

Abstract
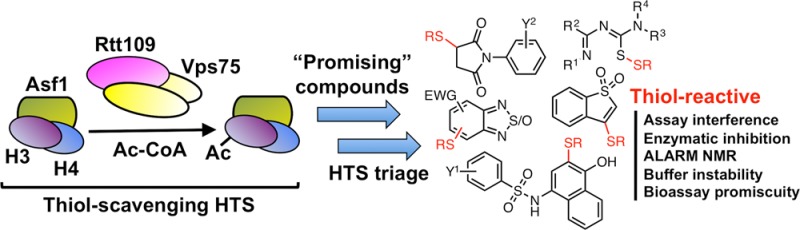
Significant resources in early drug discovery are spent unknowingly pursuing artifacts and promiscuous bioactive compounds, while understanding the chemical basis for these adverse behaviors often goes unexplored in pursuit of lead compounds. Nearly all the hits from our recent sulfhydryl-scavenging high-throughput screen (HTS) targeting the histone acetyltransferase Rtt109 were such compounds. Herein, we characterize the chemical basis for assay interference and promiscuous enzymatic inhibition for several prominent chemotypes identified by this HTS, including some pan-assay interference compounds (PAINS). Protein mass spectrometry and ALARM NMR confirmed these compounds react covalently with cysteines on multiple proteins. Unfortunately, compounds containing these chemotypes have been published as screening actives in reputable journals and even touted as chemical probes or preclinical candidates. Our detailed characterization and identification of such thiol-reactive chemotypes should accelerate triage of nuisance compounds, guide screening library design, and prevent follow-up on undesirable chemical matter.
Introduction
The growing use of high-throughput screening (HTS) as a discovery tool in academic translational centers has resulted in the pursuit of assay artifacts, promiscuous bioactive compounds, and screening actives with major absorption, distribution, metabolism, excretion, and toxicological (ADMET) liabilities. A similar situation may exist in industry, and this observation may simply be a reflection of academic pressures to publish. In either case, the follow-up of such compounds can significantly burden the post-HTS triage and hit-to-lead stages of the discovery process. Therefore, chasing assay artifacts and promiscuous screening compounds can waste both time and other valuable resources, and failure to triage these compounds has led to many artifacts and “frequent hitters” making their way into the scientific literature, patent applications, and research funding applications.
As an example, pan-assay interference compounds (PAINS) can display apparent bioactivity and/or interfere with assay readouts across unrelated biological targets and testing methods.1−3 Multiple sources for promiscuous behavior or assay interference have been described, including: chemical aggregation,4 chelation,5 singlet oxygen production,6 compound fluorescence effects,7,8 redox activity,9 sample impurities,10−15 membrane disruption,16 cysteine oxidation,17 and nonselective compound reactivity with proteins.18 Several well-designed experiments using firefly luciferase have also shown compound–reporter interference as the most likely source of biological assay readouts in a compound that has progressed to human clinical trials.19−25 An important point with these luciferase experiments is that confounding readouts are not isolated to cell-free assays. Cell-based assays with perturbations in cell proliferation may be particularly susceptible to assay interference or off-target and confounding effects. Misleading readouts can have clinical relevance, as a recent study suggests the pharmacological activity of acamprosate (an FDA-approved drug for relapse prevention in alcoholism) may be due to the calcium cation component of its formulation rather than the long-presumed bioactive ingredient, N-acetylhomotaurinate.26
Despite the risks associated with pursuing these types of undesirable compounds, their identities and the chemical mechanisms by which they can mislead even seasoned researchers often go uncharacterized in the pursuit of identifying lead compounds. Unfortunately, this leaves open the possibility for other groups to fall into the same scientific maelstrom that most often results in costly failure. In an effort to alert the uninitiated, we describe herein the structure–interference relationships (SIR) in five series of problematic compounds we encountered in a recent HTS campaign.
