

Abstract
Pyoderma gangrenosum (PG) and Sweet's syndrome (SS) are two inflammatory skin diseases presenting with painful ulcers and erythematous plaques, respectively; both disorders have a debilitating clinical behaviour and PG is potentially life-threatening. Recently, PG and SS have been included among the autoinflammatory diseases, which are characterized by recurrent episodes of sterile inflammation, without circulating autoantibodies and autoreactive T cells. However, an autoinflammatory pattern clearly supporting this inclusion has never been demonstrated. We studied 16 patients with PG, six with SS and six controls, evaluating, using a sandwich-based protein antibody array method, the expression profile of inflammatory effector molecules in PG, SS and normal skin. The expressions of interleukin (IL)-1 beta and its receptor I were significantly higher in PG (P = 0·0001 for both) and SS (P = 0·004–0·040) than in controls. In PG, chemokines such as IL-8 (P = 0·0001), chemokine (C-X-C motif) ligand (CXCL) 1/2/3 (P = 0·002), CXCL 16 (P = 0·003) and regulated upon activation normal T cell expressed and secreted (RANTES) (P = 0·005) were over-expressed. In SS, IL-8 (P = 0·018), CXCL 1/2/3 (P = 0·006) and CXCL 16 (P = 0·036) but not RANTES were over-expressed, suggesting that chemokine-mediated signals are lower than in PG. Fas/Fas ligand and CD40/CD40 ligand systems were over-expressed in PG (P = 0·0001 for Fas, P = 0·009 for Fas ligand, P = 0·012 for CD40, P = 0·0001 for CD40 ligand), contributing to tissue damage and inflammation, while their role seems to be less significant in SS. Over-expression of cytokines/chemokines and molecules amplifying the inflammatory network supports the view that PG and SS are autoinflammatory diseases. The differences in expression profile of inflammatory effectors between these two disorders may explain the stronger local aggressiveness in PG than SS.
Keywords: autoinflammation, chemokines, cytokines, pyoderma gangrenosum, Sweet's syndrome
Introduction
Pyoderma gangrenosum (PG) and Sweet's syndrome (SS) are two rare inflammatory cutaneous diseases that present with recurrent skin ulcers having undermined violaceous borders and erythematous plaques, respectively. Both PG and SS can occur alone or in association with different conditions, notably inflammatory bowel diseases, rheumatological disorders, haematological malignancies and intake of triggering drugs 1–5. However, the two diseases differ in that PG is associated more frequently with gut autoinflammatory disorders, while SS occurs commonly in combination with haematological disorders and immunostimulatory drug intake 3. The histopathological pattern of PG and SS consists mainly of neutrophilic inflammatory infiltrates involving the skin and, rarely, internal organs, giving rise to the term ‘neutrophilic dermatoses’ 6–8. Systemic corticosteroids are considered the first-choice treatment for both PG and SS, while other immunosuppressants are reserved for refractory cases 6,7. Recently, neutrophilic dermatoses have been classified within the spectrum of autoinflammatory diseases 1,7,9, which includes genetically determined forms caused by mutations of genes regulating innate immunity 10. Autoinflammatory diseases manifest clinically as relapsing episodes of sterile inflammation in the affected organs, in the absence of high titres of circulating autoantibodies and autoreactive T cells 11–15. PG was the first to be considered an autoinflammatory disease when associated with arthritis and acne in the so-called PAPA (pyogenic arthritis, pyoderma gangrenosum and acne) syndrome. In PAPA syndrome, specific mutations involving the PSTPIP1 (proline–serine–threonine phosphatase-interacting protein 1) gene, via an increased binding affinity to pyrin, induce the assembly of inflammasomes. These are molecular platforms responsible for the activation of the caspase 1, an enzyme inducing the proteolytic cleavage of the inactive pro-interleukin (IL)-1 beta to its functionally active form, IL-1 beta, which is overproduced in PAPA syndrome 16. IL-1 beta overproduction can also occur in non-genetically determined neutrophilic dermatoses, in which it may trigger the synthesis and release of several proinflammatory cytokines and chemokines. Chemokines could, in turn, induce further neutrophil recruitment and activation 17, leading to a neutrophil-mediated inflammation regarded as the pathophysiological hallmark of the neutrophilic dermatoses 1,18,19. However, the actual occurrence of all these pathogenic pathways in neutrophilic dermatoses has never been demonstrated clearly. Thus, to support the inclusion of nutrophilic dermatoses within the spectrum of the autoinflammatory diseases, we have evaluated the cytokine expression profile in the lesional skin of PG and SS by a protein array method.
Patients and methods
Patients
Lesional skin biopsies taken from 16 patients with PG (nine men and seven women; mean age 48 years, range 15–78 years) and six patients with SS (three men and three women; mean age 44 years, range 26–60 years) were studied by a cytokine array method. All PG patients presented with the classic ulcerative variant. All patients with SS had the papulonodular presentation. The diagnosis of PG as well as SS was established on the basis of clinical, histopathological and laboratory criteria 1. Two patients with PG had IBD as associated disease, one patient had Klinefelter's syndrome and one patient had cystic fibrosis; the other 12 PG cases were idiopathic. Only one of six patients with SS had an associated disease, namely chronic B cell lymphatic leukaemia. The clinical findings of patients with PG and those of patients with SS are summarized in Tables 1 and 2, respectively.
Table 1.
