; glycosylation, bacterial cell wall, quinone, and heme a biosynthesis; as well as in formation of several bacterial virulence factors.9−12 This interest is reflected in, for example, the recent development of new classes of FPPS inhibitors13,14 including compounds such as 5 and 6, these more hydrophobic species being of interest as potential anticancer drug leads that target soft tissues.13
Here, we report the results of an investigation into the structures of two FPPS inhibitors that we find have unusual binding modes, not reported previously: the diamidine inhibitor 7 (BPH-1358; NSC50460; N-[3-(4,5-dihydro-1H-imidazol-2-yl)phenyl]-4-[4-[[3-(4,5-dihydro-1H-imidazol-2-yl)phenyl]carbamoyl]phenyl]benzamide dihydrochloride), bound to human FPPS; and the bisphosphonate inhibitor 8 (BPH-6; NE-58051; 1-hydroxy-1-phosphono-3-(pyridin-3-yl)-propyl phosphonic acid), bound to Trypanosoma brucei FPPS. We also report the structures of risedronate 2 and the aminomethylene bisphosphonate 9, bound to T. brucei FPPS (TbFPPS), as well as that of apo TbFPPS, for purposes of comparison. The diamidine 7 is of interest since it has quite potent activity against human FPPS and is hydrophobic, enabling soft tissue penetration, and is not expected to bind to bone mineral. The bisphosphonate 8 is of interest because it has been the topic of a computational investigation into the mechanism of FPPS inhibition;15 it has activity against the trypanosomatid parasite Trypanosoma brucei,16 and we report here that, as with 7, 8 has an unusual FPPS binding mode.
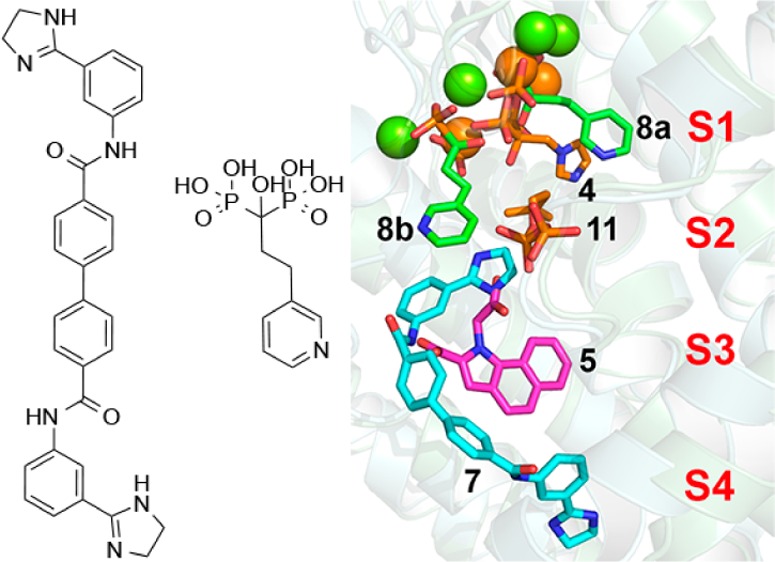
The reactions catalyzed by FPPS involve the sequential condensation of the allylic species dimethylallyl diphosphate (DMAPP, 10) with isopentenyl diphosphate (IPP, 11) to form geranyl diphosphate (GPP, 12) and thence farnesyl diphosphate (FPP, 13), Figure 1. Carbocations are transition state/reactive intermediates,17 and the side-chains in the most potent bisphosphonate FPPS inhibitors have been proposed to act as carbocation isosteres,2,9 while the bisphosphonate groups themselves act as diphosphate isosteres. Bisphosphonate inhibitors have all been found to bind to the allylic site S1, as shown in Figure 2, with the two phosphonate groups interacting with a [Mg2+]3 cluster18 in site S1, the Mg2+ in turn being bound to the protein via a cluster of highly conserved Asp residues (a so-called DDXXD motif). The homoallylic substrate 11 binds to a second site, S2, with the diphosphate group interacting with Arg/Lys residues. Much larger inhibitors (e.g., the diterpene quinone methide, taxodione, and another quinone methide, arenarone) have also recently been found19 to bind to the S2 site. A third site, S3, is occupied by the “allosteric” inhibitors developed by Novartis.13 These are very potent FPPS inhibitors, developed to target tumors in soft tissue,13 and are located below the S2 site, in S3, as shown in Figure 2.
