

Abstract

Novel acetylenyl-containing benzamide derivatives were synthesized and screened using an in vitro assay measuring increases in glucokinase activity stimulated by 10 mM glucose concentration and glucose uptake in rat hepatocytes. Lead optimization of an acetylenyl benzamide series led to the discovery of several active compounds via in vitro enzyme assays (EC50 < 40 nM) and in vivo OGTT assays (AUC reduction > 40% at 50 mg/kg). Of the active compounds tested, 3-(3-amino-phenylethynyl)-5-(2-methoxy-1-methyl-ethoxy)-N-(1-methyl-1H-pyrazol-3-yl)-benzamide (19) was identified as a potent glucokinase activator exhibiting an EC50 of 27 nM and eliciting a 2.16-fold increase in glucose uptake. Compound 19 caused a glucose AUC reduction of 47.4% (30 mg/kg) in an OGTT study in C57BL/6J mice compared to 22.6% for sitagliptin (30 mg/kg). Single treatment of the compound 19 in C57BL/6J mice elicited basal glucose lowering activity without any significant evidence for hypoglycemia risk. Compound 19 was therefore selected as a candidate for further preclinical development for the treatment of type 2 diabetes.
Keywords: Type 2 diabetes mellitus, T2DM, glucokinase, glucokinase activator, GKA, acetylenyl benzamide derivatives
Type 2 diabetes mellitus (T2DM) has established itself as a rapidly growing public epidemic now affecting over 300 million people worldwide. The first-line oral therapy for T2DM is metformin and there exist several second-line oral therapies together with which it is administrated in combination, including dipeptidyl peptidase-4 (DPP-4) and sodium-glucose cotransporter 2 (SGLT2) inhibitors. However, currently available antidiabetic agents have limited long-term efficacy and/or significant side effects. To address these clinical unmet needs for T2DM patients, great efforts have been made to develop new therapeutics, focusing on safety and efficacious glycemic control.
Glucokinase (GK, also called hexokinase IV or hexokinase D) is a hexokinase isozyme of 465 amino acids (molecular weight = 50 kDa). GK facilitates phosphorylation of glucose to glucose-6-phosphate and is expressed in cells of the liver, pancreas, gut, and brain of humans and many other vertebrates. GK activity1−3 varies substantially with the concentration of glucose present, unlike other hexokinases. Glucokinase activator (GKA) is associated with a dual mechanism for lowering blood glucose concentrations by enhancing glucose uptake in the liver and increasing insulin secretion from pancreatic beta cells. Therefore, GK has become an attractive target for antidiabetic therapy over the last two decades. Several GKA candidates have advanced to clinical studies4 and have been shown to lower both fasting and postprandial glucose levels in healthy subjects and T2DM patients. Hypoglycemia and liver or testicular toxicity5 have been revealed as the primary adverse effects of concern for GKAs. To address the hypoglycemia issue, several clinical strategies have been utilized including dose titration, more frequent dosing times, and the design of partial or liver-selective glucose activators.6,7 Although significant progress in GKA development has not been achieved to date,8 several small molecule GKAs are currently in clinical studies (phase I and II).
A number of allosteric small molecule GKAs have been investigated by numerous research groups over the past decade4 (representative small molecule GKAs are shown in Figure 1). Since the first report of small molecule allosteric GKAs in 2003,9 a phenylacetamide series (110 and 2(11)) a number of benzamides (3,124,13 and 5(4)) and an imidazolylacetamide (6)6 have also been identified as potent GKAs. GKAs putatively bind to allosteric sites of the protein to achieve antihyperglycemic effects, according to structural information derived from GK-GKA complexes published in 2009.14,15 More recently, Park et al. identified novel phenylethyl benzamide GKAs, 7a (YH-GKA)16 and pyridyl benzamide 7b,17 which exhibit good biological and pharmacological activities with favorable pharmacokinetics. Herein, we report the lead optimization of an acetylenyl containing benzamide series for the development of novel glucokinase activators for the treatment of type 2 diabetes mellitus.
Figure 1.

Representative structures of GKAs.
