

Abstract

Purine-rich foods have long been suspected as a major cause of hyperuricemia. We hypothesized that inhibition of human concentrative nucleoside transporter 2 (hCNT2) would suppress increases in serum urate levels derived from dietary purines. To test this hypothesis, the development of potent hCNT2 inhibitors was required. By modifying adenosine, an hCNT2 substrate, we successfully identified 8-aminoadenosine derivatives as a new class of hCNT2 inhibitors. Compound 12 moderately inhibited hCNT2 (IC50 = 52 ± 3.8 μM), and subsequent structure–activity relationship studies led to the discovery of compound 48 (IC50 = 0.64 ± 0.19 μM). Here we describe significant findings about structural requirements of 8-aminoadenosine derivatives for exhibiting potent hCNT2 inhibitory activity.
Keywords: 8-Aminoadenosine derivatives, human concentrative nucleoside transporter 2, dietary purines, hyperuricemia, gout
Hyperuricemia is a condition in which serum urate levels are abnormally elevated. It is widely accepted that sustained hyperuricemia is the primary risk factor for urate deposition diseases, such as gouty arthritis, tophi, urinary stones, and renal damage.1 Prospective cohort studies conducted in healthy men or patients with asymptomatic hyperuricemia demonstrated that serum urate levels exceeding 7.0 mg/dL are closely associated with an increased risk for developing gouty arthritis.2,3 Moreover, a retrospective study of patients with gout revealed that the frequency of recurrent gouty arthritis decreases when serum urate levels are kept low.4 These findings suggest the importance of proactively reducing serum urate levels to prevent the onset of gouty attacks.
Purine-rich foods have long been suspected as a major cause of hyperuricemia. Recent epidemiologic studies reported that high levels of meat and seafood consumption are associated with high levels of serum urate and an increased risk of developing gout.5,6 Another study demonstrated that ingesting yeast RNA for several days induces hyperuricemia in healthy subjects.7 Conversely, a low purine diet effectively lowers serum urate levels in patients with hyperuricemia.8 Together, these findings suggest that dietary purines increase serum urate levels and that suppression of the absorption of dietary purines may be a promising approach for treating hyperuricemia. Maintaining a strict diet with low purine foods over a long period is difficult, however, because nutrition is impaired and quality of life is decreased. Therefore, we attempted to develop a therapeutic drug as an alternative to restricting the intake of dietary purines.
We focused on the absorption paths of purines in the gastrointestinal tract. Dietary purines comprise various forms, such as DNA, RNA, nucleoproteins, oligonucleotides, purine nucleotides, purine nucleosides, and purine bases. Most of these forms are poorly absorbed directly via the plasma membrane because of their high molecular weights and/or highly hydrophilic nature. Thus, carrier-mediated transport likely plays an important role in the gastrointestinal absorption of dietary purines. Nucleoside transporter proteins have been extensively studied as potential carriers.9−13 Currently, they are classified into roughly two groups in mammals.14,15 One group contains concentrative nucleoside transporters (CNT1–3), which mediate unidirectional sodium- and/or proton-dependent transport.16 The second group contains equilibrative nucleoside transporters (ENT1–4), which mediate bidirectional sodium-independent transport (facilitated diffusion).17 Although distribution of the seven transporters varies depending on the tissue, in epithelia, CNTs and ENTs are mostly localized in the apical and basolateral membranes, respectively.14,15,18,19 Therefore, CNTs are thought to be more important carriers for the absorption of purine nucleosides in the gastrointestinal tract.
