

Abstract
Elevating GABA levels in the synaptic cleft by inhibiting its reuptake carrier GAT1 is an established approach for the treatment of CNS disorders like epilepsy. With the increasing availability of crystal structures of transmembrane transporters, structure-based approaches to elucidate the molecular basis of ligand–transporter interaction also become feasible. Experimental data guided docking of derivatives of the GAT1 inhibitor tiagabine into a protein homology model of GAT1 allowed derivation of a common binding mode for this class of inhibitors that is able to account for the distinct structure–activity relationship pattern of the data set. Translating essential binding features into a pharmacophore model followed by in silico screening of the DrugBank identified liothyronine as a drug potentially exerting a similar effect on GAT1. Experimental testing further confirmed the GAT1 inhibiting properties of this thyroid hormone.
Introduction
Imbalances in the levels of excitatory and inhibitory neurotransmitters, such as serotonin, dopamine, and GABA, can lead to severe CNS disorders like epilepsy, schizophrenia, anxiety, and depression. Tackling CNS diseases related to the GABAergic system is most commonly achieved by using drugs of the benzodiazepine family (e.g., diazepam), which allosterically modulates the pentameric GABAA receptor (GABAA-R).1 However, an alternative way of enhancing GABA action is inhibition of the corresponding neurotransmitter uptake system.2−4 In the case of the GABA transporter (GAT) family, four GABA reuptake transporter subtypes (GAT1–3, BGT1) and one vesicular carrier exist in mammalian organisms.5 The GAT family belongs to the neurotransmitter:sodium symporters (NSS) which is organized as oligomers at the plasma membrane6 while, in contrast to the GABAA-R, functions as a monomer.7 Usually, NSS transporters use a sodium gradient for uphill transport of neurotransmitters out of the synaptic cleft. In certain cases, a reverse transport mode is also known, releasing neurotransmitter in a nonvesicular way.8 At present, only one drug targeting this receptor, the anticonvulsant tiagabine, is on the market. Tiagabine selectively inhibits GAT1, the most abundant GAT subtype in the human brain.8 An X-ray crystallographic structure is not yet available for any member of the GAT family, but a number of homology models have been constructed. Further docking studies indicated distinct modes of drug–transporter interaction.9−15 The molecular basis of tiagabine action, however, remains elusive, as experimental evidence for proposed binding modes is still lacking. Furthermore, ligand-based exploration of inhibitor scaffolds is limited by the low tolerance of this transporter for inhibitor modification. On the basis of a set of tiagabine analogs from literature sources, we recently investigated ligand-based structure–activity relationships of the compound class.16 Briefly, binary QSAR allowed classification of GABA uptake inhibitors into active and inactive bins by using the degree of rigidity and polarity distribution as main descriptors. With the increasing knowledge provided by the X-ray structures of analogous transport proteins,17 structure-based approaches for elucidating the molecular basis of drug–transporter interaction also become feasible. In the present study, we describe a binding hypothesis of tiagabine in GAT1 and its successful validation by in silico screening.
Results and Discussion
Comparative Modeling
The closest transporter proteins related to hGAT1 for which structures are available are the bacterial leucine amino acid transporter protein, LeuTAa, and the drosophila dopamine transporter, dDAT. Despite its lower overall sequence identity, closer substrate relationship and significantly higher resolution of 2.00 vs 2.95 Å favored the use of LeuT as template structure.18,19 Several sequence alignments between hGAT1 and LeuTAa have been published, and all alignments are almost identical within the conserved central substrate binding cavity.11,20,21 Both template candidates were available in an open-to-out conformation, thus granting access to bulky inhibitor molecules. Suitable templates for the intracellular N- and C-terminal domains of hGAT1 are not available and thus were not included in the final homology model. Because of the differing stoichiometry of eukaryotic NSS family members for Cl–, the LeuT structure (PDB code: 3F3A) was modified by engineering a chloride binding site using structural information from crystal structure of the dDAT and topological information from the literature.22−24 On the basis of a combination of low B-values and proximity to the binding site or stabilization of adjacent domains, several water molecules were selected and kept in the template file. Finally, a known disulfide bridge between C164 and C173 of EL-2 was defined.3 Modeller25 was used to generate 100 models, which were ranked according to their respective discrete optimized protein energy (DOPE) score for estimating the geometric quality.26 For the 10 highest ranked models, additional quality checks were performed using the model assessment tools of the SWISS-MODEL server.27−29 Models with core residues showing disallowed geometry according to the Ramachandran plot were omitted. The remaining models were visually inspected for their ability to reflect residue proximity and accessibility data from literature.30−34 In addition, the models were evaluated regarding the orientation of nonconserved polar residues in TM regions. Subsequently, the best structure was selected according to aforementioned criteria and subjected to a soft minimization protocol for relaxation of the system.
Binding Site Sampling
Focused sampling of the conformational space in the putative tiagabine interaction site can be achieved by molecular dynamics (MD) simulations. Tiagabine cannot be accommodated in the occluded state of the transporter, as both the extracellular gate between R69 and D451 as well as the upper lid of the binding side, formed by the bulky F294 side chain, are limiting the available space.11 In addition, preliminary MD simulations of the transporter model in the apo open-to-out state led to rearrangement of the gating residues impeding subsequent placement of compounds larger than substrates like GABA, guvacine, or nipecotic acid. Thus, tiagabine was placed into the central cavity using Glide35 prior to 30 ns of molecular dynamics simulations, which was used for validating and equilibrating the model.
Subsequently, 10 representative snapshots for the last 10 nanoseconds of the run were extracted based on maximum RMSD diversity of binding pocket residues. Thus, focused sampling of the conformational space in the binding site could be achieved, using the snapshots as input structures for subsequent docking experiments.
Docking Studies
The constrained GABA analogs, nipecotic acid and guvacine (2, 3), are potent uptake inhibitors in vitro but are unable to penetrate the blood–brain barrier.36 In addition, these compounds act as GAT1 substrates.8 In contrast, tiagabine and the selective GAT-1 inhibitor SK&F 89976-A (4, 5) that contain bulky aromatic substituents are pure inhibitors that are not transported (Figure 1). A large number of systematically modified derivatives of the basic tiagabine scaffold have since been synthesized and tested.37−41 These derivatives contain a conformationally restricted GABA-mimetic nipecotic acid or guvacine moiety, a 4–8 atom linker, and a large, mostly diaromatic, hydrophobic moiety. These analogs provide a rich data source to construct structure–activity relationships.
Figure 1.

GABA, conformationally restricted analogs, and lipophilic aromatic derivatives.
A total of 162 compounds were extracted from the literature, spanning an activity range from low nanomolar to millimolar IC50 values, each of them tested under comparable assay conditions.42 Ligands exhibiting substantial activity differences linked to distinct structural changes in the key regions shown in Figure 2 were selected for subsequent experimental data guided docking.1,43
Figure 2.

Chemical structures and literature IC50 values of ligands with key modifications in linker length, polarity, and rigidity of the aromatic moiety.
