

Abstract
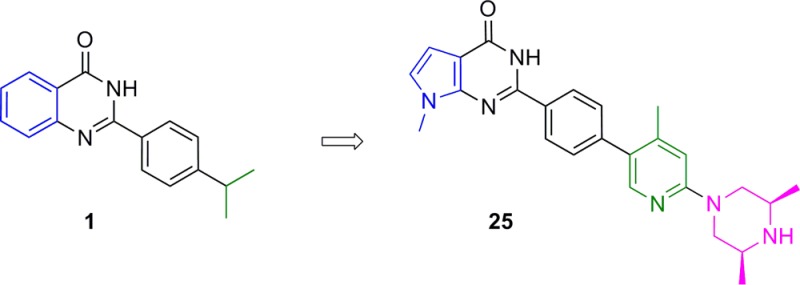
The canonical Wnt pathway plays an important role in embryonic development, adult tissue homeostasis, and cancer. Germline mutations of several Wnt pathway components, such as Axin, APC, and ß-catenin, can lead to oncogenesis. Inhibition of the poly(ADP-ribose) polymerase (PARP) catalytic domain of the tankyrases (TNKS1 and TNKS2) is known to inhibit the Wnt pathway via increased stabilization of Axin. In order to explore the consequences of tankyrase and Wnt pathway inhibition in preclinical models of cancer and its impact on normal tissue, we sought a small molecule inhibitor of TNKS1/2 with suitable physicochemical properties and pharmacokinetics for hypothesis testing in vivo. Starting from a 2-phenyl quinazolinone hit (compound 1), we discovered the pyrrolopyrimidinone compound 25 (AZ6102), which is a potent TNKS1/2 inhibitor that has 100-fold selectivity against other PARP family enzymes and shows 5 nM Wnt pathway inhibition in DLD-1 cells. Moreover, compound 25 can be formulated well in a clinically relevant intravenous solution at 20 mg/mL, has demonstrated good pharmacokinetics in preclinical species, and shows low Caco2 efflux to avoid possible tumor resistance mechanisms.
Keywords: Wnt, oncogenesis, pharmacokinetics, Caco2, tankyrase, PARP
The discovery and development of small molecule inhibitors of the canonical Wnt pathway has been a long-standing pursuit within oncology.1 Under normal circumstances, the Wnt signaling pathway plays a key role in embryonic development and the regeneration of adult tissue such as intestinal epithelial cells.2 Human tumors frequently harbor gene mutations that impact important proteins along the pathway such as APC, Axin, and ß-catenin.3 In most cases, these mutations lead to constitutive activation of Wnt signaling, which results in a Wnt ligand independent increase in the transcription of Wnt genes and leads to heightened proliferation.4 Approximately 85% of colorectal cancer (CRC) harbors APC mutations.5 A plethora of small molecules has been discovered that can modulate this pathway by targeting various proteins (e.g., casein kinases and porcupine).6,7 Despite this, there are currently no commercial drugs that target the Wnt pathway in oncology.
In 2009, Huang et al. reported that inhibition of the poly(ADP-ribose) polymerase (PARP) catalytic domain of tankyrases (TNKS1 and TNKS2) can inhibit the Wnt pathway via Axin stabilization.8 Using the small molecule tankyrase inhibitor XAV939, they demonstrated that tankyrase interacts with and PARsylates Axin. Tankyrase inhibition prevents PARsylation of Axin, which in turn leads to increased Axin protein levels in the cell. Higher levels of Axin lead to sequestration and/or destruction of ß-catenin, preventing translocation of ß-catenin to the nucleus and a reduction in transcription of Wnt pathway genes. Moreover, they showed evidence that XAV939 can inhibit soft agar colony formation of DLD-1 cells grown under low serum conditions.
