

Abstract
Purpose
To identify the genetic defect in a consanguineous Israeli Muslim Arab family with juvenile retinitis pigmentosa (RP).
Methods
DNA samples were collected from the index patient, her parents, her affected sister, and two non-affected siblings. Genome-wide linkage analysis with 250 K single nucleotide polymorphism (SNP) arrays was performed using DNA from the two affected patients. Owing to consanguinity in the family, we applied homozygosity mapping to identify the disease-causing gene. The candidate gene SPATA7 was screened for mutations with PCR amplifications and direct Sanger sequencing.
Results
Following high-density SNP arrays, we identified several homozygous genomic regions one of which included the SPATA7 gene. Several mutations in SPATA7 have been reported for various forms of retinal dystrophy, including Leber congenital amaurosis (LCA) and juvenile RP. PCR-based sequence content mapping, long-distance PCR amplifications, and subsequent sequencing analysis revealed a homozygous 63.4 kb large deletion that encompasses the 5′ part of the SPATA7 gene including exons 1–5. The mutation showed concordant segregation with the phenotype in the family as expected for autosomal recessive mode of inheritance and is consistent with a diagnosis of juvenile RP.
Conclusions
We report a novel homozygous large deletion in SPATA7 associated with juvenile RP in a consanguineous Israeli Muslim Arab family. This is the first larger deletion mutation reported for SPATA7.
Introduction
Leber congenital amaurosis (LCA; OMIM 204000) and juvenile retinitis pigmentosa (RP) are closely related inherited retinal dystrophies with largely overlapping clinical characteristics. LCA is the most severe form of inherited retinal degeneration characterized by severe visual loss at or shortly after birth and nystagmus [1]. The incidence of LCA has been variably described as ranging from 1.2 [2] to 3.0 [3] per 100,000 live births. Fundus may be normal at diagnosis or present various abnormalities, including pigmentary retinopathy as seen in patients with retinitis pigmentosa [4]. In most cases, the electroretinogram (ERG) responses are non-detectable, a fact that accentuates the early loss of rod and cone photoreceptor function [5]. Usually, LCA shows an autosomal recessive pattern of inheritance. However, a small number of autosomal dominant LCA cases have been reported [6-8]. In approximately 20% of patients with LCA, mental retardation or behavioral disorders are observed [9]. Juvenile RP has an overall milder phenotype with disease onset occurring in the first years of a child’s life, night blindness, nystagmus, and severely reduced or extinguished ERG responses.
LCA was first described as an intrauterine form of RP [10]. A later classification by Foxman et al. enabled a clear differentiation between LCA and early-onset RP based on the age at onset of symptoms, severity of visual loss, and associated non-ocular abnormalities [11]. The distinction between LCA and juvenile or early-onset RP can become challenging, as the clinical features overlap. It is therefore also expected that the diseases might be caused by mutations in the same gene [12].
Previous work showed that, consistent with its clinical heterogeneity, LCA may be caused by mutations in at least 19 different genes: GUCY2D (Gene ID: 3000; OMIM 600179) [13], RPE65 (Gene ID: 6121; OMIM 180069) [14], SPATA7 (Gene ID: 55812; OMIM 609868) [12], AIPL1 (Gene ID: 23746; OMIM 604392) [15], LCA5 (Gene ID: 167691; OMIM 611408) [16], RPGRIP1 (Gene ID: 57096; OMIM 605446) [17,18], CRX (Gene ID: 1406; OMIM 602225) [7,19], CRB1 (Gene ID: 23418; OMIM 604210) [20], NMNAT1 (Gene ID: 64802; OMIM 608700) [21], CEP290 (Gene ID: 80184; OMIM 610142) [22,23], IMPDH1 (Gene ID: 3614; OMIM 146690) [8], RD3 (Gene ID: 343035; OMIM 180040) [24], RDH12 (Gene ID: 145226; OMIM 608830) [25,26], LRAT (Gene ID: 9227; OMIM 604863) [27,28], TULP1 (Gene ID: 7287; OMIM 602280) [29], KCNJ13 (Gene ID: 3769; OMIM 603208) [30], IQCB1 (Gene ID: 9657; OMIM 609237) (also NPHP5) [31,32], MERTK (Gene ID: 10461; OMIM 604705) [4,33], and OTX2 [34] (Gene ID: 5015; OMIM 600037). Mutations in some LCA genes, such as RPE65 [35], SPATA7 [12], CRB1 [36], RDH12 [26], LRAT [27], and TULP1 [37], are also known to cause autosomal recessively inherited juvenile RP. By contrast, heterozygous mutations in AIPL1 might cause an autosomal dominant form of juvenile RP [38].
Spermatogenesis associated protein 7 (SPATA7) was identified in 2003 by cloning and characterization of full-length cDNA from rat testes, and therefore designated as RSD-3; accordingly, the homologous human gene was called HSD-3.1 [39]. Human SPATA7 consists of 12 exons spanning a 52.9 kb genomic sequence and maps to chromosome 14q31.3 (Ensembl Genome Browser, transcript ID ENST00000393545, GRCh37 / hg19). SPATA7 encodes a protein of 599 amino acids that is conserved from sea urchins to humans but is absent in lower eukaryotes, such as insects and fungi [12]. In addition to expression in spermatocytes, Spata7 has been reported to be expressed in multiple retinal layers in the adult mouse retina, including the inner segments of photoreceptors as well as the ganglion cell and inner nuclear layers [12]. Expression patterns in the developing and mature mouse retina suggested that Spata7 is important for normal retinal function rather than development [12]. Two isoforms resulting from alternative splicing of exon 3 are predominantly expressed in the testis (isoform lacking exon 3) or in the brain and retina, respectively, proposing specific functions of the two SPATA7 isoforms in spermatozoa and neurons [9]. According to the Human Gene Mutation Database (HGMD), four nonsense mutations, three missense mutations, three splicing mutations, and seven small indel (insertions/deletions) mutations have been identified in SPATA7. Thus, this minor gene for LCA or juvenile RP accounts for only a small percentage of cases.