An important observation was the dramatic activity loss caused by introduction of a direct link between the two aromatic moieties (5 vs 7). In contrast, enhanced activity had been reported for introduction of a polar region in the linker. This is exemplified by compound pair 8 and 9, as well as compound 6, being the closest available derivative to reference compound 5. In terms of activity, extending the linker length was well tolerated because compounds 5, 8, and 10 gave potent inhibitors. Finally, exchange of a benzene by a pyridine leads to a dramatic loss of activity (10 vs 11). These activity differences should also be reflected by respective differences in the ligand/protein interaction pattern and thus aid in the prioritization of the docking poses.
Docking into 10 snapshots derived from the hGAT-1–tiagabine complex was performed in a sequential ensemble-like manner using GOLD,44,45 thereby allowing for minor movements of the backbone and focused sampling of the binding site side chain orientations. The binding site was defined within a 10 Å radius around the simulated tiagabine coordinates. Two water molecules were kept optional, as they had turned out to be stably involved in the hydrogen bonding network during the previous MD simulation. However, in the subsequent docking runs, no direct contribution of these two water molecules to the binding of the selected ligands was observed.
Side chain orientations of possible interaction partners for polar linker compounds 6, 8, 10, and 11 were addressed individually. Conformational sampling of the binding site had been performed with tiagabine as ligand, which lacks a corresponding electronegative moiety in the linker. Hence, the full range of conformational flexibility of the R69, Y139, Y140, and S452 side chains was explored using the internal rotamer library of the GOLD software package.
For each of ligands 6–11, 100 docking poses per snapshot were generated and ranked by ChemScore, which provides parameters for a putative interaction with sodium, in analogy to LeuT.21 However, relying on just a single scoring function bears the risk of missing relevant poses, especially when no experimentally derived complexes for redocking studies are available. Thus, all poses were reranked using rank-by-rank consensus scoring that included GoldScore, ChemPLP, London dG, GBVI, and XScore scoring functions.45−50 The top 10 consensus score poses for each ligand were subsequently visually analyzed.
Binding Mode
Analysis of the 10 top ranked poses per ligand among the ensemble docking results clearly indicated a common binding mode for tiagabine analogs (Figure 3). As already expected from literature results, the most prominent interaction was coordination of the Na1 sodium cation by the negatively charged acid moiety, which fulfills an octahedral geometry, together with side chain atoms of N66, S295, and N327, as well as the backbone carbonyl oxygens of A61 and S295. The majority of observed poses showed an interaction between the positively charged nipecotic acid nitrogen and the backbone of F294, while the carboxylate group interacting with Na1 was in an equatorial conformation. In contrast, poses with the R-configured carboxy group sampled in an axial conformation tended to form an intramolecular hydrogen bond with the charged nitrogen atom. While no clear preference for one of the two carboxylate orientations could be deduced from scoring values, X-ray and NMR studies of nipecotic acid indicated a preferred equatorial configuration, which would be more pronounced by adding a bulky moiety like the biaromatic tail.51,52 In addition, Skovstrup et al. reported less stable behavior of axial-configured tiagabine poses in molecular dynamics simulations.11 Thus, the poses with the axial carboxylate configuration were considered less plausible.
Figure 3.
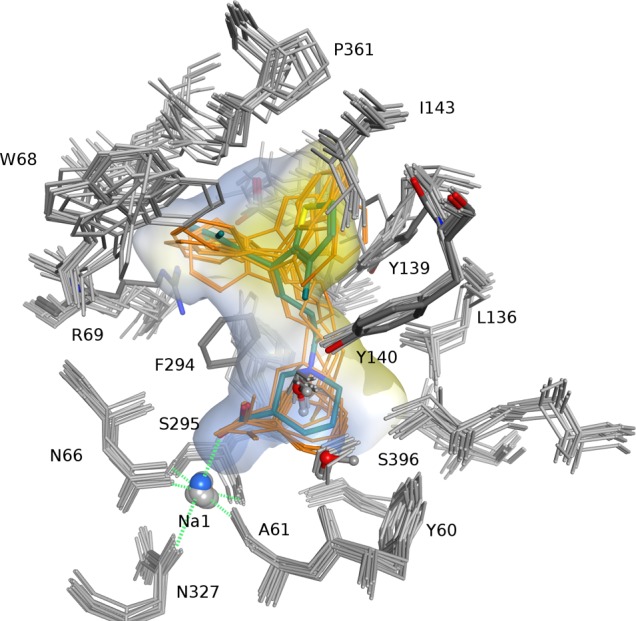
Docking poses of tiagabine (turquoise) and analogs (orange) in 10 MD snapshots of the hGAT-1 model. Polar regions in blue, hydrophobic areas in yellow.
The upper boundary of the binding pocket is formed by a bent region of EL-4, extending into the hydrophobic cavity with the backbone of G360 as a ‘ceiling beam’, hence separating it into two pockets, each of which is able to accommodate a single hydrophobic aromatic rings. As it can be seen in Figure 3, the ligand–transporter interaction in one hydrophobic pocket is stabilized by a π–π interaction with the side chain of Y139, as well as by a cavity able to accommodate a small o-substituent as present in 5 and 6. This cavity is confined by the side chains of I143 and Y140, the latter being a residue known to be also important for ligand recognition (see Figure 3).33
The second cavity is mainly shaped by the hydrophobic side chains of W68, F294, and A358 (not shown).
Because of the relative torsion of the two pockets, poses of 7 tended to encounter an initial steric clash that was relieved after energy minimization of the complexes, whereas compounds with a terminal bis-5-methyl-thienyl (5, 6) or diphenyl (8–10) group were able to bind in a conformation near their global energy minimum (see Figure 4), thus explaining the activity cliff between 5 and 7.
Figure 4.
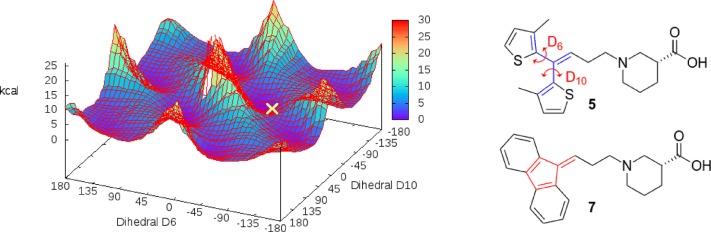
Dihedral energy landscape of 5-methyl-thiophen dihedral angles; configuration of the docking pose of 5 is marked in yellow. Constrained aromatic system of 7 is highlighted in red.
The positive effect of a polar atom in the linker moiety on binding seems to be the result of several factors. Transient interactions with residues in the entry path might play a significant role but are not reflected by the docking poses. Earlier steered MD studies of tiagabine in GAT1 by Skovstrup et al. had indicated a transient interaction with the R69 side chain upon entry in the binding site.53 Hence, docking poses biased toward an interaction between this residue and one of the electronegative linker atoms in 6, 8, 10, and 11 were generated, turning out to be possible but rather short-lived in short MD runs due to reset forces of the basic side chain (data not shown).