Clinical findings in 16 patients with pyoderma gangrenosum
| N | Sex | Age (years) | Lesion number | Sites | Associated conditions | Treatment | Course/follow-up |
|---|---|---|---|---|---|---|---|
| 1 | M | 65 | 3 | Legs | Klinefelter | Pred; Cyc; Infliximab | Chronic-relapsing/exitus due to sepsis |
| 2 | F | 70 | 3 | Legs | – | Pred | Single episode/complete remission |
| 3 | M | 15 | 9 | Trunk | – | Pred | Chronic-relapsing/complete remission |
| 4 | M | 61 | 2 | Legs | – | Pred | Chronic-relapsing/complete remission |
| 5 | F | 28 | 3 | Legs | Cystic fibrosis | Pred | Chronic-relapsing/complete remission |
| 6 | M | 30 | 3 | Legs | IBD | Pred; Adalimumab | Single episode/complete remission |
| 7 | F | 72 | 2 | Legs | – | Pred | Single episode/complete remission |
| 8 | F | 35 | 4 | Legs | – | Pred | Chronic-relapsing/complete remission |
| 9 | F | 30 | 7 | Legs | IBD | Pred; Aza | Chronic-relapsing/complete remission |
| 10 | M | 67 | 3 | Trunk | – | Pred; Dapsone | Chronic-relapsing/partial remission |
| 11 | F | 78 | 3 | Scalp | – | Pred; Cyc | Chronic-relapsing/partial remission |
| 12 | M | 62 | 4 | Legs | – | Pred | Single episode/partial remission |
| 13 | M | 57 | 1 | Legs | – | Pred | Chronic-relapsing/complete remission |
| 14 | M | 42 | 8 | Legs, Arms | – | Pred; Cyc | Chronic-relapsing/complete remission |
| 15 | F | 44 | 4 | Legs | – | Pred; Dapsone | Chronic-relapsing/complete remission |
| 16 | M | 18 | 12 | Trunk, Legs | – | Pred | Chronic-relapsing/complete remission |
Aza = azathioprine; Cyc = oral cyclosporin; IBD = inflammatory bowel diseases; Pred = oral prednisone; M = male; F = female.
Table 2.
Clinical findings in six patients with Sweet's syndrome
| N | Sex | Age (years) | Lesion number | Sites | Associated conditions | Treatment | Course/follow-up |
|---|---|---|---|---|---|---|---|
| 1 | F | 44 | 5 | Face; trunk | – | Pred | Single episode/complete remission |
| 2 | M | 30 | 3 | Face | – | Pred | Single episode/complete remission |
| 3 | M | 58 | 9 | Face; trunk Legs; arms | cB-CLL | Pred | Chronic-relapsing/exitus due to sepsis |
| 4 | M | 26 | 4 | Face; hands | – | Pred | Chronic-relapsing/complete remission |
| 5 | F | 60 | 3 | Face; neck | – | Pred | Single episode/complete remission |
| 6 | F | 46 | 6 | Neck; hands | – | Pred | Chronic-relapsing/complete remission |
cB-CLL = chronic B cell lymphatic leukaemia; Pred = oral prednisone; M = male; F = female.
Skin biopsies were obtained from patients with PG and patients with SS before both systemic and topical treatment. In PG patients, skin specimens were taken from the undermined edge surrounding the ulcerative lesion to the centre of the ulcer. In SS cases, specimens were obtained from lesional skin. The controls were normal skin tissue specimens taken from six patients who underwent excision of benign skin tumours.
The protocol was approved by our Institutional Review Board and all the subjects gave their informed consent before participating in the study.
Protein array
Each tissue sample was weighed and diced into very small pieces using a clean razor blade. Frozen tissue were sliced very thinly and thawed in radioimmunoprecipitation assay (RIPA) buffer (sc-24948) containing protease- and phosphatase-inhibitors using 3 ml of ice-cold RIPA buffer per gram of tissue. Samples were incubated on ice for 30 min, transferred to microcentrifuge tubes and centrifuged at 10 000 g for 10 min at 4°C. The supernatant was collected and the sample was centrifuged again. The new supernatant fluid was added to the previous one, this mixture representing the total cell lysate. In order to standardize the cell lysate of each tissue sample, we measured the total proteins in each sample by a microBCA kit (ThermoScientific, Waltham, MA, USA). For each sample, we loaded a volume containing 100 μg of proteins in a glass-slide format of cytokine antibody array (RayBio®, Norcross, GA, USA). The volume to be loaded was calculated by the following formula: volume (expressed in μl) = 100 μg/protein concentration (expressed in μg/μl). Each glass-slide array contained 14 subarrays and was suitable for 14 samples. Each subarray allowed the evaluation of cytokine expression levels in a sample. Normalization of data at the end of the experiment provided semi-quantitative results. The subarray was composed of specific antibodies against target molecules coated on the glass-slide. After hybridization of the tissue lysate, each antibody bound its target molecule and unbound proteins were washed out. The slide was then incubated with biotin-conjugated antibodies against the same target cytokines, washed and then incubated with cyanin 3 (Cy3)-conjugated streptavidin, creating a biotin–streptavidin–Cy3 complex detectable using a microarray laser scanner. Using a data extraction software we could convert fluorescent signals into numerical data and, after normalization, we obtained an expression value of signal intensity for each molecule in each sample. The molecules tested were the following: IL-1 beta, IL-1RI (IL-1 receptor I), IL-1RII, tumour necrosis factor (TNF)-alpha, TNF-receptor I (TNF-RI), TNF-RII, IL-17, IL-17R, leucocyte selectin (L-selectin), IL-8, regulated on activation, normal T cell expressed and secreted (RANTES), [chemokine (C-X-C motif) ligand 1,2,3, (C = cysteine, X = any amino acid) (CXCL 1,2,3)], CXCL 16, matrix metalloproteinase-2 (MMP-2), MMP-9, tissue inhibitor of metalloproteinase 1 (TIMP-1), TIMP-2, sialic acid-binding immunoglobulin-type lectin 5 (Siglec 5), Siglec 9, Fas protein also known as CD95 (Fas), Fas ligand also known as CD178 (FasL); cluster of differentiation 40 (CD40) and CD40 ligand (CD40L).
