

Abstract
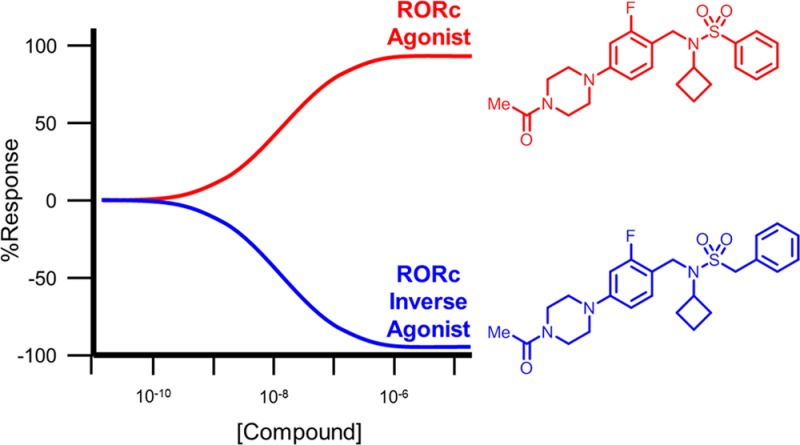
A minor structural change to tertiary sulfonamide RORc ligands led to distinct mechanisms of action. Co-crystal structures of two compounds revealed mechanistically consistent protein conformational changes. Optimized phenylsulfonamides were identified as RORc agonists while benzylsulfonamides exhibited potent inverse agonist activity. Compounds behaving as agonists in our biochemical assay also gave rise to an increased production of IL-17 in human PBMCs whereas inverse agonists led to significant suppression of IL-17 under the same assay conditions. The most potent inverse agonist compound showed >180-fold selectivity over the ROR isoforms as well as all other nuclear receptors that were profiled.
Keywords: RORc, RORγ, TH17, IL-17, PBMC, agonist, inverse agonist
The retinoic acid receptor-related orphan receptor gamma (RORγ or RORc)1 is a key nuclear receptor (NR) of the ROR family that has been implicated in the production and regulation of interleukin-17 (IL-17).2 This cytokine is required for the differentiation of naïve pro-inflammatory CD4+ T cells and for the production of T helper-17 (TH17) cells,2 which results in the expression of diverse pro-inflammatory cytokines such as IL-17,2 IL-22,3 and GM-CSF.4 Interleukin-17 is a critical driver of autoimmune diseases such as psoriasis,5 rheumatoid arthritis (RA),6 multiple sclerosis (MS),7 and inflammatory bowel disease (IBD).8 Therefore, the modulation of IL-17 expression through small molecule inhibitors of RORc9−11 has gained significant momentum as a molecular target since the first disclosure of a direct link between RORc signaling and IL-17 production by Littman et al. in 2006.2
Small molecule NRs may modulate transcription through several mechanisms.12,13 Ligands can induce a conformational change in the NR that results in the reduction of coactivator protein recruitment to the NR. Such ligands are termed inverse agonists.14 Inverse agonists decrease gene transcription, and in the case of RORc, this results in a reduction of IL-17 production. Conversely, ligands that promote coactivator protein recruitment and formation of the [NR·coactivator] complex generally result in increased gene transcription and are termed agonist ligands.14 A third method of modulation involves ligands that bind to the ligand-binding domain (LBD) of the NR but do not affect the basal transcriptional activity of the protein. Such molecules may be termed silent ligands or neutral antagonists.15
In addition to numerous small molecule RORc modulators previously reported,9−11 compound 1 was recently disclosed as a potent biaryl sulfonamide inverse agonist that was identified in a biochemical screen of the Genentech/Roche corporate collection (Figure 1).16 Co-crystallization of 1 with the human RORc-LBD [PDB: 4QM0]17 revealed that the benzylic phenyl ring of 1 interacted with His479, thus preventing His479 from forming a crucial hydrogen bond with Tyr502 in the protein. The interaction between His479 and Tyr502 is required to stabilize the secondary structure of helices 11–12 and induce coactivator recruitment.18 The disruption of this interaction may account for the inverse agonist mechanism of action (MOA)19,20 observed in our biochemical coactivator recruitment assay with compound 1. Further replacement of the biaryl motif in 1 with an acylpiperazine (2) led to significant improvement of the physicochemical properties, plasma–protein binding, and permeability of the series while preserving RORc inverse agonist potency.17
Figure 1.

Evolution of Genentech’s tertiary sulfonamide RORc modulators.
Additional optimization of benzylsulfonamide replacements was explored using parallel synthesis and resulted in the discovery of the matched pair of phenyl sulfonamide 3 and benzyl sulfonamide 4. While benzyl analog 4 showed the typical inverse agonist behavior for the series, phenyl sulfonamide 3 was a high affinity ligand of RORc that showed strong agonist efficacy in the biochemical coactivator recruitment assay. Herein, we disclose the discovery of additional phenylsulfonamide and benzylsulfonamide pairs that demonstrate that a minor change in the structure of the ligand (i.e., one methylene group) can lead to agonist and inverse agonist MOAs, respectively.
