

Abstract
We propose a new approach for calculating
the change of the absorption spectrum of a molecule when moved from
the gas phase to a crystalline morphology. The so-called gas-to-crystal
shift Δ m is mainly caused by dispersion effects and depends
sensitively on the molecule’s specific position in the nanoscopic
setting. Using an extended dipole approximation, we are able to divide
Δ
m is mainly caused by dispersion effects and depends
sensitively on the molecule’s specific position in the nanoscopic
setting. Using an extended dipole approximation, we are able to divide
Δ m= −QWm in two factors, where Q depends only on the
molecular species and accounts for all nonresonant electronic transitions
contributing to the dispersion while Wm is a geometry factor expressing the site dependence of the shift
in a given molecular structure. The ability of our approach to predict
absorption spectra is demonstrated using the example of polycrystalline
films of 3,4,9,10-perylenetetracarboxylic diimide (PTCDI).
m= −QWm in two factors, where Q depends only on the
molecular species and accounts for all nonresonant electronic transitions
contributing to the dispersion while Wm is a geometry factor expressing the site dependence of the shift
in a given molecular structure. The ability of our approach to predict
absorption spectra is demonstrated using the example of polycrystalline
films of 3,4,9,10-perylenetetracarboxylic diimide (PTCDI).
Introduction
Thin films of conjugated organic molecules are essential building blocks in organic and hybrid optoelectronics. These films are typically characterized by a nanocrystalline structure. Planar or bulk heterojunctions are realized either by different organic molecules or by combination with inorganic semiconductors or metal oxides such as ZnO and TiO2.1−3 The energetic alignment of the ground and excited states of the different materials governs the efficiency of charge separation and the open-circuit voltage in photovoltaic cells and, similarly, the injection of charge carriers and energy transfer dynamics in light-emitting devices.4−6 Hence, it is essential to understand in detail how structural disorder and site-dependent interactions affect the energetic properties of organic thin films.
When a molecule m is moved from the gas phase into a crystalline morphology, its optical transition energies undergo the so-called gas-to-crystal shift. This shift is due to (i) Coulomb coupling of static charge distributions, (ii) inductive polarization shifts, (iii) excitonic shifts, (iv) dispersion shifts caused by the interaction between higher excited transition densities of the molecule m and surrounding molecules, and (v) charge transfer states coupling to Frenkel exciton states.
In this manuscript, we show that absorption spectra of nanocrystalline molecular films are dominated by dispersive shifts (iv). These shifts are typically assumed to be constant for all molecules of the system which is true in a bulk crystal where all constituents feel the same infinitely large environment. However, in a nanoscopic morphology, the dispersion becomes site-dependent. It will strongly depend on whether the molecule is situated close to the surface or deep within the nanocrystallite. We will introduce in the following an efficient computation scheme to calculate the site-dependent level shift and demonstrate its capability by explaining optical absorption spectra of a series of thin nanocrystalline films of 3,4,9,10-perylenetetracarboxylic diimide (PTCDI) of different crystallite size. Figure 1 shows a typical absorption spectrum of a thin PTCDI film grown by molecular beam deposition. The spectral shape is characterized by a distinct double-structure that does not resonate with the features of the isolated molecule in solution (see Figure 1). We will show in the following that the two features are due to molecules experiencing distinctively different environments.
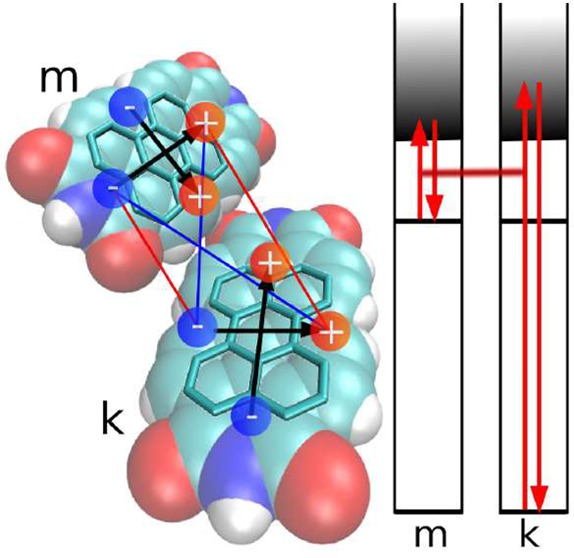
Figure 1.