Extensive studies have revealed the inherent substrate selectivity of CNTs (Table 1).14,15,18 CNT1 recognizes pyrimidine nucleosides and adenosine, and adenosine is transported in a high-affinity and low-capacity manner.16,20,21 Hence, CNT1 is a pyrimidine nucleoside-specific transporter. CNT2 is the preferred purine nucleoside transporter because it transports purines and uridine.16,22,23 CNT3 has much broader substrate selectivity, transporting both purine and pyrimidine nucleosides.16,24 On the basis of these findings, CNT2 and/or CNT3 are likely to be the key transporters for the uptake of purine nucleosides in the gastrointestinal tract. Therefore, we performed quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) to confirm the expression profiles of human CNT2 (hCNT2) and CNT3 (hCNT3) in the gastrointestinal tract.25 In the small intestine, hCNT2 was abundantly expressed, whereas hCNT3 was only weakly expressed. Furthermore, analysis of the distribution patterns in the gastrointestinal tract revealed that hCNT2 was expressed mostly in the duodenum of the upper small intestine, followed by the jejunum; and hCNT3 and hCNT2 expression were comparable in the ileum, though the expression was weak overall. These findings suggest that hCNT2 is a central carrier responsible for the absorption of purine nucleosides in the gastrointestinal tract. Hence, we selected hCNT2 as our drug target candidate and hypothesized that its inhibitors would suppress the increase in serum urate levels derived from dietary purines.
Table 1. Substrate Selectivities of hCNTs14,15,18.
| nucleoside transportersa |
||||
|---|---|---|---|---|
| substrates | CNT1 | CNT2 | CNT3 | |
| purine nucleosides | adenosine | T | T | T |
| guanosine | NT | T | T | |
| inosine | NT | T | T | |
| pyrimidine nucleosides | cytidine | T | NT | T |
| thymidine | T | NT | T | |
| uridine | T | T | T | |
| nucleobases | NT | NT | NT | |
T, transported; NT, not transported.
Unlike human ENT inhibitors, there were few reports about CNT inhibitors at the beginning of our research. Among them, 2′-deoxy-5-fluorouridine,26 phlorizin,27,28 and 7,8,3′-trihydroxyflavone28 are reported to be strong or moderate hCNT2 inhibitors (Figure 1). Evaluation of the inhibitory effects of these compounds on sodium-dependent inosine uptake in COS-7 cells transiently expressing hCNT2,25 however, revealed unexpectedly weak inhibitory effects (IC50 > 1000 μM). Thus, to test our hypothesis, the development of more potent hCNT2 inhibitors was required. Here we describe the discovery of a new class of potent hCNT2 inhibitors along with significant findings about structural requirements for exhibiting potent hCNT2 inhibitory activity.
Figure 1.

Structures of known hCNT2 inhibitors.
We investigated the conversion of adenosine, an hCNT2 substrate, into an inhibitor by its derivatization. In an initial study, we synthesized a series of adenosine derivatives modified at the 2-, 8-, 5′-, or N6-position (Figure 2) and evaluated their inhibitory effects on sodium-dependent inosine uptake in COS-7 cells transiently expressing hCNT2. Among the base-modified adenosines 1–12, only 8-(benzylamino)adenosine 12 exhibited an inhibitory effect greater than 50% at 100 μM (IC50 = 52 ± 3.8 μM). However, the sugar-modified adenosines 13–24 were inactive or almost inactive (IC50 > 100 μM), consistent with a previous report demonstrating that the 5′-hydroxy group in the ribofuranosyl moiety is essential for recognition by hCNT2.29 Thus, we performed structure–activity relationship (SAR) studies of the 8-modified adenosines.
Figure 2.

Structures of modified adenosines evaluated in an initial study.
The 8-substituted adenosines evaluated in this study were synthesized from commercially available 8-bromoadenosine 25 as a common starting material (Scheme 1). Aromatic nucleophilic substitution reactions of compound 25 at the 8-position with appropriate amines and BnSH gave 8-aminoadenosine derivatives 9–12, 27, 28, 32–48, and 8-(benzylsulfanyl)adenosine 31, respectively.30O-Silylation of compound 25 and subsequent Pd-catalyzed coupling reaction with PhNH2 followed by treatment with NH4F gave 26. O-Acetylation of compound 25 and subsequent Sonogashira coupling with ethynylbenzene gave 2′,3′,5′-tri-O-acetyl-8-(phenylethynyl)adenosine, which was then reduced under catalytic hydrogenation conditions before treatment with NaOMe to afford 29. O-Silylation of compound 25 and subsequent aromatic nucleophilic substitution reaction at the 8-position with BnOH followed by treatment with NH4F gave 30.
Scheme 1. Synthesis of 8-Substituted Adenosines.

