

Abstract
The availability of structures and linked bioactivity data in databases is powerfully enabling for drug discovery and chemical biology. However, we now review some confounding issues with the divergent expansions of public and commercial sources of chemical structures. These are associated with not only expanding patent extraction but also increasingly large vendor collections amassed via different selection criteria between SciFinder from Chemical Abstracts Service (CAS) and major public sources such as PubChem, ChemSpider, UniChem, and others. These increasingly massive collections may include both real and virtual compounds, as well as so-called prophetic compounds from patents. We address a range of issues raised by the challenges faced resolving the NIH probe compounds. In addition we highlight the confounding of prior-art searching by virtual compounds that could impact the composition of matter patentability of a new medicinal chemistry lead. Finally, we propose some potential solutions.
Chemistry and Bioactivity Data: From Famine to Feast to Overload
It is hard to imagine now that in the early 2000s there was a dearth of chemistry and bioactivity data that were publicly accessible. Yet in the decade since the appearance of the large publically accessible PubChem1 and ChEBI databases2 we are arguably approaching an era of drug-discovery-related data overload as data generation, with high-throughput methods, is used to populate increasingly large databases.3 Having just passed 53 million compounds, PubChem4 has undoubtedly made the largest aggregated contribution to public or open chemistry and biology data, collating thousands of assay results against cells or biological targets for 2 million compounds. This will soon be complemented not only by the European Lead Factory,5 which will focus on high throughput screening (HTS) and data generation, but also by a Knowledge Management Center that will capture data from the National Institutes of Health (NIH) “Illuminating the Druggable Genome” (IDG) program.6 When we consider the availability of additional large chemical or biology related databases such as ChemSpider,7 ChEMBL,8 UniChem,9 BindingDB,10 and BARD,11 as well as the emergence of Google as a de facto merged chemistry source,12 two aspects come into focus. The first is that the era of the aforementioned “multistop datashops” (and the essential big data integration challenges this presents) is here to stay. The second is that public and commercial chemistry and bioactivity data sources will increasingly diverge. Users are thus faced with the necessity to compare content between the former but also to guess the proportion of unique structures in the latter (since, by definition, the latter do not openly benchmark themselves against each other or the former). Consequently, it is our view that commercial chemistry databases like SciFinder13 from Chemical Abstracts Service (CAS) will be unable to keep pace with the totality of public chemistry data. It should however be noted that they ensure high curation quality14 of their largely manually extracted data, with the assistance of software tools. The public domain resources, however, beyond their submission filtration pipelines, are dependent on the quality of depositing sources (analogous to the case with GenBank for primary sequence data). Multiple reviews of public domain data sources indicate that, in the main, data quality issues arise that are independent of the submitter.15 Logically, some kind of comparative database quality metrics need to be generated and reported by a completely independent party. This would need a sampling strategy agreed by all those sources (commercial and public) prepared to participate in such a bench marking study.
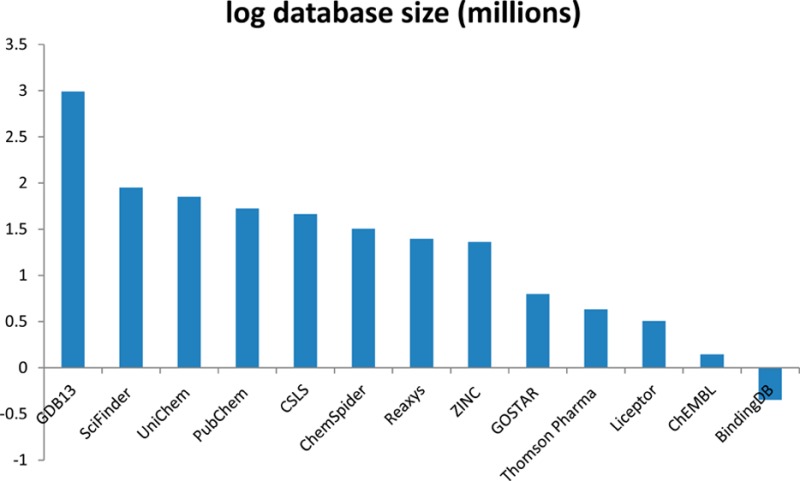
Our view that commercial chemistry databases will be unable to keep pace is especially validated as patent data continue to become openly available. For example IBM has deposited 2.6 million extracted patent compounds, SCRIPDB 6.6 million, and SureChem 9.4 million into PubChem. The European Bioinformatics Institute (EBI) recently acquired the SureChem operation and will expand the extraction pipeline to populate SureCHEMBL at EBI.16 Efforts by a number of groups to extract chemical structures and content from patents and uncurated scientific papers17 open up even more automated data flows whose scale precludes human verification.18 In addition there are close to a billion virtual molecules in databases like the chemical universe database, GDB,19 and at the other end of the scale are relatively small repositories of real molecules that may never appear in the pages of a journal.20 A basic survey of some of the larger chemistry and biology data resources we are aware of is shown in Table 1 and highlights some of the differences in the content (vendor compounds, virtual compounds, etc.). It is important to put the scale of these big databases into context by considering that we are likely far from having a database of all possible chemistry, since a single simple empirical formula could potentially result in hundreds of millions of molecules with the same atomic composition.21
Table 1. Summary Statistics for the Public and Commercial Chemistry Databases above or near Half a Million Structures (at the Time of Writing), Most of Which Include Linkages to Bioactivity and Biological Dataa.