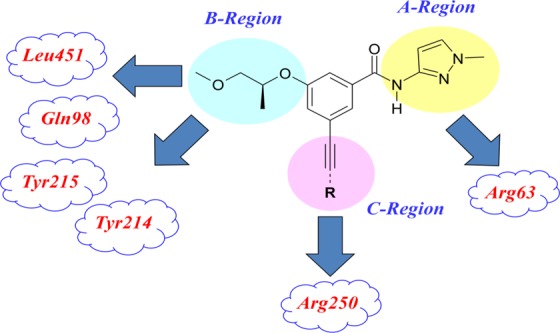
The benzamide scaffold was chosen as a starting point for the synthesis due to the existence of unambiguous structural information from the known cocrystal complexes.14 Through initial diversification and structure–activity relationship (SAR) analysis of the acetylenyl containing benzamide scaffold, the 1-methyl-1H-pyrazol moiety was selected in the A-region for a strong hydrogen bond interaction with ARG63, and the (R)-(−)-2-methoxy-1-methyl-ethoxy moiety was fixed in the B-region for a hydrophobic interaction with TYR214, TYR215, and GLY97 (Figure 2).
Figure 2.
Synthetic strategy for benzamide GKAs.
The pocket in the C-region of the enzyme is relatively large and the end portion of the C-moiety has the potential for a hydrogen bond interaction with Arg250, which may increase binding affinity. Therefore, the C-region was analyzed systematically by introducing a variety of acetylenyl containing alkyl, aryl, or heteroaryl groups, which led to the identification of several active moieties in the C-region as targets for GKA compounds, as summarized in Tables 1 and 2. The synthesis of acetylenyl containing benzamide derivatives is described in Schemes 1–3.
Table 1. In Vitro Glucokinase Activity of the Acetylenyl Benzamide Derivatives 13a–b, 16a–c, 18a–d, 19, and 21.
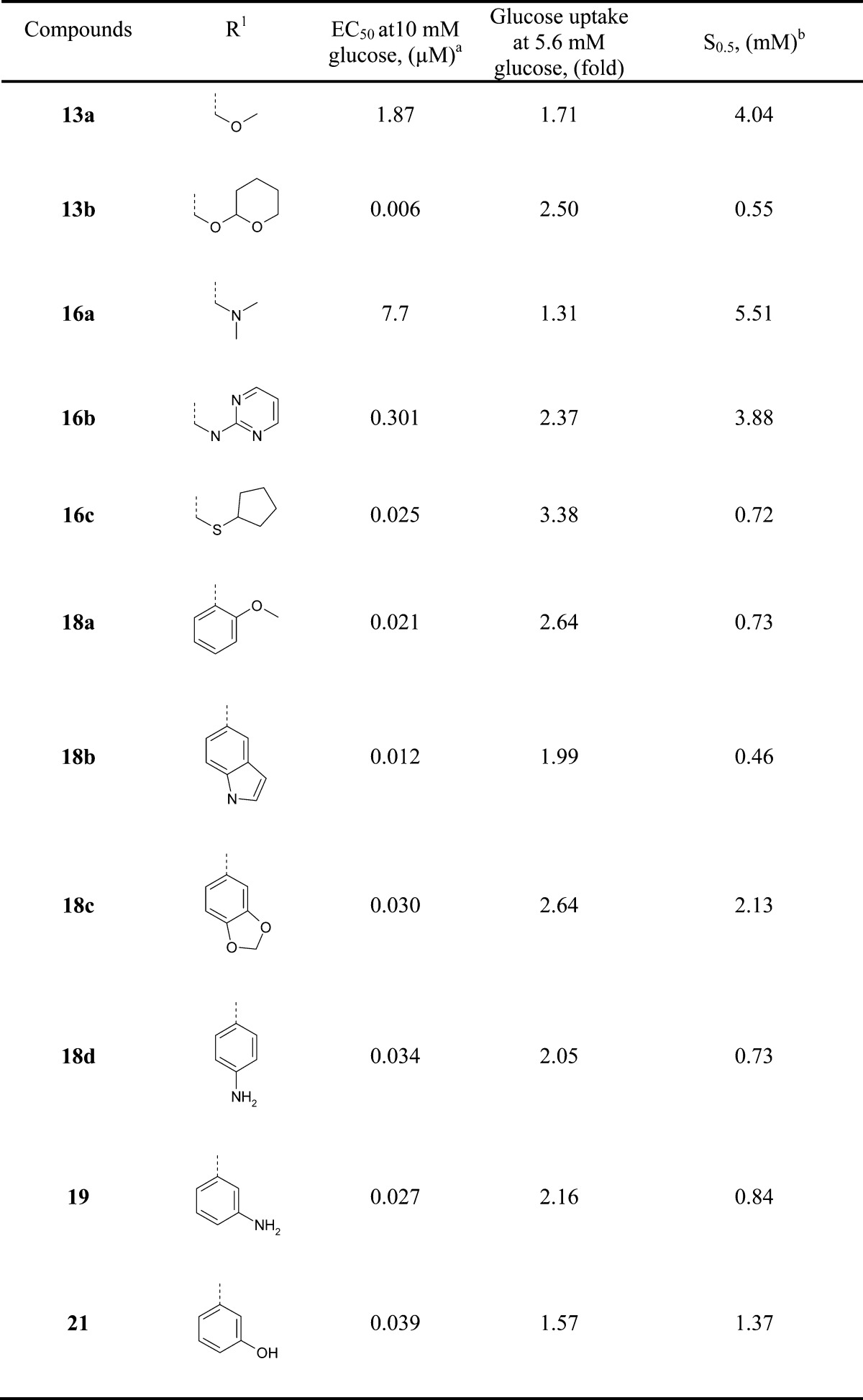
Data represented as the mean values of data obtained from triplicate runs.
