

Abstract
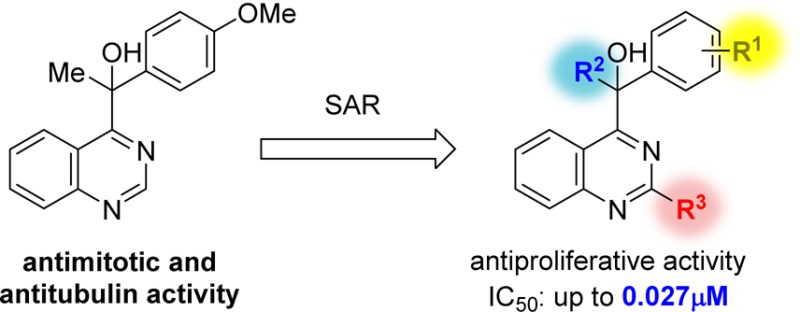
A quinazoline derivative PVHD121 (1a) was shown to have strong antiproliferative activity against various tumor-derived cell lines, including A549 (lung), NCI-H460 (lung), HCT116 (colon), MCF7 (breast), PC3 (prostate), and HeLa (cervical) cells with IC50 values from 0.1 to 0.3 μM. A structure–activity relationship (SAR) study at the 2- and 4-position of the quinazoline core lead to the discovery of more potent anticancer agents (14, 16, 17, 19, 24, and 31). The results of an in vitro tubulin polymerization assay and fluorescent-based colchicine site competition assay with purified tubulin indicated that 1a inhibits tubulin polymerization by binding to the colchicine site.
Keywords: Quinazoline, anticancer, tubulin inhibitor, colchicine binding site, structure−activity relationship study
Cancer is one of the most common causes of death worldwide.1 According to the World Health Organization, cancer killed 7.6 million people in 2008, representing approximately 13% of all deaths. The number of cancer deaths is estimated to reach 13.1 million by 2030. Many bioactive compounds have been shown to possess anticancer activity.2 However, their use is limited due to side effects or limited scope of activity. Thus, the identification of novel, potent drugs with fewer side effects and a broad spectrum of activity is desirable to improve cancer treatment. Among the available anticancer drugs, antimitotic agents, which interact with microtubules, are one of the most successful therapeutics.3−8 Microtubules are a component of the cytoskeleton in eukaryotic cells and consist of tubulin protein polymers. These proteins can polymerize and depolymerize. Antimitotic agents interact with binding sites on tubulin, such as the colchicine site and the vinblastine site,9 and inhibit or promote microtubule polymerization, thereby affecting their function.3−8 A number of antimitotic compounds have been synthesized as promising drug candidates in many laboratories.10
In the course of random screening for anticancer agents, we discovered a quinazoline derivative, PVHD121 (1a), which had strong antiproliferative activity against the cervical cancer cell line, HeLa. Recently, there have been reports of quinazoline-based anticancer agents with amino groups at the 4-position of the core, rather than the 1-hydroxyethyl group in 1a.11−13 In the present study, we describe the results of a structure–activity relationship (SAR) study for the development of novel anticancer drugs and demonstrate the biological activity of 1a and its derivatives for the first time.
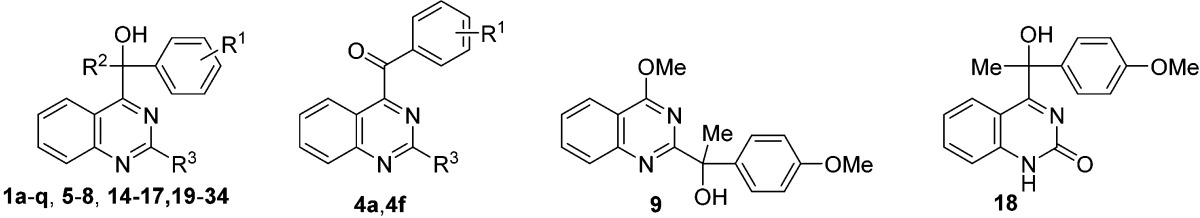
The hit compound 1a was originally prepared as a substrate in the development of a new synthetic catalysis.14 According to the reported procedure, the synthesis of derivatives 1a–n, which contain various aromatic rings on the substituents at the 4-position of the quinazoline nucleus, was initiated by NHC-catalyzed aroylation using an aldehyde, followed by a Grignard reaction with methylmagnesium bromide. Derivatives 4o–q, which have methylamino, dimethylamino, and methylthio groups, were synthesized via nucleophilic aromatic substitution (SNAr) of the fluoro group of 4b by N- or S-nucleophiles and a subsequent Grignard reaction (Scheme 1).