Methods
Clinical evaluation and DNA specimens
A two-generation family with autosomal recessive juvenile RP was ascertained from the Triangle Regional Research and Development Center (TRDC), Kfar Qari’, Israel. A detailed patient history of the index patient and her affected sister was recorded, including disease onset, symptoms, and progression. Visual acuity and visual field testing, full-field electroretinogram (ERG) recordings, and fundus examination followed by fundus photography and optical coherence tomography (OCT) imaging were performed in the index patient. Peripheral blood was taken from all family members using EDTA-containing vials. The blood samples were stored at 4°C for one week at longest before DNA was extracted using the MasterPure DNA Purification Kit for Blood Version II (Epicentre, Madison, WI) according to the manufacturer’s protocol. The study was conducted in accordance with the principles of the Declaration of Helsinki. This study was conducted in accordance with the principles of the Declaration of Helsinki and adhered to the ARVO statement on human subjects. It was further approved by the TRDC ethic committee. Moreover, written informed consent was obtained from each study participant or from the parents in the case of minor study subjects.
SNP genotyping and homozygosity mapping
Genome-wide single nucleotide polymorphism (SNP) genotyping was performed for DNA samples of the two affected siblings by applying Affymetrix GeneChip Human Mapping 250 K NspI SNP arrays (Affymetrix, Inc., Santa Clara, CA). SNP chip genotypes were called with the Affymetrix Genotyping Console (GTC) Software v. 2.1 and used to assess homozygous regions by applying the online version of the HomozygosityMapper Software [40]. The threshold for the identification of homozygous regions was set at 2 Mb.
PCR analyses
DNA fragments of the SPATA7 gene (reference sequence ENST00000393545, Ensembl Genome Browser) were amplified from genomic DNA with PCR. Standard PCRs were performed with 80 ng of genomic DNA in a 25 µl volume containing 0.2 µM of each primer, 200 µM of each dNTP, 10 mM Tris pH 8.9, 50 mM KCl, 3 mM MgCl2, and 0.5 U DNA Taq Polymerase (ATG Biosynthetics, Merzhausen, Germany) using the following cycling conditions: 4 min at 94 °C followed by 35 cycles of 15 s at 94 °C, 15 s at 60 °C, 30 s at 72 °C, and one final additional elongation for 5 min at 72 °C.
Mapping of deletion breakpoints
The absence of PCR products for exons 1–5 of SPATA7 in the index patient pointed to the presence of a homozygous deletion with the telomeric breakpoint located within intron 5. To refine this breakpoint, we performed sequence tagged site (STS) content mapping by applying a fixed reverse primer (SPATA7_Ex6–1_R; located in exon 6) and a set of ten forward primers located at increasing distance in intron 5. For amplification, we applied a long distance (LD) PCR protocol using 80 ng of genomic DNA in a total volume of 25 µl containing 0.2 µM of each primer, 400 µM of each dNTP, LA Buffer (1X, without MgCl2; ATG Biosynthetics), 0.5 mM MgCl2, and 2.5 U TaKaRa LA Taq DNA polymerase (Takara Bio Europe, Saint-Germain-en-Laye, France). Thermal cycling was performed with the following conditions: 1 min at 94 °C followed by 14 cycles of 10 s at 98 °C, 30 s at 55 °C and 12 min at 68 °C, a further 16 thermal cycles with an increment of 12 s/cycle for the elongation step, and a final add-on elongation for 10 min at 68 °C.
Initial attempts to amplify a breakpoint junction fragment with LD-PCR using primer SPATA7_Ex6–1_R and a set of forward primers located 1, 5, 10, or 22 kb upstream of SPATA7 failed to result in a proper PCR product. We therefore refined the centromeric breakpoint with STS content mapping of three amplicons located 28, 33, and 38 kb upstream of SPATA7. Successful amplification of all three STS prompted us to pursue another breakpoint junction LD-PCR using SPATA7-up-28kb-F and SPATA7_Ex6–1_R as the forward and reverse primers, respectively. A junction fragment of approximately 6.5 kb was obtained and further refined with restriction fragment analysis. We finally developed a 283 bp breakpoint junction PCR (primers: SPATA7_Del-F and SPATA7_Del-R) that was used for breakpoint sequencing and segregation analysis. The primer pairs used in the deletion mapping, their sequences, and physical coordinates are given in Appendix 1.
Sanger sequencing
PCR fragments were purified with ExoSAP-IT treatment (USB, Cleveland, OH), sequenced using the BigDye Terminator v. 1.1 Cycle Sequencing Kit (Applied Biosystems [ABI], Weiterstadt, Germany), and the sequencing products were subsequently separated on a DNA capillary sequencer (ABI 3130 Genetic Analyzer; ABI). All sequences were processed with the Sequencing Analysis v. 5.2 Software (Applied Biosystems) and aligned with the reference sequence using SeqMan (LaserGene Software Package, DNAStar, Inc., Madison, WI).
