

Abstract
Complement convertases are enzymatic complexes that play a central role in sustaining and amplification of the complement cascade. Impairment of complement function leads directly or indirectly to pathological conditions, including higher infection rate, kidney diseases, autoimmune- or neurodegenerative diseases and ischaemia–reperfusion injury. An assay for direct measurement of activity of the convertases in patient sera is not available. Existing assays testing convertase function are based on purified complement components and, thus, convertase formation occurs under non-physiological conditions. We designed a new assay, in which C5 blocking compounds enabled separation of the complement cascade into two phases: the first ending at the stage of C5 convertases and the second ending with membrane attack complex formation. The use of rabbit erythrocytes or antibody-sensitized sheep erythrocytes as the platforms for convertase formation enabled easy readout based on measurement of haemolysis. Thus, properties of patient sera could be studied directly regarding convertase activity and membrane attack complex formation. Another advantage of this assay was the possibility to screen for host factors such as C3 nephritic factor and other anti-complement autoantibodies, or gain-of-function mutations, which prolong the half-life of complement convertases. Herein, we present proof of concept, detailed description and validation of this novel assay.
Keywords: complement system, convertase, eculizumab, OmCI
Introduction
The complement system, as a part of innate immunity, ideally targets the recognition and elimination of pathogens, but not the constituents of our own body. Three different activation pathways, initiated by several pattern-recognition molecules as well as multiple effector mechanisms, including opsonization, anaphylaxis and direct cell lysis, ensure a wide spectrum of direct and indirect cytocidal activities 1. All activation pathways converge at the level of C3. During activation, C3 and C5 convertases are formed. These are enzymatic complexes, which amplify the cascade by cleaving C3 and C5 molecules to their active, cell-bondable forms, C3b and C5b, and to the smaller anaphylatoxins C3a and C5a. In order to protect our cells and tissues from excessive or misguided complement attack, numerous soluble and membrane-bound complement inhibitors operate at different levels. The fact that the majority of inhibitors act on the C3 and C5 convertases underlines the importance of these enzymes for the activity of the complement cascade.
There are two main mechanisms of convertase inhibition; the first is a cleavage of C3b and C4b by the serine protease factor I in the presence of co-factors: factor H (FH), C4b-binding protein (C4BP), CD35 and CD46, and the second mechanism is a decay-acceleration activity supported independently by CD35, FH, C4BP and decay-accelerating factor (DAF, CD55) 2. Dysregulation of the complement cascade at the level of the convertases by autoantibodies such as those preventing convertase decay (i.e. C3 nephritic factor, C3NeF), deficiencies or mutations in inhibitors and complement factors contribute to a number of diseases, such as dense deposit disease (DDD) 3, glomerulonephritis 4, age-related macular degeneration (AMD) 5, atypical haemolytic uraemic syndrome (aHUS) 6, systemic lupus erythematosus (SLE) 7 and many more (reviewed in 8). Furthermore, autoantibodies against the inhibitors, such as FH (FHaAbs) are associated with aHUS, DDD and early stages of lung cancer 9.
Straightforward assessment of activity of complement convertases in whole serum is currently not available. Existing assays that are designed to detect factors influencing convertase activity are based on purified components and enzyme-linked immunosorbent assay (ELISA) 10 and/or haemolytic assays 11,12. Recently, we described a new method to test the influence of putative regulators on the formation and stability of the convertases 13. The undoubted advantage of this method is the assembly of classical or alternative complement convertases in the physiological milieu of whole serum; however, only purified components can be tested. Therefore, we designed and validated a similar system which can measure the activity and stability of convertases directly in serum samples. Addition of C5 blockers, Ornithodoros moubata complement inhibitor (OmCI) 14 or eculizumab 15 to human serum, followed by complement activation on the surface of sensitized sheep erythrocytes (classical pathway) or rabbit erythrocytes (alternative pathway) results in activation of the complement cascade up to the stage of C5 convertases (C4b2aC3b or C3bBbC3b) (shown in Fig. 1), but no further. In this system, the relative activity of C5 convertases depends directly on the formation and activity of C3 convertases (C4b2a or C3bBb), which cleave C3 to C3a and C3b. Because transition of C3 to C5 convertase is achieved by the addition of a single C3b molecule to the C3 convertase complex, all events influencing activity of C3 convertase are reflected in the activity of C5 convertase. To this end, we have shown previously that C3 and C5 convertases are regulated by the same inhibitors present in serum 13, and thus we conclude that measurement of convertase activity at the C5 stage includes all physiologically and functionally important events.
Fig 1.
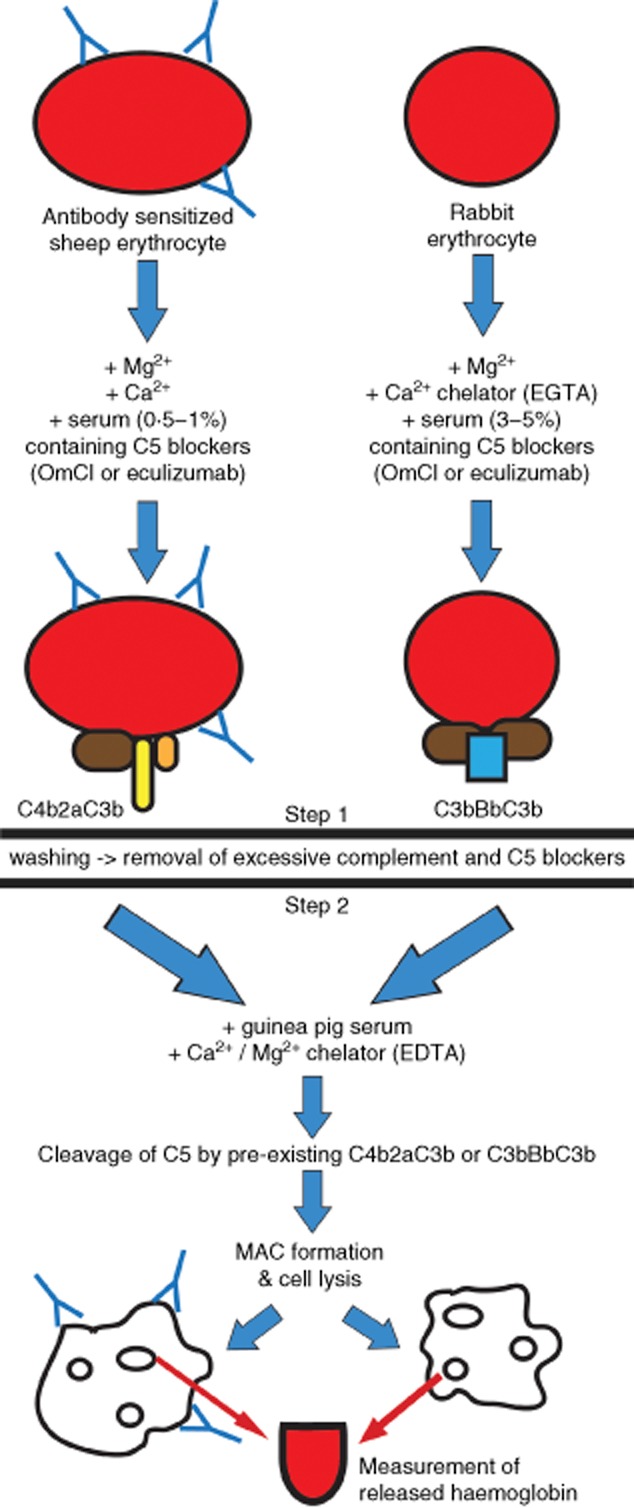
The principle of the two-step method of complement activation. Step 1 (upper part) terminates with the formation of the alternative C5 convertase (C3bBbC3b) or classical C5 convertase (C4b2aC3b), respectively. Step 2 (bottom part) begins after removal of excessive serum, C5 blockers and complement inhibitors by washing. Guinea pig serum diluted in ethylenediamine tetraacetic acid (EDTA) buffer is added as a source of membrane attack complex (MAC) components. These conditions allow initiation of MAC formation only by convertases formed in step 1 and disable de-novo convertase formation.
