

Abstract
Purpose
A spontaneous frameshift mutation, c.3481delC, in the Crb1 gene is the underlying cause of dysplasia and retinal degeneration in rd8 mice. The rd8 mutation is found in C57BL/6N but not in C57BL/6J mouse sub-strains. The development of ocular pathology in single knockout Ccl2−/−, Cx3cr1−/− and in double knockout Ccl2−/−/Cx3cr1−/− mice raised on a C57BL/6 background has been reported to depend on the presence of a rd8 mutation. In this study, we investigated the influence of the rd8 mutation on the retinal pathology that we previously described in the late-onset retinal degeneration (L-ORD) mouse model with a heterozygous S163R mutation in the C1q-tumor necrosis factor-related protein-5 (Ctrp5+/−) gene that was generated on a C57BL/6J background.
Methods
Mouse lines carrying the Ctrp5 S163R and rd8 mutations (Ctrp5+/−;rd8/rd8), corresponding controls without the rd8 mutation (Ctrp5+/−;wt/wt), and wild-type mice with and without the rd8 mutation (Wtrd8/rd8 and Wtwt/wt, respectively) were generated by systematic breeding of mice in our L-ORD mouse colony. Genotyping the mice for the rd8 (del C at nt3481 in Crb1) and Ctrp5 S163R mutations was performed with allelic PCR or sequencing. Retinal morphology was studied with fundus imaging, histology, light microscopy, electron microscopy, and immunohistochemistry.
Results
Genotype analysis of the mice in L-ORD mouse colony detected the rd8 mutation in the homozygous and heterozygous state. Fundus imaging of wild-type mice without the rd8 mutation (Wtwt/wt) revealed no autofluorescence (AF) spots up to 6–8 months and few AF spots at 21months. However, the accumulation of AF lesions accelerated with age in the Ctrp5+/− mice that lack the rd8 mutation (Ctrp5+/−;wt/wt). The number of AF lesions was significantly increased (p<0.001), and they were small and uniformly distributed throughout the retina in the 21-month-old Ctrp5+/−;wt/wt mice when compared to the age-matched controls. Wild-type and Ctrp5+/− mice with the rd8 mutation (Wtrd8/rd8 and Ctrp5+/−;rd8/rd8, respectively) revealed an integrated retinal architecture with well-defined outer segments/inner segments (OS/IS), outer nuclear layer (ONL), outer plexiform layer (OPL), and inner nuclear layer (INL). The presence of pseudorosette structures reported in the rd8 mice between the ONL and the INL in the ventral quadrant of the retina was not observed in all genotypes studied. Further, the external limiting membrane was continuous in the Ctrp5+/−;rd8/rd8 and Wtrd8/rd8 mice. Evaluation of the retinal phenotype revealed that the Ctrp5+/−;wt/wt mice developed characteristic L-ORD pathology including age-dependent accumulation of AF spots, development of sub-retinal, sub-RPE, and basal laminar deposits, and Bruch’s membrane abnormalities at older age, while these changes were not observed in the age-matched littermate WTwt/wt mice.
Conclusions
The Wtrd8/rd8 and Ctrp5+/−;rd8/rd8 mice raised on C57BL/6J did not develop early onset retinal changes that are characteristic of the rd8 phenotype, supporting the hypothesis that manifestation of rd8-associated pathology depends on the genetic background. The retinal pathology observed in mice with the Ctrp5+/−;wt/wt genotype is consistent with the L-ORD phenotype observed in patients and with the phenotype we described previously. The lack of rd8-associated retinal pathology in the Ctrp5+/−;wt/wt mouse model raised on the C57BL/6J background and the development of the L-ORD phenotype in these mice in the presence and absence of the rd8 mutation suggests that the pathology observed in the Ctrp5+/−;wt/wt mice is primarily associated with the S163R mutation in the Ctrp5 gene.
Introduction
The Crb complex, first identified in Drosophila, is required for polarity and adhesion in embryonic epithelia, stalk membrane morphogenesis in photoreceptors and for the formation of adherens junctions between cells in the retina [1-3]. In mice and humans, the Crb complex is involved in retinal integration and organization [4,5].
The human crumbs homolog 1 (CRB1; Gene ID: 23418, OMIM: 604210) gene encodes a protein containing a signal peptide, 19 epidermal growth-factor (EGF) domains, three A-globular-like domains, and a transmembrane domain [6]. CRB1 is part of the protein complex at adhesion junctions (AJ) and localized to the external limiting membrane (ELM) in the retina. Mutations in the CRB1 gene are associated with phenotypically diverse retinal disorders including Leber congenital amaurosis (LCA), early onset retinitis pigmentosa (e.g., RP12), retinitis pigmentosa with coats-like exudative vasculopathy, and pigmented paravenous retinochoroidal atrophy [7-13]. Four percent of patients with autosomal recessive retinitis pigmentosa (arRP) and 10–15% of patients with autosomal recessive Leber congenital amaurosis (arLCA) carry mutations in the CRB1 gene [9-11]. The phenotypic diversity of the patients who carry CRB1 mutations has led to the hypothesis that environmental and genetic modifiers influence the severity and presentation of CRB1-associated retinal degeneration [11,14,15].
The naturally occurring retinal degeneration 8 (rd8) mouse model carries a homozygous single base deletion (c.3481delC) in Crb1 (Crb1rd8/rd8) causing early onset retinal degeneration, retinal folds, pseudorosettes, focal retinal dysplasia, and ocular spots [5]. The ocular spots correspond to the retinal folds and pseudorosettes that form due to abnormality in the adherens junctions between the photoreceptors and the Müller glia (MG) cells. The outer nuclear layer (ONL) and the inner nuclear layer (INL) are affected in rd8 mice. The deletion causes a frameshift resulting in a truncated Crb1 protein that consists of only the N-terminal extracellular domain [16].