Bioinformatics analysis of junction sequences
For bioinformatics analysis of the breakpoint junctions, we downloaded 300 bp of reference sequence around both breakpoints (i.e., 150 bp of upstream and 150 bp of downstream sequences) and applied the following online bioinformatics tools: BLAST2seq to detect sequence (micro-)homology, RepeatMasker open-4.0.5 to search for repetitive elements, non-B DB search to identify non-B-DNA structures (which are susceptible to DNA breakage), and MEME Suite to search for shared sequence motifs. For convenience, all bioinformatics tools were applied with the default settings, and if not set by default, the cDNA sequences were also analyzed.
Results
In an Israeli Muslim Arab family (TR 11) with four children (three female, one male), two children (17- and 4-year-old sisters) were clinically diagnosed with autosomal recessive juvenile RP (Figure 1). Their parents were first-degree cousins and otherwise healthy. The affected siblings were born after normal pregnancies and normal deliveries. When the older affected daughter was 1 year old, the parents noticed nystagmus and vision abnormalities. Upon clinical examination, the diagnosis of juvenile RP was made. The younger affected daughter showed similar symptoms at about the same age, and clinical examination revealed retinal degeneration similar to that in the older sister. The visual deterioration showed considerable progression, resulting in reduced visual acuity (6/24 and 6/60 in the right and left eyes, respectively), severely constricted visual fields, and extinguished full-field ERGs in the older sister (at the age of 17). Funduscopic examination revealed areas of hypopigmentation due to atrophic RPE changes with pigmentary alterations from pigment migration (“salt and pepper fundus”), attenuated arterioles, and optic disc pallor, all characteristic of juvenile RP. OCT imaging showed an overall decrease in retinal thickness with shortening of the photoreceptor outer segments, reduction in the outer nuclear layer, and RPE atrophy. The inner retinal layers were less affected. The morphological findings for the older sister are presented in Figure 2. The two affected siblings were otherwise healthy; neurologic examination demonstrated normal findings without dysmorphic features or skin lesions.
Figure 1.

Pedigree of family TR 11. The parents were first-degree cousins. Affected members are indicated with filled symbols, and unaffected members are represented by open symbols. The arrow indicates the index patient.
Figure 2.
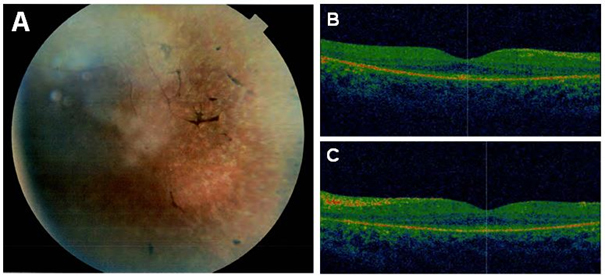
Retinal imaging findings. A: Funduscopy revealed the salt-and-pepper appearance of the retina, characteristic of juvenile retinitis pigmentosa (RP). B: Optical coherence tomography (OCT) image of the right eye. C: OCT image of the left eye. OCT imaging showed an overall decrease in retinal thickness with shortening of the photoreceptor outer segments, reduction in the outer nuclear layer, and RPE atrophy. The foveal photoreceptor layer was more preserved.
Homozygosity mapping performed with DNA samples of the two affected subjects revealed two larger homozygous regions on chromosome 8 (96.8M–116.1M) and chromosome 14 (84.9M–96.0M) and a few smaller homozygous regions on chromosomes 3, 17, and 22. The only known RD gene that localizes within these homozygous regions was SPATA7 that maps on chromosome 14 (at about 88.8M).
SPATA7 mutations have been reported for various forms of retinal dystrophy, including LCA and juvenile RP. Therefore, we thought to perform a mutation screening of SPATA7 by applying PCR amplification and direct Sanger sequencing. During this screening, we consistently failed to amplify exons 1–5 in the affected index patient, while further downstream amplicons were readily amplified. We thus hypothesized the presence of a homozygous deletion in the affected patients with a telomeric breakpoint in intron 5 of SPATA7. Taking advantage of the homozygous state of the affected patients, we further refined the localization of the telomeric and centromeric deletion breakpoint with PCR-based STS content mapping and LD PCR-based breakpoint junction amplifications (for details, see Methods), using this strategy, we mapped the centromeric deletion breakpoint to a 6 kb interval within the intergenic sequence between SPATA7 and its upstream gene potassium channel, subfamily K, member 10 (KCNK10; Gene ID 54207; OMIM 605873).
With refined breakpoint mapping, an approximately 6.5 kb deletion junction fragment was successfully amplified. We then used restriction site content mapping to further narrow the localization of the deletion breakpoint within the junction fragment and to design a novel primer pair directly flanking the deletion breakpoints. The reduced size of this PCR product facilitated subsequent Sanger sequencing to cover and define the exact breakpoints (Figure 3). This approach revealed a deletion at the SPATA7 locus that encompasses 63,411 bp and is accompanied by a small insertion of three extra nucleotides between the centromeric and telomeric breakpoints (g.88.828.457_88.891.867delinsTGG; c.1–23706_372+8679delinsTGG, ex. 1–5; GRCh37 / hg19). Since the centromeric breakpoint is located 23,417 bp upstream of SPATA7 5′ untranslated region (UTR) and the telomeric breakpoint is located 708 bp upstream of SPATA7 exon 6, the deletion included exons 1, 2, 3, 4, and 5 of the SPATA7 gene.
Figure 3.

Breakpoint sequence of the homozygous deletion at the SPATA7 locus. Electropherogram of the breakpoint sequence was obtained from PCR amplification covering the deletion. The deletion allele sequence is given in red, and the left and right junction sequences are indicated by black and gray letters. While the centromeric junction of the deletion is located within the intergenic sequence between SPATA7 and its upstream gene KCNK10, the telomeric junction is located within the intronic sequence between exons 5 and 6 of SPATA7. Sequencing of the deletion junction showed that the deletion is accompanied by a small insertion of three extra nucleotides (TGG) at the deletion site.