Figure 1.

Structures of FPPS inhibitors, substrates, the GPP intermediate, and the FPP product.
Figure 2.
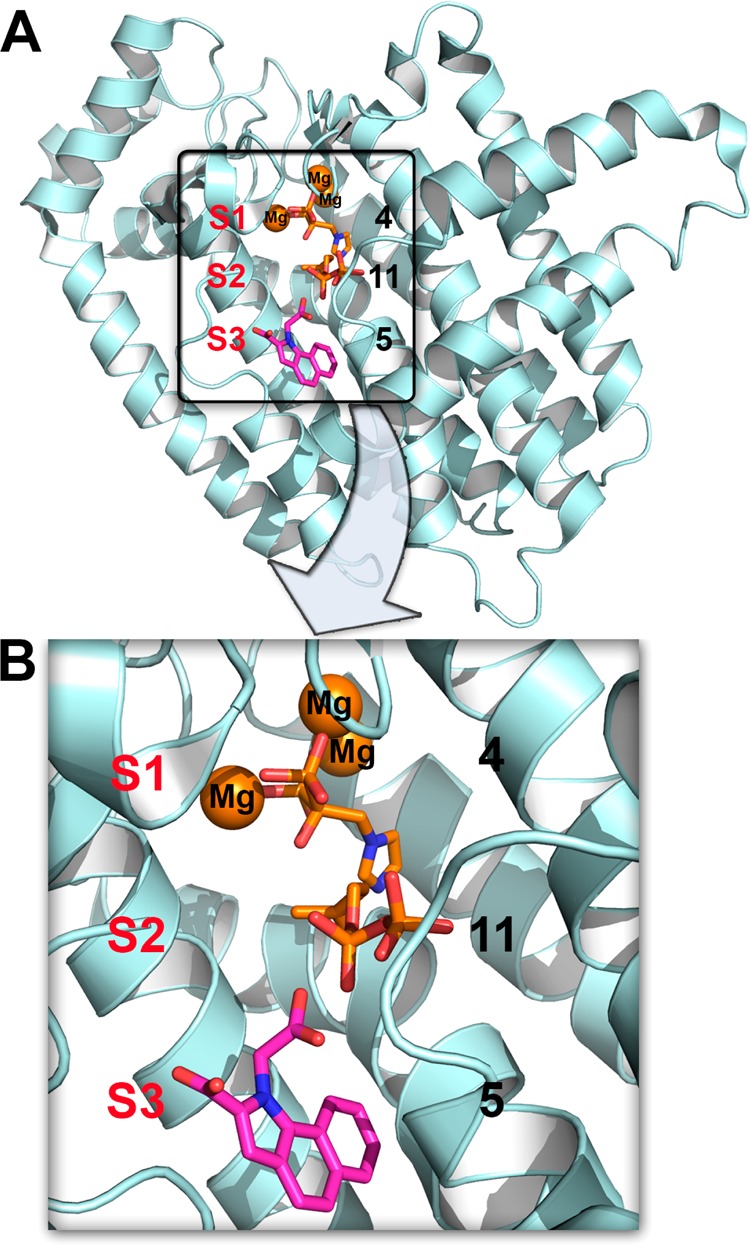
FPPS structures. The structures of 4 + 11 (PDB ID code 2F8Z) and 5 (PDB ID code 3N6K) are shown superimposed. Compound 4 binds in S1, 11 binds in S2, and 5 binds in S3.