Similar to patients carrying CRB1 mutations, phenotypic variations are observed between and within mice strains homozygous for the rd8 mutation [17]. It has also been reported that the C57BL/6 lines provided by several commercial vendors harbor the rd8 mutation and present the characteristic Crb1-associated phenotype [17]. Although rd8 mice of the naturally occurring strain (Crb1rd8/rd8) exhibit ocular spots in the inferior nasal quadrant, the eyes of mice with the rd8 mutation on the CAST/EiJ background (CAST/EiJrd8/rd8) are reported to have fewer spots than the naturally occurring strain [5]. About 19% of the CAST/EiJrd8/rd8 mice do not develop ocular spots. Furthermore, N7 generation of Crb1rd8/rd8 backcrossed with C57BL/6J did not develop retinal spots [5]. The retinal histology of the C57BL/6J mice homozygous for the rd8 mutation are normal, lacking retinal folds and photoreceptor inner segment (IS) disorganization suggesting that the C57BL/6J background is a strong modulator of the rd8 phenotype. In contrast, C57BL/6N, a sub-strain derived from C57BL/6J after 1951, harbors the rd8 mutation and invariably expresses the characteristic Crb1-associated phenotype [17]. The discrepancies in the rd8 phenotype between and within mouse strains provide clear evidence that genetic modifiers influence the development of rd8 pathology. Two genetically related homozygous rd8 mouse lines obtained from a backcross with C57BL/6JOlaHsd mice display variable phenotypes, and a genetic factor on chromosome 15 has been suggested to be responsible for this variation [18].
The early onset ocular phenotypes observed in HLA-A29 transgenic mice and CCDKO mice are associated with presence of the rd8 mutation and are not due to mutations in the HLA-A29, Ccl2, or Cx3cr1 gene [17]. HLA-A29 transgenic and CCDKO mice carrying the rd8 mutation in the homozygous state exhibit retinal dysplasia. Interestingly, about 20–25% of the HLA-A29 transgenic mice with the rd8 mutation in the heterozygous state also develop dysplasia [17,19]. The genetic background of the rd8 mutation differentially modulates the retinal phenotype in the original CCDKO and the rederived Ccl2−/−, Cx3cr1−/−, and Ccl2−/−/Cx3cr1−/− double-knockout mice. The early onset retinal degeneration seen in Ccl2−/−/Cx3cr1−/− double-knockout mice are dependent on the rd8 mutation, rather than the synergistic effect of both mutations [19].
We previously reported the ocular phenotype of a knock-in mouse model for late-onset retinal degeneration (L-ORD) caused due to a heterozygous S163R mutation in the Ctrp5 gene [20]. These mice develop a late-onset retinal degeneration phenotype resembling the clinical phenotype observed in patients with L-ORD, including slower rod b-wave recovery, RPE abnormalities, drusen, Bruch’s membrane abnormalities including basal laminar deposits, predominant cone photoreceptor loss with onset around age 13 months, followed by rapid progression of retinal degeneration to near complete loss of cones by 21 months, and an age-dependent accumulation of hyperfluorescent spots [21-23]. Because the L-ORD mouse model (with Ctrp5+/− genotype) was developed using embryonic stem (ES) cells from mixed C57BL/6J and C57BL/6N strains and maintained on a C57BL/6J background, we investigated if the rd8 mutation is present in our colonies and whether it influenced the retinal phenotype associated with the Ctrp5 S163R mutation. The L-ORD mouse model (Ctrp5+/−;rd8/rd8 and Ctrp5+/−; wt/wt) and the wild-type littermate controls (Wt rd8/rd8 and Wt wt/wt) were evaluated for the presence of the rd8 mutation and its associated phenotype.
Methods
Animals
The L-ORD mice were generated using ES cells derived from mixed C57BL/6J and C57BL/6N strain background mice [20]. These mouse lines were expanded by crossing with the C57BL/6J strain obtained from Jackson Laboratories (Bar Harbor, ME) to maintain the line. Mice with the desired genotype were generated for the current study by selectively breeding the mice in our L-ORD mice colony to generate the required genotypes. All studies were performed using L-ORD mice and wild-type (WT) littermate mice of age groups 6–8, 9–11, 12–14, and 20–21 months. Five mice of each group were used for autofluorescent-scanning laser ophthalmoscopy (AF-SLO) imaging analyses; three to five mice were used for light microscopy and immunohistochemistry (as described below in the Funduscopy section). All mice were maintained according to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and with protocols approved by UCSD Institutional Animal Care and Use Committee.
Detection of the rd8 mutation with allele-specific PCR and sequencing
The mice were genotyped, as described previously [17,20]. Mouse tail DNA was isolated with Qiagen reagents following the manufacturer’s instructions (Gentra Puregene Mouse Tail Kit, Qiagen, Valencia, CA). Genotyping and sequencing for the Crb1 c.3481delC and Ctrp5 S163R mutations were performed using the primers described in Table 1.
Table 1. PCR primer for genotyping Wt wt/wt,Wtrd8/rd8, Ctrp5+/−,rd8/rd8 mice and for amplification of rd8 locus for sequencing.