To better understand how the changes to the sulfonamide groups affected the binding mode of the ligands and resulted in divergent MOAs, we cocrystallized inverse agonist ligand 2 and agonist ligand 3 with the human RORc-LBD and obtained 2.0 and 2.2 Å resolution X-ray structures, respectively.21 In both cases, the ligands were bound within the ligand binding pocket of RORc (Figure 2), and approximately one-third of the ligands (from the acetyl moiety through the piperazine ring) superimposed nearly identically. The trajectory of the compounds then diverged slightly, projecting their terminal phenyl rings from diverging vectors toward helix 11. The agonist ligand 3 (Figure 2, purple ribbon) packed against Trp317, His479, and Tyr502, stabilized the agonist conformation of helix 12, and recruited the coactivator peptide. On the other hand, the benzyl moiety of the inverse agonist ligand 2 (Figure 2, yellow ribbon) displaced His479, and prevented formation of the hydrogen bond with Tyr502 of helix 12. The disruption of the hydrogen bond between His479 and Tyr502 dislodged helix 12 entirely in the costructure with 2 and eliminated the binding site for coregulator proteins. This observation was consistent with the RORc inverse agonist activity of 2. The C-terminal half of helix 11 bent 16° inward and filled in the gap created while multiple aromatic side chains reoriented in a switch-like fashion, and repacked the apex of the pocket in the absence of helix 12. Similarly, concerted motions of Trp317 and Phe486 rotated, and filled pockets created by these changes. Both 2 and 3 formed a single hydrogen bond from the oxygen of their terminal acetyl groups to an ordered water molecule at the opposite end of the pocket from helix 12 (Figure S2). The rest of the contacts made to the compounds were van der Waals interactions with the predominantly hydrophobic binding site residues.
Figure 2.
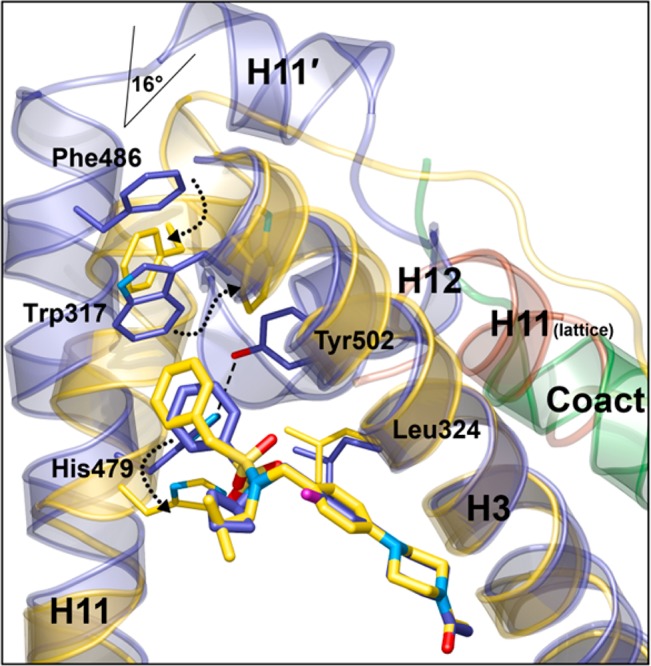
Binding modes of agonist ligand 3 and inverse agonist ligand 2 within the ligand binding pocket of RORc. The crystal structure of the agonist ligand 3 (purple ribbon) stabilized helix 12 and revealed a coactivator peptide (green) bound to the receptor. The crystal structure of the inverse agonist ligand 2 (yellow ribbon) did not have resolved density for helices 11–12. Concerted motions of Trp317, Phe486 and His479 are depicted with arrows.
To further evaluate the effects of different sulfonamide substituents on the MOA, we synthesized a series of benzyl- and arylsulfonamides using a late-stage diversification synthetic approach (Scheme 1). Benzaldehyde intermediate 7 was obtained using a Buchwald–Hartwig amination reaction22 between acetylpiperazine 5 and acetal-protected bromoarene 6, followed by acetal removal under acidic conditions. Subsequent reductive amination of aldehyde 7 with cyclobutylamine gave the advanced intermediate amine 8. This late-stage intermediate allowed for the incorporation of a diverse set of sulfonyl chlorides and gave sulfonamides 3, 4, and 9–33.23
Scheme 1. Synthesis of Sulfonamides 3, 4, and 9–33.

Reagents and conditions: (a) Pd(OAc)2, RuPhos, NaOt-Bu, 1,4-dioxane, 100 °C; (b) HCl, H2O, THF, 23 °C, 60% (2 steps); (c) cyclobutylamine, NaBH(OAc)3, AcOH, 1,2-DCE, 23 °C, quant.; (d) R-SO2Cl, DIPEA, CH2Cl2, 23 °C, 29–57%.