| name | total (million) | URL | notes |
|---|---|---|---|
| GDB13 | 977 | http://www.gdb.unibe.ch/gdb/home.html | Virtual compounds, no bioactivity data |
| SciFinder | 89 | http://www.cas.org/products/scifinder | Includes 28 million vendor compounds |
| UniChem | 71 | https://www.ebi.ac.uk/unichem/ | Includes 15 million SureChEMBL from patents |
| PubChem | 53 | https://pubchem.ncbi.nlm.nih.gov/ | Includes 42 million vendor compounds and 15 million from patents |
| CSLS | 46 | http://cactus.nci.nih.gov/cgi-bin/lookup/search | Update status unclear |
| ChemSpider | 32 | http://www.chemspider.com/ | Includes 12 million vendor compounds |
| Reaxys | 25 | http://www.elsevier.com/online-tools/reaxys | 5.1 million medicinal chemistry data |
| ZINC | 23 | http://zinc.docking.org/ | All vendor compounds, 8.1 million in PubChem |
| GOSTAR | 6.3 | https://gostardb.com/gostar/ | Activity linked |
| Thomson Pharma | 4.3 | http://www.thomson-pharma.com/ | Counted inside PubChem |
| Liceptor | 3.2 | http://www.evolvus.com/products/databases/liceptordatabase.html | |
| ChEMBL | 1.4 | https://www.ebi.ac.uk/chembl/ | 0.94 million inside PubChem |
| BindingDB | 0.45 | http://www.bindingdb.org/bind/index.jsp |
Note that apart from the three sources that have update cycles within PubChem (Thomson Pharma, ChEMBL, and BindingDB) all the others are likely to have at least a proportion of unique content (e.g., extractions from different journal articles).
A previous study has compared the content of several public and commercial databases.22 This showed that the commercial databases captured a significant proportion of unique content and suggested they were complementary. However, even with the massive amount of public and commercial chemistry and bioactivity data now available in the various databases, finding the necessary information effectively remains difficult. As an example there are challenges in using molecular structures alone to search for and ascertain whether there is already biology or screening data associated with them, whether they are desirable as chemical probes or lead compounds, and for assessment of novelty for patent claims.23 Differentiating between those molecules (the “usual suspects”) known to have liability or reactivity issues,24 approved drugs,25 useful probes,26 prophetic compounds,27 text abstracted compounds, and nominal probes with no provenance links in database records, is certainly now more complicated (Figure 1). This difficulty can be seen for even small defined sets of compounds, such as the National Center for Advancing Translational Sciences (NCATS) molecules for repurposing28 or the NIH Molecular Libraries Program (MLP) probes.29 The NIH MLP probes were initially the subject of a crowdsourcing analysis in which 11 scientists scored an initial set of 64 probes based on their own criteria of being acceptable or not.26 This work has recently been greatly extended to 322 NIH MLP probes (at the time of this study) using the selection criteria of a single medicinal chemist.30
Figure 1.
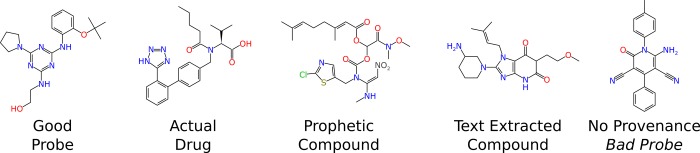
The “usual suspects” lineup, representing molecules of different classes from public and commercial databases, illustrating the difficulty of selecting desirable ones. From left to right, the documented probe is ML010 (CID 17757274), the drug is valsartan (CID 60846), a prophetic compound is from CAS 1164083-19-5 from WO 2001056358 (not in PubChem or ChemSpider),42 a text extracted compound is from US2012004098217 (CID 57498937), and one of the probes with incomplete data linkage is ML160 (CID 824820).
The data needs for medicinal chemistry differ from those of biology. While both medicinal chemists and biologists seek high quality biology data to support their target choices, medicinal chemists also require information on freedom to operate by searching the literature for compounds identical to, similar to, or that are substructures of their leads, a search process we call “medicinal chemistry due diligence”. The NIH MLP probes are stored on PubChem and describe one or several probes with detailed rich biology but lack sufficient information for the medicinal chemist. We explored what a medicinal chemist might do in the early stages of medicinal chemistry due diligence using, as an example, the NIH MLP probes. Currently the most widely used and complete source of literature relevant to a putative lead resides in the CAS databases, very often accessed through the SciFinder software. We uncovered significant obstacles that a medicinal chemist would face trying to translate public sector probe discovery into a typical medicinal chemistry due diligence search.
Attempting to track the status and provenance of this set of NIH MLP probes30 (which we have clustered to simplify the visualization, Figure 2) exemplifies the complexity of linking current biology and chemistry data30 and led directly to this review. For this reason we have used this set to discern trends that are reflected in the wider database “ecosystem”, which will now be described as examples. To many that are not seasoned explorers of these databases, we hope that this will be enlightening prior to your future quest to find information that is relevant. To those readers that have encountered these same issues, we hope that this increased attention will bring awareness to those involved in curating and funding such databases and that solutions will follow in due course.
Figure 2.

Chemical structures for 322 NIH MLP probes (http://molsync.com/demo/probes.php) have been clustered into 44 groups for visualization purposes, using ECFP_6 fingerprints58 and using a Tanimoto similarity threshold of >0.11 for cluster membership. The threshold was chosen empirically in order to show a representative selection of the kinds of molecules found within the set of probes. For each cluster, a representative molecule is shown (selected by picking the structure within the cluster with the highest average similarity to other structures in the same cluster). The clusters are decorated with semicircles which are colored blue for compounds that were considered high confidence based on our medicinal chemistry due diligence analysis. This analysis suggests that there is not an obvious correlation between structural composition and whether they pass the medicinal chemist’s logic.30 Red is for those that are not. Circle area is proportional to cluster size, and singletons are represented as a dot.