| Gene | Allele | Primer name | Sequence (5′-3′) |
|---|---|---|---|
|
Crb1 |
Wild-type |
3663-mCrb1 mF1 |
GTGAAGACAGCTACAGTTCTGATC |
| 3665-mCrb1 mR |
GCCCCATTTGCACACTGATGAC |
||
|
Crb1 |
Mutant |
3664-mCrb1 mF2 |
GCCCCTGTTTGCATGGAGGAAACT
TGGAAGACAGCTACAGTTCTTCTG |
| 3665-mCrb1 mR |
GCCCCATTTGCACACTGATGAC |
||
|
Crb1 |
rd8 |
3666-Crb-F |
GGTGACCAATCTGTTGACAATCC |
| 3667-Crb-R |
GCCCCATTTGCACACTGATGAC |
||
|
Ctrp5 |
Knock-in |
2855-Ctrp5-F |
CCCCTACCTTTCGACCGT |
| 2856-Ctrp5-R | GAAGAAAGAGGCGATGGACTG |
The allele-specific PCR was performed using mCrb1mF1, mCrb1mF2, and mCrb1mR primers as described earlier [5]. The PCR reaction was carried out in a 20 μl reaction volume containing 1.5 mM MgCl2, 100 μM of each dNTP, 1.6 μM each of forward and reverse primer for Wt allele and 0.8 μM of forward and 1.6 μM of reverse primer for rd8 mutate allele with 0.026U AmpliTaq DNA polymerase. Approximately 25 ng of DNA was taken for the reaction. The PCR reaction was initially denatured at 94 °C for 5 min followed by 35 cycles at 94 °C for 30 s, 56 °C for 30 s, 72 °C for 30 s with a final extension of 72 °C for 7 min. Amplification of the wild-type allele produces a 220 bp fragment, and the mutant allele produces a 240 bp fragment. Confirmation of the rd8 mutation was performed by sequencing the amplicons [17]. Genotyping for the Ctrp5 S163R knock-in mutation was performed as described earlier [20].
Fundoscopy
Fundoscopy and autofluorescence (AF) imaging were performed using a SPECTRALIS high-resolution, using a SPECTRALIS high-resolution, combination scanning laser ophthalmoscope and spectral domain optical coherence tomography (HRA+OCT) imaging system (Heidelberg Engineering, Inc., Carlsbad, CA) as previously described [20,24-26]. The number of AF spots per eye was counted, and the average number of AF spots per fundus in each group were calculated.
Retinal histology
L-ORD (n=5) and WT (n=5) mice with and without the rd8 mutation were enucleated and the eyes fixed with the immersion fixation method using 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M phosphate buffer. Eyecups were postfixed in osmium-tannic acid-paraphenylenediamine to preserve neutral lipids [27] before being embedded in Epon 812 per our published methods [28]. Thin sections were examined on a JEM 1200EX II electron (JOEL USA, Inc. Peabody, MA) microscope. Additional eyecups were embedded in methacrylate [17]. Sections (8–10 µm) were stained with hematoxylin and eosin and examined under a light microscope at 20X magnification. Retina cryosections (8–10 μm), obtained from the eyes fixed in 2% paraformaldehyde, were stained for β-catenin using specific antibodies and images were captured with an Olympus FV1000 confocal microscope.
Antibodies
S-opsin antibodies, goat polyclonal (1:200 Chemicon, Temecula, CA), and M-opsin antibody rabbit polyclonal (1:200, Chemicon) were obtained from a commercial source. The anti-mouse secondary antibody conjugated to Alexa fluor 555 and Alexa fluor 488 (1:1,000, Life Technologies, Grand Island, NY) were used to detect the protein expression with immunohistochemistry. β-catenin antibody (1:200; Santa Cruz Biotechnology Inc., Santa Cruz, CA) was used to check the integrity of the external limiting membrane (ELM).
Statistical analysis
Statistical significance between the number of AF spots in the mutant and wild-type mice was determined using the two-tailed, independent Student t test.
Results
Detection of the rd8 mutation in L-ORD and WT mice
Analysis of mice in the L-ORD mouse model colony revealed the presence of the rd8 mutation. The location and identity of the mutation were confirmed with dideoxy sequencing (Figure 1A). In the L-ORD mouse model colony, the rd8 mutation was detected in the L-ORD mice and the WT littermate controls (Figure 1B). The majority of the mice carried the rd8 mutation in the homozygous or heterozygous state, while a minor proportion were negative for the rd8 mutation.
Figure 1.

Detection of the rd8 mutation with sequencing and agarose gel analysis. A: Sequence of the Crb1 locus showing the wild-type and the c.3481delC mutant allele in the homozygous state. The asterisk denotes the deletion. B: Agarose gel electrophoresis showing the migration of PCR amplicons for the rd8 locus from Wt mice (220 bp), mice heterozygous (244 bp and 220 bp) and homozygous (244 bp) for the rd8 mutation. The DNA ladder was included to indicate the size of amplicons. Wtwt/wt, Crb1 wild-type; Wtwt/rd8, heterozygous for the Crb1 mutation; Wtrd8/rd8, homozygous for the Crb1 mutation.
Ocular phenotype of L-ORD (Ctrp5+/−;wt/wt) and WT littermate control (Wtwt/wt) mice
The presence of ocular spots is a common finding in L-ORD and rd8 mice strains. AF-SLO was used to document the ocular phenotype of Ctrp5+/−;wt/wt mice (n=5) and age-matched littermates Wtwt/wt (n=5; Figure 2). Autofluorescent spots were not observed in the 6- to 8-month-old and 9- to 11-month-old WTwt/wt mice (0±0). Few spots were visualized in the 12- to 14-month-old and 20- to 21-month-old WTwt/wt mice (23±7 and 70±26, respectively). In contrast, the Ctrp5+/−;wt/wt mice displayed more spots by 12–14 months (116±72). The accumulation of AF spots in the Ctrp5+/−;wt/wt mice was accelerated with age and increased several fold by 20–21 months (1569±105). The AF spots were small and round and distributed evenly throughout the retina in the Ctrp5+/−;wt/wt mice.
Figure 2.

Autofluorescence SLO fundus images of L-ORD mice lacking the rd8 mutation. A: Representative autofluorescent-scanning laser ophthalmoscopy (AF-SLO) images of C57BL/6 wild-type control mice free of the rd8 mutation (Wtwt/wt) and late-onset retinal degeneration (L-ORD) mice free of the rd8 mutation (Ctrp5+/−;wt/wt). The Wtwt/wt mice showed few AF spots by 21 months. However, the AF spots were significantly increased in the Ctrp5+/−;wt/wt mice by 21 months of age. Magnification bar=200 μm. B: Quantification of the AF spots in Wtwt/wt (black triangle) and Ctrp5+/−;wt/wt (black diamond) per the AF-SLO images. The number of AF spots is shown as the mean number of spots ± SD. There was an age-related increase in AF spots (linear regression plot) in both genotypes. However, the AF spots accumulated to a high number in L-ORD mice by 21 months when compared to the age-matched control mice (p<0.001).