Scheme 1.

Reagents and conditions: (i) N,N′-dimethylimidazolium iodide (catalyst), NaH, THF, reflux; (ii) MeMgBr, THF, rt; (iii) MeNH2, 1,4-dioxane, reflux; (iv) Me2NH, 1,4-dioxane, reflux; (v) NaSMe, 1,4-dioxane, reflux.
The synthesis of compounds 5–8, which have ethyl, benzyl, trifluoromethyl, and a hydrogen atom at the benzyl position instead of a methyl moiety, is shown in Scheme 2. The Grignard reaction between 4a and ethylmagnesium iodide produced ethyl-substituted derivative 5. Benzyl-substituted derivative 6 was similarly synthesized from 4a. Trifluoromethylation of the keto group of 4a using TMS-CF3 and TBAF15 yielded derivative 7. The reduction of the keto group of 4a using NaBH4 afforded 8.
Scheme 2.
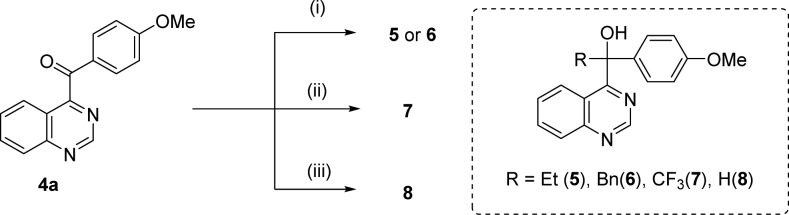
Reagent and conditions: (i) RMgI, THF, rt; (ii) TMS-CF3, TBAF, THF, 0 °C to rt; (iii) NaBH4, ethanol, rt.
Derivative 9, which has a 1-(4-anisyl)-1-hydroxyethyl group at position 2 of the quinazoline core, instead of the 4-position, was also prepared from 2,4-dichloroquinazoline 13a via substitutions of the 2- and 4-chloro groups and a Grignard reaction with methylmagnesium bromide (see Scheme S-1, Supporting Information).
The compounds having methyl, phenyl, trichloromethyl, and chloro groups at position 2 of the quinazoline (14–17) were synthesized using the same procedure as 1a–n (Scheme 3). Specifically, NHC-catalyzed aroylation of 2-substituted 4-chloroquinazoline 10a–13a12,16−18 was performed, followed by Grignard reaction, yielding 14–17 (Scheme 3).
Scheme 3.

Reagents and conditions: (i) N,N′-1,3-dimethylimidazolium iodide (catalyst), p-anisaldehyde, NaH, THF/DMF, reflux; (ii) MeMgI, THF, rt.
Synthesis of 18–34 from the 2-chloro derivative 17 was performed by SNAr or Suzuki–Miyaura cross coupling (Scheme 4).19,20
Scheme 4.

Reagent and conditions: (i) NaOH, THF, reflux/R′ONa, MeOH, rt/MeSNa, 1,4-dioxane, reflux/amine, 1,4-dioxane, reflux (see Supporting Information for details); (ii) ArB(OH)2, K2CO3, Pd(OAc)2, 1,4-dioxane, reflux.
Hit compound 1a induced G2/M accumulation in human lung carcinoma A549 cells (Figure 1A). In addition, 1a showed a broad spectrum of antiproliferative activity against various solid tumor-derived cell lines including A549 (lung), NCI-H460 (lung), HCT116 (colon), MCF7 (breast), PC3 (prostate), and HeLa (cervical) cells, with IC50 values ranging from 0.1 to 0.3 μM (see Figure S-1, Supporting Information).