Materials and methods
Sera, proteins and antibodies
Normal human serum (NHS) was prepared from blood of healthy volunteers after written informed consent had been obtained and according to the permit by the ethics committee of Lund University (permit number 418/2008). Blood was collected and kept at room temperature for 30 min to coagulate, then on ice for another 60 min followed by centrifugation for 7 min at 700 g. The serum fraction was collected, centrifuged again to remove residual erythrocytes, aliquoted and stored at −80°C until use. Purified complement components C1q, C6 and factor B (FB) as well as FB, C6 and C1q-depleted sera were purchased from Complement Technologies (Tyler, TX, USA). Guinea pig serum was purchased from Harlan Laboratories (Horst, the Netherlands). Antibody OX24, which inhibits the interaction of FH with surface-bound C3b 16, was obtained from hybridoma cells purchased from Health Protection Agency Culture Collections (Salisbury, UK) and purified from conditioned medium by binding to 5 ml protein G-sepharose column (GE Healthcare, Uppsala, Sweden) following washing with phosphate-buffered saline (PBS) and elution with 0·1 M glycine pH 2·5. Antibody C18/3, which binds to the C3b recognition domain located on the C-terminus of FH 17, was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Antibody mk54 against protein S (used here as isotype control) was produced in our laboratory and purified using protein A affinity chromatography, as described previously 18. Eculizumab was purchased from Alexion Pharmaceuticals (Lausanne, Switzerland).
The tick protein OmCI was recombinantly produced in Escherichia coli, as follows. A codon optimized OmCI-TEV-His6 sequence, based on information in the NCBI database but excluding the signal sequence, was manufactured by GeneArt (Regensburg, Germany). E. coli strain BL21(DE3) was transformed with the constructed vector and protein expressed during growth of the cells in a fermentor (Belach Biotech, Stockholm, Sweden) at 30°C, pH 7·0, using a fed-batch protocol. Harvested cells were resuspended in 25 mM sodium phosphate, pH 7·5, lysed by sonication and inclusion bodies were isolated by centrifugation. Proteins were solubilized in 6 M guanidine hydrochloride and refolded by dilution 100-fold into a buffer containing 100 mM Tris, 0·2 mM oxidizied glutathione, 1 mM reduced glutathione, pH 8. The solution was gently stirred overnight at 4°C. OmCI was then purified by anion exchange chromatography on a Q Sepharose HP column (GE Healthcare) using gradient elution in 20 mM Tris, pH 7 with 0–1 M sodium chloride. Pooled fractions containing OmCI were diluted 1:1 in 20 mM sodium phosphate, 500 mM sodium chloride, 20 mM imidazole, pH 7·5 before being loaded onto a Ni Sepharose HP column (GE Healthcare). The column was washed with 50 mM imidazole before elution with an imidazole gradient from 50–300 mM. After a final polishing step using size exclusion chromatography (Superdex 200 prep grade; GE Healthcare) in 10 mM sodium phosphate, 145 mM sodium chloride, pH 7·5, the purified OmCI protein was concentrated and stored at −70°C.
DAF and Coxsackie adenovirus receptor (CAR) were cloned into the pTorsten vector (kind gift of Dr Brad Spiller, Cardiff University, UK) in frame with a C-terminal human Fc tag and expressed in Chinese hamster ovary (CHO) cells, as described previously 19. The whole immunoglobulin (Ig)G fraction from the plasma of a female patient (affected with DDD) carrying a very high titre of C3 nephritic factor (C3NeF) was isolated using a protein G affinity chromatography column and, after dialysis against PBS, used as a source of C3NeF autoantibodies.
Clinical samples
Serum samples were collected from patients suffering from diseases linked to the complement system. We obtained four samples from the cohort of patients presenting histological and/or clinical evidence of DDD, membranoproliferative glomerulonephritis I/III and other renal pathologies as well as signs of alternative complement pathway activation (reduced C3 and FB levels). The presence of C3 nephritic factor (C3NeF) in four serum samples (labelled GN39, GN23, GN32 and IND4) was confirmed with modified Rother's assay to be over the cut-off value of 20% haemolysis, as described by Paixao-Cavalcante et al. 10. Eleven samples were also collected from patients with atypical haemolytic uraemic syndrome (aHUS), six (labelled 1–6) were positive for FH autoantibodies (FHaAb), as measured by ELISA, described by Dragon-Durey et al. 20. Patient 5 carried both FHaAb and heterozygous mutation in C3 p.Lys65Gln. Three additional samples (patients 7–9) were from the patients carrying only mutations in complement genes; namely: C3 p.Arg161Trp (patient 7), FH p.Arg1206Cys (patient 8) and FB p.Lys323Glu (patient 9). Patient sample 10 carried a p.Arg1215Gln mutation in FH, and no FHaAb 21. Patient sample 11 contained FHaAb as assessed by the ELISA described by Moore et al. 21 and quantified further using the FHaAb ELISA assay described by Watson et al. 22; this sample also has a p.Lys633Arg mutation in C3 21. All samples were obtained upon informed consent and in the framework of ethical permit issued by respective local ethic committees. Detailed description of samples is given in Supporting information, Table S1.
Erythrocytes
Sheep erythrocytes were purchased from Hatunalab AB (Bro, Sweden). Erythrocytes (3·75 × 108, corresponding to 150 μl of stock solution) were washed with dextrose gelatin veronal buffer (DGVB++) buffer (2·5 mM veronal buffer, pH 7·3, 72 mM NaCl, 140 mM glucose, 0.1% gelatin, 1 mM MgCl2, and 0·15 mM CaCl2), pelleted and mixed with 1 ml of DGVB++ containing amboceptor (Complement Technologies) diluted 1:200. Samples were then incubated for 10 min at 37°C with 210 rpm shaking. Afterwards, sensitized sheep erythrocytes were washed twice, pelleted and resuspended in 500 μl of DGVB++. Ten μl of such suspension was used per single experimental point.