The tankyrases are members of the PARP enzyme superfamily of proteins, all of which contain a PARP catalytic domain that can catalyze the attachment of ADP ribose to the surface of proteins using the cofactor nicotinamide adenine dinucleotide (NAD+).9 As a consequence of their linkage to the Wnt pathway, association with telomeres,10 and spindle structure and function,11 the tankyrases have attracted a high level of interest from the drug discovery community. Numerous research groups have published reports of tankyrase inhibitors with varying degrees of potency and PARP enzyme family selectivity.12−14
The goal of our medicinal chemistry effort was to discover a potent dual TNKS1 and TNKS2 enzyme inhibitor that demonstrated potent inhibition of the Wnt pathway as demonstrated in a TCF TOPflash reporter assay in DLD-1 cells and that had sufficient DMPK properties for hypothesis testing in preclinical in vivo xenograft models. In order to have confidence that the biological effects of these compounds were due to TNKS inhibition and not other PARPs, selectivity was desired against other PARP family members, primarily PARP1.9 To avoid transporter mediated resistance mechanisms in CRC, we sought compounds with low efflux potential as measured in a standard Caco2 permeability assay at pH 7.4. Finally, we strove to optimize this series for IV dosing, to allow greater control over compound exposure and possibly mitigate expected on-target intestinal toxicity.15
A high-throughput screen (HTS) of the AstraZeneca screening collection identified the 2-phenyl quinazolinone 1 (Table 1), which had a 7 nM IC50 as a TNKS1 enzyme inhibitor16 and was 2 μM in our DLD-1 Wnt reporter assay. 2-Phenyl quinazolinones have been previously described in the literature as tankyrase inhibitors.17,18 The potency, small size, and synthetic accessibility of this compound class encouraged us to explore the series further. Modeling and systematic exploration of substituents around the quinazolinone ring revealed that the 8-position could accommodate small groups. We soon found that a hydroxymethyl moiety greatly improved DLD-1 Wnt reporter potency. Further optimization of the 2-phenyl ring suggested that the isopropyl group could be replaced with 5- and 6-membered aromatic rings. Exploration of various aryl rings gave compound 2a (Table 1), which had improved cell potency (27 nM Wnt reporter).
Table 1. Optimization of HTS Hit Compound 1 to Lead Optimization Starting Compound 2a.
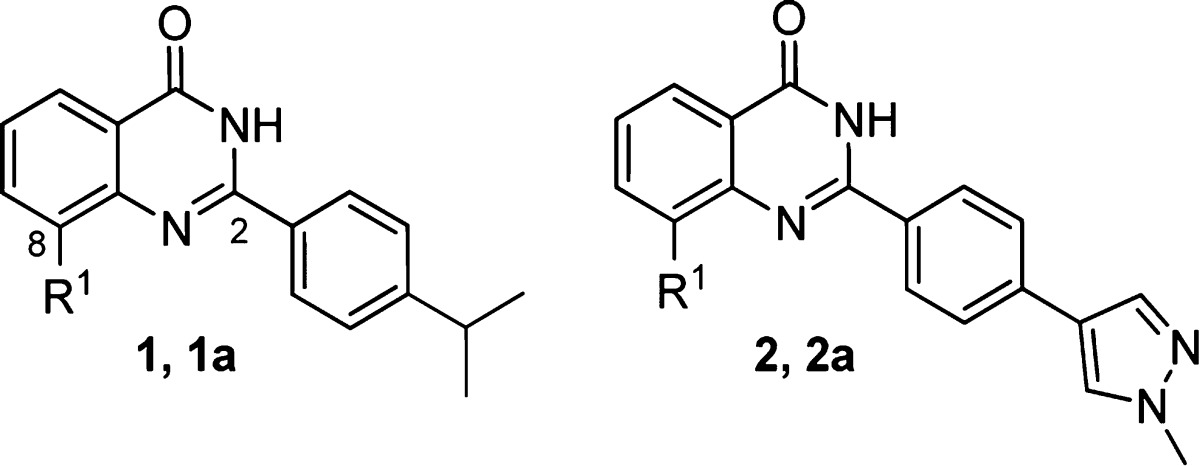
| Cmpd | R1 | TNKS1 enz IC50 (μM) | Wnt cell IC50 (μM) |
|---|---|---|---|
| 1 | H | 0.007 | 2.0 |
| 1a | CH2OH | 0.006 | 0.145 |
| 2 | H | 0.007 | 1.25 |
| 2a | CH2OH | 0.004 | 0.027 |
Intrigued by the cell potency of compound 2a, we obtained a crystal structure of this molecule bound to TNKS1 (Figure 1). Compound 2a (yellow) binds in the nicotinamide pocket of TNKS1 (gray), making critical H-bonds to the backbone NH and carbonyl of Gly1185. The quinazolinone ring is sandwiched between Tyr1224 and Tyr1213 making π-stacking interactions. The hydroxymethyl moiety points toward Glu1291. Upon binding, the compound displaces Tyr1203 (from apo structure in cyan),19 which then reorganizes around the 2-phenyl linker, while the pyrazole ring partially fills the newly vacated space. The pyrazole sits at a ∼90° dihedral angle to the linking phenyl, while its methyl group is solvent exposed.