Example 1. Complexities in Finding the NIH MLP Probes in PubChem
With just a few exceptions as we shall describe, NIH MLP probe compounds can be identified from NIH’s PubChem Web-based book29 summarizing 5 years of probe discovery efforts. A probe compound is defined as essentially an excellent lead compound: very high binding affinity and ideally a well understood binding mode, high selectivity, good solubility, and low toxicity.26 MLP probes are identified by a Molecular Libraries (ML) number and by a PubChem compound identification (CID) number that can be readily found by searching the NIH probe book.29 Compared to many peer reviewed published formats, the NIH probe book is exemplary in being concise, but also information rich in both chemistry and biology. Subheadings across probe reports illuminate the importance and utility of each compound. Extensive out-linking (provided these do not decay) also adds to the user-friendliness of the reports. However, while some reports cover the medicinal chemistry aspects well, others are only designated by the PubChem substance identification (SID) number, which requires added effort to find the salient chemistry details. In this case, the probe is primarily characterized by a biological activity and SID link. Also, it was found that searching certain ML numbers listed in the book would not retrieve a CID in PubChem. In addition, a detailed Excel spreadsheet summary (WebTable 121012.xlsx) found on the NIH MLP Web site31 contained data on only about two-thirds of the probes.32 It appears that the concise organization found in the more recent probe reports may have been lacking at the outset. A few compounds were also identified that were originally described as NIH MLP probes but for which there is no probe report. We have recently compiled and shared the available information on the 322 NIH MLP probes we were able to resolve in an easily searchable collection available on Collaborative Drug Discovery (CDD)’s public database33 as a free resource for the community34 as well as elsewhere.35
Example 2. Identifier and Structure Searches in SciFinder Reveals an Extreme Disclosure
As we move beyond the NIH MLP probes to other databases to find more data on these or other compounds, we encounter further issues. The process of converting CID identifiers to CAS registry numbers can be used to obtain a summary of the number of literature references in SciFinder, and this identifier conversion is essential to medicinal chemistry due diligence. For example, when a high throughput screening (HTS) hit becomes of potential value in lead optimization, it is essential to conduct exact, substructure, and similarity searches on it.
Literature descriptions of structure–function relationships are of value even if the prior literature report on the chemistry is in a very different field of biology to the current interest. There is a fundamental explanation for this observation, as diverse targets are under evolutionary pressure to interact with common signaling ligands.36 In this sense ligand chemistry (at least for orthosteric ligands) is more conserved than target structure. This finding, coupled with the known conservation of biology target motifs,37 is consistent with the knowledge that similar chemistry motifs tend to recur across varied biology. Computationally, this observation is also found in the RECAP technology38 in which known drugs are fragmented and chemistry motifs are reconnected in new patterns to give new and often unexpected biological activities. These connected observations are also relevant to the behavior of medicinal chemists, who have been characterized, we think incorrectly, as conservative because they often tend to use and reuse the same chemical motifs in the compounds they make.39 Rather, we think this medicinal chemistry behavior is better characterized as pragmatic as professional survival depends on creating compounds to meet project goals, and the use and reuse of chemical motifs previously shown to have useful biological activity are a proven successful strategy.
SciFinder’s use of SMILES input rather than InChI or InChIKey preserves chemistry structure tautomeric information, which could be important for medicinal chemistry analysis and patent law, where tautomer structure can be critical. It is interesting that the SciFinder choice is consonant with the same selection for the Journal of Medicinal Chemistry digital structure capture.40 Structure searches within SciFinder are subject to the well-known issues more broadly associated with chemistry structure drawing and include problems with stereochemical depiction, unclear double bond geometry, and unclear links between free base and salt forms.15,41 When SciFinder refuses a structure search because of stereo bond depiction problems, the structure can be edited to remove stereo information from offending bonds, and the correct structure must be deduced from the pattern of literature citations.
It should be noted that if the structure search within SciFinder fails to find a CAS registry number, the search can be repeated as a similarity search to ensure that the registry number was not missed because of a salt form. Once the CAS registry number is found, the total number of literature references with biological activity captured in SciFinder can be retrieved. It is at this point that any reference to the 2009 Goldfarb U.S. patent application on life extension in eukaryotic organisms (US 2009016354542) should be noted.
US2009016354542 contains a data table (Figure 16 in the patent ref (42)) on 499 compounds with PubChem substance IDs. However, SciFinder abstracts 6018 substances. How can this be? The patent includes the phrase, referring explicitly to (PubChem assay ID) 775, “the contents of which are herein incorporated in their entirety by reference”. This is full data disclosure taken to an extreme via subsummation of public HTS data into a patent by reference. While only 5796 substances from the HTS were referenced as “use” substances in SciFinder, 132 781 compounds were specified in the HTS (i.e., 32% of the entire Molecular Libraries screening collection, MLSMR). Thus, while this may be an exceptional patent abstraction example in SciFinder, it nonetheless illustrates how intellectual property (IP) due diligence searching can be confounded. Across the set of 322 NIH MLP probes, 72 intersect with the CIDs from AID 775, so a significant proportion will also intersect with the US20090163545 exemplifications. We were initially worried that a reference to this patent application was somehow an indicator for a flawed or promiscuous compound. We now believe the prevalence of references to this single patent application is an example of how complete data disclosure can lead to unexpected and potentially harmful consequences when performing IP due diligence.
Example 3. The Parallel Worlds of Commercial and Public Database Disclosure Do Not Completely Intersect
We expected that the chemical structures of all the NIH MLP probes would be abstracted by SciFinder. This proved not to be the case, raising the possibility of two parallel worlds of disclosure: the proprietary commercial database world of chemistry data abstracted by SciFinder and another data rich world of publically available and predominantly NIH funded chemistry and biology screening data, largely in Web format but not abstracted by SciFinder. Three CID examples are provided in Table 2, including one of the NIH MLP probes and two Web-only provenanced bioactive structures.