Rabbit erythrocytes suspended 1:1 in Alsever's solution (114 mM glucose, 28 mM Na-citrate dihydrate, 68 mM NaCl, 0·2 mM citric acid) were washed with Mg-ethylene glycol tetraacetic acid (EGTA) buffer (2·075 mM veronal buffer pH 7·3, 10 mM EGTA, 7 mM MgCl2, 0·083% gelatin, 116 mM glucose, 60 mM NaCl) until there was no visible haemoglobin in the supernatant. Then, 3·75 × 108 of rabbit erythrocytes (corresponding to 150 μl of Alsever's solution) were suspended in 1 ml of Mg-EGTA and used as a master stock for the assays. Ten μl of suspension was used per single experimental point.
Haemolytic assay
Sensitized sheep (classical) or non-sensitized rabbit erythrocytes (alternative pathway) were incubated in buffers permissive for the given pathway (DGVB or Mg-EGTA) for 30 min in the presence or absence of C5-blocking agents and different concentrations of NHS or NHS. Thereafter, erythrocytes were centrifuged and released haemoglobin was measured at 405 nm using Cary50 MPR microplate reader (Varian, Mulgrave, Australia).
Analysis of convertase activity
Analysis was performed in V-bottomed 96-well plate format on sensitized sheep erythrocytes (for classical pathway) or non-sensitized rabbit erythrocytes (alternative pathway). Step 1 of the convertase assay is the formation of convertase in the presence of C5 inhibitor. Concentrations of OmCI and eculizumab, which effectively blocked membrane attack complex (MAC) formation and subsequently used in convertase assays, were determined using haemolytic assay. Formation of the classical convertase was performed in DGVB buffer and serum concentration not exceeding 1%, as sensitization of sheep erythrocytes with antibodies activates the classical pathway and low serum concentration eliminates the occurrence of the alternative pathway. Formation of the alternative convertase was performed in Mg-EGTA buffer, as the presence of magnesium ions and chelation of calcium disables the classical and lectin pathways 13,23. We used a serum concentration not less than 2·5%, as a lower serum concentration was not able to support the alternative pathway. After the indicated time, erythrocytes were washed with 150 μl of GVB-ethylenediamine tetraacetic acid (EDTA) buffer (40 mM EDTA, 5 mM veronal buffer, 0·1% gelatin, 145 mM NaCl). Supernatant was aspirated and cells were overlaid with 50 μl of GVB-EDTA containing guinea pig serum diluted 1:40 and another 50 μl of DGVB or Mg-EGTA buffer, respectively. The aim of the second step of our assay is the formation of MAC using the platform of convertases preformed in step 1, as EDTA disables de-novo convertase formation. After 20 min incubation the cells were pelleted by centrifugation, 80 μl of supernatant was collected and haemolysis was assessed by measurement of absorbance at 405 nm. All steps were carried out at 37°C and shaking at 650 rpm (Thermomixer Comfort, Eppendorf, Hamburg, Germany).
Analysis of MAC formation
MAC formation supported by tested serum was analysed in DGVB buffer containing 50 mM trisodium citrate. Convertase was formed on sensitized sheep erythrocytes subjected to 1% normal human serum for 1 min (Tmax: time of maximal convertase activity) in the presence of 50 μM OmCI. Afterwards cells were washed with 150 μl of DGVB-50 mM citrate and subjected to 5% serum diluted in 50 μl DGVB-50 mM trisodium citrate for 60 min. The erythrocytes were overlaid with 50 μl DGVB, cells were pelleted and the supernatant (80 μl) was measured at 405 nm to detect the presence of released haemoglobin.
Results
Effect of OmCI and eculizumab on the classical and alternative complement pathways
We first determined the effective concentration of OmCI and eculizumab (both block C5 cleavage) on haemolysis in our models of classical and alternative complement pathway activation. Hindering C5 function stops the complement cascade at the level of the C5 convertases. This allows measurement of the overall activity of preformed convertases in the second step of the assay. OmCI effectively blocked haemolysis on sensitized sheep erythrocytes at 12·5 nM (Fig. 2a), whereas eculizumab did so at 2 nM (Fig. 2b). Rabbit erythrocytes used for the alternative pathway haemolytic assay were more vulnerable to non-specific lysis, as evidenced by 30% background lysis by heat-inactivated (Δ) NHS (Fig. 2c,d). None the less, we found concentrations of OmCI (100 nM) and eculizumab (60 nM), at which the inhibitory effect on haemolysis in the alternative pathway was saturated. As the lower serum concentration (2·5%) gave almost no distinguishable haemolysis, and using higher (5%) serum concentration resulted in incomplete inhibition by C5-blocking agents, we concluded that in our current settings 3·75% serum concentration was optimal for further experiments on this batch of rabbit erythrocytes. It should be noted that both non-specific lysis by Δ NHS and inhibition by C5 blocking varied from batch to batch of rabbit erythrocytes used in these assays, and while we never observed parameters worse than those presented in Fig. 2c,d, the quality of erythrocytes should always be determined prior to carrying out this assay.
Fig 2.
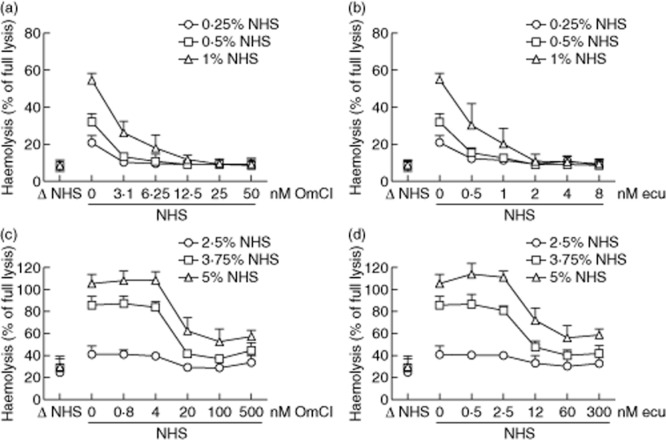
Inhibition of complement-mediated haemolysis by C5 inhibitors. The upper panel shows a titration of Ornithodoros moubata complement inhibitor (OmCI) (a) or eculizumab (b) for blocking of haemolysis mediated by the classical complement pathway. The bottom panel shows analogous data for the alternative complement pathway (c, OmCI; d, eculizumab). Heat-inactivated normal human serum (Δ NHS) was used as a negative control. Data were collected from three independent experiments and error bars show standard deviation.
Evaluation of Tmax in the two-step assay and validation of the new method
The purpose of the second step of this assay was to form MAC exclusively on the pre-existing convertase platforms formed during step 1 of the assay. Therefore, we applied guinea-pig serum diluted in an EDTA-containing buffer as a source of MAC components. Guinea-pig serum has been used historically for the development of convertase assays as it is fully compatible with human convertases, but even more efficient than human components in the haemolysis of sheep and rabbit erythrocytes 24. The presence of EDTA, which sequesters calcium and magnesium, prevents de-novo convertase formation from the guinea pig serum. The time of convertase formation in step 1 required for maximal attainable enzymatic activity, Tmax, is an important parameter and crucial for assay optimization. Previously, we showed that Tmax depends upon serum concentration, the content of soluble complement inhibitors (factor I, FH, C4BP) and also on spontaneous convertase decay. These features may vary in a particular serum; additionally, the Tmax curve also depends on the quality of the erythrocytes used in experiments 13. As experiments described in the current publication were performed on several different stocks of erythrocytes and serum, we present representative Tmax profiles for classical (Fig. 3a) and alternative pathways (Fig. 3b) performed on the same batch as data presented in Fig. 3c–f. Importantly, inhibition of convertase formation by either OmCI or eculizumab in step 1 always resulted in a comparable Tmax profile when the same serum concentration was used (Supporting information, Fig. S1). This implied that either C5 blocker can be used in these experiments.