To test segregation of the deletion allele, all family members were analyzed with diagnostic PCR amplifications with (1) primers directly flanking the deletion breakpoints (deletion specific assay) and (2) with primers amplifying exon 2 of SPATA7 (wild-type specific assay). In doing so, we observed concordant segregation of the deletion in the family as expected for autosomal recessive mode of inheritance (Figure 4). The index patient and her affected sister were homozygous deletion carriers, whereas the unaffected parents and the two unaffected siblings were heterozygous mutation carriers.
Figure 4.
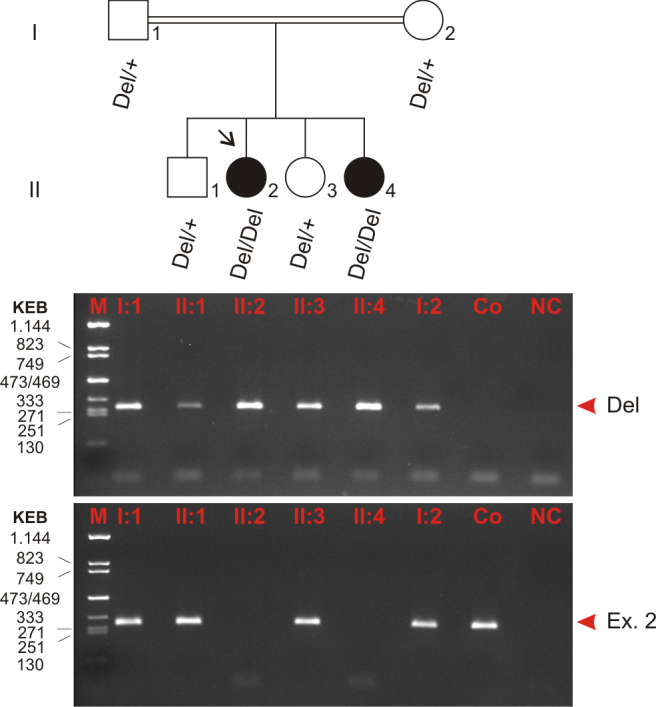
Segregation analysis of the SPATA7 mutation in family TR 11. Affected members are indicated with filled symbols, and unaffected members are represented by open symbols. The arrow indicates the index patient. Segregation analysis was performed with all family members as denoted by the genotype data (underneath). Two distinct PCR amplifications were used to analyze deletion and wild-type alleles, respectively: (1) one PCR with primers directly flanking the deletion breakpoints (top panel, Del) and (2) one PCR with primers amplifying exon 2 of SPATA7 located within the deletion (bottom panel, Ex. 2). The latter indicates the presence of the wild-type allele. All PCR products were analyzed on a 2% agarose gel. According to autosomal recessive inheritance, both parents and both unaffected siblings are heterozygous, and the two affected sisters are homozygous mutation carriers. Del, deletion allele; +, wild-type allele; Ex. 2, exon 2 (in place of wild-type allele); M, size marker (KEB, pcDNA 3.1zeo/TaqI-digested, fragment sizes given in bp); Co, control (DNA of an unaffected, not related person); NC, negative control (water control).
To obtain clues about the mechanism underlying the occurrence of this deletion, bioinformatics analyses of the sequences flanking the deletion breakpoints were performed. However, except for a few unspecific short motifs distant to the actual breakpoints, we did not detect repetitive sequences or other segments of sequence homology that may indicate a homology-based mechanism underlying the deletion. We therefore propose non-homologous end joining (NHEJ), a mechanism that accurately rejoins double-strand breaks, as the most likely mechanism underlying the occurrence of this deletion. In line with this assumption, NHEJ is frequently accompanied by the insertion of one to four nucleotide(s) at the breakpoint junction site [41] as also seen in our family.
Discussion
Since recessive diseases in consanguineous families are usually caused by homozygous mutations due to identity-by-descent, homozygosity mapping serves as an efficient tool for defining candidate chromosomal loci for diseases. After homozygosity mapping, we picked the correct candidate gene in a consanguineous Israeli Muslim Arab family. Using PCR and subsequent Sanger sequencing, we identified a homozygous large deletion in SPATA7 that caused juvenile RP in this family. We determined the centromeric and telomeric breakpoints of the deletion that is accompanied by a small insertion of three extra nucleotides at the site of deletion (g.88.828.457_88.891.867delinsTGG; c.1–23706_372+8679delinsTGG). The deletion includes exons 1–5 of SPATA7 as well as about 23.4 kb of upstream sequences that likely contain important regulatory sequence elements such as the proximal promoter and proximal transcription factor binding sites. We therefore argue that the mutation (g.88.828.457_88.891.867delinsTGG; c.1–23706_372+8679delinsTGG, ex. 1–5) results in a complete lack of SPATA7 gene or protein expression. Analysis of the breakpoint junctions did not provide evidence of a homology-based mechanism or non-B DNA structures underlying the occurrence of this deletion. Therefore, NHEJ seems to be the most likely mechanism.
SPATA7 is a known LCA gene also reported to cause juvenile RP. Nonetheless, SPATA7 is a minor gene for LCA and juvenile RP that accounts for only a small percentage of cases. We observed concordant segregation of the deletion in the family as expected for the autosomal recessive mode of inheritance. The two affected sisters showed similar clinical manifestations, and their functional and morphological findings support the diagnosis of juvenile RP. Both affected siblings were otherwise healthy with no non-ocular abnormalities. The observed clinical features were in line with previously reported cases [12,42]; the SPATA7 mutation caused an infantile-onset severe rod-cone dystrophy with early degeneration of the peripheral retina but comparatively preserved foveal structure, as seen on the OCT images.