To address the relatively low activity of compound 11, per-atom contributions to ΔG in the binding pose were evaluated using HYDE.54 An unfavorable effect of the pyridine nitrogen in the hydrophobic receptor environment was indicated (see Supporting Information, Figure S3). In addition, repulsive forces between negative partial charges of the aromatic nitrogen atom and the oxime moiety might force the pyridine ring in a sterically unfavorable orientation.
Taken together, the common orientation of the compound class was in agreement with the structure–activity relationships of the ligand set and the topology of the extended substrate binding pocket.
Virtual Screening
To further probe the proposed binding mode against pharmacologically relevant chemical space, a pharmacophore-based screening strategy was applied. The 3D orientation of the main tiagabine binding features was extracted from the docked complex and encoded in a four-feature pharmacophore model using LigandScout.55 Two hydrophobic features were placed in the respective cavities occupied by the thiophene moieties. Groups capable of complexing sodium were described by a negatively ionizable property. Finally, a positively ionizable feature was placed on the basic nitrogen to mirror the compound’s zwitterionic character (Figure 5, the model is available for download at http://pharminfo.univie.ac.at).
Figure 5.
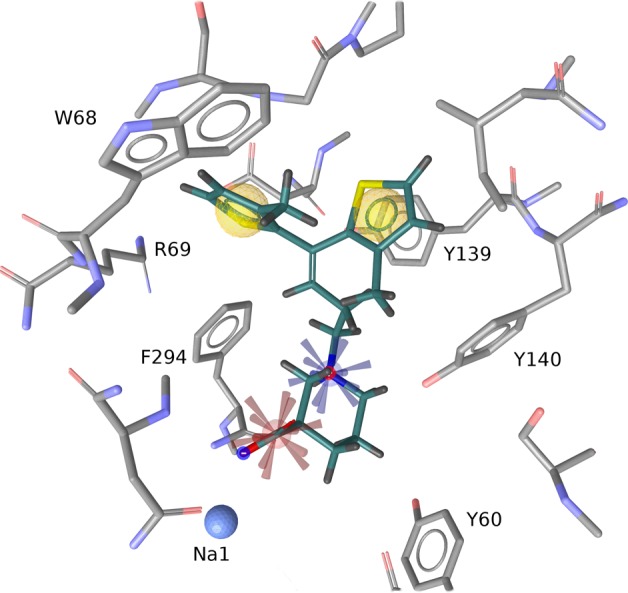
Pharmacophore model of tiagabine: hydrophobic (yellow), positive (blue), and negative (red) ionizable features in context with the GAT1 substrate binding site.
The sensitivity of the pharmacophore model was validated by a decoy set generated using the DUD-E platform (http://dude.docking.org),56 retrieving just the compounds with known GAT-1 activity.
To test the predictive value of the model, a commercial vendor database57,58 consisting of 1.7 million compounds, as well as the Drugbank Index59 covering 1491 marketed drugs, were screened. A total of 79 and eight compounds, respectively, matched the pharmacophore query and passed the PAINS filter for frequent hitters.60
Subsequently, the virtual hits were docked into a representative MD snapshot of the homology model, from which the tiagabine pharmacophore model had been derived. Protein–ligand interaction fingerprints (PLIF) for the calculated poses were retrieved using MOE.61 The interaction fingerprints were used to filter out compounds, which did not show an interaction with Na1. This reduced the hit list to 13 compounds for the Enamine database available in sufficient purity, and seven compounds for DrugBank, respectively (see Figure 6). For the latter, tiagabine, three thyroid hormones (liothyronine, levothyroxine, dextrothyroxine), two angiotensin conversion enzyme (ACE) inhibitors (ramipril, perindopril), and an antihistaminergic drug (bepotastine) were retrieved. On the basis of pharmacophoric fit and docking performance (details in Supporting Information, Table S2), bepotastine, ramipril, and liothyronine were selected for biological testing.
Figure 6.

Compounds tested in the [3H]-GABA uptake assay: Enamine (red frame) and Drugbank hits (blue frame), and reference compounds (black frame).
Experimental Testing
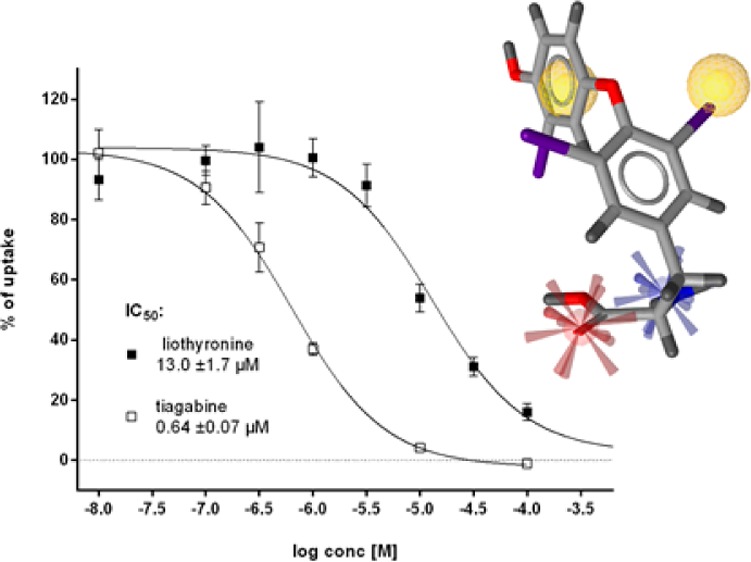
Inhibitory potency of the selected compounds was evaluated by an uptake inhibition assay of radiolabeled GABA in HEK cells stably expressing rGAT1. First, the IC50 value for tiagabine (5) in the test system was determined to be 0.64 ± 0.07 μM. This was about a factor of 10 higher than the value reported for the unspecific rat synaptosome assay used by Andersen et al. but is in accordance with data reported for mouse GAT1.37,62 Subsequently, compounds were measured at a concentration of 100 μM against 5 as standard. Diazepam (28) and tiagabine (5) were used as negative and positive control.
As illustrated in Figure 7, one of the commercial screening compounds, 18, weakly reduced uptake to just below 80% of saline. One DrugBank substance, 27a (liothyronine, a thyroid hormone also known as T3), turned out to significantly inhibit radioligand uptake, which prompted the acquisition and testing of other commercially available derivatives 27b–d. Among those, the other bioactive hormone levothyroxine (27b) showed reduced uptake, albeit weaker than 27a. The representative of the ACE inhibitors and the antihistaminergic drug bepotastine were essentially inactive.
Figure 7.
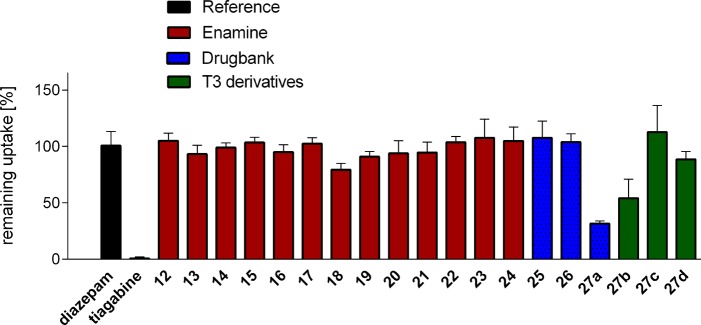
Remaining uptake of [3H]-GABA in the presence of 100 μM of the respective compound (n = 3).