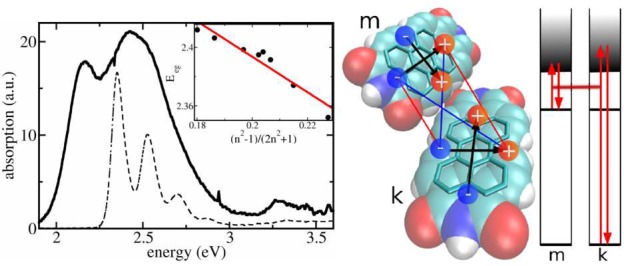
Left panel: Room temperature absorption spectra of different PTCDI systems. Solid line: PTCDI film vacuum-deposited on Al2O2. The nominal thickness is 12 nm. Dashed line: PTCDI monomer in toluene solution. The inset shows the S0,ν=0 → S1,ν=0 transition energy for the PTCDI derivate N,N′-bis(1-hexylheptyl)perylene-3,4:9,10-bis(dicarboximide) in nonpolar solvents with different refractive indices n: pentane (n = 1.36), hexane (n = 1.38), nonane (n = 1.41), dodecane (n = 1.42), cyclohexane (n = 1.43), hexadecane (n = 1.43), tetrachloromethane (n = 1.46), and benzene (n = 1.50). Red line: linear fit. Right panel: Space-filling model of two PTCDI molecules. The carbon bondings establishing the π-electron system are highlighted. The positive and negative partial charges of an extended dipole model for higher electronic transitions of type ξ are indicated. Their interaction represents the position and orientation dependent excitonic coupling between molecule m and molecule k. The depicted interaction constitutes V(m∥,k⊥). A scheme of coupled virtual transitions giving rise to the dispersive shift ΔEme(k), eq 1, is presented as well. Shown is the transition of molecule m into a higher level f and the transition of a molecule k of the surrounding from its ground state to the higher level f′.
Model
We start the description of the system by defining the electronic
states φma and related energies Ema of an isolated molecule
at site m and in state a. In the
most naive picture, singly excited states now follow from the standard
Frenkel exciton Hamiltonian Hexc = ∑m,n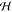 mn|ϕm⟩⟨ϕn|, where the ground state energy E0 = ∑mEmg = 0 is shifted to the origin
of the energy scale.
mn|ϕm⟩⟨ϕn|, where the ground state energy E0 = ∑mEmg = 0 is shifted to the origin
of the energy scale.
Here, we have defined the molecular product
states ϕm with molecule m in the first excited state φme and all other molecules in the ground state φng. The overall ground state ϕ0 is
given by the product of all molecular ground states φng. The diagonal and off-diagonal part of the Hamiltonian
matrix 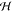 mn represent the excitation energies
mn represent the excitation energies 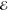 m = Eme −Emg and the
resonant excitonic coupling terms
m = Eme −Emg and the
resonant excitonic coupling terms 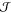 mn, respectively. A proper rescaling of all
elements of
mn, respectively. A proper rescaling of all
elements of 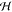 mn may account for the dispersion effects, but a more
microscopic picture is desirable. We note that site-dependent dispersive
energy shifts have been calculated for aggregates of Rydberg atoms
in ref (10). Here,
we suggest a method that is capable of calculating those shifts for
molecular aggregates.
mn may account for the dispersion effects, but a more
microscopic picture is desirable. We note that site-dependent dispersive
energy shifts have been calculated for aggregates of Rydberg atoms
in ref (10). Here,
we suggest a method that is capable of calculating those shifts for
molecular aggregates.