The analogs were then evaluated in two RORc biochemical assays.24 The affinity of the analogs for the RORc-LBD was measured using a radiometric binding assay that monitored the displacement of [3H2]25-hydroxycholesterol from the RORc-LBD. Compounds were also evaluated in a time-resolved fluorescence biochemical assay that monitored the ability of the human RORc-LBD to bind to a coactivator peptide derived from steroid receptor coactivator-1 (SRC1). Compounds disrupting the recruitment of the SCR1 peptide were determined to be inverse agonists whereas compounds that enhanced peptide recruitment were agonists. Alternatively, compounds that did not modulate the level of recruitment but showed affinity in the radiometric binding assay were identified as silent ligands. Full inverse agonism (−100% efficacy) was experimentally measured and corresponded to complete suppression of basal activity. Full agonism (+100% efficacy) was calculated as the additive inverse of a complete suppression of basal activity, since the experimental maximum % efficacy of a RORc full physiological agonist is currently undefined.
Matched pair analysis of the aryl- and benzylsulfonamides (Table 1) revealed that phenylsulfonamide 3 was a RORc agonist (EC50 = 69 nM, +35% efficacy) while the corresponding benzyl analog 4 was a potent full inverse agonist (EC50 = 11 nM, −99% efficacy). The phenethylsulfonamide (9) was also an inverse agonist, albeit with a significantly lower potency and efficacy (EC50 = 310 nM, −64% efficacy). We then sought to improve the potency of our ligands by introducing small nonpolar substituents around the arene to provide favorable van der Waals interactions with the ligand-binding pocket. The ligand-lipophilicity efficiency (LLE)25,26 was used as a metric to ensure the effective use of nonpolar interactions. Incorporation of a 2-fluorine substituent in compound 10 resulted in an equipotent and equi-efficacious agonist (EC50 = 63 nM, +46% efficacy) when compared to the phenyl analog 3. However, the same 2-fluoro substituent provided a more substantial potency improvement with inverse agonist benzyl compound 11 (EC50 = 3 nM, −98% efficacy). The LLE of compound 11 was also improved compared to compound 4. Similar trends were observed with 3-fluorophenylsulfonamide 12 and 3-fluorobenzylsulfonamide 13, with 12 being a slightly more potent agonist (EC50 = 41 nM, +46% efficacy) than 10. Interestingly, 4-fluoro substitution (14) led to a loss of effect on peptide recruitment and weak inverse agonism (EC50 > 10 μM, −37% efficacy), although this ligand demonstrated significant affinity for the receptor in the radioligand binding assay (RORc IC50 = 220 nM). The 4-fluorobenzylsulfonamide 15 was a potent inverse agonist (EC50 = 7 nM, −97% efficacy).
Table 1. Structure–Activity Relationships of Phenyl- and Benzylsulfonamide Matched Pairsa.
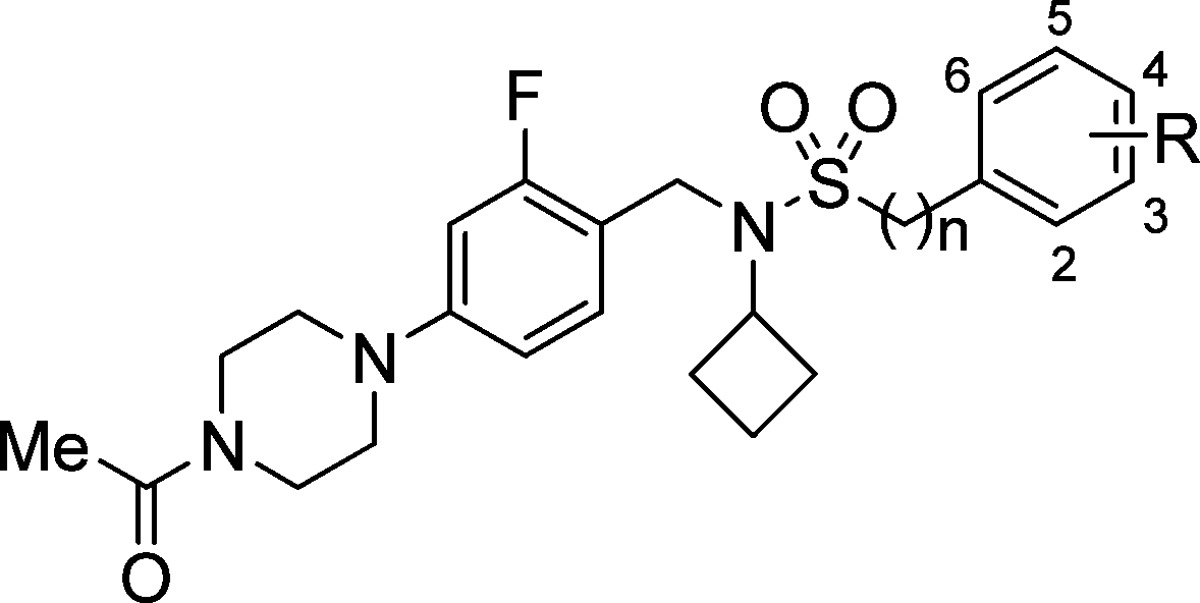
| Compd | n | R | RORc IC50b (μM) | RORc SRC1 EC50c (μM) [% efficacy] | cLogPd | LLEe | MOAf |
|---|---|---|---|---|---|---|---|
| 3 | 0 | H | 0.25 | 0.069 [+35%] | 3.7 | 3.5 | Agonist |
| 4 | 1 | H | 0.015 | 0.011 [−99%] | 3.3 | 4.7 | Inverse Agonist |
| 9 | 2 | H | 0.11 | 0.31 [−64%] | 3.7 | 2.8 | Inverse Agonist |
| 10 | 0 | 2-F | 0.076 | 0.063 [+46%] | 3.9 | 3.3 | Agonist |