Recombinant human pancreatic glucokinase was used, and the activity was measured at 1 μM concentration.
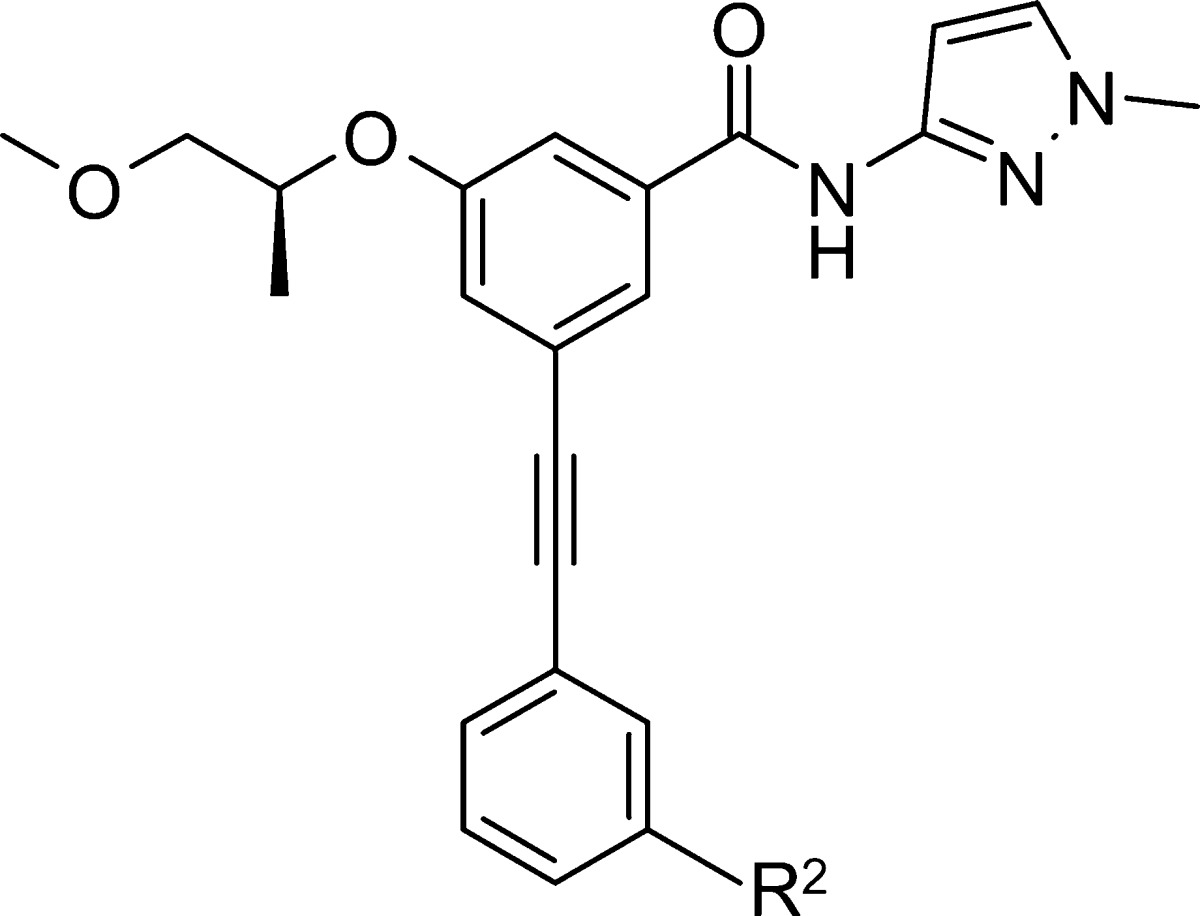
Table 2. In Vitro Glucokinase Activity of m-Substituted Ethynylphenyl Benzamide Derivatives 20a–h and 22a–e.