Statistics
Because the signal intensity data were positively skewed, they were log-transformed before analysis. The results are reported as anti-log values of means with standard deviation (s.d.). Student's t-test for unpaired values was used to assess statistical significance of differences between normal controls, patients with PG and patients with Sweet's syndrome. The significance level was set at P < 0·05.
Results
Cytokine expression
Pyoderma gangrenosum
IL-1-beta and its receptors (IL-1RI and -RII) were significantly more expressed in PG lesional skin (34·74 ± 26·89, 3·68 ± 2·67 and 11·00 ± 8·79, respectively) than in normal skin (3·42 ± 1·87, 2·00 ± 0·82 and 4·49 ± 1·73; P = 0·0001 for all) (Fig. 1). The proinflammatory cytokine TNF-alpha was also over-expressed in PG (3·24 ± 0·64 versus 2·60 ± 0·19; P = 0·01), as well as its receptors TNF-RI (12·51 ± 7·83 versus 5·87 ± 3·43; P = 0·015) and TNF-RII (13·58 ± 7·83 versus 6·25 ± 3·31; P = 0·023) (Fig. 2). Finally, we observed an overproduction of IL-17 (5·80 ± 3·62 versus 2·17 ± 0·44; P = 0·008) and its receptor IL-17R (5·47 ± 1·24 versus 3·98 ± 1·33; P = 0·010) in PG (Fig. 3).
Fig 1.
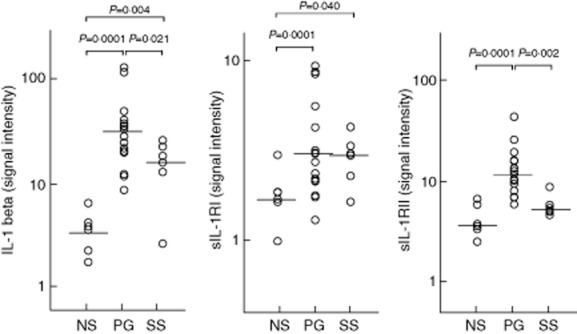
Expression of interleukin (IL)-1 beta and its soluble receptors I and II (sIL-1RI and sIL-1RII) in homogenate samples of lesional skin from 16 patients with pyoderma gangrenosum (PG) and six patients with Sweet's syndrome (SS). Six normal subjects (NS) served as controls. Numerical values represent signal intensity in a cytokine array assay.
Fig 2.
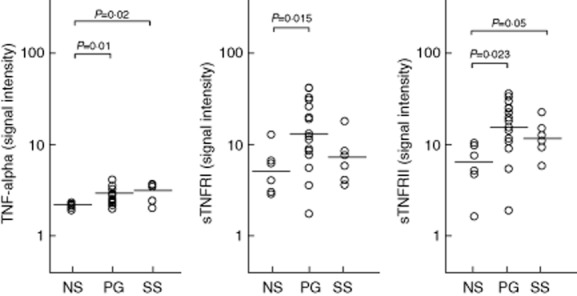
Expression of tumour necrosis factor (TNF)-alpha and its soluble receptors I and II (sTNF-RI and sTNF-RII) in homogenate samples of lesional skin from 16 patients with pyoderma gangrenosum (PG) and six patients with Sweet's syndrome (SS). Six normal subjects (NS) served as controls. Numerical values represent signal intensity in a cytokine array assay.
Fig 3.
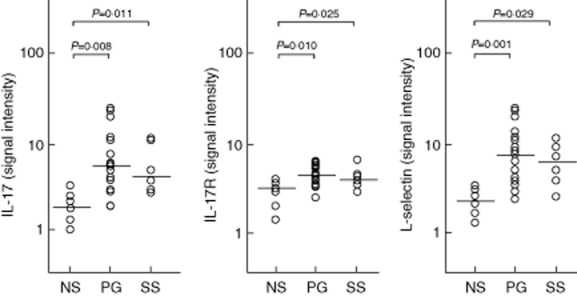
Expression of interleukin (IL)-17, its soluble receptor (sIL-17R) and leucocyte selectin (L-selectin) in homogenate samples of lesional skin from 16 patients with pyoderma gangrenosum (PG) and six patients with Sweet's syndrome (SS). Six normal subjects (NS) served as controls. Numerical values represent signal intensity in a cytokine array assay.
Sweet's syndrome
The expression of IL-1 beta was significantly higher in SS lesional skin (15·70 ± 7·88) than in normal skin (3·42 ± 1·87; P = 0·004), but significantly lower than in PG lesional skin (P = 0·021) (Fig. 1). IL-1RI (3·59 ± 1·76) was significantly over-expressed compared to normal skin (2·00 ± 0·82; P = 0·040); a trend towards an increase in IL-1RII expression was also observed (5·06 ± 2·51 versus 4·49 ± 1·73). Statistically significant over-expression of TNF-alpha (3·46 ± 0·85 versus 2·60 ± 0·19) and its receptor TNF-RII (11·83 ± 4·55 versus 6·25 ± 3·31), but not its receptor TNF-RI (7·50 ± 4·50 versus 5·87 ± 3·43), was found (P = 0·02 and P = 0·05, respectively) (Fig. 2). An expression significantly higher than in normal skin was detected for both IL-17 (4·93 ± 1·02 versus 2·17 ± 0·44; P = 0·011) and its receptor IL-17R (4·54 ± 0·72 versus 3·98 ± 1·33; P = 0·025) (Fig. 3).