Epigenetic enzymes, such as histone deacetylases, methyltransferases, and histone acetyltransferases (HATs), are an important emerging class of therapeutic targets. Epigenetic chemical probes and enzymatic modulators are sought for a variety of human diseases including cancers.27 Our group and others have focused on a series of enzymes unique to fungi, Rtt109 HATs, that are critical for DNA replication-coupled nucleosome assembly and genomic stability and therefore may represent a novel antifungal therapeutic approach.28−31 Several types of HTS technologies have been adapted to screening epigenetic targets. Antibody-based approaches can probe for specific histone modifications such as methylation and acetylation.32 Another more indirect approach probes for reaction byproducts via chemical probes or reporter enzymes. One chemical probe, N-[4-(7-diethylamino-4-methylcoumarin-3-yl)phenyl]maleimide (CPM), readily reacts with free thiols to form highly fluorescent adducts.33,34 Several CPM-based assays and screens have been reported for multiple biological targets, including some epigenetic enzymes.29,35−41
Recently, our group screened approximately 225K small molecules for their ability to inhibit Rtt109-catalyzed histone acetylation using a cell-free CPM-based HTS.42 PAINS were computationally filtered at the beginning of our triage and were not initially evaluated in our post-HTS counter-screens. Post-HTS triage of approximately 1.5K primary screening hits demonstrated only a few confirmed actives. In retrospect, this indicated a significant portion of the screening hits were either false positives or assay artifacts resulting from fluorescence quenching, compound–reagent interference, and/or other mechanisms. On the basis of the chemical structures of the triaged compounds, we speculated many chemotypes from the primary HTS campaign were reacting with the CoA byproduct to produce an interfering assay readout mimicking enzymatic inhibition. We also speculated many of these thiol-reactive compounds, including several series of PAINS we had previously triaged, could also inhibit enzymatic activity by reacting with protein cysteines, a recognized source of promiscuous enzyme inhibition and metabolic liability. We observed chemotypes that were enriched among the actives that appeared chemically similar to many of the published PAINS substructures but were not flagged by our cheminformatic PAINS filters.1,42
There is a growing interest in both assay interference and promiscuous enzymatic inhibition, including nonspecific thiol reactivity.18,43,44 Therefore, identifying thiol-reactive chemotypes in compound screening libraries is important for enhancing library design and post-HTS decision-making. Additionally, the characterization of the chemical mechanisms of thiol reactivity may also be useful for reactivity prediction, compound optimization, and the avoidance of follow-up on compounds that may have metabolic or selectivity liabilities further downstream in the drug discovery pipeline. The observation that some PAINS-like compounds may escape cheminformatics filters may have significant consequences for screening centers without experienced HTS triage personnel, especially if those performing HTS triage are overly reliant on cheminformatics filters.45
Herein, we report the identification of multiple chemotypes (“chemical structural motifs”) that showed publication-quality IC50 values in our primary assay but through a series of orthogonal assays and counter-screens showed thiol-reactive assay interference and also promiscuous enzymatic inhibition. Using SIR and structure–activity relationships (SAR) from our Rtt109 post-HTS triage, along with multiple analytical techniques, we provide evidence supporting several chemical mechanisms of assay interference relating to thiol reactivity. We show these chemotypes can form covalent adducts with other biologically relevant thiols such as glutathione (GSH) and cysteine residues on multiple structurally unrelated proteins. The chemical mechanisms we propose contributing to assay promiscuity include addition–elimination reactions, nucleophlic aromatic substitution, buffer instability, disulfide bond formation, and H2O2 production. Our findings may be more broadly applicable, as several compounds containing these chemotypes formed covalent adducts with the La antigen (ALARM NMR), demonstrating potentially broad-spectrum thiol reactivity. Furthermore, compounds with these chemotypes showed evidence of assay promiscuity in analyses of PubChem and the HTS database of a major pharmaceutical company. Despite these red flags, several such compounds have been reported in the patent literature and reputable scientific journals with varying claims of target specificity and utilities as either chemical probes or therapeutic leads. It is hoped that the identification and detailed characterization of these thiol-reactive chemotypes can accelerate post-HTS triage, enhance lead identification, and prevent follow-up on unpromising chemical matter by other researchers.
Results
Identification of Artifact Chemotypes in a Fluorescence-Based HTS
We previously reported the use of a CPM-based method to screen approximately 225K compounds for their abilities to inhibit Rtt109-catalyzed histone acetylation in vitro.42 In a HAT reaction, an acetyl group is enzymatically transferred from acetyl-CoA to the ε-amino group of a histone lysine side chain, resulting in the production of an acetylated lysine and a CoA byproduct. The free thiol on CoA can then react with suitable substrates, such as maleimide-based probes like CPM, to form highly fluorescent adducts that can indirectly assay HAT activity (Figure 1A). In practice, we and others have found this method to be low-cost and relatively robust.46 We were also aware that this method is subject to assay interference by thiol-containing compounds.47 However, we soon learned that this assay is highly susceptible to other mechanisms of reactive compound interference especially when testing potentially heterogeneous chemical matter like HTS libraries. In principle, compounds can interfere with the CPM fluorescence intensity by fluorescence quenching. Also, compounds with nucleophilic or electrophilic reactivity can react with either the CPM probe or the CoA reaction product, respectively.
Figure 1.

Susceptibility of CPM-based HTS to screening compound-based interference. (A) Assay schematic for the CPM-based HTS used in this study. The assay measures the HAT activity of the Rtt109–Vps75 complex, which catalyzes the transfer of an acetyl moiety from acetyl-CoA to specific lysine residues on the Asf1–dH3–H4 substrate complex to produce acetylated histone residues and coenzyme A (CoA). Addition of the thiol-scavenging probe CPM leads to a highly fluorescent adduct by reacting with the CoA byproduct, which is used to quantify HAT activity via fluorescence intensity measurement. (B) Representative assay interference chemotypes identified during post-HTS triage.