Table 2. CIDs from Selected Sources without Exact Structure Matches in SciFinder (November 2014).
| CID | source |
|---|---|
| CID 56593118 | ML226 probe inhibitor of lysophospholipase 1 [AID: 2202] |
| CID 46905036 | ML233 probe agonist of the APJ receptor [AID: 2580] |
| CID 53301938 | ML258 probe inhibitor of Bcl-B [AID: 720677] |
| CID 45100448 | ML179 probe inverse agonist of LRH-1 [AID: 504933] |
| CID 70789094 | ML353 probe modulator of mGlu5 [AID: 686927] |
| CID 71819646 | http://opensourcemalaria.org/, open source antimalarial active |
| CID 71819647 | http://opensourcemalaria.org/, open source antimalarial active |
| CID 77014274 | http://www.chemotion.net/, open chemistry publishing |
| CID 78243694 | http://www.chemotion.net/, open chemistry publishing |
If this trend were to continue, intellectual property due diligence would be rendered even more difficult, requiring searching of multiple parallel disclosure formats at the same time.23 Other intellectual property/legal due diligence issues may arise from the parallel worlds of public and commercial data. Much of the data input into public chemistry databases comes from deposition of massive numbers of compounds from chemical vendors (previously termed “vendor dilution effect” because only a minority of these compounds can be linked to bioactivity data43), many of them suffering from significant quality issues in structure representation as evidenced by our experiences. For example the ChemSpider7,44 database required processing millions of chemical compounds for deposition, some of which had quality issues that required removal. Such data quality issues continue to plague chemistry databases and require vigilance.15,41b From previous work with a chemistry compound vendor, we estimate that at least half of “commercially available” compounds have never been made but rather are compounds that suppliers think can be made and that are listed as available in an attempt to elicit customer interest. These are commonly known as “make-on-demand” (MOD) compounds and are segregated in databases such as the ZINC database.45 Most such compounds are identified by a chemical structure depiction and are annotated with some type of database identifier, but no other experimental data on the chemical depicted by the chemical structure drawing exist. On the basis of spot checks, about one-third of such low data value compounds found in PubChem do not appear in the CAS registry system.46 For low data value compounds, the lack of abstraction by CAS can be viewed in a positive light, since abstracting such compounds could dilute the value of those abstracted real compounds, which are associated with experimental data. SciFinder had previously initiated abstraction of data from the ChemSpider database and had deposited over 300 000 chemicals from the database into the registry,47 and this was discouraged by the hosts of ChemSpider because they had no way of distinguishing MOD compounds from synthesized and fully characterized chemicals. To our knowledge, CAS has not taken any ChemSpider data since the Royal Society of Chemistry (RSC) acquisition in 2008 (i.e., that is credited as such in SciFinder) and there has not been any agreement between RSC and CAS regarding ChemSpider data.
Example 4. Integration and Intersections of Databases and the Need for Bioassay Ontology Adoption
Understanding associations between chemical structures and biological assays is a further challenge, since there is essentially no standardization for describing the protocols for obtaining activity metrics (IC50, Ki, Kd, etc.) against a biological target, besides plain English text with scientific jargon. Because this form is intractable to software, it is impossible to determine whether two measurements of activity from different research groups are comparable, other than to have an expert read the full text for both descriptions. The use of a standard ontology, such as BioAssay Ontology,48 across such databases would be helpful. This would enable enhanced searching and comparison, allowing for the automated aggregation and organization of assays to do sophisticated structure–activity relationship analysis and identify artifacts. Despite the benefits to the community, it currently requires substantial time and expert ontology understanding to correctly annotate each bioassay, so it has not been widely adopted. Efforts are currently underway to design a hybrid manual/automated method for making it relatively fast and easy for scientists to add semantic annotations to their bioassay protocols, which could improve the current situation.49
This discussion leads us to ask whether compounds in databases without any experimental data and without any link to potential utility should be considered as prior art. This class of compounds is growing dramatically, especially in the public databases, and the utility is arguably markedly less than for prophetic compounds (defined in the Glossary) in patents, which may not be real compounds in an experimental sense but for which the relationship to experimentally tested compounds is at least clear. Such prophetic compounds have been abstracted in SciFinder since December 2007. As we have described earlier, the days when one could assume SciFinder had captured everything relevant to the entire global realm of bioactive chemistry are perhaps well passed. By definition, no quantitative assessment (such as the statistics of structure matching) across databases is possible without access to all of them, and to our knowledge this has not been undertaken to date. As the largest commercial source (Table 1) SciFinder contains organics, inorganics, organometallics, and “tabular inorganics”. Their reported (September 2014) total of 89 million substances would merge to a smaller collection of unique organic molecules if converted to InChiKeys followed by tautomer collapse (i.e., using just the 14-character connectivity layer). We can also estimate somewhere between 50 and 60 million InChiKeys are “in the wild”12 mainly via the Google indexing of PubChem and ChemSpider, but there could be other sources of unique structures (including virtual compounds as described earlier). The intersections and differentials between SciFinder, PubChem, and ChemSpider and other databases (Table 1) are, to date, unknown and require quantification. In the future, with SciFinder opening up an STN application programming interface (API) for pharmas,50 assessment of this overlap may become feasible. Other databases such as SureChEMBL may also overlap with PubChem (12.5 million compounds, of which 9.4 million are in PubChem). The ContentMine initiative51 extracting molecules from documents could also further emphasize that SciFinder is perhaps no longer the definitive site for chemistry prior art checking. As SciFinder is based primarily on abstraction of compounds from the chemical literature and patents, it should be noted that the distinction of the public compound databases to host data that may never be published means that these databases will also continue to deviate until the commercial databases determine how to extract quality data from the public platforms.