L-selectin expression
Pyoderma gangrenosum
The expression of L-selectin was significantly higher in PG lesional skin (7·19 ± 6·31) than in normal skin (2·14 ± 1·21; P = 0·001) (Fig. 3).
Sweet's syndrome
The expression of L-selectin was significantly higher in SS lesional skin (6·23 ± 3·81) than in normal skin (2·14 ± 1·21; P = 0·029) (Fig. 3).
Chemokine expression
Pyoderma gangrenosum
Compared to controls, PG patients showed over-expression of chemokines promoting neutrophil transendothelial migration into inflamed tissues, such as IL-8 (14·05 ± 62·85 versus 2·73 ± 1·05; P = 0·0001), CXCL 1,2,3 (36·74 ± 118·58 versus 7·61 ± 7·65; P = 0·002), CXCL 16 (4·91 ± 5·73 versus 2·50 ± 0·74; P = 0·003) and RANTES (9·75 ± 19·07 versus 3·41 ± 1·56; P = 0·005) (Fig. 4).
Fig 4.
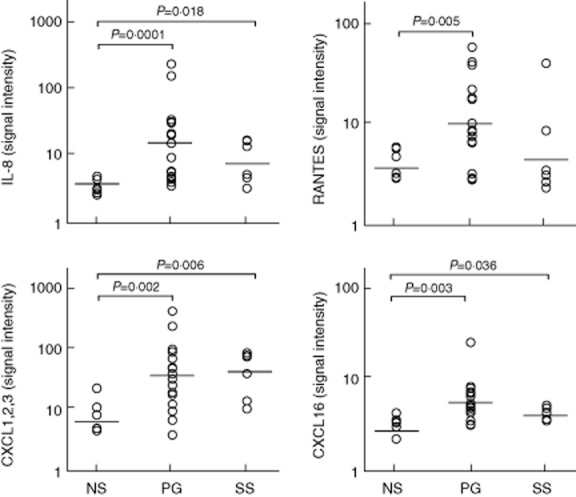
Expression of interleukin (IL)-8, regulated on activation, normal T cell expressed and secreted (RANTES), chemokine (C-X-C motif) ligand (CXCL) 1,2,3 (C = cysteine, X = any amino acid) and CXCL 16 in homogenate samples of lesional skin from 16 patients with pyoderma gangrenosum (PG) and six patients with Sweet's syndrome (SS). Six normal subjects (NS) served as controls. Numerical values represent signal intensity in a cytokine array assay.
Sweet's syndrome
In SS patients, statistically significant over-expression was observed for IL-8 (7·79 ± 7·03 versus 2·73 ± 1·05; 0·018), CXCL 1,2,3 (40·25 ± 37·14 versus 7·61 ± 7·65; P = 0·006) and CXCL 16 (3·62 ± 0·71 versus 2·50 ± 0·74; P = 0·036), but not for RANTES (4·45 ± 17·25 versus 3·41 ± 1·56) (Fig. 4).
MMP and TIMP expression
Pyoderma gangrenosum
In PG patients, we observed significant over-expression of molecules involved in tissue damage such as MMP-2 (5·72 ± 3·16 versus 2·49 ± 0·90; P = 0·001) and MMP-9 (230·94 ± 137·95 versus 24·70 ± 68·32; P = 0·0001) (Fig. 5). Overproduction of molecules responsible for inhibitory signals aimed at attenuating MMP-mediated inflammation was also demonstrated: TIMP-1 (79·78 ± 155·65 versus 4·45 ± 1·63; P = 0·0001) and TIMP-2 (208·16 ± 149·67 versus 48·69 ± 24·40; P = 0 0001) (Fig. 5).
Fig 5.
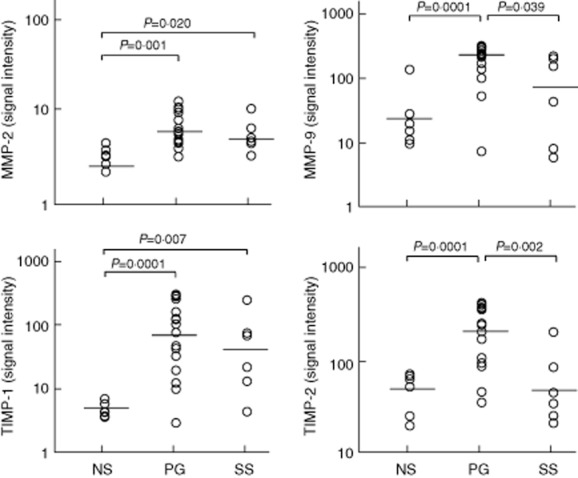
Expression of matrix metalloproteinase (MMP) 2, MMP-9, tissue inhibitor of metalloproteinase 1 (TIMP)-1 and TIMP-2 in homogenate samples of lesional skin from 16 patients with pyoderma gangrenosum (PG) and six patients with Sweet's syndrome (SS). Six normal subjects (NS) served as controls. Numerical values represent signal intensity in a cytokine array assay.