Until now, no larger deletion as observed in this study has been reported for SPATA7. The Human Gene Mutation Database (HGMD) lists only seven small indel mutations, predominantly resulting in a frameshift and a premature stop codon (for instance the c.265_268delCTCA / p.L89KfsX3 mutation in Mackay et al.’s study [42]). In addition, three missense mutations, three splicing mutations, and four nonsense mutations (with c.253C>T / p.R85* [42] causing the earliest premature transcription termination codon in exon 5 and c.1183C>T / p.R395* causing the latest premature transcription termination codon in exon 11 [12]), are assumed to cause disease. Wang et al. [12] suggested a correlation between the severity of mutant alleles and the resulting clinical phenotype (i.e., the mutant alleles associated with LCA are likely to be more severe loss-of-function alleles than those associated with juvenile RP). However, this was not confirmed in the family reported here, since the large deletion detected at the SPATA7 gene locus was the cause of the overall milder clinical phenotype of juvenile RP.
The Database of Genomic Variants (DGV), a catalog of human genomic structural variations of clinically unaffected, healthy individuals, lists two deletions of 1442 bp and 1486 bp, respectively, in SPATA7 in a single Yoruban individual (NA18507) as reported by McKernan et al. [43]. The smaller of these two deletions maps to intron 5 and coincides with several smaller deletions in this region observed in several population studies. The telometric breakpoint of the large deletion reported in our family is also located in intron 5, however >1,000 bp downstream of the cluster of deletions annotated in DGV. The larger 1,486 bp deletion encompasses the alternatively spliced exon 3 of the SPATA7 gene. The consequence of such a loss of exon 3 is unknown; at least the reading frame of the coding sequence is maintained and results in the shortened SPATA7 isoform. Interestingly, DGV also lists a series of small sequence duplications in intron 2 that overlap with the deduced 5′ breakpoint of the 1,486 bp deletion in NA18507. These findings indicate the existence of two clusters of small copy number variations (CNVs) in SPATA7 that are with the exception of the 1,486 bp deletion in NA18507 restricted to intronic sequences and most likely without functional consequence.
SPATA7 was first identified in the rat testis. Then, researchers showed that Spata7 is also expressed in multiple layers of the mature mouse retina [12]. Since two different isoforms were shown to be expressed in the testis (transcript lacking exon 3) or in the brain and retina (transcript containing exon 3), specific functions of the two SPATA7 isoforms in spermatozoa and neurons were proposed [9]. Analyzing the mouse retina, Wang et al. [12] showed that Spata7 is present in the ganglion cell layer, the inner nuclear layer, and the inner segment of the photoreceptors at P21. Concerning the uniform distribution of the protein in the cytoplasm of the inner segment, it seems not to be a typical cilium protein and might be involved in a novel pathway [12]. The relatively late onset of Spata7 expression in the retina further suggested that Spata7 is important for normal retinal function rather than development [12]. Abulimiti et al. [44] showed that homozygous knockout mice exhibit impaired rhodopsin transportation to the outer segments due to the absence of Spata7 protein function. Nonetheless, additional studies are needed for a detailed understanding of the SPATA7 function and how different mutant alleles cause either LCA or the overall milder phenotype of juvenile RP, especially since the mutation reported here most likely results in a complete lack of SPATA7 protein function but causes the overall milder phenotype of juvenile RP. Thus, our findings are contrary to the suggestion of Wang et al. [12] implying a correlation between the severity of mutant alleles and the resulting clinical phenotype.
In conclusion, a genome-wide screen for homozygosity combined with a candidate gene evaluation within one homozygous region on chromosome 14 resulted in the identification of a pathogenic mutation in SPATA7. The homozygous large deletion encompasses exons 1–5 and is suggested to result in a complete lack of SPATA7 gene or protein expression. Nonetheless, the mutation was found to cause the overall milder clinical phenotype of juvenile RP, in preference to the more aggressive progression of LCA.
Acknowledgments
We would like to give thanks to the family members for participation in this study. This work was supported by funds of the ‘Deutsche Forschungsgemeinschaft’ (Wi1189/8–1 to B.W. and SCHO 754/5–2). Dr. Muhammad Mahajnah (mohamedm@hy.health.gov.il), Dr. Rajech Sharkia (rajach.sharkia@beitberl.ac.il) and Dr. Bernd Wissinger (wissinger@uni-tuebingen.de) contribute equally to this paper.
Appendix 1. Primer pairs, primer sequences and their genomic localization.
To access the data, click or select the words “Appendix 1.”