We first determined the structure of the FPPS inhibitor 7, which is of interest since it has an IC50 = 1.8 μM against human FPPS20 under assay conditions where the IC50 for the potent FPPS inhibitor 4 (zoledronate) is 0.2 μM.20 Since it is also quite lipophilic (clogP = 4.3), 7 could be a new lead for anticancer therapeutics (e.g., against Ras-bearing tumors).21
We expressed, purified, and crystallized human FPPS as described previously19 and obtained crystals with 7 by cocrystallization. Full sample preparation, data acquisition, and data processing details are given in the Supporting Information and Table 1. The basic structure of FPPS is now well-known,22−25 and as noted above, there are three main inhibitor binding sites, S1–S3, Figure 2. As can be seen in Figure 3A, with the diamidine inhibitor 7, we find that about one-half of the molecule (three rings) binds to sites S2/S3, while the second half of the molecule (again, three rings) binds to a new site, called here S4, that is outside the S1–S3 site regions reported previously with all other FPPS inhibitors. The 2Fo–Fc electron density is shown contoured at 1σ in Figure 3A, and all rings are well resolved. The discovery map used for initial inhibitor refinement is shown in Figure 3B. Specific ligand–protein interactions are shown in the ligand interaction diagram26 in Figure 3C and indicate the presence of electrostatic, hydrogen bonding, cation–pi, and hydrophobic (Supporting Information Figure S1A) interactions. In addition, as can be seen in Supporting Information Figure S1B, the central biphenyl ring is quite solvent-exposed. This structure thus represents a new FPPS inhibitor binding mode and may be of interest in the development of more potent compounds.
Table 1. Data Collection and Refinement Statistics.
| crystal (PDB ID) | HsFPPS·7 (4RXA) | TbFPPS (4RYP) | TbFPPS·8 (4RXC) | TbFPPS·2 (4RXD) | TbFPPS·9 (4RXE) |
|---|---|---|---|---|---|
| Data Collection | |||||
| radiation source | 21-ID-G | 21-ID-G | 21-ID-G | 21-ID-G | 21-ID-G |
| wavelength (Å) | 0.97857 | 0.97857 | 0.97857 | 0.97857 | 0.97857 |
| space group | P41212 | C2 | C2 | C2 | C2 |
| a (Å) | 110.83 | 134.34 | 133.01 | 133.21 | 134.15 |
| b (Å) | 110.83 | 119,439 | 120.92 | 119.55 | 119.15 |
| c (Å) | 77.95 | 62.04 | 63.34 | 62.44 | 62.82 |
| β (deg) | 90 | 117.18 | 117.18 | 111.93 | 112.24 |
| resolution (Å)a | 50.0–2.20 | 50.0–2.20 | 50.0–2.30 | 50.0–2.30 | 50.0–2.30 |
| (2.24–2.20) | (2.28–2.20) | (2.38–2.30) | (2.38–2.30) | (2.38–2.30) | |
| no. of reflections | 23767 (1173) | 42016 (3334) | 37381 (3082) | 41878 (2595) | 49429 (5184) |
| completeness (%) | 94.4 (95.8) | 96.4 (76.7) | 97.1 (80.4) | 91.0 (56.5) | 94.2 (98.4) |
| Rmerge (%) | 8.2 (75.3) | 9.6 (21.7) | 12.9 (28.9) | 7.3 (42.4) | 7.3 (57.6) |
| I/s(I) | 30.8 (3.1) | 20.9 (4.4) | 16.1 (3.0) | 10.1 (3.3) | 23.9 (3.1) |
| Refinement | |||||
| resolution (Å)a | 50.0–2.20 | 30.0–2.20 | 50–2.30 | 50.0–2.30 | 50.0–2.50 |
| (2.25–2.20) | (2.28–2.20) | (2.38–2.30) | (2.38–2.30) | (2.57–2.50) | |
| Rwork (%) | 22.5 (34.3) | 18.0 (22.8) | 20.0 (24.7) | 21.1 (23.7) | 19.7 (23.0) |
| Rfree (%) | 25.9(34.6) | 24.1 (27.4) | 25.4 (27.4) | 25.1 (32.0) | 25.5 (33.4) |
| Geometry Deviations | |||||
| bond lengths (Å) | 0.018 | 0.016 | 0.002 | 0.010 | 0.015 |
| bond angles (deg) | 0.881 | 1.669 | 0.684 | 1.203 | 1.826 |
| Mean B-Values (Å2)/Number of Non-H Atoms | |||||
| protein atoms | 33.8/2659 | 52.2/5596 | 43.0/5639 | 28.3/8549 | 48.7/5737 |
| compound atoms | 63.4/40 | 51.4/72 | 18.1/51 | 34.4/34 | |
| PO4 ions | 65.4/5 | ||||
| Mg ions | 46.4/8 | 19.1/9 | 41.0/6 | ||
| water | 52.3/104 | 48.5/176 | 42.9/153 | 32.8/484 | 41.7/158 |
| Ramachandran Plot (%) | |||||
| most favored | 91.8 | 91.9 | 92.2 | 93.6 | 90.9 |
| additionally allowed | 7.9 | 7.8 | 7.8 | 6.1 | 9.1 |
| generously allowed | 0 | 0.3 | 0 | 0 | 0 |
| disallowed | 0 | 0 | 0 | 0 | 0 |
Values in parentheses represent the highest resolution shell.