Data represented as the mean values of data obtained from triplicate runs.
Recombinant human pancreatic glucokinase was used, and the activity was measured at 1 μM concentration.
Scheme 1.

Reagents and conditions: (a) (i) MeOH, c.H2SO4, reflux; (ii) (R)-(−)-1-methoxy-2-propanol, DIAD, PPh3, THF, 0 °C → RT; (b) NaOH, THF/MeOH/H2O; (c) 1-methyl-1H-pyrazol-3-amine, EDAC, HOBT, DIEA, CH2Cl2; (d) TBAF, PdCl2(PPh3)2, tetrahydro-2-(2-propynyloxy)-2H-pyran, 85 °C; (e) p-toluenesulfonic acid, MeOH; (f) PBr3, THF; (g) R1OH, Cs2CO3, DMF; (h) PdCl2(PPh3)2, substituted aryl acetylene (R2CCH), TBAF, 85 °C; (i) PdCl2(PPh3)2, Boc-propargylamine, TBAF, 85 °C; (j) (i) TFA, CH2Cl2, 0 °C → RT; (ii) R3Cl, K2CO3, KI, DMF, 80 °C.
Scheme 3.

Reagents and conditions: (a) PdCl2(PPh3)2, 3-ethynylaniline, TBAF, 85 °C; (b) method (A) R2Cl, DIEA, NMP, 160 °C; method (B) R2Cl, TEA, CH2Cl2, rt.; (c) PdCl2(PPh3)2, 3-ethynylphenol, TBAF, 85 °C; (d) method (A) R4Cl, DIEA, NMP, 160 °C; method (B) R4Cl, TEA, CH2Cl2, rt.
Various propargyl or acetylenyl groups containing benzamides were synthesized as shown in Scheme 1. The synthesis began with a Mitsunobu reaction of 3-bromo-5-hydroxy benzoic acid with (R)-(−)-1-methoxy-2-propanol, known for an optimized moiety in the A-region. Then, ester 8 was hydrolyzed to give acid 9, and subsequent amide coupling of acid 9 with 1-methyl-1H-pyrazol-3-amine provided the key intermediate benzamide 10. The acetylenyl group was introduced to a meta-position on the benzamide ring using Pd-mediated coupling reactions. The coupling reaction between compound 10 and tetrahydro-2-(2-propynyloxy)-2H-pyran followed by removal of the pyran protection group gave 11. The resulting propargyl alcohol 11 was treated with PBr3 to afford propargyl bromide 12. Various propargyl alkoxy analogues 13 were synthesized by simple alkylation reactions of 12. Compound 10 was also coupled with substituted aryl acetylene compounds or Boc-propargylamine to give 14 and 15, respectively. Removal of the Boc protection group followed by alkylation reactions of the resulting amine with a variety of alkyl chloride provided analogues 16.
More substituted aryl acetylenyl benzamide derivatives 18 were prepared from the key intermediate 10 in two steps as shown in Scheme 2. A Sonogashira coupling reaction of 10 with trimethylsilylacetylene followed by Pd-catalyzed coupling reactions of acetylene 17 with a variety of aryl iodides yielded aryl acetylenyl benzamides 18.
Scheme 2.

Reagents and conditions: (a) (i) trimethylsilylacetylene, PdCl2(dppf), TEA, CuI; (b) substituted aryl iodide (RPhI), PdCl2(PPh3)2, TBAF, 85 °C.
meta-Acetylenyl N-substituted anilines 20 and 1,3-alkoxy acetylenly benzene 22 were prepared by Pd-catalyzed coupling reactions with 3-ethynylaniline or 3-ethynylphenol followed by SN2 alkylation reaction of the resulting aniline 19 or phenol 21 with appropriate alkyl chlorides in the presence of a base (Scheme 3).