In the following, we will directly compute the
site-dependent shifts Δ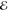 m = ΔEme – ΔEmg of the excitation energy
m = ΔEme – ΔEmg of the excitation energy 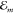 . Here,
the individual level shifts ΔEma with a = g, e arise from the nonresonant Coulomb coupling of molecule m with all its surrounding molecules. They can be written
as ΔEma = ∑kΔEma(k), where the contribution due to the coupling
to molecule k is given in second order perturbation
theory by13,14
. Here,
the individual level shifts ΔEma with a = g, e arise from the nonresonant Coulomb coupling of molecule m with all its surrounding molecules. They can be written
as ΔEma = ∑kΔEma(k), where the contribution due to the coupling
to molecule k is given in second order perturbation
theory by13,14
| 1 |
The expression includes the transition energies Emfa = Emf – Ema and Ekf′g = Ekf′ – Ekg. f and f′ count all higher excited energy levels (f, f′ > e), and Jmk(ag, f′f) is the Coulomb coupling between the electronic transition density ρfa(m)(r) of molecule m and the electronic transition density ρf′g(r′) of molecule k (cf. Figure 1).8 Equation 1 illustrates that the site dependence of the dispersion originates from the dependence of the Jmk(ag, f′f) on the position of molecule m relative to molecule k. Since the summation of the two energy differences in the denominator is always more than 1 order of magnitude larger than the coupling of transition densities, eq 1 represents a good approximation for the energy shift ΔEma(k).
The rigorous evaluation of eq 1 for a large molecule like PTCDI in a complex environment like a nanocrystalline film according to eq 1 is a formidable task. Instead, we utilize a simplified treatment based on the well-established extended dipole approximation for computation of the Jmk(ag, f′f). This allows us to derive a formula for the level shifts that contains only quantities that are, at least in principle, accessible by experiment. In the extended dipole approximation, the actual transition densities are replaced by transition dipoles represented by a single negative and a single positive charge ∓q placed at a fixed distance that resembles the spatial extension of the single-molecule excitation (see Figure 1). As a consequence, the resulting distance dependence of the Jmk(ag, f′f) is more realistic as in the point dipole approximation.
The PTCDI excited states can be classified into states with transition dipoles parallel to the long molecular axis (ξ(a,f) = ∥) and perpendicular to it (ξ(a,f) = ⊥) (Supporting Information part A). The basic idea of our simplified treatment is to place the charges at the same distance for all transitions f with polarization (ξ(a,f) = ∥) and likewise for the charges corresponding to the transitions of type ξ(a,f) = ⊥. In both cases, the q(a,f) is positioned at the boundary of the π-electron system (cf. Figure 1). As a consequence, the charge q(a,f) depends on the initial state a and the excited state f and can be derived from the ratio of the respective transition dipole moment and the intercharge distance.
We verified this approximation by performing extensive electronic structure computations for a large number of excited states by comparing the Coulomb coupling matrix elements Jmk obtained by the extended dipole approximation with the exact value based on atomic-centered partial transition charges (Supporting Information part A). The mean error is below 5% for all excited states that have been considered. The extended dipole approximation allows us to factorize the interaction matrix element Jmk(ag, f′f) = q(a,f)q(g,f′)V(mξ(a,f), kξ′(g,f′)) into two contributions, the effective charges q(a,f), q(g,f′) and the distance dependence of the interaction of extended dipoles V(mξ(a,f), kξ′(g,f′)), i.e., the interaction of a pair of unit charges of opposite sign at molecule m and k. The V(mξ(a,f), kξ′(g,f′)) still depend on the transitions af, gf′ via ξ(a,f), ξ′(g,f′).
If the different states f, f′ lie sufficiently close in energy, we may average over the two different transition dipole directions by introducing |Vmk|2 = ∑ξ, ξ′|V(mξ, kξ′)|2/4. Our electronic structure calculations of the higher excited states support this approximation. As a consequence, the Vmk becomes independent of the states f, f′ and hence Jmk(ag,f′f) ≈ q(f)q(f′)Vmk.
Finally, the site-dependent transition energy shift of molecule m can be written as
| 2 |
with
| 3 |
and Wm = ∑kVmk2.
While Wm can be interpreted as a geometry factor
determined by the structure of the nanocrystalline film, the Q-factor is specific to the molecule under consideration
and accounts for transition energies and strengths of all its excited
states f. The Q-factor cannot directly
be calculated; however, it can be obtained experimentally from an
independent absorption measurement of an amorphous film of randomly
orientated molecules. The obtained value of Δ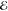 m is identified with Q⟨Wm⟩, where the angle
brackets represent the disorder average. The thus obtained Q-factor can then be used to calculate site-dependent energy
shifts for any given nanostructured environment.
m is identified with Q⟨Wm⟩, where the angle
brackets represent the disorder average. The thus obtained Q-factor can then be used to calculate site-dependent energy
shifts for any given nanostructured environment.