Retinal morphology of Ctrp5+/−;wt/wt, Wtwt/wt, and Wtrd8/rd8 mice
Retinal morphology was studied to evaluate the integrity of the retinal tissue. The gross retinal morphology of the Wtrd8/rd8 mice was compared with the Wtwt/wt mice in our L-ORD mouse colony (Figure 3). Both sets of wild-type mice showed normal retinal morphology and lacked the changes associated with the rd8 genotype at 8 months of age. The INL, ONL, outer plexiform layer (OPL), and outer segments (OS) were normal in these mice. In addition, pseudorosette structures in the OPL and sub-retinal space that are prominent characteristic features of the rd8 phenotype were absent in all mice.
Figure 3.
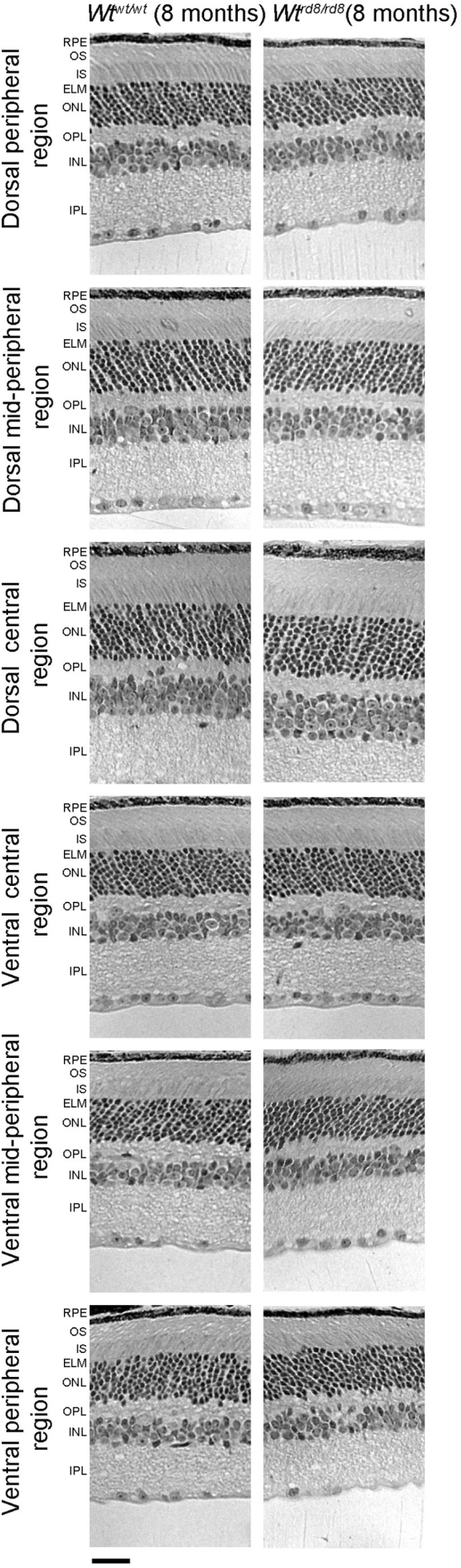
Histological analysis of the retina structure of 8-month-old Wtwt/wt and Wtrd8/rd8 mice. The retinas from 8-month-old Wtwt/wt and Wtrd8/rd8 mice showed no dysplasia. mo, month; RPE, retinal pigment epithelium; OS, outer segment; IS, inner segment; ELM, external limiting membrane; ONL, outer- nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, glial cell layer. Magnification bar=2 μm.
The retinal architecture of the Ctrp5+/−;wt/wt mice was studied up to 21 months and compared with the phenotype of the age-matched Wtwt/wt littermate control mice (Figure 4). Under light microscopy, these mice did not show significant morphological changes including the abnormalities typically associated with the rd8 phenotype.
Figure 4.

Histological analysis of the retinal structure of the L-ORD mice. Retinal changes typically associated with the rd8 phenotype were not observed in either the Ctrp5+/−;wt/wt or Wt wt/wt mice at 20–21 months. In addition, the retinal morphology of these mice lacked gross abnormalities that can be detected with light microscopy. Magnification bar=20 μm.
Ultrastructural analysis of the RPE was performed on Ctrp5+/−;wt/wt mice (n=3) at 9, 14, and 21 months. The Wtwt/wt mice (n=3) aged 21 months were used as a control to compare the pathology (Figure 5). The Wtwt/wt mice had normal RPE with well-organized basal infoldings and an intact Bruch’s membrane. RPE abnormalities were prominent as early as 9 months in the Ctrp5+/−;wt/wt mice. The RPE basal infoldings were disorganized and displaced from Bruch’s membrane. Numerous vacuoles and phagolysosomes were found throughout the cytoplasm, and several packets of undigested membranous debris were found in the basal RPE. Focal basal laminar deposits and basal linear deposits were found in Bruch’s membrane by 14 months. The same abnormalities were also found in the 21-month-old Ctrp5+/−;wt/wt mice.
Figure 5.

Ultrastructural analysis of RPE and sub-RPE areas from Wtwt/wt and Ctrp5+/− ;wt/wt mice. Areas enclosed in boxes in A–D are magnified in panels E–H. RPE from control mice (A, E) showed no abnormalities at 21 months of age. The RPE basal infoldings are organized (star), the cytoplasm is homogenous, and Bruch’s membrane is intact (bracket). In contrast, the RPE of the Ctrp5+/−,wt/wt mice (B, C, D, F, G, H) has structural aberrations beginning at 9 months of age. The RPE basal infoldings are disorganized (stars). Numerous packets of undigested membranous debris are found in the basal RPE (black arrowheads). Multiple vacuoles (white arrows) and phagolysosomes (black arrows) are found throughout the cytoplasm. Focal basal laminar deposits (white arrowheads) are present in Bruch’s membrane, as are basal linear deposits (asterisks). All marking are identical to those used in all panels. Magnification bars=200 μm in the top row and 500 μm in the bottom row.