Biological activity data for the selected acetylenyl benzamide derivatives have been summarized in Tables 1 and 2. Among the compounds in Table 1, 13b showed excellent potency with an EC50 of 6 nM. Several compounds including 16c, 18a, 18d, 19, and 21, which contain similar-sized moieties to the moiety of 13b in the R1 position, exhibited favorable enzyme activity (EC50 < 40 nM) presumably due to the van der Waals interaction in the C-pocket of glucokinase. In addition, the compounds 18b and 18c containing a bicyclic moiety such as indole and benzo[1,3]dioxole in the R1 position showed favorable activity with EC50 values of 12 and 30 nM, respectively. However, a small R1 group showed a tendency to decrease enzyme potency as seen in compounds 13a and 16a. On the basis of the potency and physicochemical properties of the compounds, more derivatives of aniline 19 and phenol 21 were synthesized for further establishment of the structure–activity relationship. For the aniline series shown in Table 2, N1,N1-dimethylethane-1,2-diamine (20c), 2-pyrrolidin-1-yl-ethylamine (20d) and 2-piperidin-1-yl-ethylamine (20e) in the R2 position led to an improvement in the potency of the enzyme (EC50 = 6 nM) by 4.5-fold compared to a simple amine (NH2) moiety in the same position (19, EC50 = 27 nM), while compound 20f containing a 2-morpholin-1-yl-ethylamine moiety (EC50 = 20 nM) and compound 20a with an N1,N1-dimethyl moiety (EC50 = 37 nM) in the R2 position maintained similar potency to 19. In addition, heterocyclic ethylamine such as 2-(2-methyl-imidazol-1-yl)-ethylamine (20h) as an R2 group improved enzyme potency by 3-fold (EC50 for 20h = 9 nM). In contrast, N1,N1-diethyl amine (20b) in the R2 position slightly decreased the potency by 6-fold (EC50 = 163 nM). Furthermore, a similar SAR trend has been observed in a 3-phenol series (21) compared to a 3-anline series (19). 2-Pyrrolidin-1-yl-ethyloxy (22c) and 2-piperidin-1-yl-ethyloxy (22d) as an R2 moiety improved the potency of enzyme activity by 6-fold (EC50 = 6–7 nM) compared to an OH moiety in the R2 position (21, EC50 = 39 nM), while a 2-morpholin-1-yl-ethyloxy moiety in the R2 position (22e) slightly increased the potency by 2.4-fold (EC50 = 16 nM). The compounds 22a–b containing relatively small groups in the R2 position maintained similar potency to compound 21. Other derivatives of 4-aminophenylethynyl series (18d) were synthesized for further investigation, but no significant increases in enzyme potency were observed (data not shown).
The improvement in potency appears to result from the size, ionic, hydrophobic, and hydrogen bonding effects of the moiety in the C-region of the GK pocket and/or the electrostatic interaction with the pocket. According to a binding mode analysis of 19 with the binding site of glucokinase generated by Surflex-Dock18 (Figure 3), the 1-methyl-1H-pyrazol moiety elicits a strong hydrogen bond with ARG63 and the (R)-(−)-2-methoxy-1-methyl-ethoxy moiety is oriented toward the hydrophobic pocket surrounded by TYR214, TYR215, and GLY97. 3-Amino-phenylethynyl group orients to the C-region in the GK pocket, which is relatively larger than the A- or B-region, and the amine group can be extended to the solvent-exposed region for hydrogen bonding with ARG250.
Figure 3.
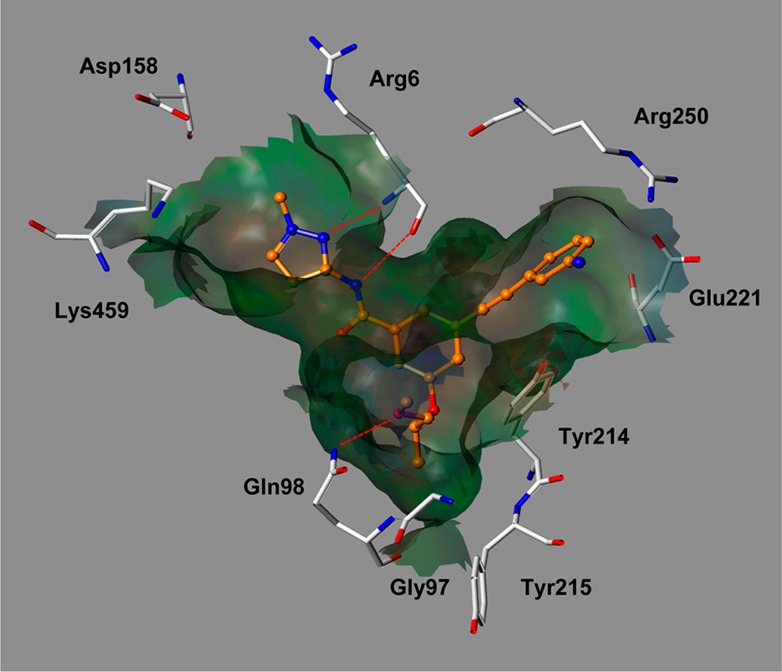
Docked conformer of 19 (at the active site of glucokinase as generated by Surflex-Dock), based on the X-ray structure of the known cocrystallized ligand. Specific binding of 19 to glucokinase shows potential hydrogen bonds in red dotted lines.