Reagents and conditions: (a) aqueous R3R5NH, EtOH, 100 °C, in a sealed tube; (b) R3R5NH, i-Pr2NEt, EtOH or PrOH, 120 °C, in a sealed tube; (c) R3R5NH, i-Pr2NEt, EtOH, 150 °C, in a sealed tube, microwave irradiation; (d) tert-butyldimethylsilyl chloride, imidazole, DMF, ambient temp.; (e) PhNH2, tris(dibenzylideneacetone)dipalladium(0), tert-BuONa, (±)-BINAP, toluene, 80 °C; (f) NH4F, MeOH, reflux; (g) Ac2O, Et3N, 4-dimethylaminopyridine, MeCN, ambient temp.; (h) ethynylbenzene, (Ph3P)4Pd, CuI, Et3N, DMF, 80 °C; (i) H2, 10% Pd–C, AcOEt, ambient temp.; (j) NaOMe, MeOH, ambient temp.; (k) BnOH, tert-BuOK, 40 °C then (l) NH4F, MeOH, reflux; (m) BnSH, i-Pr2NEt, EtOH, 120 °C, in a sealed tube.
The inhibitory effects of analogues of 12 were investigated (Table 2). Both deletion and elongation of the methylene linker between the nitrogen and benzene ring decreased the inhibitory activity (compound 12 vs 26–28). Replacement of the nitrogen with carbon, oxygen, or sulfur was also disadvantageous (compound 12 vs 29–31). Methylation of the nitrogen or at the benzylic position led to a complete loss of activity (compound 12 vs 32–34). These findings indicate that, in the 8-substituent of compound 12, the methyleneamino (−CH2NH−) structure was essential for the inhibitory effect, and therefore, we performed SAR studies on derivatives of compound 12 by modifying the terminal phenyl moiety (Table 3). Introduction of a substituent at the 3- or 4-position on the benzene ring improved the inhibitory activity regardless of the electronic nature (compound 12 vs 36, 37, 39, 40, 42, and 43). In contrast to the 2-chloro derivative 38, both the 2-methyl derivative 35 and 2-methoxy derivative 41 were less active than compound 12, suggesting a spatial limitation around the position in the binding mode. An electron-withdrawing group on the benzene ring might be preferred because all chloro derivatives exhibited more potent activity than the corresponding methyl or methoxy derivatives. However, replacement of the phenyl group with a 1- or 2-naphthyl group also enhanced the inhibitory activity (compound 44 and 45), regardless of the substituted position. On the basis of these results, a space around the phenyl moiety that accommodates rather large substituents might be present in the binging mode. Therefore, we designed biphenyl derivatives 46–48. The biphenyl-2-yl derivative 46 was inactive, as expected. The biphenyl-3-yl derivative 47 exhibited 3-fold more potent inhibitory activity than compound 12, but the activity did not surpass naphthyl derivatives 44 and 45. The introduction of a biphenyl-4-yl group, however, was highly effective, and the biphenyl-4-yl derivative 48 exhibited 81-fold more potent inhibitory activity than the parent compound 12. In addition, compound 48 exhibited inhibitory activity 1500-fold more potent than that of 2′-deoxy-5-fluorouridine, phlorizin, and 7,8,3′-trihydroxyflavone, which are well-known hCNT2 inhibitors.
Table 2. Inhibitory Effects on Sodium-Dependent Inosine Uptake in COS-7 Cells Transiently Expressing hCNT2.