Before simulating PTCDI absorption
spectra, we provide an experimental estimate of the expected magnitude
of the level shift. To this end, we measured absorption spectra placing
the molecules in various nonpolar environments (see Figure 1). Since PTCDI is not well soluble, we perform the
experiment with the related molecule N,N′-bis(1-hexylheptyl)perylene-3,4:9,10-bis(dicarboximide) whose
UV/vis spectrum perfectly coincides with that of PTCDI, as the alkyl
substituents do not affect the π-electron system. The shift
of the transition energy 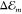 with respect to the gas phase
excitation energy Egp is related to the
solvent’s refractive index n at optical wavelengths
via
with respect to the gas phase
excitation energy Egp is related to the
solvent’s refractive index n at optical wavelengths
via 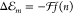 with f(n) = (n2 – 1) /(2n2 + 1).14,15 The expression originates from the solvation energy of a point dipole
in a spherical cavity surrounded by a homogeneous continuum.15 We will refer to this relation as the continuum
solvent approximation (CSA). A fit of the measured S0,ν=0 → S1,ν=0 transition energy as a function
of the solvent’s refractive index (cf. inset of Figure 1) yields
with f(n) = (n2 – 1) /(2n2 + 1).14,15 The expression originates from the solvation energy of a point dipole
in a spherical cavity surrounded by a homogeneous continuum.15 We will refer to this relation as the continuum
solvent approximation (CSA). A fit of the measured S0,ν=0 → S1,ν=0 transition energy as a function
of the solvent’s refractive index (cf. inset of Figure 1) yields 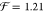 eV and Egp = 2.64 eV.
For a thin polycrystalline PTCDI film, a refractive index nPTCDI = 1.96 is reported.16,17 Application of CSA predicts a rather substantial level shift of
eV and Egp = 2.64 eV.
For a thin polycrystalline PTCDI film, a refractive index nPTCDI = 1.96 is reported.16,17 Application of CSA predicts a rather substantial level shift of 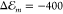 meV with respect to Egp which is in
the range of the observed spectral changes (cf. Figure 1).
meV with respect to Egp which is in
the range of the observed spectral changes (cf. Figure 1).
Experimental
The optical constants of PTCDI are measured in situ during deposition on KBr substrates by differential reflectance spectroscopy (DRS) in a thickness range between 0.1 and 5 monolayers (ML) using a home-built setup comprising a tungsten lamp and an Ocean Optics HR 4000 spectrometer connected to the deposition chamber via optical fibers. For thin films deposited on transparent substrates the DRS signal under normal incidence is proportional to the imaginary part of the films dielectric function Im(ϵ):18 DRS = ΔR/R ≈ −{8πd/λ}{Im(ϵ)/(1 – ϵKBr)}.
Information on the thin film morphology is obtained by atomic force microscopy performed in situ with an Omicron UHV AFM/STM controlled by a Matrix system. PTCDI films deposited on KBr(100) reveal steps with heights of 7 Å at sub-ML coverage (see Figure 2), indicating that the molecules do not lie flat on the surface but are tilted like in the bulk crystal.11 The arrangement of the molecules in these planes as obtained by MD simulations is depicted in Figure 2. Increasing the coverage, the thin film morphology is characterized by formation of small, slightly elongated crystallites with no preferred single in plane orientation (see Figure 2).
Figure 2.
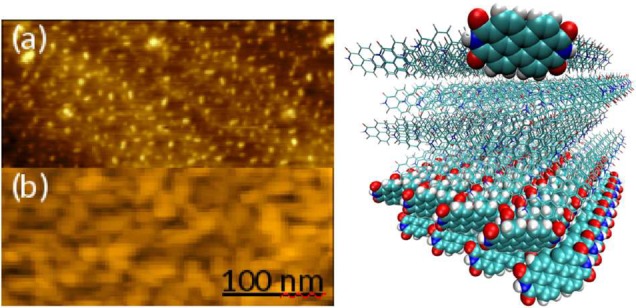
Left panel: AFM images of PTCDI deposited on KBr(100). The film thicknesses are (a) 0.2 ML and (b) 4.9 ML. The height scales are 1.5 and 6 nm, respectively. Right panel: Cross section of PTCDI monolayers. The PTCDI structure was obtained from a 1 ns MD simulation of a cube of 4.5 nm edge length, starting from the structure according to crystallographic data.11 Highlighted are one PTCDI molecule at the crystallite surface and the first two monolayers (shown via space-filling models). In a single monolayer there is one molecule per unit cell; the PTCDI unit cell of the bulk crystal containing two molecules11 is observed when at least two monolayers are present. All figures were produced with VMD.12
In order to calculate the Wm and the excitonic coupling matrix elements 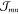 , a precise
knowledge of the morphology (size distribution and orientation of
grains) and arrangement of PTCDI molecules in nanocrystalline domains
is required. However, the experimental determination of the exact
structure of the present PTCDI film is beyond the scope of this work.