Sweet's syndrome
The expression of MMP-2 but not of MMP-9 was significantly higher than in normal skin (4·88 ± 2·87 versus 2·49 ± 0·90; P = 0 020) (Fig. 5); MMP-9 expression in SS was significantly lower than in PG (60·58 ± 136·89 versus 230·94 ± 137·95; P = 0·039) (Fig. 5). The expression of TIMP-1 but not of TIMP-2 was significantly higher than in normal skin (41·16 ± 128·93 versus 4·45 ± 1·63; P = 0·007) (Fig. 5); TIMP-2 expression in SS was significantly lower than in PG (53·59 ± 75·17 versus 208·16 ± 149·67; P = 0·002) (Fig. 5).
Siglec, FAS/FASL and CD40/CD40L expression
Pyoderma gangrenosum
Siglec 5 (61·21 ± 96·94; P = 0·0001) and Siglec 9 (50·33 ± 75·71; P = 0·0001), which carry inhibitory signals dampening inflammation, were more expressed in PG than in normal skin (4·74 ± 3·09 and 7·65 ± 3·55, respectively) (Fig. 6). Fas and its ligand were also over-expressed in PG (16·47 ± 9·15 versus 8·75 ± 2·07 with P = 0·0001 for Fas; 7·38 ± 4·40 versus 3·86 ± 1·48 with P = 0·009 for Fas ligand) (Fig. 6). CD40 and its ligand were expressed significantly more in PG than in normal skin (6·16 ± 3·57 versus 3·68 ± 1·04 with P = 0·012 for CD40; 5·77 ± 6·27 versus 2·32 ± 0·78 with P = 0·0001 for CD40L) (Fig. 6).
Fig 6.
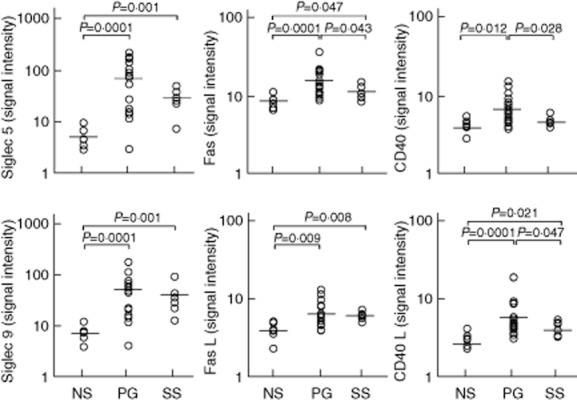
Expression of sialic acid-binding immunoglobulin-type lectin (Siglec) 5, Siglec 9, Fas protein also known as CD95 (Fas), Fas ligand also known as CD178 (FasL), cluster of differentiation (CD)40 and CD40 ligand (CD40L) in homogenate samples of lesional skin from 16 patients with pyoderma gangrenosum (PG) and six patients with Sweet's syndrome (SS). Six normal subjects (NS) served as controls. Numerical values represent signal intensity in a cytokine array assay.
Sweet's syndrome
Siglec 5 (29·21 ± 18 40; P = 0·001) and Siglec 9 (40·45 ± 38·43; P = 0·001) were expressed more in SS than in normal skin (4·74 ± 3·09 and 7·65 ± 3·55, respectively) (Fig. 6). Fas and its ligand were also over-expressed in SS (11·92 ± 3·15 versus 8·75 ± 2·07 with P = 0·047 for Fas; 7·20 ± 1·16 versus 3·86 ± 1·48 with P = 0·008 for FasL) (Fig. 6). CD40L was expressed more significantly in SS (3·75 ± 1·06) than in normal skin (2·32 ± 0·78; P = 0·021) (Fig. 6). Differences between SS and PG were found for Fas (11·92 ± 3·15 versus 16·47 ± 9·15; P = 0·013), CD40 (4·22 ± 3·77 versus 6·16 ± 3·57; P = 0·028) and CD40L (3·75 ± 1·06 versus 5·77 ± 6·27; P = 0·047).
Discussion
Although a few reports have previously shown an over-expression of cytokines such as IL-8 and IL-23 in PG 18,20,21 and of granulocyte–macrophage colony-stimulating factor (GM-CSF) in SS 22, our study, for the first time, analysed systematically the cytokine and chemokine expression profile on lesional skin of PG and SS. A cardinal finding in our study was the over-expression of IL-1β and its receptors in both PG and SS, linked theoretically to dysregulation of inflammasome function. As emphasized previously, this highly active pleiotropic cytokine is a leading actor in the autoinflammation 12,17 and plays a pivotal role in triggering the neutrophilic inflammation of the skin. IL-1 is produced mainly by macrophages, T lymphocytes, endothelial cells and fibroblasts. In particular, T lymphocytes have been reported to be predominant in the wound edge of ulcerative PG 18, where they contribute to neutrophil recruitment paving the way to ulcer formation. Conversely, several lines of evidence suggest that activated keratinocytes are also an important source of IL-1 in the skin 23,24.
IL-1 is a well-known inducer of cytokines, notably including proinflammatory cyotokines (such as IFN-γ) and chemokines, forming a network of cytokine-induced cytokines 17. It is of note that IL-1β over-expression was significantly higher in PG than in SS; moreover, IL-1β receptor II was significantly more expressed in PG than SS. IL-1 promotes the production and release of both classic proinflammatory cytokines, such as TNF-α and IFN-γ, and a number of chemokines, notably IL-8 and RANTES 17.
In fact, TNF-α, which also acts as a key regulator of other proinflammatory cytokines and chemokines, including the same IL-1β and IL-8 25, was over-expressed in the present study. Overproduction of TNF-α in lesional skin of idiopathic PG has been demonstrated previously by immunohistochemistry 18, suggesting that TNF-α plays an important role in the development of the inflammatory process in this disorder and providing the rationale for the use of anti-TNF-α therapy in refractory PG or in PG associated with inflammatory bowel diseases 26,27. In these resistant cases, on the basis of the very high levels of IL-1, the use of IL-1 blockers may be even more appropriate as in classic autoinflammatory diseases, such as familial Mediterranean fever 28.