Despite identifying 1.5K actives in the primary screen, a post-HTS triage consisting of computational filtering, experimental counter-screens, and orthogonal assays demonstrated only three compounds that could inhibit Rtt109-catalyzed histone acetylation. Therefore, significant portions of this collection of experimental and computationally filtered (“filtrand”) compounds were either false positives or assay artifacts. In addition, many of the computationally filtered actives were flagged as PAINS, which we believed could inhibit HAT activity by nontherapeutically useful mechanisms. In all, several prominent chemotypes were identified among the primary actives during the course of the post-HTS triage (Figure 1B).
On the basis of their chemical structures, it appeared likely that each chemotype could interfere with our HTS assay readout through different and, perhaps, multiple thiol-trapping mechanisms. Many of these compounds were flagged as PAINS, but still many were not verified as “bad actors” until relatively late in our triage process. To further confound matters, these electrophilic compounds could conceivably inhibit enzymatic activity by nonspecific reactivity with the protein components in the assay, further scrambling the assay readout. To better understand the chemical mechanism(s) behind this form of assay behavior and to assess whether this interference could have implications beyond our HTS, we examined five prominent subclasses of compounds in more detail.
Orthogonal and Counter-Screens Identify Inhibitors among Interference Compounds
Several classes of compounds demonstrated low micromolar IC50 values in the CPM-based HTS method (Figure 2). Compounds with steep Hill slopes (e.g., >2) have been associated with cooperativity and anomalous binding behaviors, such as chemical aggregation.42,48,49 Most of the compounds showed slightly elevated Hill slopes, but this was not an immediate concern, as we had included a detergent (Triton X-100) in our assay buffers to mitigate micelle formation.
Figure 2.

Dose–responses of select screening compounds in the Rtt109 HTS and an assay interference counter-screen. Shown are representative examples from chemotypes 1, 2, 3, 4, and 6, which displayed promising low micromolar IC50 values by the primary HTS assay (solid lines). A counter-screen that replaced the acetyl-CoA substrate with the CoA reaction product produced similar dose–response curves by the same assay readout (dashed lines). Data are mean ± SD for three replicates.
Reaction aliquots from the active compounds (1a, 2a, 3a, 4a, 6a, and 6b) all showed decreases in histone acetylation at 125 μM when they were analyzed by an orthogonal slot blot assay that uses H3K56ac- and H3K27ac-specific antibodies to probe for the acetylated histone lysine product rather than the CoA byproduct (Tables 1–5). We examined both histone modifications because the Rtt109–Vps75 complex is capable of acetylating multiple histone H3 residues. Overall, reaction aliquots showed similar levels of histone acetylation, regardless of whether H3K27ac or H3K56ac was examined, strongly suggesting that any observed enzymatic inhibition was not specific to one particular histone modification. This assay also detected decreases in histone acetylation with garcinol, a natural product previously shown to inhibit Rtt109 activity and other HATs in vitro at low micromolar compound concentrations.42,50 When the same assay was used to examine reaction aliquots at lower compound concentrations (8 μM), we observed more discrepancies between the HTS and slot blot readouts (data not shown). Overall, these observations showed that representatives of these five compound classes could inhibit acetylation activity at high concentrations, but their HTS assay readouts were confounded by assay interference near their apparent IC50 values.
Table 1. Benzothiophene 1,1-Dioxide Seriese.