Most of the submicromolar active compounds in the GK enzyme assay elicited favorable glucose uptake activity with >1.7-fold increase under glucose stimulated conditions (glucose of 0 mM → 5.6 mM). Compounds that exhibited EC50 values of <300 nM in the GK activity assay, >2.0-fold increase in the glucose uptake assay, and good physicochemical properties were chosen for in vivo oral glucose tolerance test (OGTT) experiments (Table 3). Unsurprisingly, many compounds measured by in vivo OGTT showed significant area under the curve (AUC) reduction (>40%) at 30 or 50 mg/kg including several compounds that exhibited >50% AUC reduction (18d, 19, 20c–e, and 22c). Of the active compounds in the in vivo OGTT, compound 19 was chosen for dose-dependent in vivo OGTT and the fasted glucose tolerance test. Compound 19 exhibited human pancreatic glucokinase activity of EC50 = 27 nM at 10 mM glucose with a maximum reaction rate (Vmax) of 126% and a half maximal saturation concentration (S0.5)3 of 0.84 mM. Compound 19 also enhanced glucose uptake in rat primary hepatocytes by 2.16-fold, did not affect hexokinase I or II, and increased insulin secretion by 2.13-fold from rat pancreatic islets under glucose stimulated conditions (glucose of 0 mM → 5.6 mM). In addition, compound 19 exhibited favorable pharmacokinetics19 and improved oral glucose tolerance in C57BL/6J mice in a dose-dependent manner (Figure 4). In vivo OGTT values for 19 at 30 mg/kg in C57Bl/6J mice revealed a blood glucose AUC reduction of 47.4%, which was significantly greater than that of 22.6% for sitagliptin at 30 mg/kg. The fasted glucose tolerance test of 19 due to fasting without glucose challenge indicated that the mean value of glucose level after 60 min was maintained above 55 mg/dL, the minimum glucose level at which symptoms of hypoglycaemia do not occur, and showed very similar results as for GKA50,20 a potent GKA compound from AstraZeneca (Figure 5).
Table 3. In Vivo OGTT Results for the Selected Acetylenyl Benzamide Derivatives.
| Compounds | OGTT at 50 mg/kg, (AUC reduction, %) |
|---|---|
| 13b | 44.7 |
| 18a | 49.1a |
| 18d | 53.5 |
| 19 | 52.1, 47.4a |
| 20a | 43.7 |
| 20c | 56.0a |
| 20d | 51.4 |
| 20e | 54.0 |
| 20f | 47.9 |
| 20g | 41.0a |
| 22c | 51.6 |
| 22d | 46.0 |
Measured OGTT at 30 mpk (AUC reduction, %).
Figure 4.
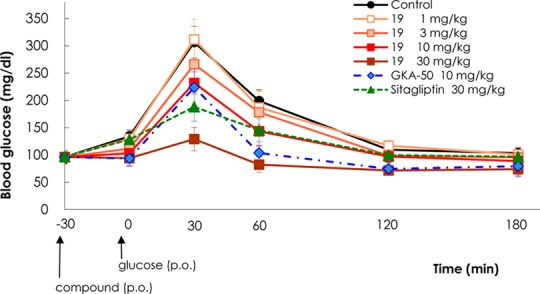
Plasma blood glucose lowering effect of 19 in C57BL/6J mice OGTT; po, per os (oral administration).
Figure 5.
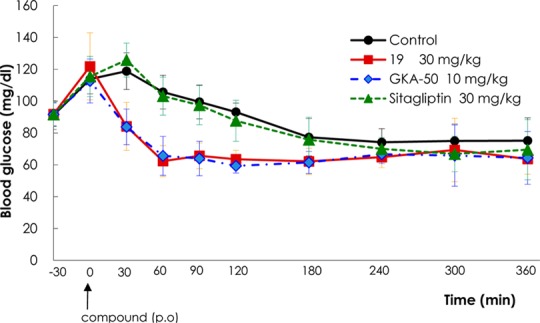
Fasted glucose tolerance test of 19 in C57BL/6J mice.