Instead, we performed extensive MD simulations of a set of 216 PTCDI
molecules forming periodic cubic crystallites of maximal edge length
of 4.5 nm in order to obtain possible PTCDI crystal configurations
(see Supporting Information part B).
, a precise
knowledge of the morphology (size distribution and orientation of
grains) and arrangement of PTCDI molecules in nanocrystalline domains
is required. However, the experimental determination of the exact
structure of the present PTCDI film is beyond the scope of this work.
Instead, we performed extensive MD simulations of a set of 216 PTCDI
molecules forming periodic cubic crystallites of maximal edge length
of 4.5 nm in order to obtain possible PTCDI crystal configurations
(see Supporting Information part B).
Results
We now apply the new methodology to explain the optical spectra of nanocrystalline PTCDI films. It is important to note that both the resonant excitonic coupling (iii) (which causes a mixing of states) and the nonresonant interactions (iv) (which are responsible for the level shifts) contribute to the observed changes in the spectrum when going from the isolated molecule to the polycrystalline thin film. It was shown in ref (7) that charge transfer states coupling to Frenkel exciton states (v) change the absorption curve only slightly for PTCDA crystals. Such a small effect was reproduced in our own calculations using the parameters given in ref (7). For the sake of clarity we will neglect charge transfer states in this work. Also, Coulomb coupling of static charge distributions (i) is found to be small (<5 meV) and thus inductive polarization shifts (ii) have been neglected (Supporting Information parts A and C).
Thus, for the derivation of absorption spectra,
three more factors have to be accounted for, that is, the computation
of the resonant excitonic coupling 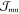 , the screening of
, the screening of 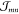 , and the vibronic
coupling from an MD generated molecular conformation. The screened
, and the vibronic
coupling from an MD generated molecular conformation. The screened 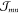 have been computed
via transition partial charges and respective screening factors9 as is explained in detail in Supporting Information part D. With the MD derived geometry,
we obtain maximal values of
have been computed
via transition partial charges and respective screening factors9 as is explained in detail in Supporting Information part D. With the MD derived geometry,
we obtain maximal values of 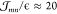 meV. As
resonant excitonic coupling is rather weak in PTCDI nanocrystallites,
site-dependent level shifts must thus be mainly responsible for the
large observed spectral changes (see Figure 1).
meV. As
resonant excitonic coupling is rather weak in PTCDI nanocrystallites,
site-dependent level shifts must thus be mainly responsible for the
large observed spectral changes (see Figure 1).
To account for vibronic coupling, only a single vibrational progression per molecule (Huang–Rhys factor of 0.62) is taken into account. This has been found sufficient for modeling the PTCDI monomer spectrum.19 The single particle approximation20 is used to derive an electron–vibrational Hamilton matrix where a single vibronically (labeled by ν) excited molecule couples to surrounding molecules in the vibrational ground state.
Exciton states are constructed as Φα= ∑m,νcα(m,ν)χmeνϕm, where χmeν is the total vibrational wave function with vibrational excitations in the excited state of molecule m and the cα(m,ν) denote the respective expansion coefficients. The absorption line shape follows as the sum over all exciton levels α with transition energies Eα weighted by the respective oscillator strengths |∑m,νcα(m,ν)dm|2. Finally, Gaussian broadening of typically 0.15 eV (full width at half-maximum) is introduced for all transitions. The value is deduced from the experimental absorption line width for 0.1 ML film coverage (see Figure 3).
Figure 3.