External limiting membrane is continuous in Ctrp5+/−; rd8/rd8 mouse
To determine whether the loss of Crb1 affects ELM integrity, we studied the localization of β-catenin in the Ctrp5+/−;wt/wt and Ctrp5+/−;rd8/rd8 mice and compared its localization with the Wtwt/wt mouse retina. The anti-β-catenin antibody strongly stained the ELM in all mice including those with the rd8 mutation (Figure 6), and the pattern of staining indicated the presence of continuous ELM not only in the Wtwt/wt mice but also in the mice that had the rd8 mutation. These results demonstrate the presence of a continuous ELM.
Figure 6.

Continuous ELM staining with anti-adherence junction marker in L-ORD mice homozygous for the rd8 mutation. A–F are stained with anti-β-catenin. A, D: Retinal specimen from rd8 free wild-type control mouse (Wtwt/wt) at 9 months of age. B, E: Retinal specimen from a 5-month-old late-onset retinal degeneration (L-ORD) mouse lacking the rd8 mutation (Ctrp5+/−;wt/wt). C, F: Retinal specimen from a 12-month-old L-ORD mouse homozygous for the rd8 mutation (Ctrp5+/−;rd8/rd8). Continuous staining is observed across the ELM in all genotypes. The arrows indicate the ELM. Magnification bar=4 μm. ELM, external limiting membrane, ONL, outer nuclear layer, INL, internal nuclear layer.
Analysis of previously published Ctrp5+/− mice and WT controls for the rd8 genotype and phenotype
Systematic analysis of the Ctrp5+/− mice and their littermate controls was previously described by Chavali et al. [20]. Recent analysis of the tail DNA of the mice reported in Chavali et al. [20] for the Crb1 mutation revealed that the WT and Ctrp5+/− mice presented in that study had the rd8 genotype either in the heterozygous or homozygous state (Table 2). A review of retinal morphology of these mice presented in that publication revealed a lack of the characteristic features of rd8 pathology in the Ctrp5+/− mice and the WT littermate controls up to 21 months [20]. Specifically, pseudorosettes were absent, and morphological changes suggestive of abnormal ELM were not observed. Ultrastructure analysis showed no abnormalities in the retina of the littermate control mice carrying the rd8 mutation up to 21 months (Table 2) [20]. The retinal ultrastructure of the Ctrp5+/− mice also had no apparent pathology until 5 months of age. However, in mice older than 12 months, the IS of the rods and cones were mildly swollen.
Table 2. Analysis of mice with L-ORD phenotype presented in Chavali et al. 2011.
| Age (month) | Genotype (n=3) | Figures (Chavali et al. 2011) | Phenotype |
|||
|---|---|---|---|---|---|---|
| Pseudo rosettes | Ocular lesion | Autofluorescent spots throughout retina | Number of cones | |||
| 5 |
Wt rd8/rd8 |
7A,9A |
Absent |
Absent |
Absent |
>200 |
|
Ctrp5+/−;
rd8/rd8 |
7B,9B,9C |
Absent |
Absent |
Absent |
>200 |
|
| 8 |
Wt
ND |
6A,6B,7C,9D |
Absent |
Absent |
Absent |
>200 |
|
Ctrp5+/−;
rd8/rd8 |
6A,6B,7D,9E, 9F |
absent |
Absent |
3±1 |
>200 |
|
| 12 |
Wt
ND |
6A,6B,7E,9G,11 |
Absent |
Absent |
Absent |
>200 |
|
Ctrp5+/−;
rd8/rd8 |
6A,6B,7F,9H,9I,11 |
Absent |
Absent |
56±7 |
183±7 |
|
| 15 |
Wt wt/rd8 |
6A,6B,7G,9J,11 |
Absent |
Absent |
15±5 |
>200 |
|
Ctrp5+/−;
wt/rd8 |
6A,6B,7H,9K,9L,11 |
Absent |
Absent |
80±10 |
108±7 |
|
| 21 |
Wt
ND |
6A,6B,7I,9M,11 |
Absent |
Absent |
25±4 |
>200 |
| Ctrp5+/−; wt/rd8 | 6A,6B,7J,9N,9O,11 | Absent | Absent | >200 | 15±5 | |
ND: not determined n=number of animals per assessment
Discussion
Analysis of our L-ORD mouse colony on a C57BL/6J background revealed the presence of the rd8 mutation [20]. In contrast to the findings reported in the original rd8 strain [5], Wtwt/wt, Wtrd8/rd8, and Ctrp5+/−; rd8/rd8 mice in our L-ORD mouse colony failed to exhibit the abnormal ocular pathology associated with the rd8 mutation up to 21 months of age, the longest time point presented in this study. In addition, the Ctrp5+/− mice free of the rd8 mutation developed the characteristic L-ORD-associated pathology.
The rd8 mice were reported to develop AF lesions that were detected as early as 3 weeks of age [5]. These lesions were large, irregular, and heavily concentrated in the inferior nasal quadrant of the fundus. Clinically, these lesions correspond to the region with retinal folds and pseudorosettes that involve the photoreceptors [29]. The laminar folds and pseudorosettes in rd8 mice were visualized between the ONL and the INL. Morphologically, they represent displacement of the IS/OS material and the photoreceptor cell bodies into the inner retina due to the incomplete formation of the ELM and loss of the structural barrier [29]. The photoreceptor IS lose their orderly arrangement, and by 4 weeks of age, 25% of photoreceptors are short in the rd8 mice when compared to WT mice [5]. A significant thinning of the IS and OS in the region with dysplasia was reported in rd8 mice by 4 weeks of age. By 10 weeks, photoreceptor OS in rd8 mice start to fragment resulting in the accumulation of granular debris in the sub-retinal space [5].