Figure 1.

(A) Flow cytometric analysis of cell cycle distribution in A549 cells. (B) Immunomicroscopic analysis of tubulin destabilization in A549 cells. (C) In vitro tubulin polymerization assay. (D) Competitive displacement binding assay on the colchicine-binding site of tubulin. (E) Concentration-dependent binding of 1a to the colchicine-binding site.
The obtained derivatives 1b–q, 5–9, and 14–34, along with both enantiomers of 1a separated by chiral column chromatography were tested for cytotoxicity against the human lung cancer cell line A549 (Table 1). Growth inhibition by the quinazoline derivatives was evaluated by MTS assay.
Table 1.

Among the derivatives with various aromatic rings on the substituent at the 4-position of quinazoline, 1f (containing a 4-ethoxyphenyl group, IC50 = 0.30 μM) and 1q (containing a 4-methylthiophenyl group, IC50 = 0.34 μM) showed the same degree of antiproliferative activity as 1a (IC50 = 0.27 μM). However, none of the derivatives had a higher antiproliferative activity than 1a in A549 cells. Although their absolute configurations are not yet determined, (+)-1a showed stronger antiproliferative activity than (−)-1a (see Figure S-2, Supporting Information).
We found that several 2-substituted compounds had stronger antiproliferative activity against A549 than 1a, including 2-methyl (14), 2-trichloromethyl (16), 2-chloro (17), 2-methoxy (19), 2-methythio (24), and 2-(3-thienyl) (31) substituted derivatives. In particular, the 2-chloro-substituted analogue 17 showed the highest activity (IC50 = 0.027 μM), which is ten times more effective than the hit compound 1a. In constast, the introduction of 2-substituents, such as hydroxyl (the analogue with 2-hydroxyl group mainly exists as its tautomer 18), propoxy (21), phenoxy (23), phenylamino (26), piperidin-1-yl (27), 4-methylpiperadin-1-yl (28), 4-(4-fluorophenyl)piperadin-1-yl (30), and 3-chlorophenyl (34) significantly decreased activity.
We further investigated the biological target of the compounds. After A549 cells had been treated with three different concentrations of 1a for 24 h, the cell cycle profiles were analyzed by flow cytometry. As shown in Figure 1A, 1a clearly affected the cell cycle progression of A549 cells and accumulated cells in the G2/M phase in a concentration-dependent manner. We next performed immunofluorescent microscopy analysis of A549 cells treated with 1a. Staining with DAPI and an anti-α tubulin antibody revealed that 1a abrogated the microtubule networks like a well-known tubulin binder, colchicine (Figure 1B). We next assessed the effect of 1a on in vitro tubulin polymerization using a fluorescence-based tubulin polymerization assay. As shown in Figure 1C, 1a moderately inhibited the tubulin polymerization at 3 μM and more strongly inhibited it at 10 μM.
To gain further insight into the mechanism of microtubule destabilization, we investigated the binding site of 1a on the tubulin protein using a fluorescent-based colchicine site competition assay with purified tubulin. Colchicine is known to show clear fluorescent properties when bound to tubulin (excitation at 350 nm and emission at 430 nm), although it is nonfluorescent by itself.21 The fluorescence signal was robustly attenuated in the presence of nocodazole, which binds the colchicine-binding site (Figure 1D). In contrast, vinblastine, which preferentially binds to a noncolchicine binding site, did not diminish the fluorescence (Figure 1E). In this competition assay, the fluorescence was clearly attenuated by 1a in a concentration-dependent manner (Figure 1D,E). These results suggest that 1a has high affinity for the colchicine-binding site.
One of the derivatives (1f) had comparable antiproliferative activity to 1a and showed similar inhibitory activity in the tubulin polymerization assay. The more potent inhibitor 15 inhibited tubulin polymerization more robustly than 1a at 10 μM (see Figure S-3, Supporting Information). Furthermore, such correlation was also seen in the experiments using both enantiomers of 1a (see Figure S-2, Supporting Information).