Figure 1.
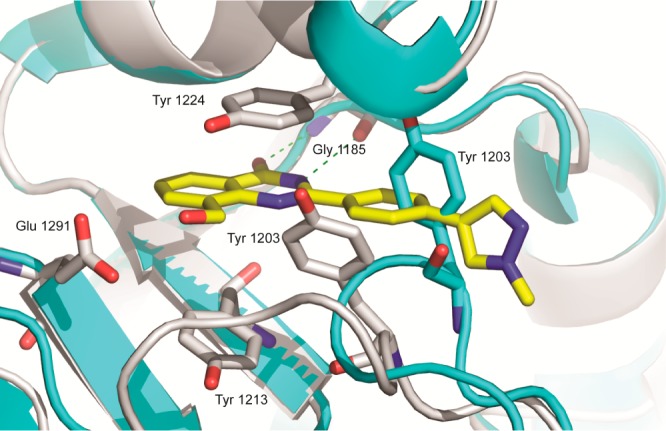
Compound 2a (yellow) bound to TNKS1 (gray, PDB ID: 4W5S) overlaid with TNKS1 apo structure (cyan, PDB ID: 2RF5).
On the basis of the crystal structure, we explored small substituents on the heteroaryl terminal ring that would bias the dihedral angle of the terminal biaryl system to 90° and fill space vacated by Tyr1203. These efforts led to compound 3 (Table 2) featuring a terminal 4-methyl-3-pyridyl ring. The next phase of optimization of the terminal aryl ring focused on exploring electron donating and withdrawing substituents at the 6-position of the 3-pyridyl ring. Introduction of σ-withdrawing groups, such as F and Cl in compounds 4 and 5, gave no significant improvement in cell potency but was accompanied by a large increase in lipophilicity as measured by logD. In contrast, π-donating groups gave an improvement in cell potency, as exemplified by 6-NH2 compound 6 and 6-OCH3 compound 7. Dimethylation of the amine was also tolerated to give compound 8, which had an IC50 < 3 nM in our cell assay, albeit with high logD and low solubility.
Table 2. 4-Methyl-3-pyridyl Moiety with π-Donating Groups at the 6-Position Improves Cell Potency.
| Cmpd | R1 | TNKS1 enz IC50 (μM) | PARP1 enz IC50 (μM) | Wnt cell IC50 (μM) | LogD pH 7.4 | Aq. Sol. pH 7.4 (μM) |
|---|---|---|---|---|---|---|
| 3 | H | 0.003 | 2.77 | 0.024 | 2.1 | 11 |
| 4 | F | 0.002 | 3.74 | 0.032 | 3.4 | <1 |
| 5 | Cl | 0.006 | 0.017 | 0.019 | 3.9 | <1 |
| 6 | NH2 | 0.017 | 0.206 | 0.004 | 2.9 | <1 |
| 7 | OCH3 | 0.006 | 0.035 | 0.004 | 2.8 | <1 |
| 8 | N(CH3)2 | 0.019 | <0.003 | >3.5 | 4 |
Assuming that dimethylamine 8 adopts a similar binding mode to compound 2a, we sought to improve the solubility and lower the logD by adding hydrophilic groups to the amine at the 6-position of the pyridyl ring, which was predicted to be solvent exposed (Table 3). Nucleophilic aromatic substitution of aryl-chloride 5 with methoxyethylamine led to compound 9, which had high Caco2 permeability and low clearance in rat but a very low volume of distribution. Hydroxyethyl compound 10 showed improved logD and solubility, but it suffered from high intrinsic clearance in rat hepatocytes due to oxidation of the terminal OH to the carboxylic acid, confirmed by metabolite identification using LC–MS/MS. The intrinsic clearance in rat hepatocytes was lowered in compound 11 by introducing geminal methyl groups alpha to the OH; however, logD, solubility, and PK parameters were no different relative to 9. A number of cyclic amines were also added to aryl-chloride 5 to yield morpholine analogue 12, which showed little difference compared to 9. A path forward was found in the isosteric, basic piperazine 13, which gave a significant improvement in solubility and a one unit drop in logD, although the Caco2 permeability of this compound was then less than 1 and the efflux ratio was above 20.