Fig 3.
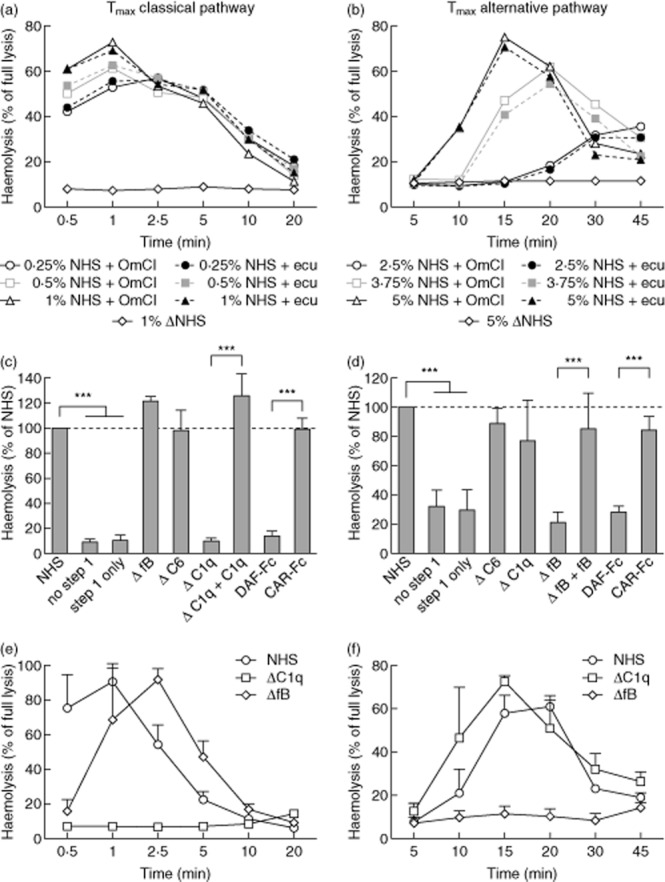
Time of maximal convertase activity (Tmax) profiles upon C5 inhibition and validation of the convertase assay. Tmax profiles were obtained for three different serum concentrations in the classical (a) and the alternative (b) pathway. Solid lines represent C5 blocking by Ornithodoros moubata complement inhibitor (OmCI) at 12·5 nM and 100 nM, whereas the dotted line represents blocking by eculizumab at 2 nM and 60 nM for the classical and alternative pathways, respectively. One representative experiment, of several experiments, performed on different batches of sera and erythrocytes, is shown. Complement-depleted sera as well as normal human serum supplemented with soluble convertase inhibitor [decay accelerating factor (DAF)-Fc] or control protein [Coxsackie adenovirus receptor (CAR)-Fc] were allowed to form classical (c) or alternative (d) convertase in the presence of OmCI. A concentration of 25 μg/ml DAF-Fc and CAR-Fc was used in the classical pathway assay, while 75 μg/ml was used in the alternative pathway assay. Convertase formation was restricted to 2·5 min in (c) and 20 min in (d), serum concentration was 0.5 and 3·75%, respectively. Mean of ≥3 independent experiments is shown. Error bars indicate standard deviation, **P < 0·01 and ***P < 0·001 according to one-way analysis of variance (anova). The bottom panels show the differences in Tmax profiles between normal human serum, C1q-depleted serum and complement factor B (FB)-depleted serum. (e) Data for the classical complement pathway and (f) shows data for the alternative complement pathway. Data were collected from three independent experiments performed on the same batch of serum and erythrocytes and error bars indicate standard deviation.
Knowing the Tmax profile of the particular batch of NHS, we chose a single time-point (at which the activity of convertases was high) to test the performance of our method on several sera depleted of complement factors or serum containing soluble inhibitors of convertases. NHS was supplemented with soluble, Fc-tagged DAF (CD55), which should interfere with convertase formation and display convertase decay-acceleration activity 25. The same serum supplemented with soluble CAR-Fc was used as negative control. Moreover, we used C1q-depleted serum (preventing assembly of classical convertase), FB-depleted serum (preventing assembly of alternative convertase) and C6-depleted serum (preventing assembly of MAC but not convertases). Loss of C1q in the classical pathway assay and loss of FB in the alternative pathway assay prevented stage 2-driven haemolysis (Fig. 3c,d). This deficit could be reversed by the inclusion of purified C1q and FB to the depleted sera. Similarly, DAF-Fc but not CAR-Fc prevented the formation of convertases in either assay. Two negative controls were included: ‘no step 1’ control, which contained erythrocytes only incubated with guinea-pig serum in 40 mM EDTA-GVB, and ‘step 1 only’ control, which contained erythrocytes incubated only with serum in pathway-permissive buffer containing C5 inhibitor during the whole period of the experiment (showing background lysis for this batch of red cells).
In the experiments presented in Fig. 3c, step 1 of the assay was performed for 2·5 min. However, in order to draw solid conclusions regarding different sera we recommend measuring whole Tmax profiles of these sera in addition to single-point haemolysis. This is especially important for the classical convertase, which reaches Tmax very quickly and therefore readout can change substantially in seconds. As an example, we show the Tmax profiles of normal human serum, FB-depleted serum and C1q-depleted serum (Fig. 3e). The time-point of 2·5 min (as performed in Fig. 3a) corresponds to the Tmax of FB-depleted serum, whereas normal human serum has already started to lose convertase activity from the 1-min time-point. Indeed, at the 2·5-min time-point, haemolysis supported by FB-depleted serum exceeded haemolysis supported by normal human serum (see Fig. 3c). C1q-depleted serum, irrespective of the time-period, supported no haemolysis. Similarly, the alternative convertase assay showed that no haemolysis was evident when erythrocytes were incubated with FB-depleted serum in step 1 (Fig. 4d). Normal human serum and C1q-depleted serum showed similar Tmax characteristics in this assay. Notably, experiments performed on the same batch of serum and erythrocytes gave reproducible results, as indicated by low standard deviations shown in Fig. 3e,f, as well as a low intra-assay coefficient of variation, which did not exceed 10% (Supporting information, Fig. S1)
Fig 4.