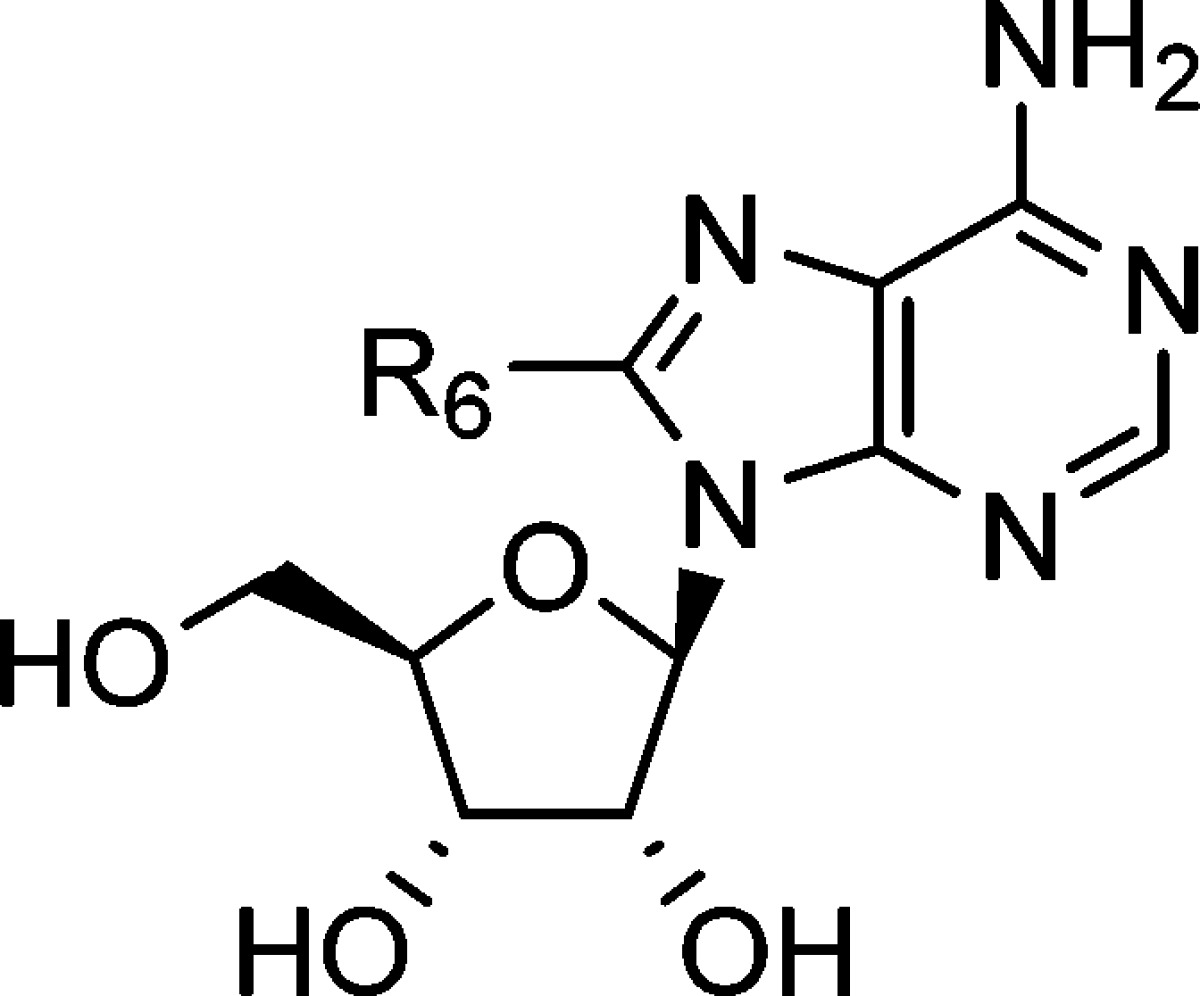
| compd | R6 | Inh.%a | IC50 (μM)b |
|---|---|---|---|
| 12 | PhCH2NH– | 58 | 52 ± 3.8 |
| 26 | PhNH– | 36 | 289 ± 87 |
| 27 | Ph(CH2)2NH– | 39 | 233 ± 19 |
| 28 | Ph(CH2)3NH– | 26 | 536 ± 89 |
| 29 | Ph(CH2)2– | <5 | |
| 30 | PhCH2O– | <5 | |
| 31 | PhCH2S– | <5 | |
| 32 | PhCH2N(Me)– | <5 | |
| 33 | (R)-PhCH(Me)NH– | <5 | |
| 34 | (S)-PhCH(Me)NH– | <5 | |
| 2′-deoxy-5-fluorouridine | 17 | ||
| phlorizin | 14 | ||
| 7,8,3′-trihydroxyflavone | <5 | ||
Inhibition % at 100 μM. If more than 20% inhibition was observed, the IC50 value was calculated.