The IC50 value for liothyronine derived from a dose–response curve was 13 ± 1.7 μM (Figure 8), providing a direct link between reported general effects of thyroid hormones on GABA uptake63−65 and the inhibitory action of 27a on the GAT-1 subtype.
Figure 8.

Inhibition curves of tiagabine (IC50 0.64 ± 0.07 μM, white squares) and liothyronine (IC50 13.0 ± 1.7 μM, black squares).
As illustrated in Figure 9, the pharmacophoric depiction of 27a reveals that one of the required hydrophobic features is not, as one would expect, the aromatic ring of the 3,5-diiodophenyl moiety, but rather a lipophilic iodine substituent, which could barely be deduced from chemical similarity measures. Relying on an appropriate position of the second ring relative to the interacting iodine atom, this type of interaction is also possible for 27b but not for 27d. The 3,3′,5′-substituted variant of the T3 layout is missing the second substituent on the proximal ring which is responsible for inducing the bioactive conformation.66 This is in line with reported structure–activity relationships of thyroid hormone derivatives both at thyroid hormone receptors and GABAergic rat brain synaptosomes.64,67 Apparently, the steric requirements in both systems are remarkably similar, relying on correct substitution pattern, stereochemistry, and degree of lipophilicity, possibly also limiting the relative efficacy of 27c. The most remarkable difference between the observed crystallographic hormone binding mode of 27a (PDB code 3UVV)68 and the GAT1 binding hypothesis can be found in the 4′ position. The presence of the phenolic hydroxyl group is crucial for hormone receptor binding but not for interaction with the transport protein.
Figure 9.
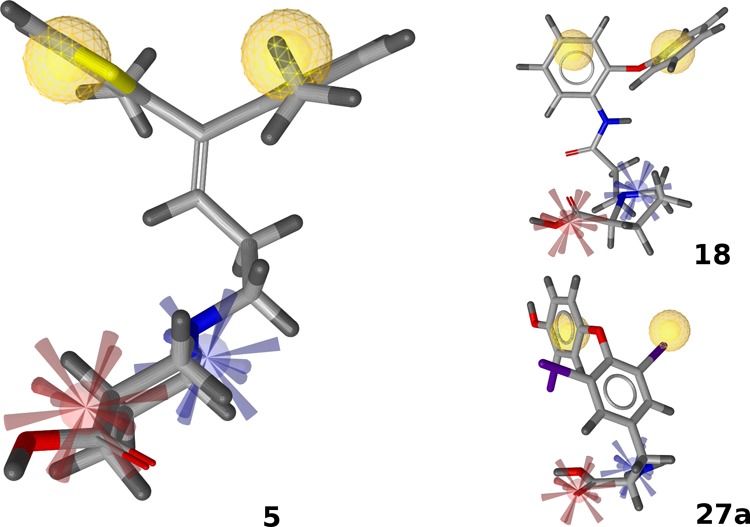
Pharmacophoric fit of 18 and 27a. Features are colored according to Figure 5.
Regarding compounds 12–24, the most crucial property among those molecules seems to be the distance between the positively ionizable group and the first occurrence of lipophilic bulk, which is in close analogy to the linker in tiagabine analogs. Distal from the nitrogen atom (as seen from the negatively ionizable group, which also can be a tetrazole moiety in a reasonable distance69), usually just one heavy atom separates the positive charge from the next aromatic moiety (12, 19, 20), or branching position (13, 21, 24). Alternatively, it is part of separate ring system not directly carrying the acidic moiety (14–17, 22). With a slightly increased distance between the aromatic ring and the carboxylate group, 18 displays some weak activity.
This observation also extends to the inactive DrugBank compounds, as the nitrogen atoms of 25 and 26 are both connected to the hydrophobic part by space demanding linkers, whereas the first aromatic ring of thyroid hormones is at a distance of two heavy atoms.
The presence of a pyridine moiety known to be unfavorable from the 10–11 compound pair could further limit the potential of 25 despite its remarkable structural similarity to the reference compounds. Just as for the bulk of a cyclopentane moiety attached to the polar part of 26, this might considerably inhibit optimal positioning in the binding site.
To further assess the degree of similarity between the compounds retrieved and the reference compound tiagabine, we calculated the Tanimoto similarity values based on chemical fingerprints (MACCS, FP2, FP4) derived from OpenBabel.70 The similarity between tiagabine and liothyronine as well as the slightly active 18, on the basis of MACCS keys, was 25.4 and 51.5%, respectively. Thus, the pharmacophore model performed independently from chemical similarity.
Conclusions
With the increasing number of X-ray structures available for transmembrane transporters, structure-based computational models have provided valuable insights into the molecular basis of ligand–transporter interaction. Within this article, we propose a binding mode of the antiepileptic drug tiagabine in GAT1 by including knowledge from ligand-based studies into the prioritization process for docking poses. Subsequent pharmacophore-based virtual screening followed by experimental testing further confirmed the validity of the pose by identifying a commonly used drug (liothyronine) as an inhibitor of GAT1. Strikingly, liothyronine has been described long ago as potential GAT inhibitor without major activity on other neurotransmitter reuptake systems (dopamine, serotonin, choline, aspartate),63 but final experimental confirmation for subtype GAT1 since has been lacking. Compounds with significantly higher chemical similarity retrieved in a commercial vendor library all prove inactive, further implying that selective transport inhibition of the protein can only be tackled from the side of steric feature arrangement.
Furthermore, the results indicate that, apart from privileged tricyclic antidepressant scaffolds known to more or less unspecifically inhibit neurotransmitter uptake,71,72 not many drugs on the market are likely to interact with GAT1.
Experimental Section
Model Building
The GAT1 models were constructed using the crystal structure of LeuT as template which shows highest available resolution in an open-to-out state of the transporter.19 When assessing the differences between available sequence alignments of LeuT and hGAT1 (UniProt entries O67854 and P30531, respectively), no differences for residues in the central binding cavity were observed, except for a one-residue gap in the middle of LeuT-TM10, either placed over GAT1-G457,21 S456,20 or A455.11 As it has been optimized for GAT1 and also is the most recent one, the alignment of Skovstrup et al. was finally chosen to build the model. Assessment of the sequence identity between hGAT-1 and rGAT-1, the first being the effective protein of interest, the second the one used in cell assays, stated 100% sequence identity for the observed core region and 97.9% for the whole modeled sequence (see Supporting Information, Figure S4).
The crystal structure of LeuT retrieved from the PDB (www.pdb.org,73 accession code 3F3A) was mutated in silico at position 290 using MOE with a serine side chain orientation corresponding to the former glutamic acid. A chlorine ion was placed at the coordinates of the previous center of the E290 side chain, then further optimized according to interaction potential calculations, giving Cl– as probe. Tethering the backbone, S290 and its surrounding residues were carefully energy minimized for final optimization of the local coordinates.