In contrast to the findings in the original colony of rd8 mice, the Wtrd8/rd8 and Ctrp5+/−; rd8/rd8 mice in our L-ORD mouse colony did not show pseudorosette pathology even at 21 months of age, the oldest age tested in this study. The histology of the mice in the L-ORD colony was devoid of the characteristic features of the rd8 phenotype. The Wtrd8/rd8 and Ctrp5+/−; rd8/rd8 mice showed well-defined nuclear layers (the INL and the ONL). The characteristic retinal changes that are prominent in rd8 mice as early as 4 weeks were not observed in either the Wtrd8/rd8 or Ctrp5+/-; rd8/rd8 mice in our colony of L-ORD mice at least up to age 21 months.
The L-ORD mouse model develops AF lesions around 12 months with a subsequent progressive increase in the number of these lesions with age [20]. Contrary to the large number of AF lesions detected in Ctrp5+/−;wt/wt mice at older ages, a minimal number of spots were observed in the age-matched Wtwt/wt mice. Although AF lesions in 21-month-old Ctrp5+/−;wt/wt were distributed throughout the retina, the ocular spots observed in the rd8 model were larger, heavily concentrated in the inferior quadrant of the fundus, and developed as early as 3 weeks of age. The rd8 mice backcrossed onto the C57BL/6 background were reported to have a discontinuous ELM phenotype [5]. In contrast, the Wtrd8/rd8 and Ctrp5+/−;rd8/rd8 mice raised on the C57BL/6J background exhibited an uninterrupted outer limiting membrane. These observations indicate that the presence of the Crb1 mutation in either the homozygous or heterozygous state failed to result in the development of the characteristic rd8 phenotype in the Wtrd8/rd8 and Ctrp5+/-; rd8/rd8 mice in our L-ORD mouse colony. A review of retinal morphology and analysis of the genotype of previously published L-ORD mice and wild-type controls revealed the absence of the rd8-associated phenotype at least up to 21 months [20].
In contrast to the findings in the L-ORD mice, the retinal pathology in the rd8 model is clearly evident by 4 weeks with shortening and swelling of the IS and OS in rd8 mice [5]. Abnormalities in the laminar architecture and retinal degeneration were also noted [5,29]. However, abnormal drusen-like, basal laminar, and basal linear deposits similar to those observed in the L-ORD mouse model were not reported in rd8 mice [20]. The inferior retina of the rd8 mouse is more severely affected with prominent dysplasia than the superior retina. These abnormalities were absent in the L-ORD mice even at 21 months [20]. These observations indicate that the presence of the Crb1 mutation either in the heterozygous state or homozygous state did not result in the development of the rd8 characteristic phenotype in the Ctrp5+/− mice in our L-ORD mouse colony.
The absence of the rd8 retinal phenotype was noted not only in Ctrp5+/−; rd8/rd8 mice but also in the littermate Wtrd8/rd8 mice. This is consistent with the findings reported by Mehalow et al. for C57BL/6J mice [5]. In that study, a significant proportion of mice with a homozygous Crb1 mutation on the C57BL/6J background did not exhibit rd8-associated retinal pathology, including ocular spots, retinal folds, pseudorosettes, inner segment shortening, and disorganization [5]. A more recent study by Luhman et al. reported that the homozygous rd8 mutation on the C57BL/6 background leads to an increase in evenly distributed small discrete autofluorescent lesions on SLO fundus images [18].
It was hypothesized that the retinal abnormalities due to the Crb1 mutation are strongly modulated by a genetic variant expressed in C57BL/6J mice [5]. Supporting this hypothesis, Luhmann et al. showed that an autosomal recessive locus in C57BL/6 can modulate the rd8 phenotype in mice homozygous for the Crb1 mutation [18,19]. The ES cells used to develop our L-ORD mice were a mixed population obtained from mice with C57BL/6J and C57BL/6N backgrounds. Subsequently, our L-ORD mice were bred with the C57BL/6J strain to maintain the mice line. It is likely that the modifier allele in the C57BL/6J strain is responsible for the absence of the rd8 phenotype in our L-ORD mouse model colony.
The clinical phenotype of Ctrp5+/−;rd8/rd8 and Ctrp5+/−;wt/wt mice is similar with characteristic late-onset sub-RPE deposits and Bruch’s membrane abnormalities including basal laminar and basal linear deposits. This pathology is consistent with the phenotype observed in patients with L-ORD, while the retinal phenotype of age-matched Wtwt/wt and Wtrd8/rd8 mice was normal. These observations clearly show that the presence of the Crb1 mutation did not result in the development of retinal abnormalities in either Ctrp5+/−;rd8/rd8 or littermate Wtrd8/rd8 mice. In addition, these data demonstrate that the S163R mutation in Ctrp5 in the heterozygous state leads to the development of the L-ORD phenotype and that the presence of Crb1 mutation did not alter the retinal phenotype in these mice. It is likely that the presence of the Crb1 mutation in addition to the Ctrp5 mutation may result in additional changes that were not investigated in this study.
The autofluorescent spots reported by Luhman et al. in C57BL/6 mice with the Crb1 mutation are similar to the AF lesions observed in Ctrp5+/− mice [18]. It is possible that the AF lesions observed in rd8, rd6, Ctrp5, and chemokine-deficient mice with diverse genetic abnormalities may have a similar origin, and studying the pathobiology of these lesions may provide better understanding of the mechanism underlying RD.
Although this L-ORD mouse model (Ctrp5+/−) generated on the C57BL/6J background developed late-onset retinal pathology, another knock-in model with the Ctrp5 S163R mutation generated on the 129SV background (Ctrp5+/−-129) did not develop retinal pathology even when older [30]. Shu et al. speculated that the genetic background may also influence the development of L-ORD pathology in mice [20,30]. Our current studies further support this hypothesis, and the identification of the underlying cause of variation in the phenotype between these two mouse models is likely to assist in understanding the molecular pathology of L-ORD and the development of therapeutic interventions to treat this condition.