Concentration of each compound required to inhibit inosine uptake by 50%. Data represent mean ± standard error of the mean of at least three independent experiments unless otherwise stated.
Table 3. Inhibitory Effects on Sodium-Dependent Inosine Uptake in COS-7 Cells Transiently Expressing hCNT2.
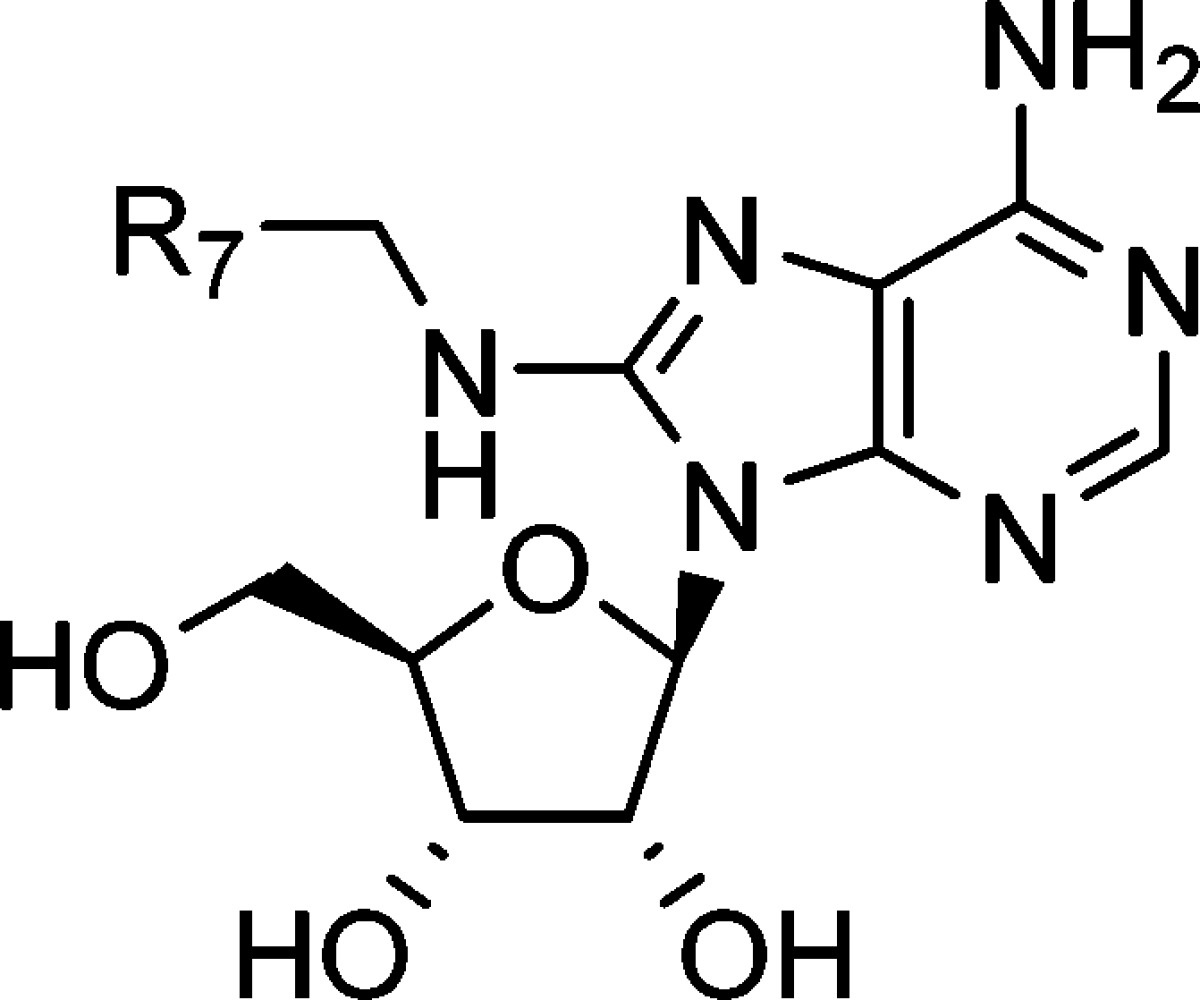
| compd | R7 | IC50 (μM)a |
|---|---|---|
| 12 | phenyl | 52 ± 3.8 |
| 35 | 2-methylphenyl | 65 ± 5.9 |
| 36 | 3-methylphenyl | 19 ± 2.3 |
| 37 | 4-methylphenyl | 11 ± 4.2 |
| 38 | 2-chlorophenyl | 5.7 ± 1.2 |
| 39 | 3-chlorophenyl | 5.4 ± 1.3 |
| 40 | 4-chlorophenyl | 11 ± 2.8 |
| 41 | 2-methoxyphenyl | >100 |
| 42 | 3-methoxyphenyl | 13 ± 5.7 |
| 43 | 4-methoxyphenyl | 36 ± 13 |
| 44 | 1-naphthyl | 5.7 ± 1.1 |
| 45 | 2-naphthyl | 5.3 ± 1.7 |
| 46 | biphenyl-2-yl | >100 |
| 47 | biphenyl-3-yl | 21 ± 9.8 |
| 48 | biphenyl-4-yl | 0.64 ± 0.19 |
| 2′-deoxy-5-fluorouridine | >1000b | |
| phlorizin | >1000b | |
| 7,8,3′-trihydroxyflavone | >1000b | |
Concentration of each compound required to inhibit inosine uptake by 50%. Data represent mean ± standard error of the mean of at least three independent experiments unless otherwise stated.
Inhibition was less than 50% at 1000 μM.
Finally, we performed a conformational analysis of the most potent compound 48 by the density functional theory (DFT) B3LYP/6-31g* method in Spartan′10 to get insight into the bioactive conformation. The calculations demonstrated that compound 48 is likely to take predominantly the anti conformation rather than the syn conformation, of which the energy difference was 3.27 kcal/mol.31 The preference for the anti conformers to the syn conformers would be due to the intramolecular hydrogen bonding formed by the N–H of the 8-substituent with the 5′-hydroxy oxygen and the ribose ring oxygen. These conformational analysis results are in accord with structure–activity relationship results that the N–H moiety in the 8-substituents is essential for exhibiting the inhibitory activity against hCNT2.
In conclusion, we identified a new class of potent hCNT2 inhibitors. Starting with a natural substrate, adenosine, we successfully identified 8-aminoadenosine derivative 12, and subsequent SAR studies led to the discovery of compound 48 (IC50 = 0.64 ± 0.19 μM). Conformational analysis data of compound 48 suggested the preference for anti conformers, and therefore, valuable information for speculating the bioactive conformation was provided. The inhibitor 48 can be a prototype compound for the development of conceptually new drugs for the treatment of hyperuricemia and gout.
Acknowledgments