To develop a novel anticancer drug, various substituents at the 2- and 4-positions of quinazoline were synthesized via NHC-catalyzed aroylation. On the basis of the SAR studies, the 2-chloroquinazoline derivatives 14, 16, 17, 19, 24, and 31 were shown to be the most potent analogues. In addition, our results suggest that this type of quinazoline derivative exhibits antiproliferative activity through binding to the colchicine-site of tubulin, thereby inhibiting microtubule polymerization in cancer cells. Further derivatization to enhance activity and the elucidation of the detailed mechanism of action are now under investigation.
Supporting Information Available
Scheme S-1, Figures S-1–3, experimental procedures, analytical data, 1H and 13C NMR charts, and chiral HPLC analysis charts. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ Department of Pharmaceutical Sciences, Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama University, 1-1-1 Tsushima-naka, Kita-ku, Okayama 700-8530, Japan.
The authors declare no competing financial interest.
This Letter was published ASAP on January 21, 2015. A new paragraph has been added as the fourth paragraph from the end of the paper. The correct version was published on February 2, 2015.
Supplementary Material
References
- WHO. Cancer. http://www.who.int/cancer/en/ (accessed Sept 16, 2014).
- DeVita V. T. Jr.; Chu E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [DOI] [PubMed] [Google Scholar]
- Tahir S. K.; Han E. K-H.; Credo B.; Jae H.-S.; Pietenpol J. A.; Scatena C. D.; Wu-Wong J. R.; Frost D.; Sham H.; Rosenberg S. H.; Ng S.-C. A-204197, a new tubulin-binding agent with antimitotic activity in tumor cell lines resistant to known microtubule inhibitors. Cancer Res. 2001, 61, 5480–5485. [PubMed] [Google Scholar]
- Prinz H.; Ishii Y.; Hirano T.; Stoiber T.; Gomez J. A. C.; Schmidt P.; Düssmann H.; Burger A. M.; Prehn J. H. M.; Günther E. G.; Unger E.; Umezawa K. Novel benzylidene-9(10H)-anthracenones as highly active antimicrotubule agents. Synthesis, antiproliferative activity, and inhibition of tubulin polymerization. J. Med. Chem. 2003, 46, 3382–3394. [DOI] [PubMed] [Google Scholar]
- Romagnoli R.; Baraldi P. G.; Remusat V.; Carrion M. D.; Cara C. L.; Preti D.; Fruttarolo F.; Pavani M. G.; Tabrizi M. A.; Tolomeo M.; Grimaudo S.; Balzarini J.; Jordan M. A.; Hamel E. Synthesis and biological evaluation of 2-(3′,4′,5′-trimethoxybenzoyl)-3-amino 5-aryl thiophenes as a new class of tubulin inhibitors. J. Med. Chem. 2006, 49, 6425–6428. [DOI] [PubMed] [Google Scholar]
- Davis A.; Jiang J.-D.; Middleton K. M.; Wang Y.; Weisz I.; Ling Y.-H.; Bekesi J. G. Novel suicide ligands of tubulin arrest cancer cells in S-phase. Neoplasia 1999, 1, 498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regina G. L.; Bai R.; Rensen W. M.; Cesare E. D.; Coluccia A.; Piscitelli F.; Famiglini V.; Reggio A.; Nalli M.; Pelliccia S.; Pozzo E. D.; Costa B.; Granata I.; Porta A.; Maresca B.; Soriani A.; Iannitto M. L.; Santoni A.; Li J.; Cona M. M.; Chen F.; Ni Y.; Brancale A.; Dondio G.; Vultaggio S.; Varasi M.; Mercurio C.; Martini C.; Hamel C.; Lavia P.; Novellino E.; Silvestri R. Toward highly potent cancer agents by modulating the C-2 group of the arylthioindole class of tubulin polymerization inhibitors. J. Med. Chem. 2013, 56, 123–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joullié