The models were built using Modeller9v8 in the automodel class, including water molecules, the chloride ion, and two cobound sodium ions as nonprotein atoms. A disulfide bridge was defined between C164 and C173 using a Modeller patch command. One hundred models were generated using very thorough VTFM (variable target function method) optimization, as provided in Modeller. Output models were ranked according to DOPE score, and the top 10 were further assessed by the SWISS-MODEL server. According to PROCHECK results and Ramachandran plots, models with disallowed backbone geometries in transmembrane regions were omitted. Hydrogen atom assignment and soft energy minimization of the raw models was performed within MOE using LigX and the Charmm27 all-atom force field, otherwise with default settings.74
Molecular dynamics simulations were performed in GROMACS 4.5.3.75,76 The selected complex was inserted into a pre-equilibrated and solvated POPC membrane by applying the program g_membed,77 using the GROMOS 53A6 united-atom force field78 and periodic boundary conditions. All simulations were performed at 310 K. The system was neutralized by adding sodium and chlorine ions to a final salt concentration of 150 mM. Gradually, position restraints on the main complex were reduced from 500 kJ mol–1 nm–2 (500 ps) to 250 kJ mol–1 nm–2 (500 ps). After an equilibration phase without restraints, a fully stable system was achieved after 20 ns. Between 21 and 30 ns of the production run, the frames were clustered according to RMSD of residues within a radius of 7 Å around the ligand. The 10 most diverse snapshots were extracted and energy minimized. Two water molecules were kept in the binding site. One showed a stable H-bond with Y60 (89% occupancy; distance ≤3.5 Å; angle ≤60°), another one was directly attached and showed an almost equally stable H-bond interaction.
Ligand Preparation
Molecules used for docking were drawn in MOE and processed with CORINA.79 Protonation states were sampled according to possible states in a physiological pH range of 7.2 ± 0.2 using LigPrep.80 These states were cross-checked with the major microspecies calculated by the ChemAxon web-tool chemicalize.org.
Docking
Primary placement of tiagabine was done using Glide in standard precision (SP) mode using default settings. The receptor grid was defined around binding site residues 60–63, 64–66, 136, 140, 294–297, 300, 396, and 400.
Docking and GoldScore/ChemPLP rescoring in the 10 MD snapshots were performed with GOLD 5.0.1, using ChemScore as primary scoring function. Early termination was disabled, keeping the 100 best solutions per ligand and snapshot. Two water molecules were set to ‘spin’ and ‘toggle’. All other settings were set to default. External rescoring was performed using XScore (XScore) and MOE (London dG, GBVI). Consensus scores were calculated by summing up indices assigned according to respective ranks within a scoring function.
Secondary docking runs for investigating side chain orientations for specific interactions with polar linker moieties were performed by (a) constraining residues Y139, Y140, and S456 to the internal library of allowed rotamers in GOLD and (b) for investigating interactions as reported by Skovstrup et al.,53 likewise rotamer rotations of R69 and F294 were allowed but with an additional distance constraint of 1.5–3.5 Å (spring constant 5) between the R69 guanidine function and the polar linker moiety.
Determination of the potential energy landscape for different dihedral angle configurations was performed with Gaussian 09.81 After initial geometry optimization with HF/3-21G implemented in the software package, configurations with an increment of 15° were calculated using the M06-2X hybrid functional and the 6-31G* basis set.82,83
Screening
Pharmacophore models were built using LigandScout 3.0.55,84,85
For assembling the customized decoy library, 50 decoys per active compound (5–11) were compiled in SMILES format using the DUD-E platform (dude.docking.org). The nonredundant compounds with similar physicochemical properties but dissimilar 2-D topology for each input line were extracted from the ZINC database. The retrieved set of decoys and the active compounds as SMILES were assembled and processed to a LigandScout screening database using the maximum number of possible conformers.
The DrugBank database was downloaded from the Web site www.drugbank.ca and consisted of 1491 entries (version of June 2013). Enamine Advanced and HTS screening collections were obtained from the download site at www.enamine.net (n = 1719682; version 032013). Counterions were removed using the Software MOE. LigandScout command line modules idbgen and iscreen were used for conformation generation and for performing the pharmacophore screening. Training set, decoy compilation, and DrugBank compounds were prepared using OMEGA-best settings (max 500 conformations), Enamine Advanced and HTS databases were compiled with OMEGA-fast settings (max 25 conformations) (www.eyesopen.com,86−88).
Path-based (FP2) and substructure-based (MACCS, FP4) similarity fingerprinting was performed using OpenBabel 2.3.1.
Pose Filtering
Primary checking for pan assay interference (PAINS) compounds60 was done by uploading retrieved virtual screening hits to the PAINS remover web service (available at http://cbligand.org/PAINS), returning no suspicious compounds. Protein–ligand interaction fingerprints of docked virtual hit compounds were calculated in MOE. Poses missing the bin for Na1 interaction were removed.
Pharmacological Testing
Screening compounds were purchased from Enamine (Enamine Ltd., Riga, Latvia), Fluka/Sigma-Aldrich (Sigma-Aldrich Co., Saint Louis, MO), and AvaChem (AvaChem Scientific, San Antonio, TX), all with a purity ≥95% (see Supporting Information, Table S4). Tiagabine was obtained from Sanofi-Synthelabo, Montpellier, France.
Cell lines of HEK293 cells stably expressing YrGAT-1 were generated as described elsewhere.8 Cloned cells (∼4 × 104 cells/well) were seeded and grown at 37° on poly(d)-lysine coated standard plasticware 24 h in advance.
Uptake of [3H]-GABA into was measured in the presence of 100 μM of the compounds, while unspecific uptake was defined as uptake in the presence of 100 μM tiagabine. For IC50 determination tested concentrations were: 5, 0.001, 0.01, 0.1, 0.3, 1, 10, 100 μM; 27a, 0.01, 0.1, 0.3, 1, 3, 10, 30, 100 μM. After 3 min preincubation, [3H]-GABA (35 Ci/mmol, PerkinElmer, Boston, MA) in a final concentration of 0.015 μM was added. Uptake was stopped by adding ice-cold Krebs-HEPES buffer (10 mM HEPES adjusted to pH 7.4 with 35.9 mM solid NaOH, 120 mM NaCl, 3 mM KCl, 2 mM CaCl2, 2 mM MgSO4, and 2 mM d-glucose as supplement). Cells were lysed with 1% SDS (sodium dodecyl sulfate) solution, taken up in 2 mL of scintillation cocktail (Rotiszint Eco Plus, Carl Roth GmbH, Karlsruhe, Germany) and counted in a standard liquid scintillation counter (Packard TriCarb 2300TR, Packard Instruments). At least three independent experiments per compound were performed, each in triplicate. Data analysis was performed by nonlinear regression using Prism 6.01.89
Acknowledgments
The computational results presented have in part been achieved using the Vienna Scientific Cluster (VSC). We thank Peter Wolschann for his support with quantum chemical calculations, Martin Kratzel for kindly providing d- and l-thyroxine samples, and Christina Dangl for additional HPLC analyses. We acknowledge financial support provided by the Austrian Science Fund, grants F35 and W1232. We are grateful to Inte:Ligand and OpenEye for providing us with licenses for their software packages.