Screening for factors prolonging stability of the alternative convertase. (a) Time of maximal convertase activity (Tmax) profiles of the alternative complement convertase obtained for heat-inactivated normal human serum (Δ NHS, negative control) or normal human serum (NHS) supplemented with 30 μg/ml of C3 nephritic factor (C3NeF) (NHS+C3NeF) or human immunoglobulin (Ig) [NHS+human immunoglobulin (hIg)]. (b) Tmax profiles of sera positive for C3NeF (black connecting lines) and control, individual serum from healthy volunteer (grey connecting line), both mixed 1:1 with NHS at 2·5%. (c) Experiments similar to those in (a) but NHS was supplemented with 25 μg/ml of mouse monoclonal antibodies (mAbs) OX24 or C18/3 (both are FH function-blocking mAbs) or mouse control antibody mk54. (d) Tmax profiles of sera positive for autoantibodies against complement factor H (FHaAbs) (black connecting lines) and control, individual serum from healthy volunteer (grey connecting line). (e.f) Tmax profiles of convertases formed by sera containing mutated complement components, when tested unmixed at 5% concentration. Data shown are the average of at least three independent experiments, error bars represent standard deviation and significance is indicated by *P < 0·05, **P < 0·01 and ***P < 0·001, according to two-way analysis of variance (anova), respectively. Originally, three more individual sera from healthy volunteers were included to the data presented in (b) and (d) but they did not differ significantly from each other at any time-points, so were removed for better visibility of the graphs.
Screening for factors prolonging convertase half-life in clinical samples
The activity of complement convertases must be tightly regulated with regard not only to the place of activation but also to the duration or stability of the convertases; prolonged activity of alternative complement convertase is especially linked with pathogenic events. To model this, we supplemented normal serum with 30 μg/ml (final concentration) of IgG purified from C3NeF-positive serum (Fig. 4a) or 25 μg/ml of monoclonal antibodies (mAbs) (OX24 or C18/3), which bind FH and impair its complement-regulatory function 16,17 (Fig. 4c). C3NeF is an autoantibody which binds and stabilizes C3bBb complexes, making them resistant to decay. FH is the main soluble inhibitor of the alternative pathway and acts as a co-factor to FI-mediated cleavage of C3b as well as possessing decay-acceleration activity. In our experimental settings, the addition of C3NeF or mAb blocking the function of FH, but not human immunoglobulins or control mouse mAb of the same isotype (mk54), significantly prolonged activity of alternative convertases (Fig. 4a,c). These results suggest that, using our method, identification of sera containing factors prolonging convertase stability can be achieved. However, analysis of serum samples collected from patients can be problematic. The necessary condition for successful analysis requires that haemolytic activity is retained, and clinical samples are often collected or stored in a way which does not preserve complement activity. Furthermore, some patient sera will have lower complement activity as a result of their disease; e.g. those containing C3NeF that will constantly deplete C3 as a result of an overactive alternative complement pathway 26. In these circumstances, adding normal serum as a source of active complement to the patient sample is an option. We applied this strategy for analysis of C3NeF-containing sera. When mixed 1:1 with NHS (2·5% of serum + 2·5% NHS) all these sera, but not sera from healthy volunteers, showed significant prolongation of convertase activity (Fig. 4b), validating this approach. Next, we analysed six sera from aHUS patients containing detectable amounts of FHaAbs (1–6). One of these patients (no. 5) carried both FHaAbs and heterozygous mutation in C3 p.Lys65Gln, which was shown previously to alter binding of recombinant C3b to FH 27. These samples presented haemolytic activity comparable to each other and to NHS, and therefore we tested them unmixed at 5% concentration (Fig. 4d). Only four sera showed significantly prolonged convertase activity, namely samples 2, 3, 5 and 6, and two other sera performed similarly to control sera from healthy volunteers. These results correlate with FHaAbs assessments by ELISA (see Supporting information) to a limited extent; for instance sample 2, which performed the best in our assay, also displayed the highest level of FHaAbs. However, sample 6 has almost three times lower levels of FHaAbs than samples 1 and 4, which performed similarly to control sera. Detection of FHaAbs with purified FH on ELISA plate or even binding to a certain part of FH, however, does not necessarily determine any functional consequence of that interaction, so this result is not particularly contradictory.
We compared two other sets of serum samples collected from aHUS patients. First, we analysed sample 10 with missense mutation in C-terminus of FH (p.Arg1215Gln, which should not affect affinity to C3b, according to 28) and sample 11, which carried an as-yet uncharacterized mutation pLys633Arg in C3 in addition to FHaAbs. Sample 10 performed similarly to NHS, as theoretically predicted, whereas sample 11 showed prolonged convertase activity (Fig. 4e). A second set of samples included serum from a patient with a p.Arg161Trp mutation in C3 (sample 7). This change resulted in slightly decreased binding to FH 27 in vitro, but functional assays showed normal regulation by FH, in contrast to regulation by membrane-bound complement inhibitor CD46 29. Serum sample 8 carried a FH mutated in position 1206, which was found to be important for interaction with C3b 28 whereas serum sample 9 carried a mutation in FB, which results in an alternative pathway convertase that is more resistant to decay by FH 30. In our assay, serum sample 7 did not differ from NHS but, surprisingly, neither did serum sample 8, showing that under our experimental conditions the Arg1206Cys mutation still allows normal complement regulation. When pretested by haemolytic assay, serum sample 9 exhibited approximately 25% activity of NHS and therefore we decided to examine this sample mixed 1:1 with 2·5% NHS (similarly to C3NeF-positive sera), as mutation in FB should produce a dominant effect. The inclusion of serum sample 9 when unmixed (Fig. 4f) and mixed (Fig. 4g) into analysis of unmixed sera (samples 7 and 8) allowed clear comparison of both readouts. As expected, sample 9 without added NHS performed as serum of lower concentration (see Fig. 3b), but when mixed 1:1 with NHS resistance to convertase decay was observed, in contrast to serum sample 7 mixed with NHS at the same ratio (Fig. 4g). To confirm this, we performed an additional control, labelled ‘NHS compensation’, where we used a volume of NHS which gave the equivalent pretest haemolytic activity of serum sample 9 (i.e. 25% of normal). As expected, this control sample still performed as a serum of lower concentration, unable to generate a high level of haemolysis, which was in contrast to the mix of NHS with serum sample 9, showing a high haemolytic level over an extended period of time.
Analysis of MAC-formation ability
Separation of the complement cascade into two steps, the first ending with convertase formation and the second with MAC formation, enables studying properties of a given serum sample in both steps. Thus, a standard serum sample, in the presence of a C5 blocker, can be used to form a ‘common/identical’ convertase allowing the ability to generate MAC to be compared in individual patient or control sera in step 2. We needed to modify the conditions for step 2 as human serum, unlike guinea pig serum, is not compatible with EDTA, which blocked MAC formation even at very low concentrations. Instead, we used 50 mM trisodium citrate directly suspended in DGVB buffer, which completely blocks convertase formation in 5% serum. We demonstrate validation of the method through the use of the same depleted sera and/or complement inhibitors as used in the convertase assays above (Fig. 5). Here, all sera except C6-depleted serum supported MAC formation. Reconstitution of C6-depleted serum with purified C6 restored its haemolytic activity in this assay. The addition of soluble DAF also resulted in impairment of haemolytic activity, presumably as a result of the rapid decay of the preassembled convertase preventing MAC assembly.
Fig 5.