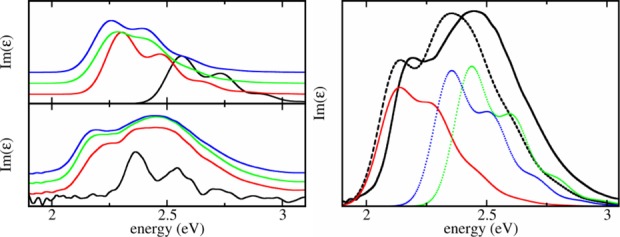
Left: Computed absorption line shapes assuming perfect two-dimensional films (upper panel) and measured (lower panel) absorption line shapes of different thicknesses of a PTCDI film (see text for details). Red: 1 ML. Green: 3 ML. Blue: 5 ML (4.9 ML for the experiment). The calculated black line shows the calculation for a single PTCDI molecule on the surface, according to the geometry of Figure 2. The measured black line shows the spectrum for 0.1 ML coverage, where the molecules are assumed to lay flat on the KBr surface. Right: A partly inhomogeneous film is assumed. Full black line: experimental spectrum for a 4.9 ML film of PTCDI on KBr. Dashed black line: calculated spectrum of a nanocrystalline film of nominally 5 ML thickness. For details on the structure, see text. Red line: calculated spectrum of a crystalline film composed of 5 ML arranged as in Figure 2. Dotted lines: calculated spectra for two types of thin films consisting of regular arranged small crystallites each containing 12 molecules; see Supporting Information part E for details.
The
calculations have been done considering the conformation shown in
Figure 2. The spectra calculated for the first
one, three, and five monolayers (MLs) of the cube, assuming a perfect
crystalline film, are depicted in Figure 3.
We checked that the exciton spectrum does not further change when
increasing the size of the system in directions parallel to the KBr
surface. The dispersive energy shifts have been computed assuming
periodic boundary conditions in the directions parallel to the KBr
surface. Setting the theoretical shift 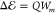 equal to the experimentally obtained
equal to the experimentally obtained 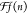 , the Q-factor
was calculated for the 5 ML thick, perfect film as Q = 3.2 eV Å2.
, the Q-factor
was calculated for the 5 ML thick, perfect film as Q = 3.2 eV Å2.
The dispersive molecular energy shifts due to interaction with the KBr surface have been approximated as follows. For a single molecule on a large PTCDI block the dispersive shift may be calculated as described above and interpreted as interaction between a single PTCDI molecule and a homogeneous dielectric which is proportional to the factor f(n). In this way, the interaction with the KBr surface can be treated. Since a single PTCDI on a PTCDI bulk is shifted by 0.1 eV, and the refractive indices of PTCDI and KBr are nPTCDI = 1.96 16 and nKBr = 1.55, respectively, we estimate an energy shift of ΔEPTCDI(KBr) = 0.075 eV for a PTCDI molecule on KBr. Since the molecules in the second ML (7 Å apart from the surface) show only a small dispersive energy shift due to interaction with the PTCDI bulk, we neglect respective energy shifts of all PTCDI molecules that are not directly located on the surface.
Calculated and experimental Im(ϵ) spectra are compared in Figure 3 for different PTCDI coverages. Both the calculated and experimental 1 ML spectra are red-shifted compared to the 0.1 ML spectra. This red-shift increases with increasing coverage. The trend is clearly seen considering the narrow feature in the experimental spectra. As already noted above, the calculations indicate that this shift is due to dispersion interaction. The large difference between experimental and calculated spectrum at 0.1 ML coverage is due to the fact that in the calculations we assume tilted molecules according to Figure 2. In the actual 0.1 ML film, the molecules most likely lay flat on the surface and receive thus a stronger red-shift.
The experimental thin film spectra display, besides the just discussed narrow feature, a broad band in the energy range between 2.3 and 2.8 eV which is absent in the calculated spectra where instead a vibrational progression similar to the monomer spectrum is observed. We conclude that the measured films consist not only of perfectly grown layers.
Instead, the line shape suggests that the spectrum is a superposition of spectra of a perfectly grown crystalline phase (feature at ∼2.1 eV) and a more loosely packed, rather disordered phase (broad band between 2.3 and 2.8 eV). The spectral weight of the two contributions implies that the proportion of well ordered and disordered phases is about the same.