Table 3. Optimization of the Solvent Group Improves Solubility and Metabolic Stability while Maintaining Permeability and Potency.
Unit: 10–6 cm/s.
Unit: mL/min·kg.
Unit: L/kg.
Unit: h.
To mitigate the efflux within this more soluble amine containing subseries, we sought to tune the amine basicity, steric environment, and H-bond potential through the addition of nearby methyl groups. The addition of a single methyl group α to the amine gave (S)-2-methylpiperazine 14, which had improved permeability but also improved intrinsic clearance, presumably due to reduced metabolism of the amine or CH2 α to the amine. Rat PK of 14 revealed that this compound had an increased volume of distribution and terminal half-life relative to 9. Permeability was improved further by N-methylation to give 15, but the drawback was a compound with poor metabolic stability in vitro due to rapid N-demethylation and N-oxidation, as confirmed by metabolite identification using LC–MS/MS. Further blocking the NH group led to the cis-2,6-dimethyl piperazine analogue 16, which had a good balance of improved solubility, low intrinsic clearance, acceptable efflux, and good rat pharmacokinetics. Interestingly, rigidification of this structure by linking the methyl groups to form the ethylene bridged piperazine in 17 gave a high efflux compound. The origin of the approximately 10-fold difference in efflux between the two compounds may lie in the accessibility of the NH group. The piperazine methyl groups in 16 are bis-equatorial, providing steric hindrance around the NH. In contrast, the alkyl groups are locked in a bis-axial position in bicyclic compound 17, further exposing the NH toward H-bonding with efflux transporters.
A TNKS1-bound crystal structure was obtained for 15 (Figure 2). The terminal pyridine ring sits roughly perpendicular to the linker phenyl ring, placing the methyl group below the plane of the core quinazolinone and filling the hydrophobic space near Tyr1203. The pyridine lone pair makes a hydrogen bond with an ordered water molecule. The (S)-1,2-dimethyl piperazine moiety is coplanar with the pyridine ring. This group is not completely solvent exposed as initially hypothesized; there are hydrophobic contacts between Phe1188 and the inner half of the piperazine ring. The piperazine adopts a conformation that places the 2-methyl group axial to avoid A1,2 strain with the N-Me group. We also observed an H-bond interaction network between the core hydroxymethyl group, a water molecule, and Glu1291. In published quinazolinone–TNKS2 structures,19 a water at this position is highly conserved even in the absence of an 8-position substituent (15/18 structures); however, with 15, the Glu1291 carboxylate shifts ∼2.6 Å to replace an additional water molecule observed in 11 of these structures.
Figure 2.
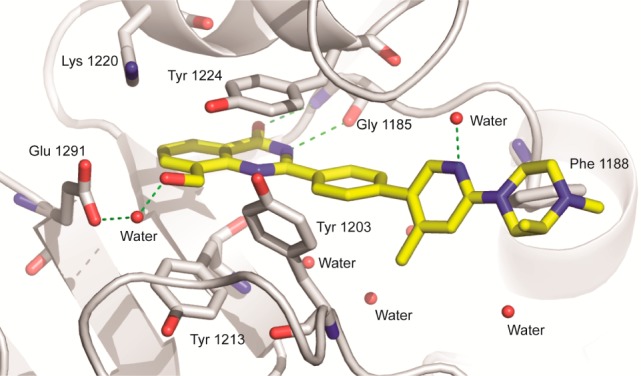
Crystal structure of compound 15 bound to TNKS1 (PDB ID: 4W6E).
We next turned our attention to the linker phenyl group. Initial SAR during the hit-to-lead phase indicated that even small substituents around the phenyl, such as F or CH3, were deleterious to TNKS1 enzyme potency. However, we postulated that solubility could be improved by introducing saturated linkers that would maintain a similar geometry.20 A number of phenyl isosteres were employed, with piperazine and piperidine distinguishing themselves (Table 4). Four matched pairs were made, with either a piperidine or piperazine linker and either the cis-2,6-dimethyl piperazine or the (S)-1,2-dimethyl piperazine solvent group. All four compounds showed a significant improvement in solubility, lower logD, reduced human plasma protein binding, and reduced PARP1 enzyme potency relative to phenyl. The piperidine linked compounds 18 and 20 had better selectivity against PARP1, lower Caco2 efflux ratios, and lower rat clearance than the piperazine linked counterparts. Conversely, it was the terminal solvent group that drove the logD, plasma protein binding, and intrinsic clearances for these saturated linked compounds. Compounds 20 and 21 bearing the (S)-1,2-dimethyl piperazine feature an N-Me group, which increased logD and protein binding and reduced microsomal and hepatocyte stability. Overall, compound 18 had the best properties and rat PK of this set, but the improvement came at the expense of cell potency. Alternatively, compound 21 maintained cell potency comparable to phenyl-linked 16 with improved solubility, but the in vivo PK showed higher CL and shorter terminal half-life.