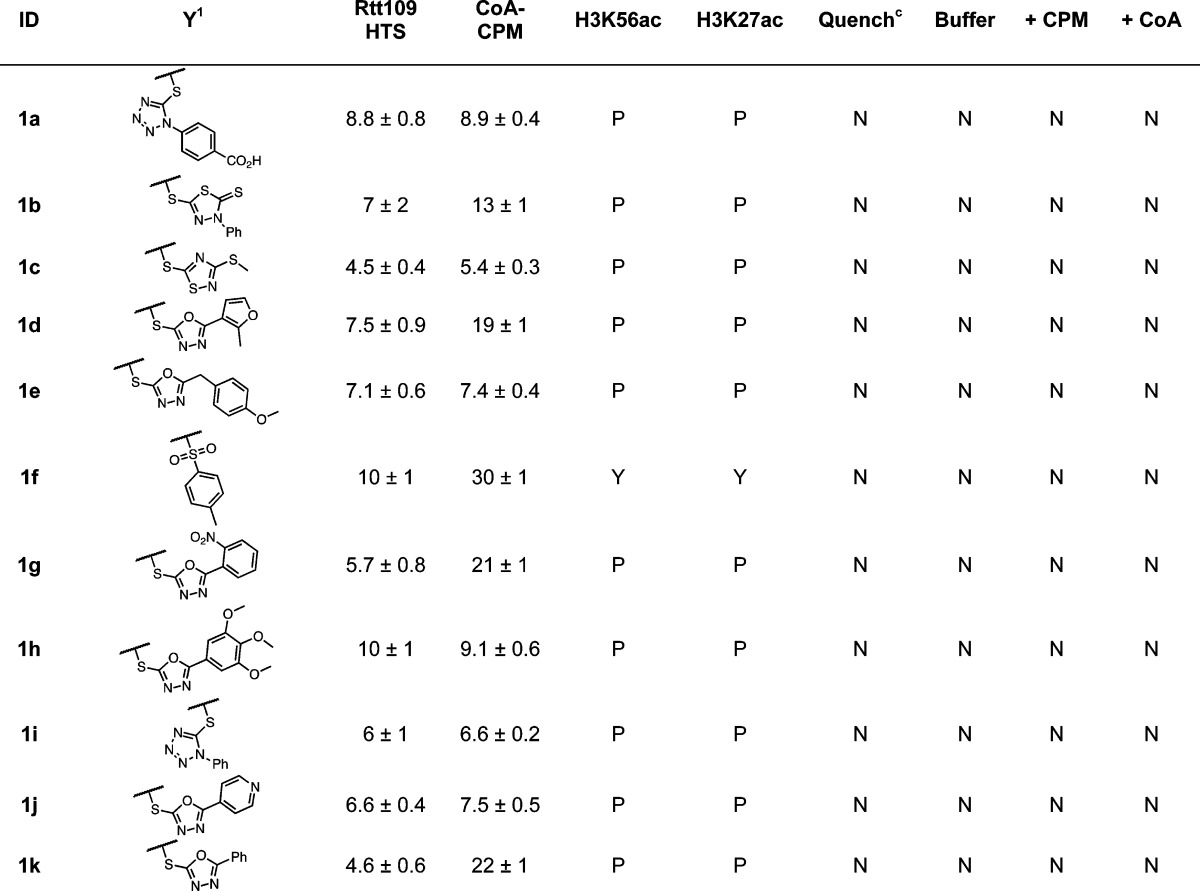
HTS refers to IC50 values calculated from the CPM-based Rtt109 HTS method, CoA-CPM refers to IC50 values calculated from the CoA-based HTS counter-screen.
HTS reaction aliquots from compounds tested at 125 μM final concentrations; Yes (“Y”), No (“N”), or Partial (“P”).
Compounds flagged as quenchers if greater than 20% assay signal reduction at 10 μM final concentrations.
Compounds flagged if fluorescence intensity greater than 20% assay signal at 125 μM final concentrations; compounds tested in either HTS buffer (“Buffer”), HTS buffer plus 20 μM CPM (“+ CPM”), or HTS buffer plus 7.5 μM CoA (“+ CoA”).
IC50 values shown are means ± SD for three replicates.
Table 5. p-Hydroxyarylsulfonamide Seriese.
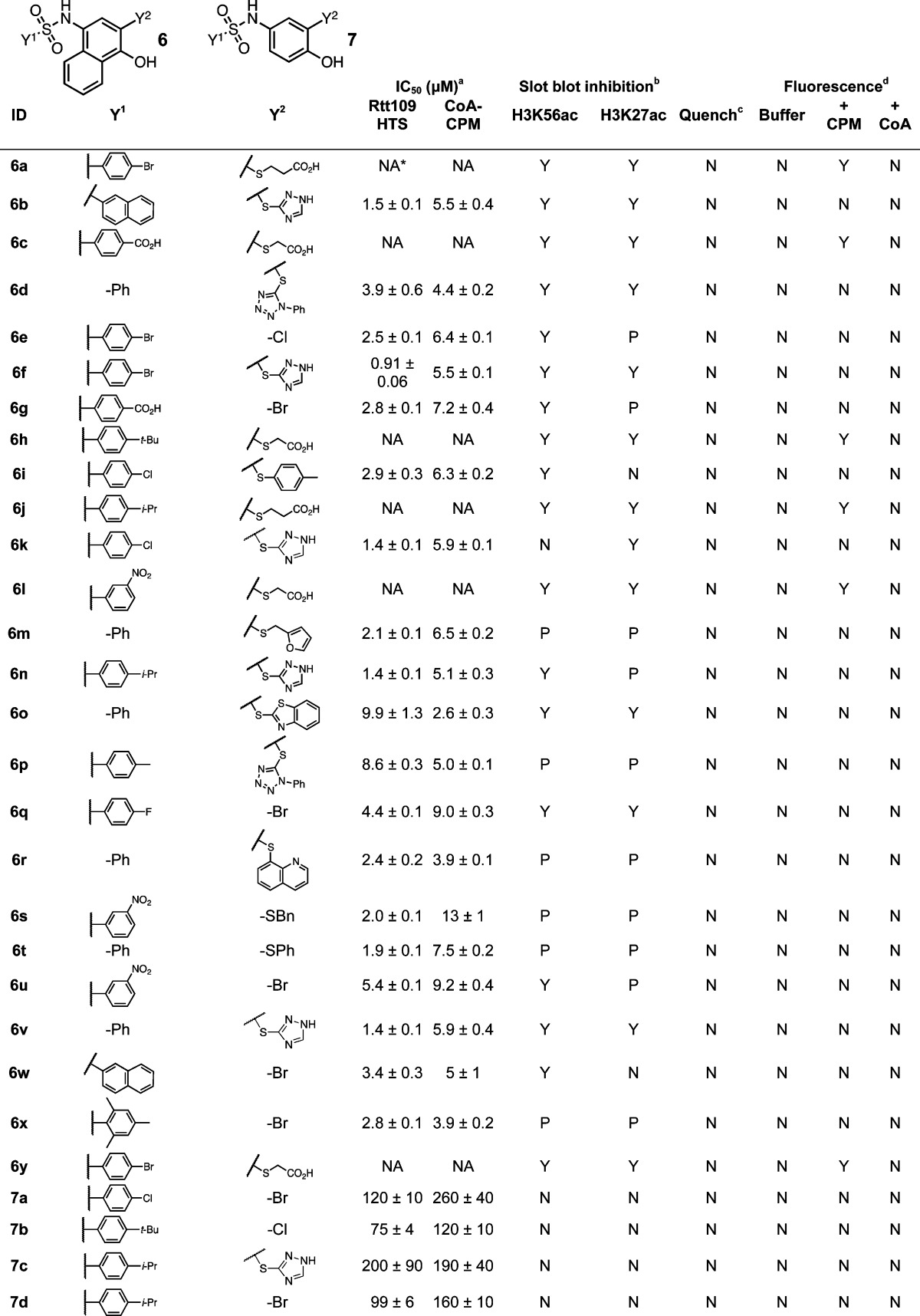
HTS refers to IC50 values calculated from the CPM-based Rtt109 HTS method, CoA-CPM refers to IC50 values calculated from the CoA-based HTS counter-screen
HTS reaction aliquots from compounds tested at 125 μM final concentrations; Yes (“Y”), No (“N”) or Partial (“P”).
Compounds flagged as quenchers if greater than 20% assay signal reduction at 10 μM final concentrations.
Compounds flagged if fluorescence intensity greater than 20% assay signal at 125 μM final concentrations; compounds tested in either HTS buffer (“Buffer”), HTS buffer plus 20 μM CPM (“+ CPM”), or HTS buffer plus 7.5 μM CoA (“+ CoA”).
NA denotes IC50 value not available due to fluorescence interference.
IC50 values shown are means ± SD for three replicates.
To further examine the mechanisms underlying this assay interference, we first established a fluorescence-quenching counter-screen to assess for fluorescence interference. This assay accurately identified the fluorescence quencher BHQ-1, a positive control (Supporting Information, Figure S1). However, none