Several chemokines, such as IL-8, CXCL 1,2,3, CXCL16 and RANTES, were over-expressed in our study, promoting neutrophil transendothelial migration into the site of inflammatory process, which was also favoured by the up-regulation of L-selectin.
Interestingly, the chemokine expression profile was different in SS, in which IL-8, CXCL1,2,3 and CXCL16 proved to be significantly more expressed than in normal skin; in contrast, RANTES expression was not significantly higher than in normal skin, suggesting that chemokine-mediated signals inducing neutrophil recruitment in SS are lower than in PG. Taken together, these findings suggest that the IL-1β-induced release of cytokines and chemokines may be lower in SS than PG, providing the physiopathological background for the different clinical presentation of these two conditions. In fact, PG presents with a skin ulcer, which is due to strong tissue damage, while SS manifests as a plaque lesion with only a tendency to forming vesicles.
In our study we found an over-expression of IL-17 and its receptor in both PG and SS, confirming the previously hypothesized role for this T helper type 17-related cytokine in the pathophisiology of the whole spectrum of neutrophilic dermatoses, similarly to psoriasis and other autoimmune diseases 18,29,30. IL-17 amplifies the recruitment of neutrophils and monocytes by increasing the local production of chemokines, most notably IL-8 31, and synergizing with various other cytokines, in particular with TNF-α 32. Moreover, IL-17, like IL-1 and TNF-alpha, induces the production of MMPs 33, a family of endopeptidases that include the so-called gelatinases MMP-2 and MMP-9. Gelatinases are major contributors to the breakdown and reconstitution of the extracellular matrix in both physiological processes, such as wound repair, and in pathological conditions, including neutrophilic dermatoses 18,34. An improper activity of MMPs synthesized by inflammatory cells, particularly neutrophils, is known to cause the destruction of tissue via degradation of components of the extracellular matrix and to influence the production of chemokines, promoting neutrophil transendothelial migration 35.
In the present study we found an over-expression of MMP-9 in the inflammatory infiltrate of PG lesions, suggesting that this proteinase and, to a lesser degree, MMP-2 may be relevantly involved in inducing both tissue damage and repair. In contrast, the role of MMPs as tissue damage effector molecules in SS seems to be less relevant, and this fits in well with the less local aggressiveness of SS.
Interestingly, we also detected overproduction of TIMP-1 and TIMP-2, which probably represents an inhibitory pathway aimed at attenuating MMP-mediated inflammation. Other important inhibitory signals that attenuate immune responses and dampen inflammation in PG, and in autoinflammation in general, are probably carried by Siglec 5 and Siglec 9, both over-expressed in our cases. In fact, Siglecs are inhibitory receptors expressed mainly by cells of the innate immune system that regulate inflammation mediated by damage-associated and pathogen-associated molecular patterns 36. Compared to PG, the TIMP- and Siglec-mediated inhibitory signals act to a lesser degree in SS, in which the skin inflammatory process is less strong. Our study suggests that two other important systems, the Fas/FasL system and the CD40/CD40L system, may contribute to tissue damage and inflammation in PG. The Fas/FasL system belongs to the TNF/TNF receptor superfamily and to date is the best-known pathway mediating apoptosis 37. Apoptosis is a form of cell death involved in many inflammatory conditions, including cutaneous disorders 38,39. The CD40/CD40L system also belongs to the TNF/TNF receptor superfamily. It represents a co-stimulatory system that amplifies the immune response and can promote inflammation via up-regulation of adhesion molecules and inducing the production of various cytokines and chemokines such as IL-1, TNF-α, IL-8 and RANTES 40. The pathogenetic role of these two systems (Fas/FasL and CD40/CD40L) in SS is less significant, and this could contribute to explaining the lack of evolution of the typical SS plaque-lesion into a frank skin ulcer, as in classical ulcerative PG.
Overall, our data clearly show high values of proinflammatory cytokines, chemokines and tissue damage effector molecules in patients with PG and, to a lesser degree, in patients with SS in which, however, the small number of cases may represent a limitation. To confirm the specificity of our findings, future studies are needed aimed at evaluating the cytokine expression profile on other inflammatory skin diseases. Moreover, to characterize the cells responsible for the increased secretion of the most important cytokines and chemokines, double-staining studies with specific cell markers should be performed.
Conclusions
This is the first systematic study showing an increased cytokine and chemokine expression profile on lesional skin of PG and SS. This finding supports the inclusion of these two disorders into the neutrophil-mediated autoinflammatory diseases and suggests that the different cytokine profiles may be responsible for the clinical differences between the two diseases. Accordingly, molecules contributing to tissue damage, such as MMPs, and mediating apoptosis, such as the Fas/FasL system, are expressed differently in PG and SS. Finally, inhibitory signals dampening inflammation act to a different degree. The different inflammatory scenarios explain the stronger local aggressiveness in PG than in SS and pave the way to the use of inflammatory cytokine blockers to treat severe PG cases unresponsive to corticosteroids.
Author contributions
A. V. M. and M. C. designed the study. A. V. M. followed the patients and collected clinical and laboratory data. D. F. performed laboratory investigations. M. C. and A. V. M. analysed the data and all the authors contributed to the interpretation of the results. A. V. M. and M. C. drafted the manuscript and P. L. M. contributed to the writing. All the authors critically reviewed the manuscript and approved the final version for submission.