To model the experimental spectra, an additional disordered
phase is considered consisting of small crystallites (12 molecules)
with a packing density reduced by 50% (see Supporting
Information part E for structural details). The Q-parameter is recalculated taking into account the size distribution
of the crystallites in the film. Because of the fact that the average
over the Wm is smaller
in a disordered film, setting Q⟨Wm⟩m equal to the experimentally obtained 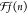 yields a larger Q = 4.26 eV Å2.
Because of the larger Q-value, the line shape for
the perfect crystalline phase in Figure 3 (right
panel) is red-shifted with respect to that in Figure 3 (left panel) where a perfect crystal was assumed. Finally,
to simulate the experimental absorption spectrum, the average is taken
over a mixture of ordered and disordered phases (Figure 3). Agreement with the experimental spectrum is obtained when
assuming that the PTCDI film consists of 53% of the orderd phase.
The small red-shift of the calculated curve by about 0.05 eV may be
due to errors when deriving the parameters
yields a larger Q = 4.26 eV Å2.
Because of the larger Q-value, the line shape for
the perfect crystalline phase in Figure 3 (right
panel) is red-shifted with respect to that in Figure 3 (left panel) where a perfect crystal was assumed. Finally,
to simulate the experimental absorption spectrum, the average is taken
over a mixture of ordered and disordered phases (Figure 3). Agreement with the experimental spectrum is obtained when
assuming that the PTCDI film consists of 53% of the orderd phase.
The small red-shift of the calculated curve by about 0.05 eV may be
due to errors when deriving the parameters 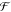 and Egp.
and Egp.
The only free “parameter” used in the calculations is the structure of the disordered film (its ratio with respect to the ordered phase is fixed by the integral over the line shapes). A reduction of the packing density by about 30% results in a blue-shift of at least 0.2 eV with respect to the spectrum of a perfect crystalline film. As obviously many different configurations contribute to the experimental spectrum in the range between 2.3 and 2.8 eV, the vibronic progression is smeared out leading to a broad and unstructured absorption band.
Conclusions
In summary, we introduced a new approach for calculating the optical absorption spectra of organic polycrystalline thin films. In particular, a novel relation for molecular excitation energy shifts due to dispersion effects of the environment has been derived. The obtained formulas allow for the determination of site-dependent level shifts of a molecule in a given nanoscale environment. On that basis, we were able to explain a distinct double structure of the S0 → S1 transition in the absorption spectrum of PDCTI films by the coexistence of two molecular phases. Large crystals where most molecules are located in the interior give rise to the component on the low-energy side, while the blue-shifted broad unstructured feature is due to a phase of loosely packed small aggregates experiencing a weaker gas-to-crystal shift, as most molecules reside at or close to the surface. We note that our results explain the absorption features as disorder induced dispersive effects due to nonresonant excitonic coupling. In previous works these features are explained by strong resonant excitonic coupling that transfers oscillator strength to the vibrational satellites (see refs (21) and (22)). While a fully quantitative analysis of the film morphology is beyond the scope of this work, our findings emphasize that an explicit consideration of the local energy structure is mandatory to understand the optical properties of molecules in nanoscaled solid-state systems.
Acknowledgments
Financial support by the Deutsche Forschungsgemeinschaft through Grant Sfb 951 and through Project ME 4215/2-1 (J.M.) and by the Austrian Science Fund (FWF), Grant P24774-N27 (T.R.), is gratefully acknowledged. We are indebted to Frank Müh (Johannes Kepler Universität Linz) and Stefan Kowarik (Humboldt-Universität zu Berlin) for fruitful discussions.
Supporting Information Available
Electronic structure calculations in part A, MD simulations in part B, comments on the neglect of inductive polarization shifts in part C, computation of excitonic coupling using partial transition charges in part D, and description of our model of the small aggregates in more detail in part E. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Author Status
∥ The authors mourn the loss of Fritz Henneberger who passed away on February 1, 2015.