of these compounds (1a, 2a, 3a, 4a, and 6b) showed evidence of fluorescence quenching, intrinsic fluorescence or the generation of fluorescent adducts with either CoA or CPM in a set of assays mimicking our HTS procedures (Tables 1–5). When the acetyl-CoA reactant was replaced by the CoA reaction byproduct, these compounds provided assay readouts that were strikingly similar to those obtained under the HTS assay conditions (Figure 2). The degree of assay signal reduction was also dependent on the levels of CoA present (data not shown). Similar assay behavior was observed for a variety of chemical analogues bearing these chemotypes (Tables 1–5). Therefore, the body of evidence strongly implicated a thiol-trapping mechanism of assay interference. Next, we sought to understand the chemical basis of this interference and potential sources of enzymatic inhibition.
Further Characterization of Compound–Thiol Adducts by UPLC-MS and LC-HRMS
To provide more direct evidence of the presumed compound–CoA adducts, we incubated the compounds with CoA under HTS-like conditions. We also tested for adducts with reduced l-glutathione (GSH), another important biological thiol, to assess if this presumed thiol reactivity was unique to CoA. UPLC–MS and LC–HRMS analyses showed that the test compounds (1a, 2a, 3a and 4a) form adducts with both CoA and GSH (Figure 3 and Supporting Information, Figure S2). We also observed the similar and expected adducts by UPLC–MS for multiple other representative compounds from each chemotype (data not shown). While the p-hydroxyarylsulfonamides 6a–6e yielded the expected compound-GSH adducts (6a′–6e′; Figures 4A,B), we also observed that 6a′–6e′ were really intermediates that converted to a common adduct in situ after only 15 min under the HTS conditions (Figure 4B,C).