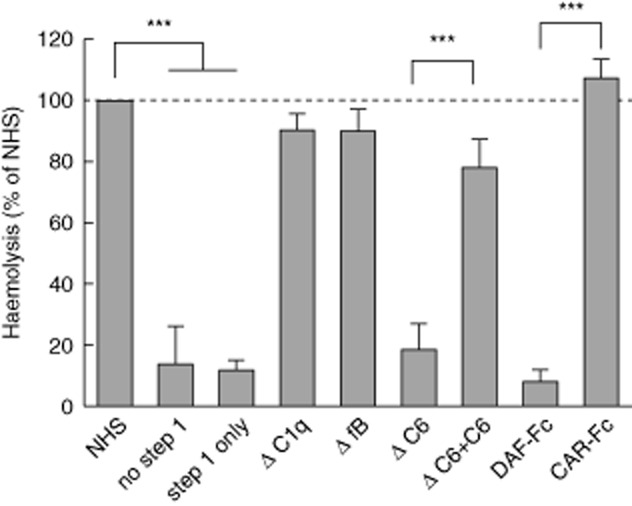
Validation of membrane attack complex (MAC) formation assay using complement-depleted sera and complement inhibitors. Complement-depleted sera as well as normal human serum supplemented with soluble convertase inhibitor [decay accelerating factor (DAF)-Fc] or control protein [Coxsackie adenovirus receptor (CAR)-Fc] at 150 μg/ml were allowed to form MAC (step 2 of the assay) on the platform of the classical C5 convertase (preformed in step 1 of the assay from normal human serum). Data were collected from three independent experiments, error bars represent standard deviation; ***P < 0·001 by one-way analysis of variance (anova).
Discussion
To the best of our knowledge, we propose for the first time an assay which is capable of directly assessing convertase and MAC formation capabilities of an individual serum sample. A definite advantage of our assay lies in the fact that it is performed in a physiological milieu of whole serum, and thus may reflect all-important relations between particular serum components. Both convertases and MAC are multi-protein complexes and their assembly depends not only on the availability of single components but on many other factors and variables. Therefore, the environment of whole serum in a functional assay is superior to attempts based on either purified or recombinant components assembled into convertases.
In its current design, this assay is a screening method for factors affecting convertases or MAC and it cannot precisely point out the effect mechanism. However, several modifications, such as the addition of individual, purified complement components, can be used to narrow the area of investigation, i.e. diminished activity is restored after the addition of a particular component. By application of C5 inhibitors, we set up our assays in a way that stopped the complement cascade at the level of C5 convertases. Given that transition from C3 to C5 convertase depends directly on the activity of the first enzyme, and given that both enzymatic complexes share the same pattern of regulation, we argue that the point of C5 convertase reflects all events important for convertase formation and separates them from the lytic, effector phase of the complement cascade. Theoretically, our assay can be modified in order to subtract the activity of the classical C3 convertase by the application of C3 blocker (such as, e.g. compstatin 31) instead of C5 blocker. However, controlling alternative pathway C3/C5 convertase transition in whole serum is impossible due to the fact that the C3b molecule is already necessary at the C3 alternative convertase (C3bBb) step.
This study demonstrates clearly that one cannot rely only upon haemolytic score at a single time-point to draw an ultimate conclusion about serum functionality. In our experimental model, immunodepletion of sera undoubtedly affected the balance between complement activators and inhibitors and therefore affected the Tmax point, but not the maximal level of attainable haemolysis (Fig. 3e,f). As well as immunodepletion, differences in the collection and subsequent handling of a serum sample may affect complement and convertase Tmax. It is therefore crucial to compare the whole Tmax profile of the serum to be studied. Furthermore, haemolysis is often affected in sera collected from individuals with pathologically hyperactive convertases (e.g. C3NeF-positive 32 or gain-of-function mutations in complement components 29), which constantly deplete complement components in vivo. Exhaustion of complement may cause a different Tmax profile when compared with control serum of the same concentration but hyperactivity of convertases should be reflected, for instance, in prolonged Tmax with a concurrent high level of overall haemolysis, when serum is mixed with unaffected serum (Fig. 4b,g). A change in Tmax is also the case for sera of lower concentration or activity (Figs 3 and 4g) but, conversely, they do not reach as high a level of haemolysis when compared to control sera or sera with hyperactive convertases.
Assessing the functional outcome of mutations in complement components on convertase activity is another advance of our assay. Of note, we observed discrepancy between predicted and obtained outcome of an Arg1206Cys mutation in FH. Studies performed on a recombinant fragment of FH carrying the Arg1206Ala mutation revealed diminished binding to purified C3b, heparin and the surface of mouse glomerular endothelial cells 28; thus, this mutation should negatively affect convertase decay by FH. In our assay FH mutated at the same site, but to a cysteine instead of an alanine performed normally (Fig. 4f). This suggests that the mutation to cysteine has a different phenotype or that the putative reduction of FH affinity to C3b observed in the system based on purified components is sufficient to mirror proper complement regulation in our system based on full serum. Notably, our system was sensitive enough to observe the effect of other abnormalities related to FH, like FHaAbs. Not all sera with detectable level of FHaAb exhibited prolonged convertase activity, and this may reflect the functionality rather than quantity of these antibodies. Not all FHaAbs necessarily affect FH–convertase interaction, and some may affect FH more than others. Additionally, an important point to consider here is the nature of cells used to test the activity of alternative complement convertase. We used rabbit erythrocytes, as they spontaneously activate the alternative complement pathway and offer haemolysis as an easy readout. However, they do not contain human membrane-bound complement inhibitors (such as, e.g. CD46, which is crucial for observing the effect of mutation C3 p.Arg161Trp present in our serum sample 7) and contain relatively low levels of sialic acid 33, which in turn diminishes the binding of soluble FH on the surface 34. Thus, FH can be less relevant in the overall protection of rabbit erythrocytes from complement when compared to other types of cells, for which the importance of surface-bound FH for protection from complement attack was described 35–37. Therefore, some physiologically important effects may need to be visualized using the concept of C5 blocking on alternative cell types. For instance, those that are used routinely to model a particular disease, e.g. activated microvessel endothelial cells as an aHUS model 28, where an alternative readout of lysis, such as 51-Cr release 38, would be needed.
Our assay was robust in detection of C3NeF, which directly binds and stabilizes convertases. All C3NeF-positive sera included in the test study were clearly distinguished and showed far more significant prolongation of convertase activity compared to sera with FHaAbs. Our experiments showed that the addition of 30 μg/ml of IgG prep containing only a minor fraction of C3NeF extended convertase activity more effectively than 25 μg/ml of FH mAbs of defined function-blocking properties. On one hand, this effect may be due to surface characteristics of rabbit erythrocytes but, on the other hand, a 3 μM concentration of FH in serum markedly exceeds the number of active convertase complexes attainable in our experimental conditions. Therefore, one should expect better performance of C3NeF than FH mAbs.