We thank Mr. Shinjiro Watanabe for providing biological activity data. We thank Dr. Tomonaga Ozawa for performing conformational analysis.
Glossary
Abbreviations
- RNA
ribonucleic acid
- DNA
deoxyribonucleic acid
- CNT
concentrative nucleoside transporter
- ENT
equilibrative nucleoside transporter
- RT-PCR
reverse transcription-polymerase chain reaction
Supporting Information Available
Synthetic procedures and characterization data of all synthesized compounds, assay method, and conformational analysis data of compound 48. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Yamanaka H. Japanese guideline for the management of hyperuricemia and gout: Second edition. Nucleosides Nucleotides Nucleic Acids 2011, 30, 1018–1029. [DOI] [PubMed] [Google Scholar]
- Campion E. W.; Glynn R. J.; DeLabry L. O. Asymptomatic hyperuricemia. Risks and consequences in the normative aging study. Am. J. Med. 1987, 82, 421–426. [DOI] [PubMed] [Google Scholar]
- Lin K.-C.; Lin H.-Y.; Chou P. The interaction between uric acid level and other risk factors on the development of gout among asymptomatic hyperuricemic men in a prospective study. J. Rheumatol. 2000, 27, 1501–1505. [PubMed] [Google Scholar]
- Shoji A.; Yamanaka H.; Kamatani N. A retrospective study of the relationship between serum urate level and recurrent attacks of gouty arthritis: Evidence for reduction of recurrent gouty arthritis with antihyperuricemic therapy. Arthritis Rheum. 2004, 51, 321–325. [DOI] [PubMed] [Google Scholar]
- Choi H. K.; Liu S.; Curhan G. Intake of purine-rich foods, protein and dairy products and relationship to serum levels of uric acid. Arthritis Rheum. 2005, 52, 283–289. [DOI] [PubMed] [Google Scholar]
- Choi H. K.; Atkinson K.; Karlson E. W.; Willett W.; Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N. Engl. J. Med. 2004, 350, 1093–1103. [DOI] [PubMed] [Google Scholar]
- Yü T.-F.; Berger L. Impaired renal function in gout; Its association with hypertensive vascular disease and intrinsic renal disease. Am. J. Med. 1982, 72, 95–100. [DOI] [PubMed] [Google Scholar]
- Emmerson B. T. Identification of the causes of persistent hyperuricemia. Lancet 1991, 337, 1461–1463. [DOI] [PubMed] [Google Scholar]
- Lang T. T.; Selner M.; Young J. D.; Cass C. E. Acquisition of human concentrative nucleoside transporter 2 (hCNT2) activity by gene transfer confers sensitivity to fluoropyrimidine nucleosides in drug-resistant leukemia cells. Mol. Pharmacol. 2001, 60, 1143–1152. [DOI] [PubMed] [Google Scholar]