Supplementary Material
References
- Kamat P. V. Meeting the Clean Energy Demand: Nanostructure Architectures for Solar Energy Conversion. J. Phys. Chem. C 2007, 111, 2834–2860. [Google Scholar]
- Imahori H.; Umeyama T. Donor-Acceptor Nanoarchitecture on Semiconducting Electrodes for Solar Energy Conversion. J. Phys. Chem. C 2009, 113, 9029–9039. [Google Scholar]
- Peumans P.; Uchida S.; Forrest S. R. Efficient Bulk Heterojunction Photovoltaic Cells Using Small-Molecular-Weight Organic Thin Films. Nature 2003, 425, 158–162. [DOI] [PubMed] [Google Scholar]
- Meyer J.; Hamwi S.; Kröger M.; Kowalsky W.; Riedl T.; Kahn A. Transition Metal Oxides for Organic Electronics: Energetics, Device Physics and Applications. Adv. Mater. 2012, 24, 5408–5427. [DOI] [PubMed] [Google Scholar]
- Koch N. Organic Electronic Devices and Their Functional Interfaces. ChemPhysChem 2007, 8, 1438–1455. [DOI] [PubMed] [Google Scholar]
- Kirchartz T.; Taretto K.; Rau U. Efficiency Limits of Organic Bulk Heterojunction Solar Cells. J. Phys. Chem. C 2009, 113, 17958–17966. [Google Scholar]
- Gisslén L.; Scholz R. Crystallochromy of Perylene Pigments: Interference between Frenkel Excitons and Charge-Transfer States. Phys. Rev. B 2009, 80, 115309. [Google Scholar]
- Madjet M. E.-A.; Abdurahman A.; Renger T. Intermolecular Coulomb Couplings from ab Initio Electrostatic Potentials: Application to Optical Transitions of Strongly Coupled Pigments in Photosynthetic Antennae and Reaction Centers. J. Phys. Chem. B 2006, 110, 17268–17281. [DOI] [PubMed] [Google Scholar]
- Megow J.; Renger T.; May V. Mixed Quantum-Classical Description of Excitation Energy Transfer in Supramolecular Complexes: Screening of the Excitonic Coupling. ChemPhysChem 2014, 15, 478–485. [DOI] [PubMed] [Google Scholar]
- Zoubi H.; Eisfeld A.; Wüster S. van der Waals-Stabilized Rydberg Aggregates. Phys. Rev. A 2014, 89, 053426. [Google Scholar]
- Klebe G.; Graser F.; Hädicke E.; Berndt J. Crystallochromy as a Solid-State Effect: Correlation of Molecular Conformation Crystal Packing and Colour in Perylene-3,4:9,10-Bis(dicarboximide) Pigments. Acta Crystallog., Sect. B 1989, 45, 69–77. [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD—Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Salam A.Molecular Quantum Electrodynamics: Long-Range Intermolecular Interactions; Wiley: Hoboken, NJ, 2010. [Google Scholar]
- Renger T.; Grundkötter B.; Madjet M. E.-A.; Müh F. Theory of Solvatochromic Shifts in Nonpolar Solvents Reveals a New Spectroscopic Rule. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 13235–13240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayliss N. S. The Effect of the Electrostatic Polarization of the Solvent on the Electronic Absorption Spectra in Solution. J. Chem. Phys. 1950, 18, 292–296. [Google Scholar]
- El-Nahhas M. M.; Abdel-Khalek H.; Salem E. Structural and Optical Properties of Nanocrystalline 3,4,9,10-Perylene-tetracarboxylic-diimide Thin Film. Adv. Condens. Matter Phys. 2012, 2012, 698934. [Google Scholar]
- We assume that the crystallite distribution in ref (16) for which the refractive index nPTCDI = 1.96 was deduced is similar to our film, since the two absorption spectra are nearly similar.
- McIntyre J. D. E.; Aspnes D. E. Differential Reflection Spectroscopy of Very Thin Surface Films. Surf. Sci. 1971, 24, 417–434. [Google Scholar]
- Scholz R.; Schreiber M. Linear Optical Properties of Perylene-Based Chromophores. Chem. Phys. 2006, 325, 9–21. [Google Scholar]
- Spano F. C. Absorption and Emission in Oligo-phenylene Vinylene Nanoaggregates: The Role of Disorder and Structural Defects. J. Chem. Phys. 2002, 116, 5877–5891. [Google Scholar]
- Vragović I.; Scholz R.; Schreiber M. Model Calculation of the Optical Properties of 3,4,9,10-Perylene-tetracarboxylic-dianhydride (PTCDA) Thin Films. EPL 2002, 57, 288. [Google Scholar]
- Our calculations of the screened excitonic couplings gave values that were too small to allow for an effective transfer of oscillator strength (see right panel of Figure 3).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.