There are several assays based on in-vitro convertase assembly from purified components, which screen for C3NeF 10,39, as well as an assay designed more than 30 years ago 11 and modified recently 12, which is based on whole serum and separation into a pre- and post-C5 step, the latter supported by exogenous rat serum in EDTA-containing buffer. However, the authors did not use C5 inhibitors, but assumed that human serum cannot support complement activation on non-sensitized sheep erythrocytes beyond the C5 level. This assumption is controversial, since haemolytic assay performed on human serum is a standard procedure in the complement field and human serum is definitely capable of lysing red blood cells of different origin. In fact, sheep erythrocytes are not optimal to activate alternative complement pathway unless desialylated by treatment with neuraminidase 40. We checked whether haemolysis of non-sensitized sheep erythrocytes cannot be obtained with normal human serum at 30°C, as suggested by other authors 11,12. Indeed, we did not observe haemolysis higher than that supported by heat-inactivated NHS after 20 min incubation, but there was obvious lysis after 1 h incubation. We conclude that the kinetics of complement activation on non-sensitized sheep erythrocytes is much slower than that seen on sensitized sheep erythrocytes or rabbit erythrocytes, the conventional target. Therefore, it is theoretically possible to extract a time-point when MAC is not yet formed on pre-existing convertases, but this method does not offer strict control on MAC formation and provides no guarantee of an optimal convertase formation. Indeed, some sera analysed in this way cause significant haemolysis in the putative pre-C5 step, thus precluding any firm conclusions. We stress that using cells which easily activate complement, together with specific C5 inhibitors, is the superior approach in such assays.
Taken together, we have presented proof-of-concept for a new assay which measures activity of complement convertase and MAC formation directly in serum samples. It is a cost- and time-effective alternative to existing assays screening for factors that cause hyperactivity of complement convertases. Full standardization of this assay may be challenging, as the readout depends upon many non-technical variables, such as functionality of control serum and quality of erythrocytes. Indeed, a test run assessing the impact of these parameters should be performed before experiments are performed. The International Complement Standardization Committee, a joint initiative of the International Union of Immunological Societies (IUIS), and the International Complement Society (ICS) have established a standard of non-activated human serum (ICS 1), which can be used as reference for positive control, thus providing the opportunity of making results of our assay comparable between different test runs and laboratories 41.
In summary, we show a new concept that strict control of C5 cleavage by OmCI or eculizumab in parallel with assessment of Tmax profile enables complete appraisal of convertase activity in whole serum. Our model gave predictable and logical results, and we suggest that researchers can modify it (regarding the type of cells and readout) to best fit their needs. Importantly, application of OmCI offers extension of our model to animal (rodent) serum, as OmCI activity is not human-specific, in contrast to eculizumab.
Acknowledgments
This work was supported by Swedish Research Council (projects K2012-66X-14928-09-5 to A. B and K2010-80X-21514-01-4 to M. O.), Cancerfonden (to A. B.), grant for clinical research (ALF) (to A. B.), grant SAF2012-38636 to M. L.-T. (Ministerio de Economía y Competitividad. Spain), grants by the Dutch Kidney Foundation to E. V. and L. P. van den H. (IP10.22, KFB 11.007) and Foundations of Österlund (A. B.), Crafoord (M. O.), Greta and Johan Kock (A. B.), Gustav V 80-years anniversary (A. B.), Tore Nilsson (M. O.), John and Augusta Persson (M. O.), Knut and Alice Wallenberg (A. B.) and Inga-Britt and Arne Lundberg (A. B.). K. M. was supported by Northern Counties Kidney Research Fund. We thank Professor Santiago Rodriguez de Cordoba and Dr David Kavanagh for their advice and help.
Author contributions
Concept of the study: M. O.; design of the experiments: M. O., A. B., T. M.; experimental work: L. B., P. S., M. O., V. F., M. V.; collection and characterization of clinical samples: E. V., L. van den H., M. L-T., T. G., K. M., manuscript writing: M. O., K. M., A. B., T. M. and P. S.
Disclosure
Authors declare no conflicts of interest.
Supporting Information
Additional Supporting information may be found in the online version of this article at the publisher's web-site:
Fig S1. Intra-assay coefficient of variation (intra-CV) of time of maximal convertase activity (Tmax) curves. Tmax curve for classical pathway (left panel) and alternative pathway (right panel) was calculated from four repetitions. Classical pathway convertase was formed from 0.5% normal human serum (NHS) blocked with 12.5 nM Ornithodoros moubata complement inhibitor (OmCI) or 2 nM eculizumab. Alternative pathway convertase was formed from 5% NHS blocked with 100 nM OmCI or 60 nM eculizumab.
Table S1. Detailed characteristics of clinical samples used in the study.
References
- Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol. 2013;190:3831–3838. doi: 10.4049/jimmunol.1203487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Barricarte R, Heurich M, Valdes-Canedo F, et al. Human C3 mutation reveals a mechanism of dense deposit disease pathogenesis and provides insights into complement activation and regulation. J Clin Invest. 2010;120:3702–3712. doi: 10.1172/JCI43343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi S, Fervenza FC, Zhang Y, et al. Atypical postinfectious glomerulonephritis is associated with abnormalities in the alternative pathway of complement. Kidney Int. 2013;83:293–299. doi: 10.1038/ki.2012.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurich M, Martinez-Barricarte R, Francis NJ, et al. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc Natl Acad Sci USA. 2011;108:8761–8766. doi: 10.1073/pnas.1019338108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warwicker P, Goodship TH, Donne RL, et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998;53:836–844. doi: 10.1111/j.1523-1755.1998.00824.x. [DOI] [PubMed] [Google Scholar]
- Daha MR, van Es LA. Relative resistance of the F-42-stabilized classical pathway C3 convertase to inactivation by C4-binding protein. J Immunol. 1980;125:2051–2054. [PubMed] [Google Scholar]
- de Cordoba SR, Tortajada A, Harris CL, Morgan BP. Complement dysregulation and disease: from genes and proteins to diagnostics and drugs. Immunobiology. 2012;217:1034–1046. doi: 10.1016/j.imbio.2012.07.021. [DOI] [PubMed] [Google Scholar]
- Amornsiripanitch N, Hong S, Campa MJ, Frank MM, Gottlin EB, Patz EF., Jr Complement factor H autoantibodies are associated with early stage NSCLC. Clin Cancer Res. 2010;16:3226–3231. doi: 10.1158/1078-0432.CCR-10-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paixao-Cavalcante D, Lopez-Trascasa M, Skattum L, et al. Sensitive and specific assays for C3 nephritic factors clarify mechanisms underlying complement dysregulation. Kidney Int. 2012;82:1084–1092. doi: 10.1038/ki.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rother U. A new screening test for C3 nephritis factor based on a stable cell bound convertase on sheep erythrocytes. J Immunol Methods. 1982;51:101–107. doi: 10.1016/0022-1759(82)90386-6. [DOI] [PubMed] [Google Scholar]