Conclusions
From our own observations, we have identified a number of barriers to performing medicinal chemistry due diligence that arise due to the lack of integration between public and private data repositories. Even obtaining structures and associated data from well-funded public efforts like the NIH MLP probes and the NCATS molecules for repurposing28 in PubChem or elsewhere is profoundly challenging. A medicinal chemist can hardly avoid being exposed to the debate calling for more data sharing and as much public exposure to primary data as possible. A rational response is enhanced by case studies of what can go wrong. For example, in our work on the NIH ML probes, we discovered a confounding case where the nominal subsummation of a public HTS screen into a patent application impacts over 20% of probes from a range of institutions. In addition, prophetic compounds in SciFinder and vendor molecules deposited in many public databases that include some proportion of probable MOD compounds complicate prior art designations. While we propose some more modest solutions for the highlighted issues, the one with the biggest potential impact would be if SciFinder generated and search-indexed the InChI identifiers (strings and keys), now effectively universally adopted by public chemical databases.52 This would need to be in addition to using SMILES which retain the tautomeric structure of value to medicinal chemists and patent lawyers alike (as described earlier).
The “multistop datashop” database challenge can be highlighted by the hypothetical novelty checking requirement for a new chemical structure proposal from a medicinal chemist or chemical biologist. This is equally important for someone in open source drug discovery who simply wants an answer to “what is out there that is similar” and who may even eschew IP on principle (e.g., their first response to a similarity match might be to make collaborative contact).53
Those who seek to stake an IP position need exactly the same answer but in the different context of prior art and freedom to operate, i.e., the competitive landscape in structure terms. The issue for both of them is that all of the big four databases (SciFinder, PubChem, ChemSpider, and UniChem) have at least some unique content via differential source selectivity (as defined by an InChI not in the other three). Ipso facto all four databases need to be searched (although currently UniChem can only be interrogated for exact matches). Add to this the many open source (online) lab notebooks on the Web, and the increasing implausibility of being able to check everything “out there” becomes clear.
Perhaps what is also needed is a shift toward more collaboration or openness in terms of availability of chemistry and biology data.53,54 At the very least there needs to be increased communication between the various databases that are both public and proprietary in order to ensure the gulf does not widen further. Additionally they need to address some of the issues raised here. This would help to resolve discrepancies we have highlighted and to make analyses on what data exist for compounds more streamlined. For example, while in review, an article by Antolin and Mestres described 178 MLP chemical probes55 that overlapped with our description of over 300 MLP chemical probes.30 We think a meeting or discussion should be convened with all interested database parties. It could very well be conducted at a future American Chemical Society National Meeting or elsewhere.
From previous public efforts to collate the data on melting point and solubility data, significant differences between different published studies56 have been described for the same compounds. Recent efforts mining patents have also shown differences in biological data for the same compounds, based on the method of dispensing used.57 These limited examples suggest there are benefits to making chemistry, biology screening, and other molecule related properties data accessible because it promotes new analyses and re-evaluation, which ultimately benefits science. We should note that despite our hopes that such a meshing of data is possible and would be of high value to the community, major hurdles exist to prevent this from happening in the short-term to middle-term future, as there is still simply too much commercial value to the hosts of the proprietary databases at present. We hope our experiences encourage the scientific community to develop creative solutions to enable a more comprehensive analysis of chemistry and related biological screening data. Clearly CAS and the other commercial vendors have to take notice and respond to the current rapidly evolving chemistry database situation; otherwise, their market may be rapidly eroded by these growing public efforts.
Acknowledgments
We dedicate this to the memory of our friend Dr. Jean-Claude Bradley, a pioneer of open-notebook science. We gratefully acknowledge various discussions with Dr. Richard Kidd (RSC). We also acknowledge the reviewers’ thoughtful feedback.
Glossary
Abbreviations Used
- CDD
Collaborative Drug Discovery
- CID
PubChem compound identifier
- MLP
Molecular Libraries Program
- NCATS
National Center for Advancing Translational Sciences
- NIH
National Institutes of Health
- HTS
high throughput screening
- RSC
Royal Society of Chemistry
Glossary
Glossary
- Compound identifier
CID is the permanent identifier for a unique chemical structure in PubChem, but the unique structure still can be a mixture of enantiomers or stereoisomers.
- Prior art
Composition of matter cannot be obtained in a patent application if the compound in question is prior art. Prior art results from disclosure of the structure of the compound as well as a method for its synthesis.
- Probe compound
An excellent lead compound: very high binding affinity and ideally with well understood binding mode, high selectivity, good solubility, and low toxicity.26
- Prophetic compound
All compounds annotated by IUPAC name for which there is no experimental (chemistry or biology) in the patent. These differ from the real compounds (which have at least some data) and from Markush structures which are derived from the usual conglomeration of generic structures and x, y, z, R1, R2, etc. In theory even Markush structures could count as prior art. In practice, patent examiners mostly ignore Markush in their novelty appraisal (>90% of U.S. patent applications eventually become issued U.S. patents). The very few patents that end up as disputes and that do have very close scrutiny end up very late in time in the federal district court system, many in Wilmington, DE.
- Substance identifier
SID identifies a depositor-supplied molecule (SID) and is assigned by PubChem to each unique external registry identification provided by a PubChem data depositor. The molecule structure may be unknown, for example, a natural product identified only by name or a compound identified only by an identifier
- Tabular inorganic
Multiple components registrations in CAS. The structure is unknown, it has a nonstoichiometric fractional composition or range of compositions, it is a 3D lattice structure, or a discrete structure does not exist.