References
- 1.den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27:391–419. doi: 10.1016/j.preteyeres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 2.Stone EM. Leber congenital amaurosis - a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am J Ophthalmol. 2007;144:791–811. doi: 10.1016/j.ajo.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 3.Heckenlively JR, Foxman SG, Parelhoff ES. Retinal dystrophy and macular coloboma. Doc Ophthalmol. 1988;68:257–71. doi: 10.1007/BF00156432. [DOI] [PubMed] [Google Scholar]
- 4.Li L, Xiao X, Li S, Jia X, Wang P, Guo X, Jiao X, Zhang Q, Hejtmancik JF. Detection of variants in 15 genes in 87 unrelated Chinese patients with Leber congenital amaurosis. PLoS ONE. 2011;6:e19458. doi: 10.1371/journal.pone.0019458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perrault I, Rozet JM, Gerber S, Ghazi I, Leowski C, Ducroq D, Souied E, Dufier JL, Munnich A, Kaplan J. Leber congenital amaurosis. Mol Genet Metab. 1999;68:200–8. doi: 10.1006/mgme.1999.2906. [DOI] [PubMed] [Google Scholar]
- 6.Sohocki MM, Sullivan LS, Mintz-Hittner HA, Birch D, Heckenlively JR, Freund CL, McInnes RR, Daiger SP. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. Am J Hum Genet. 1998;63:1307–15. doi: 10.1086/302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freund CL, Wang QL, Chen S, Muskat BL, Wiles CD, Sheffield VC, Jacobson SG, McInnes RR, Zack DJ, Stone EM. De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat Genet. 1998;18:311–2. doi: 10.1038/ng0498-311. [DOI] [PubMed] [Google Scholar]
- 8.Bowne SJ, Sullivan LS, Mortimer SE, Hedstrom L, Zhu J, Spellicy CJ, Gire AI, Hughbanks-Wheaton D, Birch DG, Lewis RA, Heckenlively JR, Daiger SP. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2006;47:34–42. doi: 10.1167/iovs.05-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perrault I, Hanein S, Gerard X, Delphin N, Fares-Taie L, Gerber S, Pelletier V, Mercé E, Dollfus H, Puech B, Defoort-Dhellemmes S, Petersen MD, Zafeiriou D, Munnich A, Kaplan J, Roche O, Rozet JM. Spectrum of SPATA7 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum Mutat. 2010;31:E1241–50. doi: 10.1002/humu.21203. [DOI] [PubMed] [Google Scholar]
- 10.Leber T. Über Retinitis pigmentosa und angeborene Amaurose. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1869;15:1–25. [Google Scholar]
- 11.Foxman SG, Heckenlively JR, Bateman JB, Wirtschafter JD. Classification of congenital and early onset retinitis pigmentosa. Arch Ophthalmol. 1985;103:1502–6. doi: 10.1001/archopht.1985.01050100078023. [DOI] [PubMed] [Google Scholar]
- 12.Wang H, den Hollander AI, Moayedi Y, Abulimiti A, Li Y, Collin RW, Hoyng CB, Lopez I, Abboud EB, Al-Rajhi AA, Bray M, Lewis RA, Lupski JR, Mardon G, Koenekoop RK, Chen R. Mutations in SPATA7 cause Leber congenital amaurosis and juvenile retinitis pigmentosa. Am J Hum Genet. 2009;84:380–7. doi: 10.1016/j.ajhg.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perrault I, Rozet JM, Calvas P, Gerber S, Camuzat A, Dollfus H, Châtelin S, Souied E, Ghazi I, Leowski C, Bonnemaison M, Le Paslier D, Frézal J, Dufier JL, Pittler S, Munnich A, Kaplan J. Retinal-specific guanylate cyclase gene mutations in Leber's congenital amaurosis. Nat Genet. 1996;14:461–4. doi: 10.1038/ng1296-461. [DOI] [PubMed] [Google Scholar]
- 14.Marlhens F, Bareil C, Griffoin JM, Zrenner E, Amalric P, Eliaou C, Liu SY, Harris E, Redmond TM, Arnaud B, Claustres M, Hamel CP. Mutations in RPE65 cause Leber's congenital amaurosis. Nat Genet. 1997;17:139–41. doi: 10.1038/ng1097-139. [DOI] [PubMed] [Google Scholar]
- 15.Sohocki MM, Bowne SJ, Sullivan LS, Blackshaw S, Cepko CL, Payne AM, Bhattacharya SS, Khaliq S, Qasim Mehdi S, Birch DG, Harrison WR, Elder FF, Heckenlively JR, Daiger SP. Mutations in a new photoreceptor-pineal gene on 17p cause Leber congenital amaurosis. Nat Genet. 2000;24:79–83. doi: 10.1038/71732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.den Hollander AI, Koenekoop RK, Mohamed MD, Arts HH, Boldt K, Towns KV, Sedmak T, Beer M, Nagel-Wolfrum K, McKibbin M, Dharmaraj S, Lopez I, Ivings L, Williams GA, Springell K, Woods CG, Jafri H, Rashid Y, Strom TM, van der Zwaag B, Gosens I, Kersten FF, van Wijk E, Veltman JA, Zonneveld MN, van Beersum SE, Maumenee IH, Wolfrum U, Cheetham ME, Ueffing M, Cremers FP, Inglehearn CF, Roepman R. Mutations in LCA5, encoding the ciliary protein lebercilin, cause Leber congenital amaurosis. Nat Genet. 2007;39:889–95. doi: 10.1038/ng2066. [DOI] [PubMed] [Google Scholar]