- King K. M.; Damaraju V. L.; Vickers M. F.; Yao S. Y.; Lang T.; Tackaberry T. E.; Mowles D. A.; Ng A. M. L.; Young J. D.; Cass C. E. A comparison of the transportability, and its role in cytotoxicity, of clofarabine, cladribine, and fludarabine by recombinant human nucleoside transporters produced in three model expression systems. Mol. Pharmacol. 2006, 69, 346–353. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Visser F.; King K. M.; Baldwin S. A.; Young J. D.; Cass C. E. The role of nucleoside transporters in cancer chemotherapy with nucleoside drugs. Cancer Metastasis Rev. 2007, 26, 85–110. [DOI] [PubMed] [Google Scholar]
- Damaraju V. L.; Sawyer M. B.; Mackey J. R.; Young J. D.; Cass C. E. Human nucleoside transporters: Biomarkers for response to nucleoside drugs. Nucleosides Nucleotides Nucleic Acids 2009, 28, 450–463. [DOI] [PubMed] [Google Scholar]
- Okayama T.; Yoshisue K.; Kuwata K.; Komuro M.; Ohta S.; Nagayama S. Involvement of concentrative nucleoside transporter 1 in intestinal absorption of trifluorothymidine, a novel antitumor nucleoside, in rats. J. Pharmacol. Exp. Ther. 2012, 340, 457–462. [DOI] [PubMed] [Google Scholar]
- Molina-Arcas M.; Casado F. J.; Pastor-Anglada M. Nucleoside transporter proteins. Curr. Vasc. Pharmacol. 2009, 7, 426–434. [DOI] [PubMed] [Google Scholar]
- Parkinson F. E.; Damaraju V. L.; Graham K.; Yao S. Y. M.; Baldwin S. A.; Cass C. E.; Young J. D. Molecular biology of nucleoside transporters and their distributions and functions in the brain. Curr. Top. Med. Chem. 2011, 11, 948–972. [DOI] [PubMed] [Google Scholar]
- Gray J. H.; Owen R. P.; Giacomini K. M. The concentrative nucleoside transporter family, SLC28. Pflugers Arch. 2004, 447, 728–734. [DOI] [PubMed] [Google Scholar]
- Baldwin S. A.; Beal P. R.; Yao S. Y. M.; King A. E.; Cass C. E.; Young J. D. The equilibrative nucleoside transporter family, SLC29. Pflugers Arch. 2004, 447, 735–743. [DOI] [PubMed] [Google Scholar]
- Kong W.; Engel K.; Wang J. Mammalian nucleoside transporters. Curr. Drug Metab. 2004, 5, 63–84. [DOI] [PubMed] [Google Scholar]
- Lai Y.; Bakken A. H.; Unadkat J. D. Simultaneous expression of hCNT1-CFP and hENT1-YEP in Madin-Darby canine kidney cells. J. Biol. Chem. 2002, 277, 37711–37717. [DOI] [PubMed] [Google Scholar]
- Huang Q.-Q.; Yao S. Y. M.; Ritzel M. W. L.; Paterson A. R. P.; Cass C. E.; Young J. D. Cloning and functional expression of a complementary DNA encoding a mammalian nucleoside transport protein. J. Biol. Chem. 1994, 269, 17757–17760. [PubMed] [Google Scholar]
- Ritzel M. W.; Yao S. Y. M.; Huang M. Y.; Elliott J. F.; Cass C. E.; Young J. D. Molecular cloning and functional expression of cDNAs encoding a human Na+-nucleoside cotransporter (hCNT1). Am. J. Phyisiol. 1997, 272, C707–C714. [DOI] [PubMed] [Google Scholar]