Disclosure
The authors declare that they have no conflicts of interest.
References
- Marzano AV, Ishak RS, Saibeni S, Crosti C, Meroni PL, Cugno M. Autoinflammatory skin disorders in inflammatory bowel diseases, pyoderma gangrenosum and Sweet's syndrome: a comprehensive review and disease classification criteria. Clin Rev Allergy Immunol. 2013;45:202–210. doi: 10.1007/s12016-012-8351-x. [DOI] [PubMed] [Google Scholar]
- Maverakis E, Goodarzi H, Wehrli LN, Ono Y, Garcia MS. The etiology of paraneoplastic autoimmunity. Clin Rev Allergy Immunol. 2012;42:135–144. doi: 10.1007/s12016-010-8248-5. [DOI] [PubMed] [Google Scholar]
- Wollina U, Haroske G. Pyoderma gangrenosum. Curr Opin Rheumatol. 2011;23:50–56. doi: 10.1097/BOR.0b013e328341152f. [DOI] [PubMed] [Google Scholar]
- Ueharaguchi Y, Kabashima K, Sasahashi M, Matsuda A, Matubara K, Matsui M. A case of pyoderma gangrenosum possibly associated with sunitinib treatment. Int J Dermatol. 2013;52:634–636. doi: 10.1111/j.1365-4632.2011.04952.x. [DOI] [PubMed] [Google Scholar]
- Kyllo RL, Parker MK, Rosman I, Musiek AC. Ipilimumab-associated Sweet syndrome in a patient with high-risk melanoma. J Am Acad Dermatol. 2014;70:e85–86. doi: 10.1016/j.jaad.2013.11.022. [DOI] [PubMed] [Google Scholar]
- Cohen PR. Neutrophilic dermatoses: a review of current treatment options. Am J Clin Dermatol. 2009;10:301–312. doi: 10.2165/11310730-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Ahronowitz I, Harp J, Shinkai K. Etiology and management of pyoderma gangrenosum: a comprehensive review. Am J Clin Dermatol. 2012;13:191–211. doi: 10.2165/11595240-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Marzano AV, Trevisan V, Lazzari R, Crosti C. Pyoderma gangrenosum: study of 21 patients and proposal of a ‘clinicotherapeutic’ classification. J Dermatolog Treat. 2011;22:254–260. doi: 10.3109/09546631003686069. [DOI] [PubMed] [Google Scholar]
- Nesterovitch AB, Gyorfy Z, Hoffman MD, et al. Alteration in the gene encoding protein tyrosine phosphatase nonreceptor type 6 (PTPN6/SHP1) may contribute to neutrophilic dermatoses. Am J Pathol. 2011;178:1434–1441. doi: 10.1016/j.ajpath.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksentijevich I, Kastner DL. Genetics of monogenic autoinflammatory diseases: past successes, future challenges. Nat Rev Rheumatol. 2011;7:469–478. doi: 10.1038/nrrheum.2011.94. [DOI] [PubMed] [Google Scholar]
- McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97:133–144. doi: 10.1016/s0092-8674(00)80721-7. [DOI] [PubMed] [Google Scholar]
- Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell. 2010;140:784–790. doi: 10.1016/j.cell.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doria A, Zen M, Bettio S, et al. Autoinflammation and autoimmunity: bridging the divide. Autoimmun Rev. 2012;12:22–30. doi: 10.1016/j.autrev.2012.07.018. [DOI] [PubMed] [Google Scholar]
- Chang C. The pathogenesis of neonatal autoimmune and autoinflammatory diseases: a comprehensive review. J Autoimmun. 2013;41:100–110. doi: 10.1016/j.jaut.2012.12.010. [DOI] [PubMed] [Google Scholar]
- Zen M, Gatto M, Domeneghetti M, et al. Clinical guidelines and definitions of autoinflammatory diseases: contrasts and comparisons with autoimmunity – a comprehensive review. Clin Rev Allergy Immunol. 2013;45:227–235. doi: 10.1007/s12016-013-8355-1. [DOI] [PubMed] [Google Scholar]
- Wise CA, Gillum JD, Seidman CE, et al. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet. 2002;11:961–969. doi: 10.1093/hmg/11.8.961. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. A clinical perspective of IL-1β as the gatekeeper of inflammation. Eur J Immunol. 2011;41:1203–1217. doi: 10.1002/eji.201141550. [DOI] [PubMed] [Google Scholar]
- Marzano AV, Cugno M, Trevisan V, et al. Role of inflammatory cells, cytokines and matrix metalloproteinases in neutrophil-mediated skin diseases. Clin Exp Immunol. 2010;162:100–107. doi: 10.1111/j.1365-2249.2010.04201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzano AV, Borghi A, Stadnicki A, Crosti C, Cugno M. Cutaneous manifestations in patients with inflammatory bowel diseases: pathophysiology, clinical features, and therapy. Inflamm Bowel Dis. 2014;20:213–227. doi: 10.1097/01.MIB.0000436959.62286.f9. [DOI] [PubMed] [Google Scholar]
- Oka M. Pyoderma gangrenosum and interleukin 8. Br J Dermatol. 2007;157:1279–1281. doi: 10.1111/j.1365-2133.2007.08202.x. [DOI] [PubMed] [Google Scholar]
- Guenova E, Teske A, Fehrenbacher B, et al. Interleukin 23 expression in pyoderma gangrenosum and targeted therapy with ustekinumab. Arch Dermatol. 2011;147:1203–1205. doi: 10.1001/archdermatol.2011.168. [DOI] [PubMed] [Google Scholar]