Table 4. Replacement of the Phenyl Linker with a Piperazine or Piperidine Improves Solubility.
| Cmpd | 18 | 19 | 20 | 21 |
|---|---|---|---|---|
| A | CH | N | CH | N |
| R2 | CH3 | CH3 | H | H |
| R3 | H | H | CH3 | CH3 |
| Wnt cell IC50 (μM) | 0.038 | 0.037 | 0.012 | 0.004 |
| TNKS1 enz IC50 (μM) | 0.004 | 0.004 | 0.003 | 0.004 |
| PARP1 enz IC50 (μM) | 11 | 3 | 11 | 3 |
| LogD pH 7.4 | 1.4 | 1.6 | 2.4 | 2.3 |
| Aq. Sol. pH 7.4 (μM) | 651 | 151 | 205 | 530 |
| Hu PPB (% free) | 8 | 8 | 2 | 3 |
| Hu Mics CLinta | 11 | 12 | 25 | 32 |
| Rat Heps CLintb | 5 | 21 | 12 | 23 |
| Caco2 AB (10–6 cm/s) | 0.8 | 0.3 | 3.5 | 0.7 |
| Caco2 efflux ratio | 17 | 41 | 10 | 44 |
| Rat IV CL (mL/min·kg) | 22 | 35 | 17 | 28 |
| Rat IV Vdss (L/kg) | 6.5 | 3.2 | 2.1 | 1.9 |
| Rat IV t1/2 (h) | 4.4 | 1.7 | 1.8 | 1.0 |
Unit: μL/min·mg.
Unit: μL/min·106.
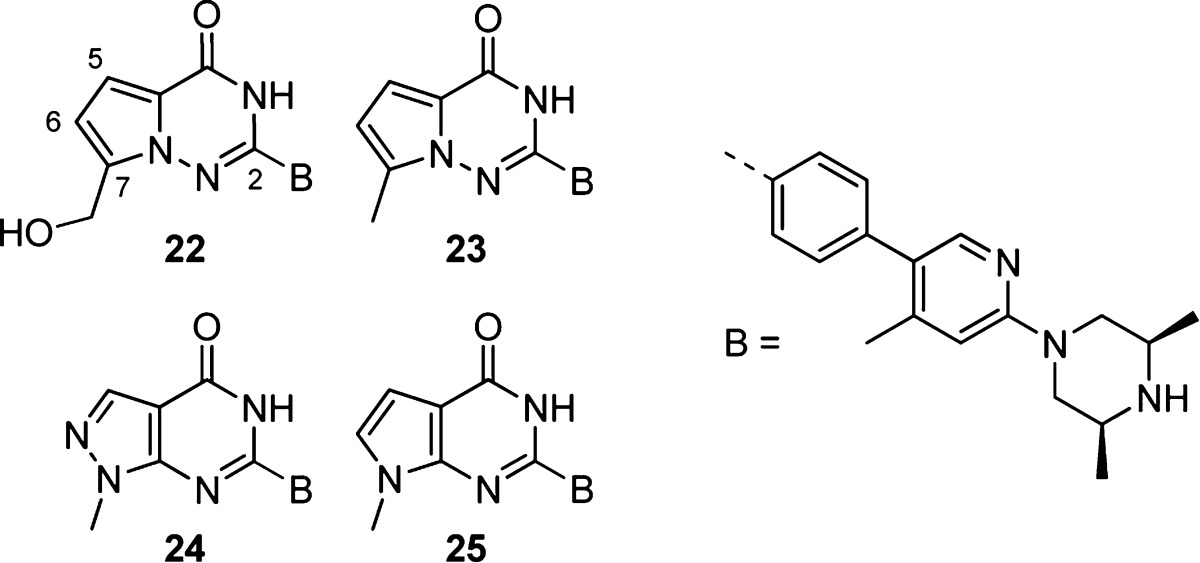
Finally, we investigated the optimization of the nicotinamide mimetic core (Table 5). We were intrigued by the possibility of morphing the quinazolinone core into a 5,6-heterocyclic ring system, but we were cognizant of the significant cell potency obtained by inclusion of the CH2OH group. We designed compound 22, featuring a pyrrolotriazinone core, which allowed stable attachment of the CH2OH group to the 7-position of a 5,6-fused ring system. The 7-CH2OH heterocycle 22 was less potent in our cell assay than 16, but it had excellent PARP1 selectivity and maintained the good DMPK properties of the quinazolinone analogue 16. However, Caco2 permeability and efflux were worse for compound 22 compared to 16. We also made the corresponding 7-CH3 pyrrolotriazinone compound 23 as a control. Encouragingly, the 7-CH3 compound was <3 nM in our cell assay with good selectivity against PARP1 and similar rat PK to 16, but it had high logD and correspondingly low solubility. Next, we synthesized 24 and 25, which were predicted to be less lipophilic (ClogP 2.5 and 3.3, respectively) relative to 23 (ClogP 3.5). The pyrazolopyrimidinone 24 was 10-fold less potent in our Wnt cell reporter