- Che M.; Ortiz D. F.; Arias I. M. Primary structure and functional expression of a cDNA encoding the bile canalicular, purine-specific Na+-nucleoside cotransporter. J. Biol. Chem. 1995, 270, 13596–13599. [DOI] [PubMed] [Google Scholar]
- Wang J.; Su S.-F.; Dresser M. J.; Schaner M. E.; Washington C. B.; Giacomini K. M. Na+-dependent purine nucleoside transporter from human kidney: Cloning and functional characterization. Am. J. Physiol. 1997, 273, F1058–F1065. [DOI] [PubMed] [Google Scholar]
- Ritzel M. W. L.; Ng A. M. L.; Yao S. Y. M.; Graham K.; Loewen S. K.; Smith K. M.; Ritzel R. G.; Mowles D. A.; Carpenter P.; Chen X.-Z.; Karpinski E.; Hyde R. J.; Baldwin S. A.; Cass C. E.; Young J. D. Molecular identification and characterization of novel human and mouse concentrative Na+-nucleoside cotransporter proteins (hCNT3 and mCNT3) broadly selective for purine and pyrimidine nucleosides (system cib). J. Biol. Chem. 2001, 276, 2914–2927. [DOI] [PubMed] [Google Scholar]
- Hiratochi M.; Tatani K.; Shimizu K.; Kuramochi Y.; Kikuchi N.; Kamada N.; Itoh F.; Isaji M. Hypouricemic effects of novel concentrative nucleoside transporter 2 inhibitors through suppressing intestinal absorption of purine nucleosides. Eur. J. Pharmacol. 2012, 690, 183–191. [DOI] [PubMed] [Google Scholar]
- Shin H.-C.; Landowski C. P.; Sun D.; Vig B. S.; Kim I.; Mittal S.; Lane M.; Rosania G.; Drach J. C.; Amidon G. L. Functional expression and characterization of a sodium-dependent nucleoside transporter hCNT2 cloned from human duodenum. Biochem. Biophys. Res. Commun. 2003, 307, 696–703. [DOI] [PubMed] [Google Scholar]
- Gupte A.; Buolamwini J. K. Synthesis and biological evaluation of phloridzin analogs as human concentrative nucleoside transporter 3 (hCNT3) inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 917–921. [DOI] [PubMed] [Google Scholar]
- Wang C.; Pimple S.; Buolamwini J. K. Interaction of benzopyranone derivatives and related compounds with human concentrative nucleoside transporters 1, 2 and 3 heterologously expressed in porcine PK15 nucleoside transporter deficient cells. Structure-activity relationships and determinants of transporter affinity and selectivity. Biochem. Pharmacol. 2010, 79, 307–320. [DOI] [PubMed] [Google Scholar]
- Cnang C.; Swaan P. W.; Ngo L. Y.; Lum P. Y.; Patil S. D.; Unadkat J. D. Molecular requirements of the human nucleoside transporters hCNT1, hCNT2, and hENT1. Mol. Pharmacol. 2004, 65, 558–570. [DOI] [PubMed] [Google Scholar]
- In the case of compound 34, an additional protection–deprotection step was needed to obtain the compound in pure form.
- Figures of the most stable anti and syn conformers are provided in the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.