- 17.Dryja TP, Adams SM, Grimsby JL, McGee TL, Hong DH, Li T, Andréasson S, Berson EL. Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am J Hum Genet. 2001;68:1295–8. doi: 10.1086/320113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerber S, Perrault I, Hanein S, Barbet F, Ducroq D, Ghazi I, Martin-Coignard D, Leowski C, Homfray T, Dufier JL, Munnich A, Kaplan J, Rozet JM. Complete exon-intron structure of the RPGR-interacting protein (RPGRIP1) gene allows the identification of mutations underlying Leber congenital amaurosis. Eur J Hum Genet. 2001;9:561–71. doi: 10.1038/sj.ejhg.5200689. [DOI] [PubMed] [Google Scholar]
- 19.Swaroop A, Wang QL, Wu W, Cook J, Coats C, Xu S, Chen S, Zack DJ, Sieving PA. Leber congenital amaurosis caused by a homozygous mutation (R90W) in the homeodomain of the retinal transcription factor CRX: direct evidence for the involvement of CRX in the development of photoreceptor function. Hum Mol Genet. 1999;8:299–305. doi: 10.1093/hmg/8.2.299. [DOI] [PubMed] [Google Scholar]
- 20.den Hollander AI, Heckenlively JR, van den Born LI, de Kok YJ, van der Velde-Visser SD, Kellner U, Jurklies B, van Schooneveld MJ, Blankenagel A, Rohrschneider K, Wissinger B, Cruysberg JR, Deutman AF, Brunner HG, Apfelstedt-Sylla E, Hoyng CB, Cremers FP. Leber congenital amaurosis and retinitis pigmentosa with Coats-like exudative vasculopathy are associated with mutations in the crumbs homologue 1 (CRB1) gene. Am J Hum Genet. 2001;69:198–203. doi: 10.1086/321263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koenekoop RK, Wang H, Majewski J, Wang X, Lopez I, Ren H, Chen Y, Li Y, Fishman GA, Genead M, Schwartzentruber J, Solanki N, Traboulsi EI, Cheng J, Logan CV, McKibbin M, Hayward BE, Parry DA, Johnson CA, Nageeb M, Finding of Rare Disease Genes (FORGE) Canada Consortium Poulter JA, Mohamed MD, Jafri H, Rashid Y, Taylor GR, Keser V, Mardon G, Xu H, Inglehearn CF, Fu Q, Toomes C, Chen R. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat Genet. 2012;44:1035–9. doi: 10.1038/ng.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.den Hollander AI, Koenekoop RK, Yzer S, Lopez I, Arends ML, Voesenek KE, Zonneveld MN, Strom TM, Meitinger T, Brunner HG, Hoyng CB, van den Born LI, Rohrschneider K, Cremers FP. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006;79:556–61. doi: 10.1086/507318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perrault I, Delphin N, Hanein S, Gerber S, Dufier JL, Roche O, Defoort-Dhellemmes S, Dollfus H, Fazzi E, Munnich A, Kaplan J, Rozet JM. Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum Mutat. 2007;28:416. doi: 10.1002/humu.9485. [DOI] [PubMed] [Google Scholar]
- 24.Friedman JS, Chang B, Kannabiran C, Chakarova C, Singh HP, Jalali S, Hawes NL, Branham K, Othman M, Filippova E, Thompson DA, Webster AR, Andréasson S, Jacobson SG, Bhattacharya SS, Heckenlively JR, Swaroop A. Premature truncation of a novel protein, RD3, exhibiting subnuclear localization is associated with retinal degeneration. Am J Hum Genet. 2006;79:1059–70. doi: 10.1086/510021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perrault I, Hanein S, Gerber S, Barbet F, Ducroq D, Dollfus H, Hamel C, Dufier JL, Munnich A, Kaplan J, Rozet JM. Retinal dehydrogenase 12 (RDH12) mutations in leber congenital amaurosis. Am J Hum Genet. 2004;75:639–46. doi: 10.1086/424889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janecke AR, Thompson DA, Utermann G, Becker C, Hübner CA, Schmid E, McHenry CL, Nair AR, Rüschendorf F, Heckenlively J, Wissinger B, Nürnberg P, Gal A. Mutations in RDH12 encoding a photoreceptor cell retinol dehydrogenase cause childhood-onset severe retinal dystrophy. Nat Genet. 2004;36:850–4. doi: 10.1038/ng1394. [DOI] [PubMed] [Google Scholar]
- 27.Thompson DA, Li Y, McHenry CL, Carlson TJ, Ding X, Sieving PA, Apfelstedt-Sylla E, Gal A. Mutations in the gene encoding lecithin retinol acyltransferase are associated with early-onset severe retinal dystrophy. Nat Genet. 2001;28:123–4. doi: 10.1038/88828. [DOI] [PubMed] [Google Scholar]
- 28.Sénéchal A, Humbert G, Surget MO, Bazalgette C, Bazalgette C, Arnaud B, Arndt C, Laurent E, Brabet P, Hamel CP. Screening genes of the retinoid metabolism: novel LRAT mutation in leber congenital amaurosis. Am J Ophthalmol. 2006;142:702–4. doi: 10.1016/j.ajo.2006.04.057. [DOI] [PubMed] [Google Scholar]
- 29.Hanein S, Perrault I, Gerber S, Tanguy G, Barbet F, Ducroq D, Calvas P, Dollfus H, Hamel C, Lopponen T, Munier F, Santos L, Shalev S, Zafeiriou D, Dufier JL, Munnich A, Rozet JM, Kaplan J. Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum Mutat. 2004;23:306–17. doi: 10.1002/humu.20010. [DOI] [PubMed] [Google Scholar]