Figure 3.

Compound–GSH adducts detected by qualitative UPLC–MS. (A) Selected interference compounds were incubated with MeOH (black traces), HTS buffer (blue traces), or HTS buffer plus GSH (red traces) and analyzed by UPLC–MS. Shown are overlays of the simultaneous ELS and 254 nm traces. Selected mass spectra are also shown for a select sample in MeOH (black spectrum) and selected adducts (red spectra). Numbers in parentheses represent the predominant ion molecular weight (“–” denotes negative ion mode). Data are representative results from one of at least two independent experiments. (B) Simplified schematics of the proposed reaction mechanisms to generate the observed adducts.
Figure 4.
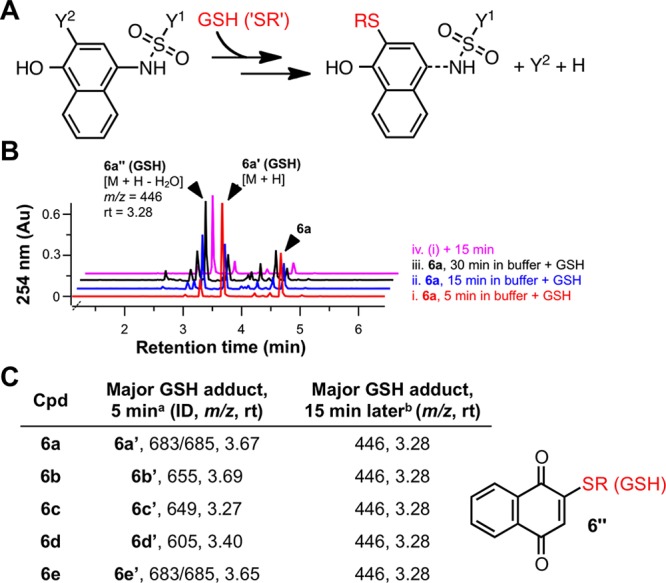
Labile adducts between p-hydroxyarylsulfonamides (6) and GSH detected by qualitative UPLC–MS. (A) Simplified scheme of adduct formation between biological thiols and chemotype 6. (B) UPLC–MS analyses of compound 6a mixed with GSH in HTS buffer. 6a was treated with GSH after varying lengths of incubation in HTS buffer (5, 15, 30 min). After 5 min, reaction aliquots were analyzed by UPLC–MS. Trace (iv) shows the same sample from trace (i) analyzed 15 min later. (C). Summary of experiments described in (B) performed with compounds 6a–6e. All test compounds initially formed the expected adducts (6a′–6e′). A common breakdown product 6″ was detected for all five sulfonamides tested (rt = 3.28 min, m/z = 446). See Supporting Information, Figures S5, S7, and S11, for additional stability studies with chemotype 6. a = compound incubated in HTS buffer for 5 min, then GSH added, then analyzed by UPLC-MS 5 min later; b = same sample from a, but analyzed by UPLC-MS 15 min later.
Together, this data is consistent with a thiol-trapping mechanism as a major contributor to the CPM-based assay signal reduction in the compound classes studied, as the tested compounds reacted with both CoA and GSH. We note the generation of compound–GSH adducts is an important consideration for certain cell-based assays, or for in situ or in vivo assays, where xenobiotic–glutathione conjugation is a major source of Phase II metabolism.
Proposed Chemical Mechanisms of Thiol Reactivity
The selected compounds interfere with the HTS assay readout and form thiol adducts by a variety of chemical mechanisms (Figures 3B and 4A). On the basis of the UPLC–MS and chemical principles, we propose the following chemical mechanisms of thiol reactivity for chemotypes 1, 2, 3, 4, and 6 (Figure 1):
Benzo[b]thiophene 1,1-dioxides 1 (“benzothiophenes”) interfere via a straightforward Michael addition–elimination reaction at the electrophilic C3-position through thiolate nucleophilic attack. The compounds with chemotype 1 most likely to interfere and form thiol adducts in our experimental conditions were those with S-linked heteroaromatic substituents (Table 1). The UPLC–MS experiments using 1a confirmed the presence of the adduct 1a′ and the leaving group 1a″ (Figure 3A). This proposed mechanism is also supported by the observations that several 2,3-dihydro analogues did not show appreciable levels of apparent Rtt109 inhibition or interference in the CoA–CPM counter-screen (Supporting Information, Figure S3). The level of assay interference is consistent with the leaving group ability of the C3 substituent, as compounds with N-, O-, or C-linked groups at this C3-position did not show as significant levels of interference or apparent inhibition (Supporting Information, Figure S3). Most of the compounds with chemotype 1 showed only partial decreases in histone acetylation at high compound concentrations (Table 1), demonstrating these compounds can weakly inhibit Rtt109 activity in our HTS, most likely by nonspecific thiol reactivity (Table 1).