- Kawakami T, Ohashi S, Kawa Y, et al. Elevated serum granulocyte colony-stimulating factor levels in patients with active phase of sweet syndrome and patients with active Behçet disease: implication in neutrophil apoptosis dysfunction. Arch Dermatol. 2004;140:570–574. doi: 10.1001/archderm.140.5.570. [DOI] [PubMed] [Google Scholar]
- Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer HD. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr Biol. 2007;17:1140–1145. doi: 10.1016/j.cub.2007.05.074. [DOI] [PubMed] [Google Scholar]
- Lee P, Lee DJ, Chan C, Chen SW, Ch'en I, Jamora C. Dynamic expression of epidermal caspase 8 simulates a wound healing response. Nature. 2009;458:519–523. doi: 10.1038/nature07687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim JJ, Zachariae CO, Mukaida N, Matsushima K. Properties of the novel proinflammatory supergene ‘intercrine’ cytokine family. Annu Rev Immunol. 1991;9:617–648. doi: 10.1146/annurev.iy.09.040191.003153. [DOI] [PubMed] [Google Scholar]
- Reguiai Z, Grange F. The role of anti-tumor necrosis factor-alpha therapy in pyoderma gangrenosum associated with inflammatory bowel disease. Am J Clin Dermatol. 2007;8:67–77. doi: 10.2165/00128071-200708020-00002. [DOI] [PubMed] [Google Scholar]
- Marzano AV, Tourlaki A, Alessi E, Caputo R. Widespread idiopathic pyoderma gangrenosum evolved from ulcerative to vegetative type: a 10-year history with a recent response to infliximab. Clin Exp Dermatol. 2008;33:156–159. doi: 10.1111/j.1365-2230.2007.02607.x. [DOI] [PubMed] [Google Scholar]
- Soriano A, Verecchia E, Afeltra A, Landolfi R, Manna R. IL-1β biological treatment of familial Mediterranean fever. Clin Rev Allergy Immunol. 2013;45:117–130. doi: 10.1007/s12016-013-8358-y. [DOI] [PubMed] [Google Scholar]
- Pene J, Chevalier S, Preisser L, et al. Chronically inflamed human tissues are infiltrated by highly differentiated Th17 lymphocytes. J Immunol. 2008;180:7423–7430. doi: 10.4049/jimmunol.180.11.7423. [DOI] [PubMed] [Google Scholar]
- Dardalhon V, Korn T, Kuchroo VK, Anderson AC. Role of Th1 and Th17 cells in organ-specific autoimmunity. J Autoimmun. 2008;31:252–256. doi: 10.1016/j.jaut.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbandian A, Crispin JC, Tsokos GC. Interleukin-17 and systemic lupus erythematosus: current concepts. Clin Exp Immunol. 2009;157:209–215. doi: 10.1111/j.1365-2249.2009.03944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruddy MJ, Wong GC, Liu XK, et al. Functional cooperation between interleukin-17 and tumor necrosis factor-alpha is mediated by CCAAT/enhancer-binding protein family members. J Biol Chem. 2004;279:2559–2567. doi: 10.1074/jbc.M308809200. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Misra R, Aggarwal A. Interleukin 17 levels are increased in juvenile idiopathic arthritis synovial fluid and induce synovial fibroblasts to produce proinflammatory cytokines and matrix metalloproteinases. J Rheumatol. 2008;35:515–519. [PubMed] [Google Scholar]
- Shapiro SD. Matrix metalloproteinase degradation of extracellular matrix: biological consequences. Curr Opin Cell Biol. 1998;10:602–608. doi: 10.1016/s0955-0674(98)80035-5. [DOI] [PubMed] [Google Scholar]
- Delclaux C, Delacourt C, D'Ortho MP, Boyer V, Lafuma C, Harf A. Role of gelatinase B and elastase in human polymorphonuclear neutrophil migration across basement membrane. Am J Respir Cell Mol Biol. 1996;14:288–295. doi: 10.1165/ajrcmb.14.3.8845180. [DOI] [PubMed] [Google Scholar]
- Pillai S, Netravali IA, Cariappa A, Mattoo H. Siglecs and immune regulation. Annu Rev Immunol. 2012;30:357–392. doi: 10.1146/annurev-immunol-020711-075018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrli P, Viard L, Bullani R, Tschopp J, French LE. Death receptors in cutaneous biology and disease. J Invest Dermatol. 2000;115:141–148. doi: 10.1046/j.1523-1747.2000.00037.x. [DOI] [PubMed] [Google Scholar]
- Caproni M, Torchia D, Schincaglia E, et al. Expression of cytokines and chemokine receptors in the cutaneous lesions of erythema multiforme and Stevens–Johnson syndrome/toxic epidermal necrolysis. Br J Dermatol. 2006;155:722–728. doi: 10.1111/j.1365-2133.2006.07398.x. [DOI] [PubMed] [Google Scholar]
- Marzano AV, Frezzolini A, Caproni M, et al. Immunohistochemical expression of apoptotic markers in drug-induced erythema multiforme, Stevens–Johnson syndrome and toxic epidermal necrolysis. Int J Immunopathol Pharmacol. 2007;20:557–566. doi: 10.1177/039463200702000313. [DOI] [PubMed] [Google Scholar]
- Brugnolo F, Annunziato F, Sampognaro S, et al. Highly Th2-skewed cytokine profile of beta-lactam-specific T cells from nonatopic subjects with adverse drug reactions. J Immunol. 1999;163:1053–1059. [PubMed] [Google Scholar]