Figure 3.
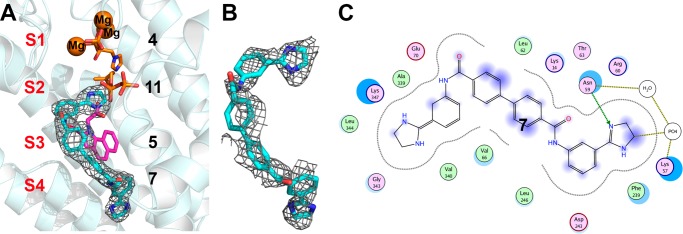
Structure of the diamidine 7 bound to HsFPPS (PDB ID code 4RXA). (A) Compound 7 contoured at 1σ, superimposed on 4, 5, and 11 (PDB ID codes 2F8Z and 3N6K) bound to HsFPPS. (B) Discovery map used for initial inhibitor refinement. (C) Ligand interaction diagram for 7 computed by using MOE.26
Next, we investigated the binding of the bisphosphonate 8 to TbFPPS. TbFPPS is a homologue of HsFPPS with 44% identity and 64% similarity overall, and 68% identity and 89% similarity in the active site region. In early work we proposed2 that bisphosphonates might bind to the FPPS allylic site S1, and this was later found to be the case.23 It was then suggested15 that some bisphosphonates might be able to bind to the homoallylic site S2, but to date, no such structures have been reported. As part of our investigation into the structures of FPPS inhibitors, we chose to investigate 8 (BPH-6; NE-58051; 1-hydroxy-1-phosphono-3-(pyridin-3-yl)propyl) phosphonic acid) since this was the subject of the earlier computational docking study.15 Plus, 8 is of interest because it is an inhibitor of trypanosomatid FPPSs (Leishmania donovani, IC50 = 5.4 μM; T. brucei IC50 ≈ 33 μM) and is also active against T. brucei rhodesiense with an EC50 of 1.7 μM.16
We crystallized T. brucei FPPS in its apo form in addition to obtaining cocrystals in the presence of 8, as well as in the presence of the potent inhibitors 2 (risedronate; IC50 = 300 nM) and 9 (IC50 = 230 nM), and solved their structures. Full crystallographic data acquisition and data processing details are given in the Supporting Information and Table 1. The structures of 2 and 9 were as expected, based on all previous FPPS–bisphosphonate structures, the inhibitors binding to S1 and interacting with 3 Mg2+, as shown in Figures 4A,B. Surprisingly, however, we found that the TbFPPS·8 structure, Figure 4C, contained two bisphosphonates, not just the one found in all other (∼50) reported bisphosphonate-containing FPPS structures. In addition, there were four , not three, Mg2+. Electron densities are shown in Figure 4C and ligand–protein interactions in Figure 4D. Figure 4E shows the structure of the complex with 8 superimposed on the structure with risedronate 2 (in S1), in stereo. Clearly, in the structure with 8, one molecule of 8, henceforth called 8a, binds in the allylic site S1 (as do all other bisphosphonates), and in this site, there are interactions with three Mg2+ (MgA2+, MgB2+ and MgC2+), as shown in Figure 4C,D. These Mg2+ are involved in diphosphate removal/carbocation formation. However, as can be seen in Figure 4C,D, there is a fourth Mg2+ present in the structure, MgD2+, and the second 8 molecule, 8b, binds with its bisphosphonate groups interacting with MgC2+ in addition to MgD2+, Figure 4C,D. The pyridine side-chain in 8b is located between the allylic site S1 and the homoallylic site S2 (Figure 5 shows the comparison with zoledronate and 11), and the pyridine ring in 8b is more solvent exposed than is found in 8a, bound to S1. This FPPS structure is interesting because it is the first to contain two bisphosphonate inhibitors bound in the active site; plus, the inhibitor has activity against two trypanosomatid FPPS enzymes as well as against T. brucei, but its activity against human FPPS is ∼1000-fold less than is found with more potent bisphosphonates such as risedronate (2),27 and under our assay conditions, 8 did not inhibit human FPPS. We used ITC to investigate the binding of the inhibitor to T. brucei FPPS, and the results are shown in Supporting Information Figure S3. There are two binding sites with similar ΔH and ΔS values, but there is only 19% and 62% occupancy, so there may be stabilization of the two-ligand structure due to lattice packing in the crystalline solid state, or binding of two ligands is just very slow.