- West CD. A hemolytic method for the measurement of nephritic factor. J Immunol Methods. 2008;335:1–7. doi: 10.1016/j.jim.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Okroj M, Holmquist E, King BC, Blom AM. Functional analyses of complement convertases using C3 and C5-depleted sera. PLOS ONE. 2012;7:e47245. doi: 10.1371/journal.pone.0047245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunn MA, Sharma A, Paesen GC, et al. Complement inhibitor of C5 activation from the soft tick Ornithodoros moubata. J Immunol. 2005;174:2084–2091. doi: 10.4049/jimmunol.174.4.2084. [DOI] [PubMed] [Google Scholar]
- Kaplan M. Eculizumab (Alexion) Curr Opin Investig Drugs. 2002;3:1017–1023. [PubMed] [Google Scholar]
- Jokiranta TS, Zipfel PF, Hakulinen J, et al. Analysis of the recognition mechanism of the alternative pathway of complement by monoclonal anti-factor H antibodies: evidence for multiple interactions between H and surface bound C3b. FEBS Lett. 1996;393:297–302. doi: 10.1016/0014-5793(96)00905-2. [DOI] [PubMed] [Google Scholar]
- Oppermann M, Manuelian T, Jozsi M, et al. The C-terminus of complement regulator Factor H mediates target recognition: evidence for a compact conformation of the native protein. Clin Exp Immunol. 2006;144:342–352. doi: 10.1111/j.1365-2249.2006.03071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlbäck B, Hildebrand B, Malm J. Characterization of functionally important domains in human vitamin K-dependent protein S using monoclonal antibodies. J Biol Chem. 1990;265:8127–8135. [PubMed] [Google Scholar]
- Okroj M, Mark L, Stokowska A, et al. Characterization of the complement inhibitory function of rhesus rhadinovirus complement control protein (RCP) J Biol Chem. 2009;284:505–514. doi: 10.1074/jbc.M806669200. [DOI] [PubMed] [Google Scholar]
- Dragon-Durey MA, Loirat C, Cloarec S, et al. Anti-Factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:555–563. doi: 10.1681/ASN.2004050380. [DOI] [PubMed] [Google Scholar]
- Moore I, Strain L, Pappworth I, et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood. 2010;115:379–387. doi: 10.1182/blood-2009-05-221549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson R, Lindner S, Bordereau P, et al. Standardisation of the factor H autoantibody assay. Immunobiology. 2014;219:9–16. doi: 10.1016/j.imbio.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Forsgren A, Quie PG. Influence of the alternate complement pathway in opsonization of several bacterial species. Infect Immun. 1974;10:402–404. doi: 10.1128/iai.10.2.402-404.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medof ME, Kinoshita T, Nussenzweig V. Inhibition of complement activation on the surface of cells after incorporation of decay-accelerating factor (DAF) into their membranes. J Exp Med. 1984;160:1558–1578. doi: 10.1084/jem.160.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lublin DM, Atkinson JP. Decay-accelerating factor: biochemistry, molecular biology, and function. Ann Rev Immunol. 1989;7:35–58. doi: 10.1146/annurev.iy.07.040189.000343. [DOI] [PubMed] [Google Scholar]
- Servais A, Noel LH, Fremeaux-Bacchi V, Lesavre P. C3 glomerulopathy. Contrib Nephrol. 2013;181:185–193. doi: 10.1159/000348654. [DOI] [PubMed] [Google Scholar]
- Volokhina E, Westra D, Xue X, Gros P, van de Kar N, van den Heuvel L. Novel C3 mutation p.Lys65Gln in aHUS affects complement factor H binding. Pediatr Nephrol. 2012;27:1519–1524. doi: 10.1007/s00467-012-2183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MJ, Rops AL, Isenman DE, van der Vlag J, Jokiranta TS. Mutations of factor H impair regulation of surface-bound C3b by three mechanisms in atypical hemolytic uremic syndrome. J Biol Chem. 2009;284:15650–15658. doi: 10.1074/jbc.M900814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumenina LT, Frimat M, Miller EC, et al. A prevalent C3 mutation in aHUS patients causes a direct C3 convertase gain of function. Blood. 2012;119:4182–4191. doi: 10.1182/blood-2011-10-383281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, et al. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci USA. 2007;104:240–245. doi: 10.1073/pnas.0603420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu A, Kay BK, Lambris JD. Inhibition of human complement by a C3-binding peptide isolated from a phage-displayed random peptide library. J Immunol. 1996;157:884–891. [PubMed] [Google Scholar]
- Dragon-Durey MA, Blanc C, Marinozzi MC, van Schaarenburg RA, Trouw LA. Autoantibodies against complement components and functional consequences. Mol Immunol. 2013;56:213–221. doi: 10.1016/j.molimm.2013.05.009. [DOI] [PubMed] [Google Scholar]
- Van Dijk H, Rademaker PM, Willers JM. Determination of alternative pathway of complement activity in mouse serum using rabbit erythrocytes. J Immunol Methods. 1980;36:29–39. doi: 10.1016/0022-1759(80)90091-5. [DOI] [PubMed] [Google Scholar]
- Ferreira VP, Pangburn MK, Cortes C. Complement control protein factor H: the good, the bad, and the inadequate. Mol Immunol. 2010;47:2187–2197. doi: 10.1016/j.molimm.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Corral P, Gonzalez-Rubio C, Rodriguez de Cordoba S, Lopez-Trascasa M. Functional analysis in serum from atypical hemolytic uremic syndrome patients reveals impaired protection of host cells associated with mutations in factor H. Mol Immunol. 2004;41:81–84. doi: 10.1016/j.molimm.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Ferreira VP, Pangburn MK. Factor H mediated cell surface protection from complement is critical for the survival of PNH erythrocytes. Blood. 2007;110:2190–2192. doi: 10.1182/blood-2007-04-083170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horl S, Banki Z, Huber G, et al. Reduction of complement factor H binding to CLL cells improves the induction of rituximab-mediated complement-dependent cytotoxicity. Leukemia. 2013;27:2200–2208. doi: 10.1038/leu.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okroj M, Eriksson I, Osterborg A, Blom AM. Killing of CLL and NHL cells by rituximab and ofatumumab under limited availability of complement. Med Oncol. 2013;30:759. doi: 10.1007/s12032-013-0759-5. [DOI] [PubMed] [Google Scholar]
- Jelezarova E, Schlumberger M, Sadallah S, Spath PJ, Schifferli JA, Lutz HU. A C3 convertase assay for nephritic factor functional activity. J Immunol Methods. 2001;251:45–52. doi: 10.1016/s0022-1759(01)00295-2. [DOI] [PubMed] [Google Scholar]
- Pangburn MK, Muller-Eberhard HJ. Complement C3 convertase: cell surface restriction of beta1H control and generation of restriction on neuraminidase-treated cells. Proc Natl Acad Sci USA. 1978;75:2416–2420. doi: 10.1073/pnas.75.5.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergseth G, Ludviksen JK, Kirschfink M, Giclas PC, Nilsson B, Mollnes TE. An international serum standard for application in assays to detect human complement activation products. Mol Immunol. 2013;56:232–239. doi: 10.1016/j.molimm.2013.05.221. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. Intra-assay coefficient of variation (intra-CV) of time of maximal convertase activity (Tmax) curves. Tmax curve for classical pathway (left panel) and alternative pathway (right panel) was calculated from four repetitions. Classical pathway convertase was formed from 0.5% normal human serum (NHS) blocked with 12.5 nM Ornithodoros moubata complement inhibitor (OmCI) or 2 nM eculizumab. Alternative pathway convertase was formed from 5% NHS blocked with 100 nM OmCI or 60 nM eculizumab.
Table S1. Detailed characteristics of clinical samples used in the study.