| 11 | 1 | 2-F | 0.010 | 0.003 [−98%] | 3.4 | 5.1 | Inverse Agonist |
| 12 | 0 | 3-F | 0.065 | 0.041 [+46%] | 3.9 | 3.5 | Agonist |
| 13 | 1 | 3-F | 0.025 | 0.010 [−98%] | 3.5 | 4.5 | Inverse Agonist |
| 14 | 0 | 4-F | 0.22 | >10 [−37%] | 3.9 | - | Inverse Agonist |
| 15 | 1 | 4-F | 0.017 | 0.007 [−97%] | 3.5 | 4.7 | Inverse Agonist |
| 16 | 0 | 2-Cl | 0.055 | 0.047 [+33%] | 4.4 | 2.9 | Agonist |
| 17 | 1 | 2-Cl | 0.009 | 0.004 [−96] | 3.9 | 4.5 | Inverse Agonist |
| 18 | 0 | 3-Cl | 0.022 | 0.050 [+53%] | 4.4 | 2.9 | Agonist |
| 19 | 1 | 3-Cl | 0.019 | 0.009 [−98%] | 4.1 | 4.0 | Inverse Agonist |
| 20 | 0 | 4-Cl | 0.092 | 0.19 [−63%] | 4.4 | 2.3 | Inverse Agonist |
| 21 | 1 | 4-Cl | 0.037 | 0.033 [−97%] | 4.1 | 3.4 | Inverse Agonist |
| 22 | 0 | 3,5-diCl | 0.006 | 0.006 [+65%] | 5.1 | 3.4 | Agonist |
| 23 | 1 | 3,5-diCl | 0.029 | 0.040 [−92%] | 4.8 | 2.6 | Inverse Agonist |
| 24 | 0 | 3,5-diF | 0.049 | 0.014 [+43%] | 4.1 | 3.7 | Agonist |
| 25 | 0 | 3-OMe | 0.036 | 0.026 [+54%] | 3.8 | 3.8 | Agonist |
All assay results are reported as the geometric mean of at least two separate runs.24
Biochemical inhibition of the RORc-LBD and [3H2]25-hydroxycholesterol interaction.
Activation or inhibition of RORc-LBD recruitment of the SRC1 coactivator; positive “%eff.” denotes agonism and negative “%eff.” denotes inverse agonism relative to the basal assay signal for apo-RORc LBD in this assay format.
Calculated logP (cLogP) value.27
Ligand-lipophilicity efficiency (LLE)25 was calculated using RORc SRC1 EC50 and the cLogP.
Mechanism of action (MOA) reported as agonist, inverse agonist, or silent ligand.
A similar scan was performed with 2-, 3-, and 4-chloro substituted phenyl- and benzylsulfonamides 16–21. The 2- and 3-chlorophenylsulfonamides (16 and 18, respectively) were both similarly potent agonists (EC50 = 47 nM, +33% efficacy and 50 nM, +53% efficacy, respectively), and their benzylsulfonamide counterparts 17 and 19 displayed full inverse agonist MOA with RORc SRC1 EC50 values of 4 nM and 9 nM, respectively. Substitution at the 4-position of arylsulfonamide 20 with the chloride atom led to a reversal of MOA, giving rise to an inverse agonist (EC50 = 190 nM, −63% efficacy), when compared to 3. The 4-chlorobenzyl (21) matched pair to 20 also showed inverse agonism (EC50 = 33 nM, −97% efficacy), although not as potent as other substituted benzylsulfonamides. Finally, the 3,5-dichloroarylsulfonamide 22 gave rise to our most potent agonist (EC50 = 6 nM, +65% efficacy), albeit with a poor LLE of 3.4 resulting from the high lipophilicity of the dichloroarene. Conversely, the 3,5-dichlorobenzyl analog 23 was a potent inverse agonist (EC50 = 40 nM, −92% efficacy). Replacing the chlorine atoms of 22 with less lipophilic fluorines (24) led to a higher LLE agonist (EC50 = 14 nM, +43% efficacy, LLE = 3.7). A further reduction in lipophilicity was accomplished by appropriately incorporating a 3-methoxy substituent (25), which led to further improvement of the LLE (EC50 = 26 nM, +54% efficacy, LLE = 3.8). This initial SAR on the right-hand side (RHS) identified single-digit nanomolar analogs with both agonist and inverse agonist profiles. Phenylsulfonamides generally behaved as agonists of RORc, while the benzylsulfonamides acted as inverse agonists.