The benzothiadiazole/benzofurazan scaffold 2 likely forms thiol adducts via nucleophilic aromatic substitution through a Meisenheimer complex intermediate between the nucleophilic thiol and the strongly electrophilic heteroaromatic core. The benzofurazan core has been previously associated with promiscuous thiol reactivity,18,43 while some related benzothiadiazoles have been reported as PAINS (e.g., substructures “diazox_B” and “diazox_sulfon_A”).1 Additionally, similar compounds have been shown to form covalent adducts with proteins.51 The related compound 4-chloro-7-nitrobenzofurazan (“NBD-Cl”) and its derivatives are widely used as probes for studying thiols in biological systems.52 UPLC–MS analysis of 2a demonstrates that the parent compound is stable to the HTS buffer but that the addition of a thiol source leads to near-complete conversion to the thiol adduct 2a′ with the thiopurine serving as the leaving group (Figure 3A). The compounds that showed the strongest apparent enzyme inhibition and interference contained electron-withdrawing substituents such as nitro groups and halogens, although there was no apparent reactivity difference between benzothiadiazoles and benzofurazans. Another important feature for interference was the presence of an S-linked aryl substituent, which serves as the leaving group even when there are other electron-withdrawing groups present (Table 2). Benzothiadiazoles without these features did not show significant levels of apparent enzyme inhibition or assay interference (Supporting Information, Figure S4). While flagged as PAINS, many sulfoxide-substituted analogues tested in our system were inactive and noninterfering, an observation we attribute to the absence of an additional strong electron-withdrawing group (e.g., nitro). Many compounds with chemotype 2 were capable of completely inhibiting Rtt109-catalyzed histone acetylation in our HTS (Table 2).
Table 2. Benzothiadiazole/Benzofurazan Seriese.
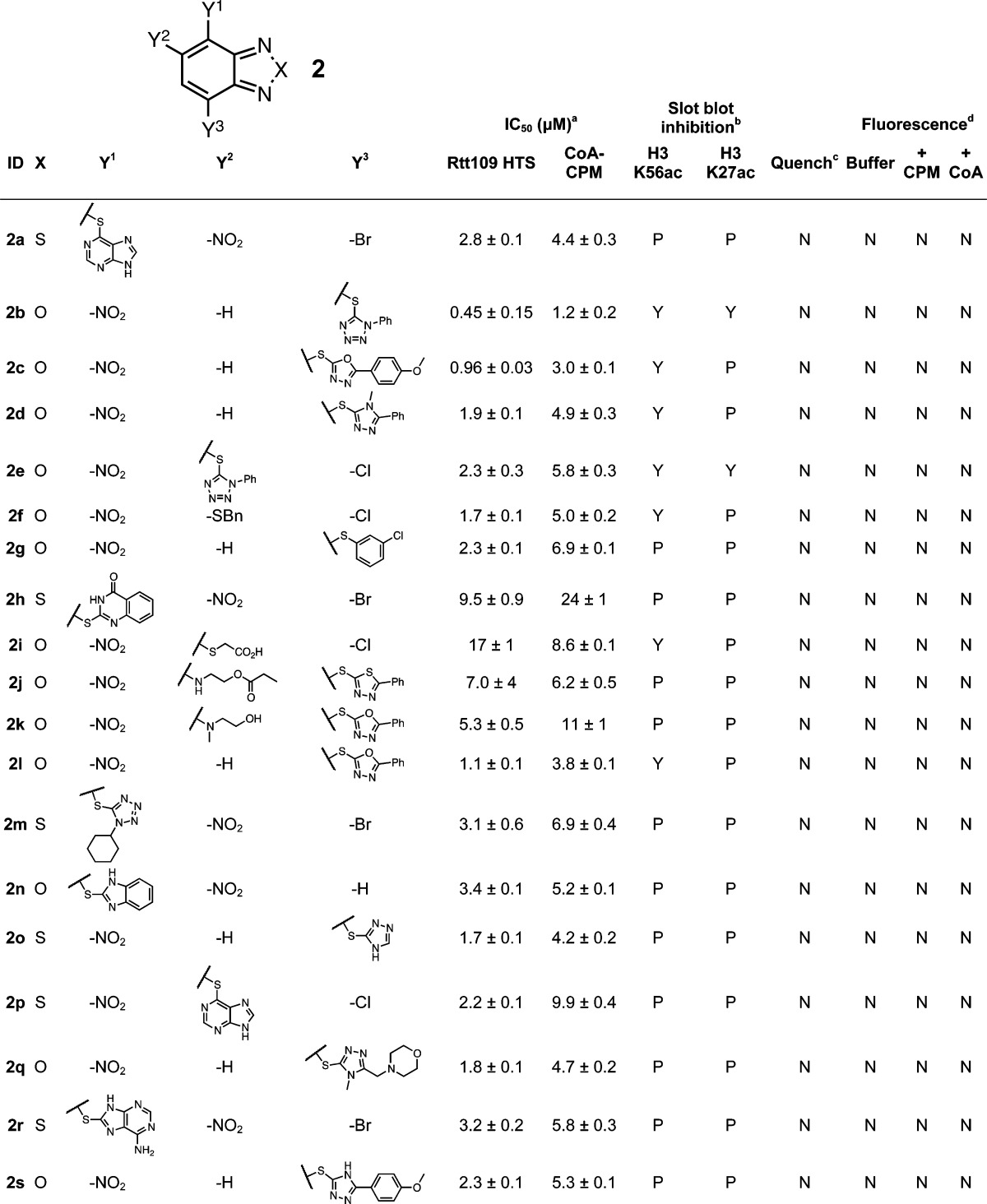
HTS refers to IC50 values calculated from the CPM-based Rtt109 HTS method, CoA-CPM refers to IC50 values calculated from the CoA-based HTS counter-screen.