assay compared to 23 but had improved efflux versus 16. In contrast, the pyrrolopyrimidinone 25 maintained single digit nanomolar potency in the cell while lowering logD to 2.5. Compound 25 had much improved PARP1 selectivity compared to core analogue 16 while maintaining good rat in vivo PK properties. Moreover, compound 25 had similarly improved Caco2 permeability and efflux ratio versus 16.
Table 5. 5,6-Heterocyclic Cores Improve PARP1 Selectivity and Caco2 Efflux.
| Cmpd | 22 | 23 | 24 | 25 |
|---|---|---|---|---|
| Wnt cell IC50 (μM) | 0.012 | <0.003 | 0.024 | 0.005 |
| TNKS1 enz IC50 (μM) | 0.004 | 0.008 | 0.005 | 0.006 |
| PARP1 enz IC50 (μM) | >30 | 5 | >30 | >30 |
| LogD pH 7.4 | 2.1 | 3.6 | 2.1 | 2.5 |
| Aq. Sol. pH 7.4 (μM) | 38 | 8 | 32 | 30 |
| Hu PPB (% free) | 1.6 | <1 | 12 | 7.2 |
| Hu Mics CLinta | 14 | 4 | 6 | 13 |
| Rat Heps CLintb | 5 | 4 | 4 | 8 |
| Caco2 AB (10–6 cm/s) | 0.1 | 4.3 | 5.5 | |
| Caco2 efflux ratio | 194 | 3.8 | 2.7 | |
| Rat IV CL (mL/min·kg) | 5 | 4 | 4 | 11 |
| Rat IV Vdss (L/kg) | 1.6 | 2.5 | 3.0 | 4.4 |
| Rat IV t1/2 (h) | 5.0 | 8.2 | 11 | 5.7 |
Unit: μL/min·mg.
Unit: μL/min·106.
Detailed PARP family enzyme data and pharmacokinetic data for compounds 9 and 25 are presented in Table 6. The enzyme assays in this table utilize a different assay technology than the enzyme assays presented Tables 1–5 (see Supporting Information for assay details). Oral lead 9 showed potent dual TNKS1/2 enzyme inhibition with moderate selectivity against PARP1 and 2 and good selectivity over PARP6.21 While the bioavailability of 59% for 9 in rat was good, the PK was characterized by very low volume of distribution and short half-life. Rat protein binding was quite high at only 0.2% unbound. Mouse PK for 9 showed good bioavailability of 69% with a higher volume and longer half-life along with reduced protein binding. Because of the poor solubility and lack of a strongly basic nitrogen, methoxyethylamine 9 was difficult to formulate for IV dosing, only achieving 0.4 mg/mL in an IV solution useful only for preclinical studies.