Intrigued by the reversal of MOA of 4-substituted arylsulfonamides 14 and 20, we sought to rationalize this phenomenon by further examining the X-ray costructure of agonist 3. The unsubstituted phenyl ring of 3 was directed at Trp317 and formed an edge-to-face π–π interaction (3.6 Å),28 which in turn formed a π stacking interaction with Phe486 (4.2 Å), stabilizing helix 11 and the bound ligand. Substitution at the 4-position on the phenyl ring in this binding mode would clash with Trp317. This could cause the pocket to adopt a different conformation and ultimately disrupt helices 11–12. We hypothesized that while no substitution at the 4-position (R = H, 3) led to the observed agonist binding mode in the RORc SRC1 assay, changes to a small substituent at the 4-position (R = F, 14) would begin destabilizing the C-terminus and prevent SRC1 peptide recruitment beyond the basal level, leading to low inverse agonist efficacy or silent MOA ligands. Further steric bulk directed toward the C-terminal region (R = Cl, 20) would fully disrupt the secondary structure of that region of the protein and prevent RORc interaction with the coactivator peptide, resulting in a stronger inverse agonist MOA.29
With this consideration in mind, we synthesized additional analogs bearing incrementally bulkier functionalities at the para position of the arylsulfonamide (Table 2) to further explore the level of efficacy of inverse agonism that could be achieved. Compound 26, bearing a 4-methyl substituent, showed significant affinity for RORc in the binding assay (RORc IC50 = 24 nM) but behaved as silent ligand in the RORc SRC1 biochemical assay. Additionally, a more polarized 4-cyano substituent30 possessed moderate RORc binding affinity, as exemplified with compound 27 (RORc IC50 = 280 nM). However, the steric bulk of the nitrile group was not sufficient to impart full RORc inverse agonism in the RORc SRC1 biochemical assay (EC50 > 10 μM, −29% efficacy). Substitution with a 4-methoxy group ultimately provided enough steric bulk and gave rise to stronger RORc inverse agonist 28 (EC50 = 3.3 μM, −52% efficacy). Further increasing the steric demand of the ligand with dichloro substitution (29) or with 4-difluoromethoxy (30), 4-trifluoromethoxy (31), 4-trifluoromethyl (32), or 4-tert-butyl (33) groups afforded similarly potent RORc inverse agonists with ranging efficacy of −37%, −75%, −82%, −39%, and −55%, respectively, with 33 being the most potent (EC50 = 120 nM). In summary, we demonstrated that arylsulfonamide agonists could be transformed into silent ligands by adding steric bulk to the 4-position of the arene. Additional steric hindrance at the 4-position eventually led to a RORc inverse agonist MOA.
Table 2. Structure–Activity Relationship of 4-Substituted Arylsulfonamidesa.
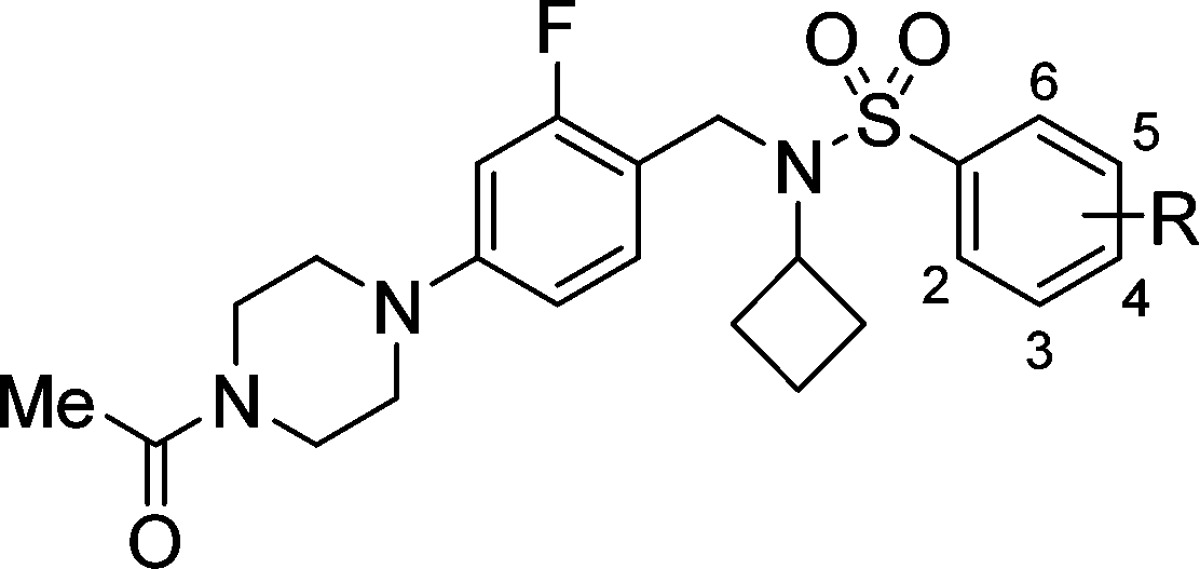
| Compd | R | RORc IC50b(μM) | RORc SRC1 EC50c (μM) [% efficacy] | cLogPd | LLEe | MOAf |