Figure 4.

Structures of 2, 8, and 9 bound to TbFPPS: (A) 2 (PDB ID code 4RXD); (B) 9 (PDB ID code 4RXE); (C) 8 (PDB ID code 4RXC). (D) Ligand–protein interactions with 8. (E) Comparison between 2 and 8 TbFPPS structures, in stereo. In panels A–C, the electron density maps are 2Fo–Fc and are contoured at 1σ.
Figure 5.
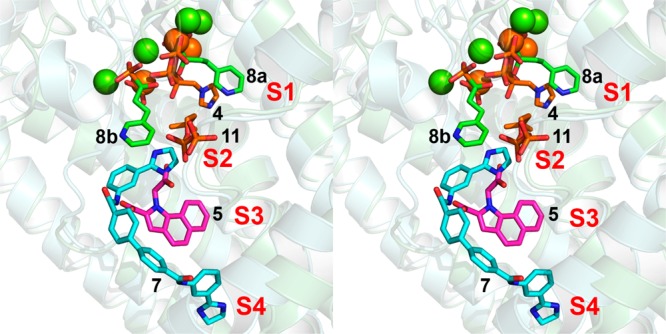
Comparison between the structures of 4, 5, 7, 8, and 11 bound to FPPS, in stereo.
When the 8 TbFPPS structure (PDB ID code 4RXC) is compared with the corresponding 2 TbFPPS structure (PDB ID code 4RXD), Supporting Information Figure S2, it is apparent that the α2 helix is closer to the risedronate ligand 2 than it is when in the presence of 8. There are also rotations of the A and B helices, not observed in the apo-FPPS structure (PDB ID code 4RYP), and the apo-FPPS and 8 structures are very similar. What these results show is that addition of a potent inhibitor causes a closing of the active site pocket, in particular, rotation of helix α2. In the absence of any preincubation with inhibitor, it has been reported that 2 and 8 have basically similar IC50 values for human FPPS inhibition (2, 82 nM; 8, 363 nM), but on preincubation, the IC50 values are very different (2, 0.36 nM; 8, 78 nM). These results are consistent with the TbFPPS structural results in which the more potent inhibitor 2 causes a closing of the active site that is not seen with 8, whose structure (even in the presence of two bisphosphonates) is very similar to that of the open, apo-form structure, Figure S2, Supporting Information.
The results we have described above are of interest for two main reasons. First, we find that the novel FPPS inhibitor 7, a member of a new class of lipophilic FPPS inhibitors, binds to a novel ligand binding site, called here S4, in addition to binding to S2/S3, Figure 5. Such lipophilic FPPS inhibitors could be of interest in the context of the development of inhibitors that inhibit prenylation in Ras-bearing tumors,21 and the availability of structural data may help guide future lead development. Second, the structure of the TbFPPS complex with 8 is of interest because while all previously reported bisphosphonate–FPPS structures contain a single bisphosphonate ligand, this complex, at least in the crystalline solid state, contains two, again as shown in the superposition in Figure 5, and 8 is a potential lead for selective T. brucei cell growth inhibitors. Overall, the results reported here are thus of general interest since they describe two new structures of FPPS–inhibitor complexes, of potential use in the design of new drug leads.