In summary, lead optimization and SAR analysis of an acetylenyl containing benzamide series led to the discovery of several novel glucokinase activators. Compound 19, 3-(3-Amino-phenylethynyl)-5-(2-methoxy-1-methyl-ethoxy)-N-(1-methyl-1H-pyrazol-3-yl)-benzamide, was found to be an active GKA with an EC50 of 27 nM and elicited an excellent AUC reduction of 47.4% (30 mg/kg) in an in vivo OGTT study with favorable pharmacokinetics. These study results strongly support the notion that GKA 19 is a promising candidate for further preclinical investigation as a potential therapeutic agent for type 2 diabetes mellitus.
Acknowledgments
The authors would like to thank Mr. Wonee Chong for his contribution to the pharmacokinetic study of 19 and Dr. Su Youn Nam for her helpful discussion at Yuhan Research Institute. We also thank Lee Farrand for proofreading the manuscript
Glossary
Abbreviations
- GK
glucokinase
- GKA
glucokinase activator
- DPP-4
dipeptidyl peptidase-4
- SGLT2
sodium-glucose cotransporter 2
- SAR
structure–activity relationship
- T2DM
type 2 diabetes mellitus
- OGTT
oral glucose tolerance test
Supporting Information Available
Description of synthesis of 19, procedure of bioassays, and pharmacokinetics protocol. This material is available free of charge via the Internet at http://pubs.acs.org.
This study was supported by a grant from the Chungcheong Leading Industry Office for Medicine and Bio Projects, Ministry of Knowledge Economy, Republic of Korea (C-2-1, 500108, and 7007618).
The authors declare no competing financial interest.
Supplementary Material
References
- Matschinsky F. M. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes 1996, 45, 223–241. [DOI] [PubMed] [Google Scholar]
- Antoine M.; Boutin J. A.; Ferry G. Binding kinetics of glucose and allosteric activators to human glucokinase reveal multiple conformational states. Biochemistry 2009, 48, 5466–5482. [DOI] [PubMed] [Google Scholar]
- Heredia V. V.; Thomson J.; Nettleton D.; Sun S. Glucose-induced conformational changes in glucokinase mediate allosteric regulation: transient kinetic analysis. Biochemistry 2006, 45, 7553–7562. [DOI] [PubMed] [Google Scholar]
- Filipski K. J.; Pfefferkorn J. A. A patent review of glucokinase activators and disruptors of the glucokinase–glucokinase regulatory protein interaction: 2011 – 2014. Expert Opin. Ther. Pat. 2014, 24, 875–891. [DOI] [PubMed] [Google Scholar]
- Waring M. J.; Brogan I. J.; Coghlan M.; Johnstone C.; Jones H. B.; Leighton B.; Mckerrecher D.; Pike K. G.; Robb G. R. Overcoming retinoic acid receptor-α based testicular toxicity in the optimization of glucokinase activators. MedChemComm 2011, 2, 771–774. [Google Scholar]
- Pfefferkorn J. A.; Guzman-Perez A.; Litchfield J.; Aiello R.; Treadway J. L.; Pattersen J.; Minich M. L.; Filipski K. J.; Jones C. S.; Tu M.; Aspnes G.; Risley H.; Bian J.; Stevens B. D.; Bourassa P.; D’Aquila T.; Baker L.; Barucci N.; Robertson A. S.; Bourbonais F.; Derksen D. R.; MacDougall M.; Cabrera O.; Chen J.; Lapworth A. L.; Landro J. A.; Zavadoski W. J.; Atkinson K.; Haddish-Berhane N.; Tan B.; Yao L.; Kosa R. E.; Varma M. V.; Feng B.; Duignan D. B.; El-Kattan A.; Murdande S.; Liu S.; Ammirati M.; Knafels J.; DaSilva-Jardine P.; Sweet L.; Liras S.; Rolph T. P. Discovery of (S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic acid as a hepatoselective glucokinase activator clinical candidate for treating type 2 diabetes mellitus. J. Med. Chem. 2012, 55, 1318–1333. [DOI] [PubMed] [Google Scholar]
- Massa M. L.; Gagliardino J. J.; Francini F. Liver glucokinase: An overview on the regulatorymechanisms of its activity. Life 2011, 63, 1–6. [DOI] [PubMed] [Google Scholar]