|---|---|---|---|---|---|---|
| 26 | 2,4-diMe | 0.024 | >10 [+9%] | 4.7 | Silent | |
| 27 | 4-CN | 0.28 | >10 [−29%] | 3.4 | Inverse Agonist | |
| 28 | 4-OMe | 0.12 | 3.3 [−52%] | 3.8 | 1.7 | Inverse Agonist |
| 29 | 2,4-diCI | 0.076 | 0.22 [−37%] | 5.2 | 1.5 | Inverse Agonist |
| 30 | 4-OCF2H | 0.031 | 0.15 [−75%] | 4.1 | 2.7 | Inverse Agonist |
| 31 | 4-OCF3 | 0.29 | 0.16 [−82%] | 4.9 | 1.9 | Inverse Agonist |
| 32 | 4-CF3 | 0.050 | 0.30 [−39%] | 4.6 | 1.9 | Inverse Agonist |
| 33 | 4-t-Bu | 0.018 | 0.12 [−55%] | 5.3 | 1.6 | Inverse Agonist |
All assay results are reported as the geometric mean of at least two separate runs.
Biochemical inhibition of the RORc-LBD and [3H2]25-hydroxycholesterol interaction.
Activation or inhibition of RORc-LBD recruitment of the SRC1 coactivator; positive “%eff.” denotes agonism, and negative “%eff.” denotes inverse agonism relative to the basal assay signal for apo-RORc LBD in this assay format.
Calculated logP (cLogP) value.27
Ligand-lipophilicity efficiency (LLE)25 was calculated using RORc SRC1 EC50 and the cLogP.
Mechanism of action (MOA) reported as agonist, inverse agonist, or silent ligand.
We also explored the effect of replacing the sulfonamide moiety in 3 and 4 with an amide functional group. Both phenyl- and benzylamide compounds showed <50% inhibition in the SRC1 peptide recruitment assay at a 10 μM concentration. Sulfonamides typically adopt a nonplanar conformation31 that was also observed in the cocrystal structures of compounds 2 and 3 with the RORc-LBD. Presumably, the planar conformation of the amide did not allow for the amide analogs to effectively bind to RORc-LBD and affect the RORc SRC1 peptide recruitment assay.
We progressed two leading agonist/inverse agonist matched pairs (3–4 and 10–11) into a human peripheral blood mononuclear cell (PBMC) cytokine production assay to evaluate their effects on IL-17 production (Table 3).24 RORc agonists 3 and 10 gave rise to an increased production of IL-17 (EC50 = 0.78 μM, +77% efficacy and 0.80 μM, +71% efficacy, respectively), which was in agreement with their MOA determined in the RORc SRC1 biochemical assay. Conversely, compounds 4 and 11, which were shown to be RORc inverse agonists in the SRC1 peptide recruitment biochemical assay, both led to a suppression of IL-17 production in the human PBMC assay (EC50 = 0.35 μM, −79% efficacy and 0.32 μM, −72% efficacy, respectively). In addition, none of the tested compounds affected interferon-γ (INF-γ) biosynthesis or cell health as assessed by CellTiter-Glo (CTG) cellular ATP measurements, confirming that the compounds did not exhibit off-target cytokine activity or gross toxicity. These results demonstrated that closely related compounds with only minor structural differences could be ligands that increased (phenylsulfonamides) or decreased (benzylsulfonamides) IL-17 production in human PBMCs.