Glossary
ABBREVIATIONS
- FPPS
farnesyl diphosphate synthase
- IPP
isopentenyl diphosphate
- DMAPP
dimethylallyl diphosphate
- GPP
geranyl diphosphate
- FPP
farnesyl diphosphate
- ITC
isothermal titration calorimetry
Supporting Information Available
Experimental details of human FPPS and TbFPPS expression, purification, and X-ray crystallography, together with a comparison of the apo and ligand bound TbFPPS structures and ligand interactions. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by grants from the United States Public Health Service (NIH grant CA158191), a Harriet A. Harlin Professorship (E.O.), and the University of Illinois Foundation/Oldfield Research Fund. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Russell R. G. Bisphosphonates: the first 40 years. Bone 2011, 49, 2–19. [DOI] [PubMed] [Google Scholar]
- Martin M. B.; Arnold W.; Heath H. T. 3rd; Urbina J. A.; Oldfield E. Nitrogen-containing bisphosphonates as carbocation transition state analogs for isoprenoid biosynthesis. Biochem. Biophys. Res. Commun. 1999, 263, 754–758. [DOI] [PubMed] [Google Scholar]
- Bergstrom J. D.; Bostedor R. G.; Masarachia P. J.; Reszka A. A.; Rodan G. Alendronate is a specific, nanomolar inhibitor of farnesyl diphosphate synthase. Arch. Biochem. Biophys. 2000, 373, 231–241. [DOI] [PubMed] [Google Scholar]
- Boissier S.; Ferreras M.; Peyruchaud O.; Magnetto S.; Ebetino F. H.; Colombel M.; Delmas P.; Delaisse J. M.; Clezardin P. Bisphosphonates inhibit breast and prostate carcinoma cell invasion, an early event in the formation of bone metastases. Cancer Res. 2000, 60, 2949–2954. [PubMed] [Google Scholar]
- Kunzmann V.; Bauer E.; Wilhelm M. Gamma/delta T-cell stimulation by pamidronate. N. Engl. J. Med. 1999, 340, 737–738. [DOI] [PubMed] [Google Scholar]
- Tsagozis P.; Eriksson F.; Pisa P. Zoledronic acid modulates antitumoral responses of prostate cancer-tumor associated macrophages. Cancer Immunol. Immunother. 2008, 57, 1451–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez N.; Bailey B. N.; Martin M. B.; Oldfield E.; Urbina J. A.; Docampo R. Radical cure of experimental cutaneous leishmaniasis by the bisphosphonate pamidronate. J. Infect. Dis. 2002, 186, 138–140. [DOI] [PubMed] [Google Scholar]
- No J. H.; de Macedo Dossin F.; Zhang Y.; Liu Y. L.; Zhu W.; Feng X.; Yoo J. A.; Lee E.; Wang K.; Hui R.; Freitas-Junior L. H.; Oldfield E. Lipophilic analogs of zoledronate and risedronate inhibit Plasmodium geranylgeranyl diphosphate synthase (GGPPS) and exhibit potent antimalarial activity. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 4058–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield E. Targeting isoprenoid biosynthesis for drug discovery: bench to bedside. Acc. Chem. Res. 2010, 43, 1216–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield E.; Lin F. Y. Terpene biosynthesis: modularity rules. Angew. Chem., Int. Ed. 2012, 51, 1124–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. I.; Liu G. Y.; Song Y.; Yin F.; Hensler M. E.; Jeng W. Y.; Nizet V.; Wang A. H.; Oldfield E. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science 2008, 319, 1391–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiezewska E.; Danikiewicz W. Polyisoprenoids: structure, biosynthesis and function. Prog. Lipid Res. 2005, 44, 235–258. [DOI] [PubMed] [Google Scholar]
- Jahnke W.; Rondeau J. M.; Cotesta S.; Marzinzik A.; Pelle X.; Geiser M.; Strauss A.; Gotte M.; Bitsch F.; Hemmig R.; Henry C.; Lehmann S.; Glickman J. F.; Roddy T. P.; Stout S. J.; Green J. R. Allosteric non-bisphosphonate FPPS inhibitors identified by fragment-based discovery. Nat. Chem. Biol. 2010, 6, 660–666. [DOI] [PubMed] [Google Scholar]
- Amstutz R.; Bold G.; Cotesta S.; Jahnke W.; Marzinzik A.; Mueller-Hartwieg C.; Ofner S.; Stauffer F.; Zimmermann J.. Quinolines as inhibitors of farnesyl pyrophosphate synthase. U.S. Patent Application PCT/EP2009/052314, September 3, 2009.