Biographies
Christopher A. Lipinski learned his medicinal chemistry skills in a 32-year career at Pfizer in Groton, CT, where he retired in 2002 at the most senior scientific position. He is currently a Scientific Advisor to Melior Discovery a drug repurposing biotechnology and carries out his medicinal chemistry consulting. Dr. Lipinski served on the scientific advisory board for academic drug discovery efforts at the Center for Drug Discovery and Development at the K. U. Leuven, Belgium. He is a member of the American Chemical Society (ACS) and the American Association of Pharmaceutical Sciences (AAPS). He is the author of the “rule of five”, and the associated publication now has over 8000 citations and is the most highly cited paper in medicinal chemistry drug discovery.
Nadia K. Litterman is Collaborations Director at Collaborative Drug Discovery, Inc. Nadia received her Bachelors in Chemistry from Princeton University, NJ, and a Ph.D. in Neuroscience from Harvard University, MA. As a postdoctoral fellow at the Department of Stem Cell and Regenerative Biology screening lab of Dr. Lee Rubin at Harvard University, she identified a small molecule that regulates the survival of motor neuron protein to promote motor neuron survival in spinal muscular atrophy. She employed stem cells to investigate molecular mechanisms of neurodegenerative disease and was engaged in numerous collaborations in areas of chemical screening and data analysis software. Nadia enjoys writing grants and collaborating with other scientists in different areas.
Christopher Southan is a Curator at the IUPHAR Database and Guide to PHARMACOLOGY Web Portal Group at the University of Edinburgh (http://www.guidetopharmacology.org/) but works from Göteborg, Sweden. Prior to this he established TW2Informatics, where he consulted on patent informatics for SureChem (2011–2012) and as a contractor for AstraZeneca Knowledge Engineering (2009–2011). In 2008–2009 he coordinated the ELIXIR Database Provider Survey at the European Bioinformatics Institute after being Principle Scientist in AstraZeneca Molecular Sciences (2004–2007), preceded by senior bioinformatics positions at Oxford Glycosciences Gemini Genomics and SmithKline Beecham. His 61 PubMed entries encompass protein target assessments, bioactive chemistry databases, bioinformatics, and protein chemistry. He has a B.Sc. from University of Dundee, U.K., an M.Sc. from University of Reading, U.K., with a Ph.D from Ludwig-Maximillian University of Munich.
Antony J. Williams is the Vice President of Strategic Development and manager of the eScience cheminformatics team for the Royal Society of Chemistry. He is one of the original founders of ChemSpider. This platform has resulted in a number of derivative projects that underpin a number of international projects for delivering chemistry related data to the community. He is widely published with almost 200 publications and book chapters and is known as the ChemConnector in the social networks. He has worked on the quality of chemistry content on Wikipedia, is a recipient of the Jim Gray award for eScience from Microsoft, and is particularly focused at this time in helping scientists understand the power of the Web for encouraging crowdsourced participation and social networking in the sciences.
Alex M. Clark graduated from the University of Auckland, New Zealand, with a Ph.D. in Synthetic Organometallic Chemistry, then went on to work in computational chemistry. His chemistry background spans both the lab bench and development of software for a broad variety of 2D and 3D computer aided molecular design algorithms and user interfaces. He is the founder of Molecular Materials Informatics, Inc., which is dedicated to producing next-generation cheminformatics software for emerging platforms such as mobile devices and cloud computing environments.
Sean Ekins divides his time between being CSO at Collaborative Drug Discovery, his work with several rare disease foundations including as CSO at the Hereditary Neuropathy Foundation, and his clients at Collaborations in Chemistry. He is also President and CEO of Phoenix Nest, Inc. and Collaborations Pharmaceuticals, Inc. He graduated from the University of Aberdeen, U.K., receiving his M.Sc., Ph.D. and D.Sc. in Clinical Pharmacology. He has worked for Lilly, Pfizer, Concurrent (now Vitae) Pharmaceuticals, Inc., and GeneGo (now Thomson Reuters). He has authored or coauthored >220 peer reviewed papers and book chapters as well as edited four books for Wiley. Sean co-developed the mobile apps ODDT (Open Drug Discovery Teams), Green Solvents, and TB Mobile with Alex Clark. He is collabchem on Twitter.
The authors declare the following competing financial interest(s): N.K.L. is an employee and S.E. is a consultant of CDD Inc. C.A.L. is on the scientific advisory board of CDD Inc. A.J.W is an employee of the Royal Society of Chemistry, and A.M.C. is an employee of Molecular Materials Informatics.
References
- Wang Y.; Xiao J.; Suzek T. O.; Zhang J.; Wang J.; Bryant S. H. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, W623–W633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley D. Public molecules: small, but perfectly formed. Nat. Rev. Drug Discovery 2004, 3, 988–989. [DOI] [PubMed] [Google Scholar]
- Villoutreix B. O.; Lagorce D.; Labbe C. M.; Sperandio O.; Miteva M. A. One hundred thousand mouse clicks down the road: selected online resources supporting drug discovery collected over a decade. Drug Discovery Today 2013, 18, 1081–1089. [DOI] [PubMed] [Google Scholar]
- Li Q.; Cheng T.; Wang Y.; Bryant S. H. PubChem as a public resource for drug discovery. Drug Discovery Today 2010, 15, 1052–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Lead Factory. http://www.europeanleadfactory.eu/# (accessed November 19, 2014).
- Illuminating the Druggable Genome—Overview. http://commonfund.nih.gov/idg/overview (accessed November 19, 2014).