Glossary
Abbreviations Used
- GABAA-R
GABA receptor subtype A
- GAT
GABA transporter
- HEPES
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)
- MACCS
molecular access system
- NSS
neurotransmitter:sodium symporter
- PAINS
pan assay interference compounds
Supporting Information Available
Additional tables and figures illustrating model selection criteria, backbone RMSD of the MD production run as well as compound inhibition and purity data (PDF and CSV). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
After this paper was published ASAP February 26, 2015, a correction was made to Figure 6. The corrected version was reposted March 2, 2015.
Supplementary Material
References
- Richter L.; de Graaf C.; Sieghart W.; Varagic Z.; Morzinger M.; de Esch I. J.; Ecker G. F.; Ernst M. Diazepam-bound GABAA receptor models identify new benzodiazepine binding-site ligands. Nat. Chem. Biol. 2012, 8, 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest L. R.; Zhang Y. W.; Jacobs M. T.; Gesmonde J.; Xie L.; Honig B. H.; Rudnick G. Mechanism for alternating access in neurotransmitter transporters. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 10338–10343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen A. S.; Andersen J.; Jorgensen T. N.; Sorensen L.; Eriksen J.; Loland C. J.; Stromgaard K.; Gether U. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol. Rev. 2011, 63, 585–640. [DOI] [PubMed] [Google Scholar]
- Clausen R. P.; Frolund B.; Larsson O. M.; Schousboe A.; Krogsgaard-Larsen P.; White H. S. A novel selective gamma-aminobutyric acid transport inhibitor demonstrates a functional role for GABA transporter subtype GAT2/BGT-1 in the CNS. Neurochem. Int. 2006, 48, 637–642. [DOI] [PubMed] [Google Scholar]
- Borden L. A. GABA transporter heterogeneity: pharmacology and cellular localization. Neurochem. Int. 1996, 29, 335–356. [DOI] [PubMed] [Google Scholar]
- Schmid J. A.; Scholze P.; Kudlacek O.; Freissmuth M.; Singer E. A.; Sitte H. H. Oligomerization of the human serotonin transporter and of the rat GABA transporter 1 visualized by fluorescence resonance energy transfer microscopy in living cells. J. Biol. Chem. 2001, 276, 3805–3810. [DOI] [PubMed] [Google Scholar]
- Scholze P.; Freissmuth M.; Sitte H. H. Mutations within an intramembrane leucine heptad repeat disrupt oligomer formation of the rat GABA transporter 1. J. Biol. Chem. 2002, 277, 43682–43690. [DOI] [PubMed] [Google Scholar]
- Sitte H. H.; Singer E. A.; Scholze P. Bi-directional transport of GABA in human embryonic kidney (HEK-293) cells stably expressing the rat GABA transporter GAT-1. Br. J. Pharmacol. 2002, 135, 93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallo A.; Bencsura A.; Heja L.; Beke T.; Perczel A.; Kardos J.; Simon A. Major human gamma-aminobutyrate transporter: in silico prediction of substrate efficacy. Biochem. Biophys. Res. Commun. 2007, 364, 952–958. [DOI] [PubMed] [Google Scholar]
- Schlessinger A.; Wittwer M. B.; Dahlin A.; Khuri N.; Bonomi M.; Fan H.; Giacomini K. M.; Sali A. High selectivity of the gamma-aminobutyric acid transporter 2 (GAT-2, SLC6A13) revealed by structure-based approach. J. Biol. Chem. 2012, 287, 37745–37756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skovstrup S.; Taboureau O.; Brauner-Osborne H.; Jorgensen F. S. Homology modelling of the GABA transporter and analysis of tiagabine binding. ChemMedChem 2010, 5, 986–1000. [DOI] [PubMed] [Google Scholar]
- Wein T.; Wanner K. T. Generation of a 3D model for human GABA transporter hGAT-1 using molecular modeling and investigation of the binding of GABA. J. Mol. Model. 2010, 16, 155–161. [DOI] [PubMed] [Google Scholar]
- Pallo A.; Simon A.; Bencsura A.; Heja L.; Kardos J. Substrate–Na+ complex formation: coupling mechanism for gamma-aminobutyrate symporters. Biochem. Biophys. Res. Commun. 2009, 385, 210–214. [DOI] [PubMed] [Google Scholar]
- Kardos J.; Pallo A.; Bencsura A.; Simon A. Assessing structure, function and druggability of major inhibitory neurotransmitter gamma-aminobutyrate symporter subtypes. Curr. Med. Chem. 2010, 17, 2203–2213. [DOI] [PubMed] [Google Scholar]
- Baglo Y.; Gabrielsen M.; Sylte I.; Gederaas O. A. Homology modeling of human gamma-butyric acid transporters and the binding of pro-drugs 5-aminolevulinic acid and methyl aminolevulinic acid used in photodynamic therapy. PLoS One 2013, 8, e65200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurik A.; Reicherstorfer R.; Zdrazil B.; Ecker G. F. Classification of high-activity tiagabine analogs by binary QSAR modeling. Mol. Inf. 2013, 32, 415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuboni S.; Hausch F. Snapshot of antidepressants at work: the structure of neurotransmitter transporter proteins. Angew. Chem., Int. Ed. 2014, 53, 5008–5009. [DOI] [PubMed] [Google Scholar]
- Penmatsa A.; Wang K. H.; Gouaux E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 2013, 503, 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S. K.; Piscitelli C. L.; Yamashita A.; Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science 2008, 322, 1655–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuming T.; Shi L.; Javitch J. A.; Weinstein H. A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol. Pharmacol. 2006, 70, 1630–1642. [DOI] [PubMed] [Google Scholar]
- Yamashita A.; Singh S. K.; Kawate T.; Jin Y.; Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl–-dependent neurotransmitter transporters. Nature 2005, 437, 215–223. [DOI] [PubMed] [Google Scholar]
- Forrest L. R.; Tavoulari S.; Zhang Y. W.; Rudnick G.; Honig B. Identification of a chloride ion binding site in Na+/Cl–-dependent transporters. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 12761–12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomot E.; Bendahan A.; Quick M.; Zhao Y.; Javitch J. A.; Kanner B. I. Mechanism of chloride interaction with neurotransmitter:sodium symporters. Nature 2007, 449, 726–730. [DOI] [PubMed] [Google Scholar]
- Stockner T.; Jurik A.; Weissensteiner R.; Freissmuth M.; Ecker G.; Sitte H.. Development of refined homology models: adding the missing information to the medically relevant neurotransmitter transporters. In Membrane Transport Mechanism; Krämer R.; Ziegler C., Eds.; Springer: Berlin, Heidelberg, 2014; Vol. 17, pp 99–120. [Google Scholar]
- Sali A.; Blundell T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [DOI] [PubMed] [Google Scholar]
- Shen M. Y.; Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar]
- Ramachandran G. N.; Ramakrishnan C.; Sasisekharan V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [DOI] [PubMed] [Google Scholar]
- Arnold K.; Bordoli L.; Kopp J.; Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [DOI] [PubMed] [Google Scholar]
- Keshet G. I.; Bendahan A.; Su H.; Mager S.; Lester H. A.; Kanner B. I. Glutamate-101 is critical for the function of the sodium and chloride-coupled GABA transporter GAT-1. FEBS Lett. 1995, 371, 39–42. [DOI] [PubMed] [Google Scholar]
- Anderson C. M.; Kidd P. D.; Eskandari S. GATMD: gamma-aminobutyric acid transporter mutagenesis database. Database 2010, 2010, baq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett E. R.; Su H.; Kanner B. I. Mutation of arginine 44 of GAT-1, a (Na(+) + Cl(−))-coupled gamma-aminobutyric acid transporter from rat brain, impairs net flux but not exchange. J. Biol. Chem. 2000, 275, 34106–34113. [DOI] [PubMed] [Google Scholar]
- Bismuth Y.; Kavanaugh M. P.; Kanner B. I. Tyrosine 140 of the gamma-aminobutyric acid transporter GAT-1 plays a critical role in neurotransmitter recognition. J. Biol. Chem. 1997, 272, 16096–16102. [DOI] [PubMed] [Google Scholar]
- Ben-Yona A.; Bendahan A.; Kanner B. I. A glutamine residue conserved in the neurotransmitter:sodium:symporters is essential for the interaction of chloride with the GABA transporter GAT-1. J. Biol. Chem. 2011, 286, 2826–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger Suite 2011 Induced Fit Docking Protocol; Schrödinger, LLC: New York, 2011. ; Glide, version 5.7; Schrödinger, LLC: New York, 2011. ; Prime, version 3.0; Schrödinger, LLC: New York, 2011.