Acknowledgments
These studies were supported by: The Foundation Fighting Blindness, Thome Foundation in AMD Research, Research to Prevent Blindness (to Departments of Ophthalmology at UCSD, La Jolla, and University of Tennessee Health Science Center), R01EY13198, R01EY21237 and P30EY022589.
References
- 1.Meuleman J, van de Pavert SA, Wijnholds J. Crumbs homologue 1 in polarity and blindness. Biochem Soc Trans. 2004;32:828–30. doi: 10.1042/BST0320828. [DOI] [PubMed] [Google Scholar]
- 2.van de Pavert SA, Kantardzhieva A, Malysheva A, Meuleman J, Versteeg I, Levelt C, Klooster J, Geiger S, Seeliger MW, Rashbass P, Le Bivic A, Wijnholds J. Crumbs homologue 1 is required for maintenance of photoreceptor cell polarization and adhesion during light exposure. J Cell Sci. 2004;117:4169–77. doi: 10.1242/jcs.01301. [DOI] [PubMed] [Google Scholar]
- 3.Wodarz A, Hinz U, Engelbert M, Knust E. Expression of crumbs confers apical character on plasma membrane domains of ectodermal epithelia of Drosophila. Cell. 1995;82:67–76. doi: 10.1016/0092-8674(95)90053-5. [DOI] [PubMed] [Google Scholar]
- 4.Jacobson SG, Cideciyan AV, Aleman TS, Pianta MJ, Sumaroka A, Schwartz SB, Smilko EE, Milam AH, Sheffield VC, Stone EM. Crumbs homolog 1 (CRB1) mutations result in a thick human retina with abnormal lamination. Hum Mol Genet. 2003;12:1073–8. doi: 10.1093/hmg/ddg117. [DOI] [PubMed] [Google Scholar]
- 5.Mehalow AK, Kameya S, Smith RS, Hawes NL, Denegre JM, Young JA, Bechtold L, Haider NB, Tepass U, Heckenlively JR, Chang B, Naggert JK, Nishina PM. CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum Mol Genet. 2003;12:2179–89. doi: 10.1093/hmg/ddg232. [DOI] [PubMed] [Google Scholar]
- 6.den Hollander AI, Johnson K, de Kok YJ, Klebes A, Brunner HG, Knust E, Cremers FP. CRB1 has a cytoplasmic domain that is functionally conserved between human and Drosophila. Hum Mol Genet. 2001;10:2767–73. doi: 10.1093/hmg/10.24.2767. [DOI] [PubMed] [Google Scholar]
- 7.Cremers FP, van den Hurk JA, den Hollander AI. Molecular genetics of Leber congenital amaurosis. Hum Mol Genet. 2002;11:1169–76. doi: 10.1093/hmg/11.10.1169. [DOI] [PubMed] [Google Scholar]
- 8.den Hollander AI, Ghiani M, de Kok YJ, Wijnholds J, Ballabio A, Cremers FP, Broccoli V. Isolation of Crb1, a mouse homologue of Drosophila crumbs, and analysis of its expression pattern in eye and brain. Mech Dev. 2002;110:203–7. doi: 10.1016/s0925-4773(01)00568-8. [DOI] [PubMed] [Google Scholar]
- 9.Bernal S, Calaf M, Garcia-Hoyos M, Garcia-Sandoval B, Rosell J, Adan A, Ayuso C, Baiget M. Study of the involvement of the RGR, CRPB1, and CRB1 genes in the pathogenesis of autosomal recessive retinitis pigmentosa. J Med Genet. 2003;40:e89. doi: 10.1136/jmg.40.7.e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.den Hollander AI, Davis J, van der Velde-Visser SD, Zonneveld MN, Pierrottet CO, Koenekoop RK, Kellner U, van den Born LI, Heckenlively JR, Hoyng CB, Handford PA, Roepman R, Cremers FP. CRB1 mutation spectrum in inherited retinal dystrophies. Hum Mutat. 2004;24:355–69. doi: 10.1002/humu.20093. [DOI] [PubMed] [Google Scholar]
- 11.den Hollander AI, Heckenlively JR, van den Born LI, de Kok YJ, van der Velde-Visser SD, Kellner U, Jurklies B, van Schooneveld MJ, Blankenagel A, Rohrschneider K, Wissinger B, Cruysberg JR, Deutman AF, Brunner HG, Apfelstedt-Sylla E, Hoyng CB, Cremers FP. Leber congenital amaurosis and retinitis pigmentosa with Coats-like exudative vasculopathy are associated with mutations in the crumbs homologue 1 (CRB1) gene. Am J Hum Genet. 2001;69:198–203. doi: 10.1086/321263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.den Hollander AI, ten Brink JB, de Kok YJ, van Soest S, van den Born LI, van Driel MA, van de Pol DJ, Payne AM, Bhattacharya SS, Kellner U, Hoyng CB, Westerveld A, Brunner HG, Bleeker-Wagemakers EM, Deutman AF, Heckenlively JR, Cremers FP, Bergen AA. Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12). Nat Genet. 1999;23:217–21. doi: 10.1038/13848. [DOI] [PubMed] [Google Scholar]
- 13.Lotery AJ, Jacobson SG, Fishman GA, Weleber RG, Fulton AB, Namperumalsamy P, Heon E, Levin AV, Grover S, Rosenow JR, Kopp KK, Sheffield VC, Stone EM. Mutations in the CRB1 gene cause Leber congenital amaurosis. Arch Ophthalmol. 2001;119:415–20. doi: 10.1001/archopht.119.3.415. [DOI] [PubMed] [Google Scholar]
- 14.Bujakowska K, Audo I, Mohand-Said S, Lancelot ME, Antonio A, Germain A, Leveillard T, Letexier M, Saraiva JP, Lonjou C, Carpentier W, Sahel JA, Bhattacharya SS, Zeitz C. CRB1 mutations in inherited retinal dystrophies. Hum Mutat. 2012;33:306–15. doi: 10.1002/humu.21653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yzer S, Fishman GA, Racine J, Al-Zuhaibi S, Chakor H, Dorfman A, Szlyk J, Lachapelle P, van den Born LI, Allikmets R, Lopez I, Cremers FP, Koenekoop RK. CRB1 heterozygotes with regional retinal dysfunction: implications for genetic testing of leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2006;47:3736–44. doi: 10.1167/iovs.05-1637. [DOI] [PubMed] [Google Scholar]