- a Pence H. E.; Williams A. J. Chemspider: an online chemical information resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar]; b Williams A. J. Internet-based tools for communication and collaboration in chemistry. Drug Discovery Today 2008, 13, 502–506. [DOI] [PubMed] [Google Scholar]
- Gaulton A.; Bellis L. J.; Bento A. P.; Chambers J.; Davies M.; Hersey A.; Light Y.; McGlinchey S.; Michalovich D.; Al-Lazikani B.; Overington J. P. Chembl: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers J.; Davies M.; Gaulton A.; Hersey A.; Velankar S.; Petryszak R.; Hastings J.; Bellis L.; McGlinchey S.; Overington J. P. UniChem: a unified chemical structure cross-referencing and identifier tracking system. J. Cheminf. 2013, 5, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.; Lin Y.; Wen X.; Jorissen R. N.; Gilson M. K. Bindingdb: a Web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza A.; Bittker J. A.; Lahr D. L.; Brudz S.; Chatwin S.; Oprea T. I.; Waller A.; Yang J. J.; Southall N.; Guha R.; Schurer S. C.; Vempati U. D.; Southern M. R.; Dawson E. S.; Clemons P. A.; Chung T. D. An overview of the challenges in designing, integrating, and delivering BARD: a public chemical-biology resource and query portal for multiple organizations, locations, and disciplines. J. Biomol. Screening 2014, 19, 614–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C. Inchi in the wild: an assessment of inchikey searching in google. J. Cheminf. 2013, 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A. B. SciFinder scholar 2006: an empirical analysis of research topic query processing. J. Chem. Inf. Model. 2006, 46, 767–774. [DOI] [PubMed] [Google Scholar]
- SciFinder. http://www.cas.org/products/scifinder (accessed November 19, 2014).
- Williams A. J.; Ekins S.; Tkachenko V. Towards a gold standard: regarding quality in public domain chemistry databases and approaches to improving the situation. Drug Discovery Today 2012, 17, 685–701. [DOI] [PubMed] [Google Scholar]
- Digital Science Transfers SureChem Patent Chemistry Data to EMBL-EBI. http://www.ebi.ac.uk/about/news/press-releases/SureChEMBL (accessed November 19, 2014).
- Southan C.; Stracz A. Extracting and connecting chemical structures from text sources using chemicalize.org. J. Cheminf. 2013, 5, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Swain M. Chemicalize.org. J. Chem. Inf. Model. 2012, 52, 613–615. [Google Scholar]; b RSC Prospect. http://www.rsc.org/Publishing/Journals/ProjectProspect/FAQ.asp (accessed November 19, 2014).; c Van Noorden R. Elsevier opens its papers to text-mining. Nature 2014, 506, 17. [DOI] [PubMed] [Google Scholar]
- Ruddigkeit L.; van Deursen R.; Blum L. C.; Reymond J. L. Enumeration of 166 billion organic small molecules in the chemical universe database gdb-17. J. Chem. Inf. Model. 2012, 52, 2864–2875. [DOI] [PubMed] [Google Scholar]
- Stefan Bräse Group . Chemotion Repository; Karlsruhe Institute of Technology: Karlsruhe, Germany; http://www.chemotion.net/.
- Williams A. J. How Many Structures Can You Generate from a Molecular Formula? http://www.chemspider.com/blog/how-many-structures-can-you-generate-from-a-molecular-formula.html (accessed November 19, 2014).
- Southan C. D.; Varkonyi P.; Muresan S. Quantitative assessment of the expanding complementarity between public and commercial databases of bioactive compounds. J. Cheminf. 2009, 1, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overington J. P. Novelty of a Chemical Structure. http://chembl.blogspot.se/2014/06/novelty-of-chemical-structure.html (accessed November 19, 2014).
- a Baell J. B.; Holloway G. A. New substructure filters for removal of pan assay interference compounds (pains) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]; b Huth J. R.; Mendoza R.; Olejniczak E. T.; Johnson R. W.; Cothron D. A.; Liu Y.; Lerner C. G.; Chen J.; Hajduk P. J. Alarm NMR: a rapid and robust experimental method to detect reactive false positives in biochemical screens. J. Am. Chem. Soc. 2005, 127, 217–224. [DOI] [PubMed] [Google Scholar]; c Metz J. T.; Huth J. R.; Hajduk P. J. Enhancement of chemical rules for predicting compound reactivity towards protein thiol groups. J. Comput.-Aided Mol. Des. 2007, 21, 139–144. [DOI] [PubMed] [Google Scholar]; d Bruns R. F.; Watson I. A. Rules for identifying potentially reactive or promiscuous compounds. J. Med. Chem. 2012, 55, 9763–9772. [DOI] [PubMed] [Google Scholar]
- Huang R.; Southall N.; Wang Y.; Yasgar A.; Shinn P.; Jadhav A.; Nguyen D. T.; Austin C. P. The NCGC pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci. Transl. Med. 2011, 3, 80ps16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea T. I.; Bologa C. G.; Boyer S.; Curpan R. F.; Glen R. C.; Hopkins A. L.; Lipinski C. A.; Marshall G. R.; Martin Y. C.; Ostopovici-Halip L.; Rishton G.; Ursu O.; Vaz R. J.; Waller C.; Waldmann H.; Sklar L. A. A crowdsourcing evaluation of the NIH chemical probes. Nat. Chem. Biol. 2009, 5, 441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a CAS Coverage of Prophetic Substances. http://www.cas.org/content/prophetics (accessed November 19, 2014).; b Belinskiy A. CAS Indexing of Prophetic Substances—Example of Megapatents, Possible Implications for Chemical Patent Searching. http://wiki.piug.org/display/PIUG/CAS+indexing+of+prophetic+substances+-example+of+megapatents,+possible+implications+for+chemical+patent+searching (accessed November 19, 2014).