Table 3. Potency and Mechanism of Action of Phenyl- and Benzylsulfonamides in hPBMC IL-17 Production Assaya.
| Compd | IL-17 PBMC EC50 (μM) | IL-17 PBMC efficacy (%) | IFNγ EC50 (μM) | CTG EC50 (μM) |
|---|---|---|---|---|
| 3 | 0.78 | +77 | >10 | >20 |
| 4 | 0.35 | –79 | >10 | >20 |
| 10 | 0.80 | +71 | >10 | >20 |
| 11 | 0.32 | –72 | >10 | >20 |
All assay results are reported as the geometric mean of at least two separate runs. All assays were conducted using peripheral blood mononuclear cells (PBMCs) isolated from human whole blood.24 Positive %efficacy denotes a potentiation of IL-17 production. Negative %efficacy denotes an inhibition of IL-17 producton. Interferon-γ (INF-γ) and CellTiter-Glo (CTG) cell measurement assays were used as positive controls to monitor for non-TH17 cell cytokine activity and adverse off-target effects on cell health, respectively.24
The potent RORc inverse agonist 11 (RORc SRC 1 EC50 = 3.1 nM, IL-17 PBMC EC50 = 320 nM) was profiled for its selectivity in a panel of HEK293 cell Gal4-ROR construct human NR-dependent transcriptional reporter assays. The panel included the three isoforms of ROR (RORa, RORb, and RORc) and monitored for suppression of basal transcription activity in the absence of any exogenous agonist.24 In addition, the following NRs were evaluated in both agonist mode (no exogenous agonist ligand added) and antagonist mode (exogenous agonist ligand added): farnesoid X receptor (FXR), liver X receptors (LXR)-α and LXRβ, and pregnane X receptor (PXR).24 Compound 11 was a potent inverse agonist of RORc in the Gal4 reporter assay (EC50 = 15 nM) and showed no detectable activity against the RORa isoform while it was 180-fold selective against RORb (EC50 = 2.7 μM). The compound also displayed favorable selectivity against other NRs in the antagonist mode. For example, it was >400-fold selective over FXR (EC50 = 6.1 μM) and >600-fold selective over LXRα (EC50 = 9.2 μM). No activity was detected against LXRβ and PXR. Additionally, 11 did not exhibit any agonist activity in all the NRs profiled.
In conclusion, we have demonstrated that a minor structural change to our tertiary sulfonamide series led to opposite mechanisms of action with RORc in both biochemical and primary human blood cell assays. We utilized X-ray cocrystal structural data to identify and characterize two distinct binding modes consistent with these biochemical and cellular MOAs. Optimized phenylsulfonamides were identified as RORc agonists while benzylsulfonamides exhibited potent inverse agonist activity. Our most potent inverse agonist showed >180-fold selectivity over all other RORc isoforms and other nuclear receptors that were profiled. Finally, compounds behaving as agonists in our biochemical assay also gave rise to an increased production of IL-17 in human PBMCs, while inverse agonists led to significant suppression of IL-17 under the same assay conditions. Thus, we were able to observe translation of biochemical MOA into the same MOA in human primary blood cells.
Supporting Information Available
Cocrystal structure data and compound characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
We thank Drs. Krista Bowman and Jiansheng Wu, and their respective Genentech research groups, for performing protein expression and purification activities. We also thank Crystallographic Consulting, LLC for diffraction data collection. Diffraction data were collected at beamline 08ID-1 at the Canadian Light Source, which was supported by the NSERC, the NRC, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan at beamline 5.0.2 of the Advanced Light Source. The Berkeley Center for Structural Biology was supported in part by the NIH, the NIGMS, and the Howard Hughes Medical Institute. The Advanced Light Source was supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hirose T.; Smith R. J.; Jetten A. Biochem. Biophys. Res. Commun. 1994, 205, 1976. [DOI] [PubMed] [Google Scholar]
- Ivanov I. I.; McKenzie B. S.; Zhou L.; Takodoro C. E.; Lepelley A.; Lafaille J. J.; Cua D. J.; Littman D. R. Cell 2006, 126, 1121. [DOI] [PubMed] [Google Scholar]
- Sabat R.; Ouyang W.; Wolk K. Nat. Rev. Drug Disc. 2014, 13, 21. [DOI] [PubMed] [Google Scholar]
- Codarri L.; Gyülvészi G.; Tosevski V.; Hesske L.; Fontana A.; Magnenat L.; Suter T.; Becher B. Nat. Immunol. 2011, 12, 560. [DOI] [PubMed] [Google Scholar]
- Rich P.; Sigurgeirsson B.; Thaci D.; Ortonne J.-P.; Paul C.; Schopf R. E.; Morita A.; Roseau K.; Harfst E.; Guettner A.; Machacek M.; Papavassilis C. Br. J. Dermatol. 2013, 168, 402. [DOI] [PubMed] [Google Scholar]
- Chabaud M.; Durand J. M.; Buchs N.; Fossiez F.; Page G.; Frappart L.; Miossec P. Arthritis Rheum. 1999, 42, 963. [DOI] [PubMed] [Google Scholar]
- Lock C.; Hermans G.; Pedotti R.; Brendolan A.; Schadt E.; Garren H.; Langer-Gould A.; Strober S.; Cannella B.; Allard J.; Klonowski P.; Austin A.; Lad N.; Kaminski N.; Galli S. J.; Oksenberg J. R.; Raine C. S.; Heller R.; Steinman L. Nat. Med. 2002, 8, 500. [DOI] [PubMed] [Google Scholar]
- Fujino S.; Andoh A.; Bamba S.; Ogawa A.; Hata K.; Araki Y.; Bamba T.; Fujiyama Y. Gut 2003, 52, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojetin D. J.; Burris T. P. Nat. Rev. Disc. 2014, 13, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauber B. P.; Magnuson S. J. Med. Chem. 2014, 57, 5871. [DOI] [PubMed] [Google Scholar]
- Yang J.; Sundrud M. S.; Skepner J.; Yamagata T. Trends Pharmacol. Sci. 2014, 35, 493. [DOI] [PubMed] [Google Scholar]
- For a description of the transcription level modulation pathways for NRs, see ref (10).