- Krogsgaard-Larsen P.; Frolund B.; Frydenvang K. GABA uptake inhibitors. Design, molecular pharmacology and therapeutic aspects. Curr. Pharm. Des. 2000, 6, 1193–1209. [DOI] [PubMed] [Google Scholar]
- Andersen K. E.; Braestrup C.; Gronwald F. C.; Jorgensen A. S.; Nielsen E. B.; Sonnewald U.; Sorensen P. O.; Suzdak P. D.; Knutsen L. J. The synthesis of novel GABA uptake inhibitors. 1. Elucidation of the structure–activity studies leading to the choice of (R)-1-[4,4-bis(3-methyl-2-thienyl)-3-butenyl]-3-piperidinecarboxylic acid (tiagabine) as an anticonvulsant drug candidate. J. Med. Chem. 1993, 36, 1716–1725. [DOI] [PubMed] [Google Scholar]
- Knutsen L. J.; Andersen K. E.; Lau J.; Lundt B. F.; Henry R. F.; Morton H. E.; Naerum L.; Petersen H.; Stephensen H.; Suzdak P. D.; Swedberg M. D.; Thomsen C.; Sorensen P. O. Synthesis of novel GABA uptake inhibitors. 3. Diaryloxime and diarylvinyl ether derivatives of nipecotic acid and guvacine as anticonvulsant agents. J. Med. Chem. 1999, 42, 3447–3462. [DOI] [PubMed] [Google Scholar]
- Andersen K. E.; Sorensen J. L.; Huusfeldt P. O.; Knutsen L. J.; Lau J.; Lundt B. F.; Petersen H.; Suzdak P. D.; Swedberg M. D. Synthesis of novel GABA uptake inhibitors. 4. Bioisosteric transformation and successive optimization of known GABA uptake inhibitors leading to a series of potent anticonvulsant drug candidates. J. Med. Chem. 1999, 42, 4281–4291. [DOI] [PubMed] [Google Scholar]
- Andersen K. E.; Sorensen J. L.; Lau J.; Lundt B. F.; Petersen H.; Huusfeldt P. O.; Suzdak P. D.; Swedberg M. D. Synthesis of novel gamma-aminobutyric acid (GABA) uptake inhibitors. 5.(1) Preparation and structure–activity studies of tricyclic analogues of known GABA uptake inhibitors. J. Med. Chem. 2001, 44, 2152–2163. [DOI] [PubMed] [Google Scholar]
- Andersen K. E.; Lau J.; Lundt B. F.; Petersen H.; Huusfeldt P. O.; Suzdak P. D.; Swedberg M. D. Synthesis of novel GABA uptake inhibitors. Part 6: preparation and evaluation of N-Omega asymmetrically substituted nipecotic acid derivatives. Bioorg. Med. Chem. 2001, 9, 2773–2785. [DOI] [PubMed] [Google Scholar]
- Fjalland B. Inhibition by neuroleptics of uptake of 3H-GABA into rat brain synaptosomes. Acta Pharmacol. Toxicol. 1978, 42, 73–76. [DOI] [PubMed] [Google Scholar]
- Klepsch F.; Chiba P.; Ecker G. F. Exhaustive sampling of docking poses reveals binding hypotheses for propafenone type inhibitors of P-glycoprotein. PLoS Comput. Biol. 2011, 7, e1002036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. [DOI] [PubMed] [Google Scholar]
- Verdonk M. L.; Cole J. C.; Hartshorn M. J.; Murray C. W.; Taylor R. D. Improved protein–ligand docking using GOLD. Proteins 2003, 52, 609–623. [DOI] [PubMed] [Google Scholar]
- Eldridge M. D.; Murray C. W.; Auton T. R.; Paolini G. V.; Mee R. P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput.-Aided Mol. Des. 1997, 11, 425–445. [DOI] [PubMed] [Google Scholar]
- Bissantz C.; Folkers G.; Rognan D. Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. J. Biol. Chem. 2000, 43, 4759–4767. [DOI] [PubMed] [Google Scholar]
- Wang R.; Lai L.; Wang S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput.-Aided Mol. Des. 2002, 16, 11–26. [DOI] [PubMed] [Google Scholar]
- Labute P. The generalized Born/volume integral implicit solvent model: estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. [DOI] [PubMed] [Google Scholar]
- Wang R.; Wang S. How does consensus scoring work for virtual library screening? An idealized computer experiment. J. Comput.-Aided Mol. Des. 2001, 41, 1422–1426. [DOI] [PubMed] [Google Scholar]
- Brehm L.; Krogsgaard-Larsen P.; Johnston G. A. R.; Schaumburg K. X-ray crystallographic and proton magnetic resonance spectroscopic investigations of nipecotic acid, a potent inhibitor of γ-aminobutyric acid (GABA) uptake. Acta Chem. Scand. 1976, B30, 542–548. [Google Scholar]
- Bostrom J.; Norrby P. O.; Liljefors T. Conformational energy penalties of protein-bound ligands. J. Comput.-Aided. Mol. Des. 1998, 12, 383–396. [DOI] [PubMed] [Google Scholar]
- Skovstrup S.; David L.; Taboureau O.; Jorgensen F. S. A steered molecular dynamics study of binding and translocation processes in the GABA transporter. PLoS One 2012, 7, e39360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reulecke I.; Lange G.; Albrecht J.; Klein R.; Rarey M. Towards an integrated description of hydrogen bonding and dehydration: decreasing false positives in virtual screening with the HYDE scoring function. ChemMedChem 2008, 3, 885–897. [DOI] [PubMed] [Google Scholar]
- LigandScout, version 3.0; Inte:ligand GmbH: Vienna, 2012.