- 30.Sergouniotis PI, Davidson AE, Mackay DS, Li Z, Yang X, Plagnol V, Moore AT, Webster AR. Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause leber congenital amaurosis. Am J Hum Genet. 2011;89:183–90. doi: 10.1016/j.ajhg.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stone EM, Cideciyan AV, Aleman TS, Scheetz TE, Sumaroka A, Ehlinger MA, Schwartz SB, Fishman GA, Traboulsi EI, Lam BL, Fulton AB, Mullins RF, Sheffield VC, Jacobson SG. Variations in NPHP5 in patients with nonsyndromic leber congenital amaurosis and Senior-Loken syndrome. Arch Ophthalmol. 2011;129:81–7. doi: 10.1001/archophthalmol.2010.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Estrada-Cuzcano A, Koenekoop RK, Coppieters F, Kohl S, Lopez I, Collin RW, De Baere EB, Roeleveld D, Marek J, Bernd A, Rohrschneider K, van den Born LI, Meire F, Maumenee IH, Jacobson SG, Hoyng CB, Zrenner E, Cremers FP, den Hollander AI. IQCB1 mutations in patients with leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2011;52:834–9. doi: 10.1167/iovs.10-5221. [DOI] [PubMed] [Google Scholar]
- 33.Coppieters F, De Wilde B, Lefever S, De Meester E, De Rocker N, Van Cauwenbergh C, Pattyn F, Meire F, Leroy BP, Hellemans J, Vandesompele J, De Baere E. Massively parallel sequencing for early molecular diagnosis in Leber congenital amaurosis. Genet Med. 2012;14:576–85. doi: 10.1038/gim.2011.51. [DOI] [PubMed] [Google Scholar]
- 34.Wang X, Wang H, Sun V, Tuan HF, Keser V, Wang K, Ren H, Lopez I, Zaneveld JE, Siddiqui S, Bowles S, Khan A, Salvo J, Jacobson SG, Iannaccone A, Wang F, Birch D, Heckenlively JR, Fishman GA, Traboulsi EI, Li Y, Wheaton D, Koenekoop RK, Chen R. Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. J Med Genet. 2013;50:674–88. doi: 10.1136/jmedgenet-2013-101558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu SM, Thompson DA, Srikumari CR, Lorenz B, Finckh U, Nicoletti A, Murthy KR, Rathmann M, Kumaramanickavel G, Denton MJ, Gal A. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet. 1997;17:194–7. doi: 10.1038/ng1097-194. [DOI] [PubMed] [Google Scholar]
- 36.den Hollander AI, ten Brink JB, de Kok YJ, van Soest S, van den Born LI, van Driel MA, van de Pol DJ, Payne AM, Bhattacharya SS, Kellner U, Hoyng CB, Westerveld A, Brunner HG, Bleeker-Wagemakers EM, Deutman AF, Heckenlively JR, Cremers FP, Bergen AA. Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12). Nat Genet. 1999;23:217–21. doi: 10.1038/13848. [DOI] [PubMed] [Google Scholar]
- 37.den Hollander AI, Lopez I, Yzer S, Zonneveld MN, Janssen IM, Strom TM, Hehir-Kwa JY, Veltman JA, Arends ML, Meitinger T, Musarella MA, van den Born LI, Fishman GA, Maumenee IH, Rohrschneider K, Cremers FP, Koenekoop RK. Identification of novel mutations in patients with Leber congenital amaurosis and juvenile RP by genome-wide homozygosity mapping with SNP microarrays. Invest Ophthalmol Vis Sci. 2007;48:5690–8. doi: 10.1167/iovs.07-0610. [DOI] [PubMed] [Google Scholar]
- 38.Sohocki MM, Perrault I, Leroy BP, Payne AM, Dharmaraj S, Bhattacharya SS, Kaplan J, Maumenee IH, Koenekoop R, Meire FM, Birch DG, Heckenlively JR, Daiger SP. Prevalence of AIPL1 mutations in inherited retinal degenerative disease. Mol Genet Metab. 2000;70:142–50. doi: 10.1006/mgme.2000.3001. [DOI] [PubMed] [Google Scholar]
- 39.Zhang X, Liu H, Zhang Y, Qiao Y, Miao S, Wang L, Zhang J, Zong S, Koide SS. A novel gene, RSD-3/HSD-3.1, encodes a meiotic-related protein expressed in rat and human testis. J Mol Med (Berl) 2003;81:380–7. doi: 10.1007/s00109-003-0434-y. [DOI] [PubMed] [Google Scholar]
- 40.Seelow D, Schuelke M, Hildebrandt F, Nürnberg P. HomozygosityMapper–an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;37:W593–9. doi: 10.1093/nar/gkp369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McVey M, Lee SE. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–38. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mackay DS, Ocaka LA, Borman AD, Sergouniotis PI, Henderson RH, Moradi P, Robson AG, Thompson DA, Webster AR, Moore AT. Screening of SPATA7 in patients with Leber congenital amaurosis and severe childhood-onset retinal dystrophy reveals disease-causing mutations. Invest Ophthalmol Vis Sci. 2011;52:3032–8. doi: 10.1167/iovs.10-7025. [DOI] [PubMed] [Google Scholar]
- 43.McKernan KJ, Peckham HE, Costa GL, McLaughlin SF, Fu Y, Tsung EF, Clouser CR, Duncan C, Ichikawa JK, Lee CC, Zhang Z, Ranade SS, Dimalanta ET, Hyland FC, Sokolsky TD, Zhang L, Sheridan A, Fu H, Hendrickson CL, Li B, Kotler L, Stuart JR, Malek JA, Manning JM, Antipova AA, Perez DS, Moore MP, Hayashibara KC, Lyons MR, Beaudoin RE, Coleman BE, Laptewicz MW, Sannicandro AE, Rhodes MD, Gottimukkala RK, Yang S, Bafna V, Bashir A, MacBride A, Alkan C, Kidd JM, Eichler EE, Reese MG, De La Vega FM, Blanchard AP. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19:1527–41. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abulimiti A, Ding Q, Xu H, Simons D, Moayedi-Esfahani Y, Gan L, Wu S, Williams D, Mardon G, Chen R. Targeted Disruption of Mouse Ortholog (Spata7) of LCA3 Gene Causes Outer Segment Dysplasia, and Progressive Photoreceptor Degeneration Triggered by Rhodopsin Mislocalization. ARVO Annual Meeting; 2012 May 6-9; Fort Lauderdale, FL. [Google Scholar]