M. M.; Berritt S.; Hamel E. Structure–activity relationships of ustiloxin analogues. Tetrahedron 2011, 52, 2136–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore S.; Pasquier E.; Braguer D. Understanding microtubule dynamics for improved cancer therapy. Cell. Mol. Life Sci. 2005, 62, 3039–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat K. M. R.; Setaluri V. Chemotherapy microtubule-associated proteins as targets in cancer. Clin. Cancer Res. 2007, 13, 2849–2854. [DOI] [PubMed] [Google Scholar]
- Wang X.-F.; Guan F.; Ohkoshi E.; Guo W.; Wang L.; Zhu D.-Q.; Wang S.-B.; Wang L.-T.; Hamel E.; Yang D.; Li L.; Qian K.; Morris-Natschke S. L.; Yuan S.; Lee K.-H.; Xie L. Optimization of 4-(N-cycloamino)phenylquinazolines as a novel class of tubulin-polymerization inhibitors targeting the colchicine site. J. Med. Chem. 2014, 57, 1390–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirisoma N.; Pervin A.; Zhang H.; Jiang H.; Willardsen J. A.; Anderson M. B.; Mather G. Discovery of N-(4-Methoxyphenyl)-N,2-dimethylquinazolin-4-amine, a Potent Apoptosis Inducer and Efficacious Anticancer Agent with High Blood Brain Barrier Penetration. J. Med. Chem. 2009, 52, 2341–2351. [DOI] [PubMed] [Google Scholar]
- Marzaro G.; Guiotto A.; Chilin A. Quinazoline derivatives as potential anticancer agents: a patent review (2007–2010). Expert Opin. Ther. Pat. 2012, 22, 223–252. [DOI] [PubMed] [Google Scholar]
- Suzuki Y.; Takemura Y.; Iwamoto K.; Higashino T.; Miyashita A. Carbon-carbon bond cleavage of a-hydroxybenzylheteroarenes catalyzed by cyanide ion: Retro-benzoin condensation affords ketones and heteroarenes and benzyl migration affords benzylheteroarenes and arenecarbaldehydes. Chem. Pharm. Bull. 1998, 46, 199–206. [Google Scholar]
- Song J. J.; Tan Z.; Reeves J. T.; Gallou F.; Yee N. K.; Senanayaka C. H. N-Heterocyclic carbene catalyzed trifluoromethylation of carbonyl compounds. Org. Lett. 2005, 7, 2193–2196. [DOI] [PubMed] [Google Scholar]
- Mei Q.; Wang L.; Guo Y.; Weng J.; Yan F.; Tiana B.; Tong B. A highly efficient red electrophosphorescent iridium(III) complex containing phenyl quinazoline ligand in polymer light-emitting diodes. J. Mater. Chem. 2012, 22, 6878. [Google Scholar]
- Smith R. F.; Kent R. A.; Quinazolines I. The structure of the polychlorinated products obtained by the phosphorus pentachloride-phosphorus trichloride chlorination of 2-methyl-4-quinazolone and 2-ethyl-4-quinazolone. J. Org. Chem. 1965, 30, 1312–1313. [Google Scholar]
- Smits R. A.; de Esch I. J. P.; Zuiderveld O. P.; Broeker J.; Sansuk K.; Guaita E.; Coruzzi G.; Adami M.; Haaksma E.; Leurs R. Discovery of quinazolines as histamine H4 receptor inverse agonists using a scaffold hopping approach. J. Med. Chem. 2008, 51, 7855–7865. [DOI] [PubMed] [Google Scholar]
- Ding S.; Gray N. S.; Wu X.; Ding Q.; Schultz P. G. A combinatorial scaffold approach toward kinase-directed heterocycle libraries. J. Am. Chem. Soc. 2002, 124, 1594–1596. [DOI] [PubMed] [Google Scholar]
- Henriksen S. T.; Sørensen U. S. 2-Chloroquinazoline. Synthesis and reactivity of a versatile heterocyclic building block. Tetrahedron Lett. 2006, 47, 8251–8254. [Google Scholar]
- Bhattacharyya B.; Wolff J. Promotion of fluorescence upon binding of colchicine to tubulin. Proc. Natl. Acad. Sci. 1974, 71, 2627–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.