- Mysinger M. M.; Carchia M.; Irwin J. J.; Shoichet B. K. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enamine Advanced Collection; Enamine Ltd.: Riga, Latvia, May2013.
- Enamine High Throughput Screening Collection; . Enamine Ltd.: Riga, Latvia, May2013.
- Law V.; Knox C.; Djoumbou Y.; Jewison T.; Guo A. C.; Liu Y.; Maciejewski A.; Arndt D.; Wilson M.; Neveu V.; Tang A.; Gabriel G.; Ly C.; Adamjee S.; Dame Z. T.; Han B.; Zhou Y.; Wishart D. S. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, D1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE), version 2012.10; Chemical Computing Group Inc.: 1010 Sherbooke Street West, Suite 910, Montreal, QC, Canada, H3A 2R7, 2012.
- Vogensen S. B.; Jorgensen L.; Madsen K. K.; Borkar N.; Wellendorph P.; Skovgaard-Petersen J.; Schousboe A.; White H. S.; Krogsgaard-Larsen P.; Clausen R. P. Selective mGAT2 (BGT-1) GABA uptake inhibitors: design, synthesis, and pharmacological characterization. J. Med. Chem. 2013, 56, 2160–2164. [DOI] [PubMed] [Google Scholar]
- Mason G. A.; Walker C. H.; Prange A. J. Jr. Modulation of gamma-aminobutyric acid uptake of rat brain synaptosomes by thyroid hormones. Neuropsychopharmacology 1987, 1, 63–70. [DOI] [PubMed] [Google Scholar]
- Mason G. A.; Walker C. H.; Prange A. J. Jr.; Bondy S. C. GABA uptake is inhibited by thyroid hormones: implications for depression. Psychoneuroendocrinology 1987, 12, 53–59. [DOI] [PubMed] [Google Scholar]
- Hashimoto H.; Walker C. H.; Prange A. J. Jr.; Mason G. A. The effects of thyroid hormones on potassium-stimulated release of 3H-GABA by synaptosomes of rat cerebral cortex. Neuropsychopharmacology 1991, 5, 49–54. [PubMed] [Google Scholar]
- Okabe N.; Fujiwara T.; Yamagata Y.; Tomita K. The crystal structure of a major metabolite of thyroid hormone: 3,3′,5′-triiodo-l-thyronine. Biochim. Biophys. Acta, Gen. Subj. 1982, 717, 179–181. [DOI] [PubMed] [Google Scholar]
- Koerner D.; Schwartz H. L.; Surks M. I.; Oppenheimer J. H. Binding of selected iodothyronine analogues to receptor sites of isolated rat hepatic nuclei. High correlation between structural requirements for nuclear binding and biological activity. J. Biol. Chem. 1975, 250, 6417–6423. [PubMed] [Google Scholar]
- Putcha B. D.; Wright E.; Brunzelle J. S.; Fernandez E. J. Structural basis for negative cooperativity within agonist-bound TR:RXR heterodimers. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 6084–6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffert E. S.; Hofner G.; Wanner K. T. Aminomethyltetrazoles as potential inhibitors of the gamma-aminobutyric acid transporters mGAT1-mGAT4: synthesis and biological evaluation. Bioorg. Med. Chem. 2011, 19, 6492–6504. [DOI] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An Open Chemical Toolbox. J. Cheminf. 2011, 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashita M.; Sasaki K.; Sakai N.; Saito N. Effects of tricyclic and tetracyclic antidepressants on the three subtypes of GABA transporter. Neurosci. Res. (Shannon, Irel.) 1997, 29, 87–91. [DOI] [PubMed] [Google Scholar]
- Cherubino F.; Miszner A.; Renna M. D.; Sangaletti R.; Giovannardi S.; Bossi E. GABA transporter lysine 448: a key residue for tricyclic antidepressants interaction. Cell. Mol. Life Sci. 2009, 66, 3797–3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salat K.; Kulig K.; Salat R.; Filipek B.; Malawska B. Analgesic and anticonvulsant activity of new derivatives of 2-substituted 4-hydroxybutanamides in mice. Pharmacol. Rep. 2012, 64, 102–112. [DOI] [PubMed] [Google Scholar]
- Van Der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; Van Der Spoel D.; Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [DOI] [PubMed] [Google Scholar]
- Wolf M. G.; Hoefling M.; Aponte-Santamaria C.; Grubmuller H.; Groenhof G. g_membed: Efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation. J. Comput. Chem. 2010, 31, 2169–2174. [DOI] [PubMed] [Google Scholar]
- Oostenbrink C.; Villa A.; Mark A. E.; van Gunsteren W. F. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [DOI] [PubMed] [Google Scholar]
- Sadowski J.; Gasteiger J.; Klebe G. Comparison of automatic three-dimensional model builders using 639 X-ray structures. J. Chem. Inf. Comput. Sci. 1994, 34, 1000–1008. [Google Scholar]
- LigPrep, version 2.5; Schrödinger, LLC: New York, 2011.
- Frisch, M. J. T G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam M. J.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision A.02; Gaussian, Inc.: Wallingford CT, 2009.
- Valero R.; Costa R.; de P. R. M. I.; Truhlar D. G.; Illas F. Performance of the M06 family of exchange-correlation functionals for predicting magnetic coupling in organic and inorganic molecules. J. Chem. Phys. 2008, 128, 114103. [DOI] [PubMed] [Google Scholar]
- Rassolov V. A.; Ratner M. A.; Pople J. A.; Redfern P. C.; Curtiss L. A. 6-31G* basis set for third-row atoms. J. Comput. Chem. 2001, 22, 976–984. [Google Scholar]
- Wolber G.; Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [DOI] [PubMed] [Google Scholar]
- Wolber G.; Dornhofer A. A.; Langer T. Efficient overlay of small organic molecules using 3D pharmacophores. J. Comput.-Aided Mol. Des. 2006, 20, 773–788. [DOI] [PubMed] [Google Scholar]
- OMEGA, version 2.2.3; OpenEye Scientific Software: Santa Fe, NM.
- Hawkins P. C.; Skillman A. G.; Warren G. L.; Ellingson B. A.; Stahl M. T. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins P. C.; Nicholls A. Conformer generation with OMEGA: learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [DOI] [PubMed] [Google Scholar]
- Prism, version 6.01; GraphPad Software: San Diego, CA, 2012.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Enamine Advanced Collection; Enamine Ltd.: Riga, Latvia, May2013.
- Enamine High Throughput Screening Collection; . Enamine Ltd.: Riga, Latvia, May2013.