- Ebetino F. H.; Roze C. N.; McKenna C. E.; Barnett B. L.; Dunford J. E.; Russell R. G. G.; Mieling G. E.; Rogers M. J. Molecular interactions of nitrogen-containing bisphosphonates within farnesyl diphosphate synthase. J. Org. Chem. 2005, 690, 2679–2687. [Google Scholar]
- Martin M. B.; Grimley J. S.; Lewis J. C.; Heath H. T. 3rd; Bailey B. N.; Kendrick H.; Yardley V.; Caldera A.; Lira R.; Urbina J. A.; Moreno S. N.; Docampo R.; Croft S. L.; Oldfield E. Bisphosphonates inhibit the growth of Trypanosoma brucei, Trypanosoma cruzi, Leishmania donovani, Toxoplasma gondii, and Plasmodium falciparum: a potential route to chemotherapy. J. Med. Chem. 2001, 44, 909–916. [DOI] [PubMed] [Google Scholar]
- Poulter C. D.; Satterwhite D. M. Mechanism of the prenyl-transfer reaction. Studies with (E)- and (Z)-3-trifluoromethyl-2-buten-1-yl pyrophosphate. Biochemistry 1977, 16, 5470–5478. [DOI] [PubMed] [Google Scholar]
- Aaron J. A.; Christianson D. W. Trinuclear metal clusters in catalysis by terpenoid synthases. Pure Appl. Chem. 2010, 82, 1585–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. L.; Lindert S.; Zhu W.; Wang K.; McCammon J. A.; Oldfield E. Taxodione and arenarone inhibit farnesyl diphosphate synthase by binding to the isopentenyl diphosphate site. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, E2530–E2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindert S.; Zhu W.; Liu Y. L.; Pang R.; Oldfield E.; McCammon J. A. Farnesyl diphosphate synthase inhibitors from in silico screening. Chem. Biol. Drug Des. 2013, 81, 742–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y.; Liu Y. L.; Xie Y.; Zhu W.; Guerra F.; Shen S.; Yeddula N.; Fischer W.; Low W.; Zhou X.; Zhang H. Y.; Oldfield E.; Verma I. M. A combination therapy for KRAS-driven lung adenocarcinomas using liphophilic bisphosphonates and rapamycin. Sci. Transl. Med. 2014, 6, 263ra161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarshis L. C.; Yan M.; Poulter C. D.; Sacchettini J. C. Crystal structure of recombinant farnesyl diphosphate synthase at 2.6-A resolution. Biochemistry 1994, 33, 10871–10877. [DOI] [PubMed] [Google Scholar]
- Hosfield D. J.; Zhang Y.; Dougan D. R.; Broun A.; Tari L. W.; Swanson R. V.; Finn J. Structural basis for bisphosphonate-mediated inhibition of isoprenoid biosynthesis. J. Biol. Chem. 2004, 279, 8526–8529. [DOI] [PubMed] [Google Scholar]
- Rondeau J. M.; Bitsch F.; Bourgier E.; Geiser M.; Hemmig R.; Kroemer M.; Lehmann S.; Ramage P.; Rieffel S.; Strauss A.; Green J. R.; Jahnke W. Structural basis for the exceptional in vivo efficacy of bisphosphonate drugs. ChemMedChem 2006, 1, 267–273. [DOI] [PubMed] [Google Scholar]
- Kavanagh K. L.; Guo K.; Dunford J. E.; Wu X.; Knapp S.; Ebetino F. H.; Rogers M. J.; Russell R. G.; Oppermann U. The molecular mechanism of nitrogen-containing bisphosphonates as antiosteoporosis drugs. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 7829–7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group Inc.: Montreal, Canada, 2014. [Google Scholar]
- Dunford J. E.; Thompson K.; Coxon F. P.; Luckman S. P.; Hahn F. M.; Poulter C. D.; Ebetino F. H.; Rogers M. J. Structure-activity relationships for inhibition of farnesyl diphosphate synthase in vitro and inhibition of bone resorption in vivo by nitrogen-containing bisphosphonates. J. Pharmacol. Exp. Ther. 2001, 296, 235–242. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.