- Matschinsky F. M. GKAs for diabetes therapy: why no clinically useful drug after two decades of trying?. Trends Pharmacol. Sci. 2013, 34, 90–99. [DOI] [PubMed] [Google Scholar]
- Grimsby J.; Sarabu R.; Corbett W. L.; Hayne N. E.; Bizzarro F. T.; Coffey J. W.; Guertin K. R.; Hilliard D. W.; Kester R. F.; Mahaney P. E.; Marcus L.; Qi L. D.; Soence C. L.; Tengi J.; Magnuson M. A.; Chu C. A.; Dvorozniak M. T.; Matschinsky F. M.; Grippo J. F. Allosteric activators of glucokinase: potential role in diabetes therapy. Science 2003, 301, 370–373. [DOI] [PubMed] [Google Scholar]
- Qian Y.; Corbett W. L.; Berthel S. J.; Choi D.-S.; Dvorozniak M. T.; Geng W.; Gillespie P.; Guertin K. R.; Haynes N.-E.; Kester R. F.; Mennona F. A.; Moore D.; Racha J.; Radinov R.; Sarabu R.; Scott N. R.; Grimsby J.; Mallalieu N. L. Identification of RO4597014, a glucokinase activator studied in the clinic for the treatment of type 2 diabetes. ACS Med. Chem. Lett. 2013, 4, 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagami T.; Moriyama N.; Kyuhara M.; Moroda A.; Uemura T.; Matsumae H.; Moritani Y.; Inoue I. Scalable synthesis of a nonracemic alpha-arylpropionic acid via ketenedesymmetrization for a glucokinase activator. Org. Process Res. Dev. 2014, 18, 437–445. [Google Scholar]
- Meininger G. E.; Scott R.; Alba R.; Shentu Y.; Luo E.; Amin H.; Davies M. J.; Kaufman K. D.; Goldstein B. J. Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes. Diabetes Care 2011, 34, 2560–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike K. G.; Allen J. V.; Caulkett P. W.; Clarke D. S.; Donald C. S.; Fenwick M. L.; Johnson K. M.; Johnstone C.; McKerrecher D.; Rayner J. W.; Walker R. P.; Wilson I. Design of a potent, soluble glucokinase activator with increased pharmacokinetic half-life. Bioorg. Med. Chem. Lett. 2011, 11, 3467–3470. [DOI] [PubMed] [Google Scholar]
- Mitsuya M.; Kamata K.; Bamba M.; Watanabe H.; Sasaki Y.; Sasaki K.; Ohyama S.; Hosaka H.; Nagata Y.; Eiki J.; Nishimura T. Discovery of novel 3,6-disubstituted 2-pyridinecarboxamide derivatives as GK activators. Bioorg. Med. Chem. Lett. 2009, 19, 2718–2721. [DOI] [PubMed] [Google Scholar]
- Iino T.; Hashimoto N.; Sasaki K.; Ohyama S.; Yoshimoto R.; Hosaka H.; Hasegawa T.; Chiba M.; Nagata Y.; Eiki J.; Nishimura T. Structure–activity relationships of 3,5-disubstituted benzamides as glucokinase activators with potent in vivo efficacy. Bioorg. Med. Chem. 2009, 17, 3800–3809. [DOI] [PubMed] [Google Scholar]
- Park K.; Lee B.-M.; Kim Y.- H.; Han T.; Yi W.; Lee D.-H.; Choi H.-H.; Chong W.; Lee C.-H. Discovery of a novel phenylethyl benzamide glucokinase activator for the treatment of type 2 diabetes mellitus. Bioorg. Med. Chem. Lett. 2013, 23, 537–542. [DOI] [PubMed] [Google Scholar]
- Park K.; Lee B. M.; Hyun K. H.; Lee D. H.; Choi H. H.; Kim H.; Chong W.; Kim K. B.; Nam S. Y. Discovery of 3-(4-methanesulfonylphenoxy)-N-[1-(2-methoxyethoxymethyl)-1H-pyrazol-3-yl]-5-(3-methylpyridin-2-yl)-benzamide as a novel glucokinase activator (GKA) for the treatment of type 2 diabetes mellitus. Bioorg. Med. Chem. 2014, 22, 2280–2293. [DOI] [PubMed] [Google Scholar]
- Jain A. N. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003, 46, 499–511. [DOI] [PubMed] [Google Scholar]
- Pharmacokinetic study of 19 in mice established clearance of 2.59 L/h/kg, distribution volume (Vdss) of 1.44 L/kg, elimination half-life (T1/2) of 1.1 h, and bioavailability (F) of 49%. Physicochemical properties of 19: solubility (pH 7.4) = 22.2 μM, Papp,PAMPA (10–6 cm/s) = 3.43, ClogP = 2.64.
- Johnson D.; Shepherd R. M.; Gill D.; Gorman T.; Smith D. M.; Dunne M. J. Glucose-dependent modulation of insulin secretion and intracellular calcium ions by GKA50, a glucokinase activator. Diabetes 2007, 56, 1694–1702. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.