Table 6. PARP Enzyme Selectivity, DMPK Profiles, and Formulations for Compounds 9 and 25.
| Cmpd | 9 | 25 |
|---|---|---|
| TNKS1 IC50 (μM) | 0.003 | 0.003 |
| TNKS2 IC50 (μM) | 0.010 | 0.001 |
| PARP1 IC50 (μM) | 0.559 | 2.0 |
| PARP2 IC50 (μM) | 0.232 | 0.5 |
| PARP6 IC50 (μM) | >3 | >3 |
| mouse IV CL (mL/min·kg) | 53 | 24 |
| mouse IV Vss (L/kg) | 2.6 | 5.5 |
| mouse IV t1/2 (h) | 3.4 | 4.3 |
| mouse oral F (%) | 69 | 12 |
| mouse PPB (% free) | 2.2 | 3.6 |
| rat oral F (%) | 59 | 18 |
| rat PPB (% free) | 0.2 | 3.7 |
| IV formulation | 0.4 mg/mL, TEG/40% SBECD 50/50, pH 5 | 20 mg/mL, 20% SBECD, pH 4 |
Compound 25 was also profiled against a panel of PARP family enzymes and showed excellent potency as a dual TNKS1 and TNKS2 inhibitor with good selectivity against PARPs 1, 2, and 6 (Table 6). In sharp contrast to 9, 25 formulated well in a clinically relevant IV solution at 20 mg/mL using SBECD as an excipient at pH 4. Dosed at 25 mg/kg IV to nude mice, 25 had a CL of 24 mL/min·kg and a half-life of 4 h. The bioavailability of 25 in mouse and rat was only moderate at 12% and 18%, respectively.
In order to confirm that the mechanism of action of this series of tankyrase inhibitors is blocking the Wnt pathway through Axin, DLD-1 cells were treated with compounds 9 and 25 at 0.3 and 0.8 μM, compound 15 at 0.3 μM, and XAV939 at 1 μM and analyzed for TNKS1, TNKS2, and Axin2 via Western blot (see Supporting Information Figure S1). At early time points (3 and 8 h), 9, 15, and 25 showed similar TNKS1/2 inhibition and Axin2 stabilization as XAV939. At longer times (24, 48, and 72 h), 9, 15, and 25 showed qualitatively stronger and longer lasting stabilization of Axin2, TNKS1, and TNKS2 at lower concentrations compared to XAV939.
Compound 9 was synthesized on multigram scale and was made available to external collaborators as an orally bioavailable small molecule inhibitor of TNKS1/2 and the Wnt pathway carrying some activity against PARP1 and 2. This compound (9 = AZ1) has demonstrated efficacy in KRAS wild type patient-derived xenograft models of CRC in combination with irinotecan.22 Optimized compound 25 has been used as an IV probe compound to explore the in vivo effects of TNKS1/2 inhibition on normal tissue and tumor xenografts. The results of these experiments will be reported in due course.
Glossary
Abbreviations Used
- TEG
tetraethylene glycol
- heps
hepatocytes
- CL
clearance
- PK
pharmacokinetics
- mics
microsomes
- aq
aqueous
- sol
solubility
- PPB
plasma protein binding
- CLint
intrinsic clearance
- SBECD
sulfobutylether-beta-cyclodextrin sodium salt
- IV
intravenous
- A1,2
1,2-allylic
- Hu
human
- enz
enzyme
- APC
adenomatous polyposis coli
- DMPK
drug metabolism and pharmacokinetics
Supporting Information Available
TNKS1/2 and Axin2 Western blot data; crystallization conditions and crystallographic data; TNKS1, PARP1, DLD-1 Wnt reporter, and Caco2 assay protocols; detailed solution formulations; and experimental details for key compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Anastas J. N.; Moon R. T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [DOI] [PubMed] [Google Scholar]
- Pinto D.; Gregorieff A.; Begthel H.; Clevers H. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev. 2003, 17, 1709–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segditsas S.; Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 2006, 25, 7531–7. [DOI] [PubMed] [Google Scholar]
- Clevers H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–80. [DOI] [PubMed] [Google Scholar]