- For a description of the nomenclature of ROR-family modulators, see:Schupp M.; Lazar M. A. J. Biol. Chem. 2010, 285, 40409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review on NR nomenclature, see:Germain P.; Staels B.; Dacquet C.; Spedding M.; Laudet V. Pharmacol. Rev. 2006, 58, 685. [DOI] [PubMed] [Google Scholar]
- Solt L. A.; Griffin P. R.; Burris T. P. Curr. Opin. Lipidol. 2010, 21, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauber B. P.; René O.; Burton B.; Everett C.; Gobbi A.; Hawkins J.; Johnson A. R.; Liimatta M.; Lockey P.; Norman M.; Wong H. Bioorg. Med. Chem. Lett. 2014, 24, 2182. [DOI] [PubMed] [Google Scholar]
- Fauber B. P.; René O.; de Leon Boenig G.; Burton B.; Deng Y.; Eidenschenk C.; Everett C.; Gobbi A.; Hymowitz S. G.; Johnson A. R.; La H.; Liimatta M.; Lockey P.; Norman M.; Ouyang W.; Wanga W.; Wong H. Bioorg. Med. Chem. Lett. 2014, 24, 3891. [DOI] [PubMed] [Google Scholar]
- For a review on the structure of NRs, see:Huang P.; Chandra V.; Rastinejad F. Annu. Rev. Physiol. 2010, 72, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita-Sato S.; Ito S.; Isobe T.; Ohyama T.; Wakabayashi K.; Morishita K.; Ando O.; Isono F. J. Biol. Chem. 2011, 286, 31409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauber B. P.; de Leon Boenig G.; Burton B.; Eidenschenk C.; Everett C.; Gobbi A.; Hymowitz S. G.; Johnson A. R.; Liimatta M.; Lockey P.; Norman M.; Ouyang W.; René O.; Wong H. Bioorg. Med. Chem. Lett. 2013, 23, 6604. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for the experimental details associated with the cocrystal structures of the RORc-LBD with compounds 2 and 3. The coordinates and structure factors for the complex between RORc and compounds 2 and 3 have been deposited with the protein data bank and assigned the accession codes 4WQP and 4WPF, respectively.
- Jiang L.; Buchwald S.. Palladium-Catalyzed Aromatic Carbon-Nitrogen Bond Formation. In Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; de Meijere A., Diederich F., Eds.; Wiley-VCH GmbH & Co. KGaA: Weinheim, 2004; Vol. 2, pp 699–760. [Google Scholar]
- For representative synthetic protocols, see:Fauber B. P.; René O.. WO 2013/092941.
- See the Supporting Information of ref (20). for the biochemical and cellular assay protocols.
- Leeson P. D.; Springthorpe B. Nat. Rev. Drug Disc. 2007, 6, 881. [DOI] [PubMed] [Google Scholar]
- Leach A. R.; Hann M. M.; Burrows J. N.; Griffen E. J. Mol. BioSyst. 2006, 2, 429. [DOI] [PubMed] [Google Scholar]
- The logP was calculated with internal software using the VolSurf approach. For additional details, see:Cruciani G.; Crivori P.; Carrupt P.- A.; Testa B. THEOCHEM 2000, 503, 17. [DOI] [PubMed] [Google Scholar]
- Salonen L. M.; Ellermann M.; Diederich F. Angew. Chem., Int. Ed. 2011, 50, 4808. [DOI] [PubMed] [Google Scholar]
- Another team has noted similar effects with RORc inverse agonists. For details, see:Yang T.; Liu Q.; Cheng Y.; Cai W.; Ma Y.; Yang L.; Wu Q.; Orband-Miller L. A.; Zhou L.; Xiang Z.; Huxdorf M.; Zhang W.; Zhang J.; Xiang J.-N.; Leung S.; Qiu Y.; Zhong Z.; Elliott J. D.; Lin X. ACS Med. Chem. Lett. 2014, 5, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming F. F.; Yao L.; Ravikumar P. C.; Funk L.; Shook B. C. J. Med. Chem. 2010, 53, 7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin A.; Collins A.; Gilmore C. J.; Wilson C. C. Acta Crystallogr., Sect. B: Struct. Sci. 2008, 64, 66. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.