- 16.Pang JJ, Chang B, Hawes NL, Hurd RE, Davisson MT, Li J, Noorwez SM, Malhotra R, McDowell JH, Kaushal S, Hauswirth WW, Nusinowitz S, Thompson DA, Heckenlively JR. Retinal degeneration 12 (rd12): a new, spontaneously arising mouse model for human Leber congenital amaurosis (LCA). Mol Vis. 2005;11:152–62. [PubMed] [Google Scholar]
- 17.Mattapallil MJ, Wawrousek EF, Chan CC, Zhao H, Roychoudhury J, Ferguson TA, Caspi RR. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci. 2012;53:2921–7. doi: 10.1167/iovs.12-9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luhmann UF, Carvalho LS, Holthaus SM, Cowing JA, Greenaway S, Chu CJ, Herrmann P, Smith AJ, Munro PM, Potter P, Bainbridge JW, Ali RR. The severity of retinal pathology in homozygous Crb1rd8/rd8 mice is dependent on additional genetic factors. Hum Mol Genet. 2015;24:128–41. doi: 10.1093/hmg/ddu424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luhmann UF, Lange CA, Robbie S, Munro PM, Cowing JA, Armer HE, Luong V, Carvalho LS, MacLaren RE, Fitzke FW, Bainbridge JW, Ali RR. Differential modulation of retinal degeneration by Ccl2 and Cx3cr1 chemokine signalling. PLoS ONE. 2012;7:e35551. doi: 10.1371/journal.pone.0035551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chavali VR, Khan NW, Cukras CA, Bartsch DU, Jablonski MM, Ayyagari RA. CTRP5 gene S163R mutation knock-in mouse model for late-onset retinal degeneration. Hum Mol Genet. 2011;20:2000–14. doi: 10.1093/hmg/ddr080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuntz CA, Jacobson SG, Cideciyan AV, Li ZY, Stone EM, Possin D, Milam AH. Sub-retinal pigment epithelial deposits in a dominant late-onset retinal degeneration. Invest Ophthalmol Vis Sci. 1996;37:1772–82. [PubMed] [Google Scholar]
- 22.Milam AH, Curcio CA, Cideciyan AV, Saxena S, John SK, Kruth HS, Malek G, Heckenlively JR, Weleber RG, Jacobson SG. Dominant late-onset retinal degeneration with regional variation of sub-retinal pigment epithelium deposits, retinal function, and photoreceptor degeneration. Ophthalmology. 2000;107:2256–66. doi: 10.1016/s0161-6420(00)00419-x. [DOI] [PubMed] [Google Scholar]
- 23.Subrayan V, Morris B, Armbrecht AM, Wright AF, Dhillon B. Long anterior lens zonules in late-onset retinal degeneration (L-ORD). Am J Ophthalmol. 2005;140:1127–9. doi: 10.1016/j.ajo.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 24.Seeliger MW, Beck SC, Pereyra-Munoz N, Dangel S, Tsai JY, Luhmann UF, van de Pavert SA, Wijnholds J, Samardzija M, Wenzel A, Zrenner E, Narfstrom K, Fahl E, Tanimoto N, Acar N, Tonagel F. In vivo confocal imaging of the retina in animal models using scanning laser ophthalmoscopy. Vision Res. 2005;45:3512–9. doi: 10.1016/j.visres.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 25.von Rückmann A, Fitzke FW, Bird AC. Distribution of fundus autofluorescence with a scanning laser ophthalmoscope. Br J Ophthalmol. 1995;79:407–12. doi: 10.1136/bjo.79.5.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koh HJ, Bessho K, Cheng L, Bartsch DU, Jones TR, Bergeron-Lynn G, Freeman WR. Inhibition of choroidal neovascularization in rats by the urokinase-derived peptide A6. Invest Ophthalmol Vis Sci. 2004;45:635–40. doi: 10.1167/iovs.03-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curcio CA, Rudolf M, Wang L. Histochemistry and lipid profiling combine for insights into aging and age-related maculopathy. Methods Mol Biol. 2009;580:267–81. doi: 10.1007/978-1-60761-325-1_15. [DOI] [PubMed] [Google Scholar]
- 28.Jablonski MM, Tombran-Tink J, Mrazek DA, Iannaccone A. Pigment epithelium-derived factor supports normal development of photoreceptor neurons and opsin expression after retinal pigment epithelium removal. J Neurosci. 2000;20:7149–57. doi: 10.1523/JNEUROSCI.20-19-07149.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aleman TS, Cideciyan AV, Aguirre GK, Huang WC, Mullins CL, Roman AJ, Sumaroka A, Olivares MB, Tsai FF, Schwartz SB, Vandenberghe LH, Limberis MP, Stone EM, Bell P, Wilson JM, Jacobson SG. Human CRB1-associated retinal degeneration: comparison with the rd8 Crb1-mutant mouse model. Invest Ophthalmol Vis Sci. 2011;52:6898–910. doi: 10.1167/iovs.11-7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shu X, Luhmann UF, Aleman TS, Barker SE, Lennon A, Tulloch B, Chen M, Xu H, Jacobson SG, Ali R, Wright AF. Characterisation of a C1qtnf5 Ser163Arg knock-in mouse model of late-onset retinal macular degeneration. PLoS ONE. 2011;6:e27433. doi: 10.1371/journal.pone.0027433. [DOI] [PMC free article] [PubMed] [Google Scholar]