- Markowitz S. D.; Bertagnolli M. M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronkov A.; Krauss S. Wnt/beta-catenin signaling and small molecule inhibitors. Curr. Pharm. Des. 2013, 19, 634–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn M. Can we safely target the WNT pathway?. Nat. Rev. Drug. Discovery 2014, 13, 513–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S. M.; Mishina Y. M.; Liu S.; Cheung A.; Stegmeier F.; Michaud G. A.; Charlat O.; Wiellette E.; Zhang Y.; Wiessner S.; Hild M.; Shi X.; Wilson C. J.; Mickanin C.; Myer V.; Fazal A.; Tomlinson R.; Serluca F.; Shao W.; Cheng H.; Shultz M.; Rau C.; Schirle M.; Schlegl J.; Ghidelli S.; Fawell S.; Lu C.; Curtis D.; Kirschner M. W.; Lengauer C.; Finan P. M.; Tallarico J. A.; Bouwmeester T.; Porter J. A.; Bauer A.; Cong F. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–20. [DOI] [PubMed] [Google Scholar]
- Rouleau M.; Patel A.; Hendzel M. J.; Kaufmann S. H.; Poirier G. G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S.; Giriat I.; Schmitt A.; de Lange T. Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science 1998, 282, 1484–7. [DOI] [PubMed] [Google Scholar]
- Chang P.; Coughlin M.; Mitchison T. J. Tankyrase-1 polymerization of poly(ADP-ribose) is required for spindle structure and function. Nat. Cell Biol. 2005, 7, 1133–9. [DOI] [PubMed] [Google Scholar]
- Lehtiö L.; Chi N. W.; Krauss S. Tankyrases as drug targets. FEBS J. 2013, 280, 3576–93. [DOI] [PubMed] [Google Scholar]
- Haikarainen T.; Krauss S.; Lehtio L. Tankyrases: structure, function and therapeutic implications in cancer. Curr. Pharm. Des. 2014, 20, 6472–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscio P.; Carotti A.; Asciutti S.; Karlberg T.; Bellocchi D.; Llacuna L.; Macchiarulo A.; Aaronson S. A.; Schüler H.; Pellicciari R.; Camaioni E. Design, synthesis, crystallographic studies, and preliminary biological appraisal of new substituted triazolo[4,3-b]pyridazin-8-amine derivatives as tankyrase inhibitors. J. Med. Chem. 2014, 57, 2807–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau T.; Chan E.; Callow M.; Waaler J.; Boggs J.; Blake R. A.; Magnuson S.; Sambrone A.; Schutten M.; Firestein R.; Machon O.; Korinek V.; Choo E.; Diaz D.; Merchant M.; Polakis P.; Holsworth D. D.; Krauss S.; Costa M. A novel tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res. 2013, 73, 3132–44. [DOI] [PubMed] [Google Scholar]
- The concentration of TNKS1 in this enzyme assay is 110 nM. Positive control compounds reproducibly show low IC50s (see Supporting Information). Since the true active concentration of the enzyme is not known, these control compounds may give a better sense of the assay sensitivity than the theoretical limit of 55 nM.
- Nathubhai A.; Wood P. J.; Lloyd M. D.; Thompson A. S.; Threadgill M. D. Design and Discovery of 2-Arylquinazolin-4-ones as Potent and Selective Inhibitors of Tankyrases. ACS Med. Chem. Lett. 2013, 4, 1173–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haikarainen T.; Koivunen J.; Narwal M.; Venkannagari H.; Obaji E.; Joensuu P.; Pihlajaniemi T.; Lehtiö L. para-Substituted 2-phenyl-3,4-dihydroquinazolin-4-ones as potent and selective tankyrase inhibitors. ChemMedChem 2013, 8, 1978–85. [DOI] [PubMed] [Google Scholar]
- Lehtiö L.; Collins R.; van den Berg S.; Johansson A.; Dahlgren L. G.; Hammarström M.; Helleday T.; Holmberg-Schiavone L.; Karlberg T.; Weigelt J. Zinc binding catalytic domain of human tankyrase 1. J. Mol. Biol. 2008, 379, 136–45. [DOI] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6. [DOI] [PubMed] [Google Scholar]
- Tuncel H.; Tanaka S.; Oka S.; Nakai S.; Fukutomi R.; Okamoto M.; Ota T.; Kaneko H.; Tatsuka M.; Shimamoto F. PARP6, a mono(ADP-ribosyl) transferase and a negative regulator of cell proliferation, is involved in colorectal cancer development. Int. J. Oncol. 2012, 41, 2079–86. [DOI] [PubMed] [Google Scholar]
- Tai W.; Quackenbush K.; Purkey A.; Bagby S.; Messersmith W.; Song E.; Pitts T.; Arcaroli J.. KRAS wild type tumors exhibit increased efficacy to the combination of AZ1 (a tankyrase inhibitor) + irinotecan in a patient-derived CRC explant model. In Proceedings of the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics, October 19–23, 2013; AACR: Philadelphia, PA, 2013; Vol.12 ( (11 Suppl), ), Abstract nr B30. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.