- Southan C.; Williams A. J.; Ekins S. Challenges and recommendations for obtaining chemical structures of industry-provided repurposing candidates. Drug Discovery Today 2013, 18, 58–70. [DOI] [PubMed] [Google Scholar]
- Probe Reports from the NIH Molecular Libraries Program. http://www.ncbi.nlm.nih.gov/books/NBK47352/ (accessed November 19, 2014). [PubMed]
- Litterman N.; Lipinski C. A.; Bunin B. A.; Ekins S. Computational prediction and validation of an expert’s evaluation of chemical probes. J. Chem. Inf. Model. 2014, 54, 2996–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probe Reports from the Molecular Libraries Program; National Center for Biotechnology Information: Bethesda, MD, 2010. [PubMed] [Google Scholar]
- MLP Probes. http://mli.nih.gov/mli/mlp-probes-2/?dl_id=1352 (accessed November 19, 2014).
- Hohman M.; Gregory K.; Chibale K.; Smith P. J.; Ekins S.; Bunin B. Novel Web-based tools combining chemistry informatics, biology and social networks for drug discovery. Drug Discovery Today 2009, 14, 261–270. [DOI] [PubMed] [Google Scholar]
- CDD. Public Access. https://www.collaborativedrug.com/pages/public_access (accessed November 19, 2014).
- Clark A. G. Molecular Libraries Probes (NIH probes dataset on Molsync). http://molsync.com/demo/probes.php (accessed November 19, 2014).
- Shoichet B. K. Drug discovery: follow your lead. Nat. Chem. Biol. 2014, 10, 244–245. [DOI] [PubMed] [Google Scholar]
- Andreeva A.; Murzin A. G. Evolution of protein fold in the presence of functional constraints. Curr. Opin. Struct. Biol. 2006, 16, 399–408. [DOI] [PubMed] [Google Scholar]
- Lewell X. Q.; Judd D. B.; Watson S. P.; Hann M. M. Recap—retrosynthetic combinatorial analysis procedure: a powerful new technique for identifying privileged molecular fragments with useful applications in combinatorial chemistry. J. Chem. Inf. Comput. Sci. 1998, 38, 511–522. [DOI] [PubMed] [Google Scholar]
- a Lin H.; Sassano M. F.; Roth B. L.; Shoichet B. K. A pharmacological organization of g protein-coupled receptors. Nat. Methods 2013, 10, 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bemis G. W.; Murcko M. A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–2893. [DOI] [PubMed] [Google Scholar]
- Gilson M. K.; Georg G.; Wang S. Digital chemistry in the journal of medicinal chemistry. J. Med. Chem. 2014, 57, 1137. [DOI] [PubMed] [Google Scholar]
- a Williams A. J.; Ekins S.; Spjuth O.; Willighagen E. L. Accessing, using, and creating chemical property databases for computational toxicology modeling. Methods Mol. Biol. 2012, 929, 221–241. [DOI] [PubMed] [Google Scholar]; b Williams A. J.; Ekins S. A quality alert and call for improved curation of public chemistry databases. Drug Discovery Today 2011, 16, 747–750. [DOI] [PubMed] [Google Scholar]
- Goldfarb D. S.Method using lifespan-altering compounds for altering the lifespan of eukaryotic organisms, and screening for such compounds. US 20090163545 A1, 2009.
- Southan C.; Varkonyi P.; Muresan S. Quantitative assessment of the expanding complementarity between public and commercial databases of bioactive compounds. J. Cheminf. 2009, 1, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A. J. Public chemical compound databases. Curr. Opin. Drug Discovery Dev. 2008, 11, 393–404. [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. Zinc—a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Zhou B.; Chen K.; Yan S. F.; King F. J.; Jiang S.; Winzeler E. A. Large-scale annotation of small-molecule libraries using public databases. J. Chem. Inf. Model. 2007, 47, 1386–1394. [DOI] [PubMed] [Google Scholar]
- Williams A. J. Chemical Abstracts Service Indexes Chemspider Content. http://www.chemspider.com/blog/chemical-abstracts-service-indexes-chemspider-content.html (accessed November 19, 2014).
- Visser U.; Abeyruwan S.; Vempati U.; Smith R. P.; Lemmon V.; Schurer S. C. BioAssay Ontology (BAO): a semantic description of bioassays and high-throughput screening results. BMC Bioinf. 2011, 12, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A. M.; Bunin B. A.; Litterman N. K.; Schurer S. C.; Visser U. Fast and accurate semantic annotation of bioassays exploiting a hybrid of machine learning and user confirmation. PeerJ 2014, 2, e524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SciFinder Now Offers API Capabilities. http://www.cas.org/news/media-releases/scifinder-offers-api-capabilities (accessed November 19, 2014).
- Contentmine. http://contentmine.org/ (accessed November 19, 2014).
- Williams A. J. InChI: connecting and navigating chemistry. J. Cheminf. 2012, 4, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson M. N.; Ylioja P. M.; Williamson A. E.; Woelfle M.; Robins M.; Badiola K. A.; Willis P.; Olliaro P.; Wells T. N.; Todd M. H. Open source drug discovery—a limited tutorial. Parasitology 2014, 141, 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilbanks J. Openness as infrastructure. J. Cheminf. 2011, 3, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antolin A. A.; Mestres J. Distant polypharmacology among MLP chemical probes. ACS Chem. Biol. 2014, 10.1021/cb500393m. [DOI] [PubMed] [Google Scholar]
- Bradley J.-C.; Guha R.; Lang A.; Lindenbaum P.; Neylon C.; Williams A.. Beautifying data in the real world. In Beautiful Data; Segaran T., Hammerbacher J., Eds; O’Reilly Media Inc.: Sebastopol, CA, 2009. [Google Scholar]
- Ekins S.; Olechno J.; Williams A. J. Dispensing processes impact apparent biological activity as determined by computational and statistical analyses. PLoS One 2013, 8, e62325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A. M. Open Source ECFP/FCFP Circular Fingerprints in CDK. https://www.collaborativedrug.com/buzz/2014/05/10/open-source-ecfpfcfp-circular-fingerprints-in-cdk/ (accessed November 19, 2014).