HTS reaction aliquots from compounds tested at 125 μM final concentrations; Yes (“Y”), No (“N”) or Partial (“P”).
Compounds flagged as quenchers if greater than 20% assay signal reduction at 10 μM final concentrations.
Compounds flagged if fluorescence intensity greater than 20% assay signal at 125 μM final concentrations; compounds tested in either HTS buffer (“Buffer”), HTS buffer plus 20 μM CPM (“+ CPM”). or HTS buffer plus 7.5 μM CoA (“+ CoA”).
IC50 values shown are means ± SD for three replicates.
While the core 1,2,4-thiadiazole heterocycle 3 may appear benign, many such compounds can react quite readily with thiols53−57 but not other functional groups like alcohols or amines.58,59 Many properties of 1,2,4-thiadiazoles have been documented.60−62 The 1,2,4-thiadiazole core can be susceptible to ring-opening reactions, recyclization side products, and nonenzymatic reductions.63−67 Notably, this scaffold is similar to the “het_thio_N_5A” PAINS substructure, although it differs by resonance and substitution at the N2-position, meaning this chemotype could bypass some PAINS filters depending on its structural representation and certain chemoinformatic parameters.1
This speculation aside, the likely chemical mechanism of interference in our assay is sulfhydryl-scavenging by the 1,2,4-thiadiazole core at the S1-position, specifically a ring-opening reaction that generates a disulfide that can then be reduced by another thiol or electron source in situ to form the corresponding thiourea.57 Indeed, we first observed the formation of the thiourea form (3a″), as evidenced by a major shift in the UPLC retention time upon the addition of thiols (Figure 3A). The parent ions for this entity (i.e., m/z = 270) were difficult to observe by UPLC–MS, and notably we did not observe any coeluting GSH ions, suggesting this peak was not the 3a′ form with an attached GSH moiety. To gain a further structural understanding of the 3a adducts, we synthesized it under HTS-like conditions and characterized its identity and structure in situ by LC–HRMS. This data further pointed toward the detectable “adduct” being the thiourea form 3a″ rather than the direct compound–GSH 3a′ adduct (Supporting Information), which is consistent with a previous report on this chemotype.57 These data, combined with our findings that compounds 3 are strongly reactive in our thiol-trapping interference screen, suggests the 3–GSH adduct forms (3′) are not stable to our characterization procedures and/or our LC-MS conditions.
Examination of close analogues showed the assay interference strongly correlates with additional alkylation at the core N2-position to generate a partially cationic nitrogen, which presumably activates the S1–N2 bond for thiol-mediated cleavage. Compounds lacking these substituents on the N2-position were inactive and showed minimal interference (Supporting Information, Figure S5). Of note, another related PAINS substructure is “het_5_inium”, which bears resemblance to the charged 1,2,4-thiadiazoles in this chemotype. Neither the nature of the R1–R4 substituents nor the particular salt composition appeared to have significant effects on thiol-trapping (Table 3). Consistent with this mechanism, many 1,2,4-oxadiazole analogues showed minimal assay activity and interference (Supporting Information, Figure S5). We observed that many of these 1,2,4-thiadiazoles inhibited Rtt109-catalyzed histone acetylation quite effectively (Table 3), suggesting these compounds can not only interfere with the HTS assay but can also inhibit enzymatic activity, again presumably by nonspecific reactivity with protein thiols.
Table 3. 1,2,4-Thiadiazole Seriese.
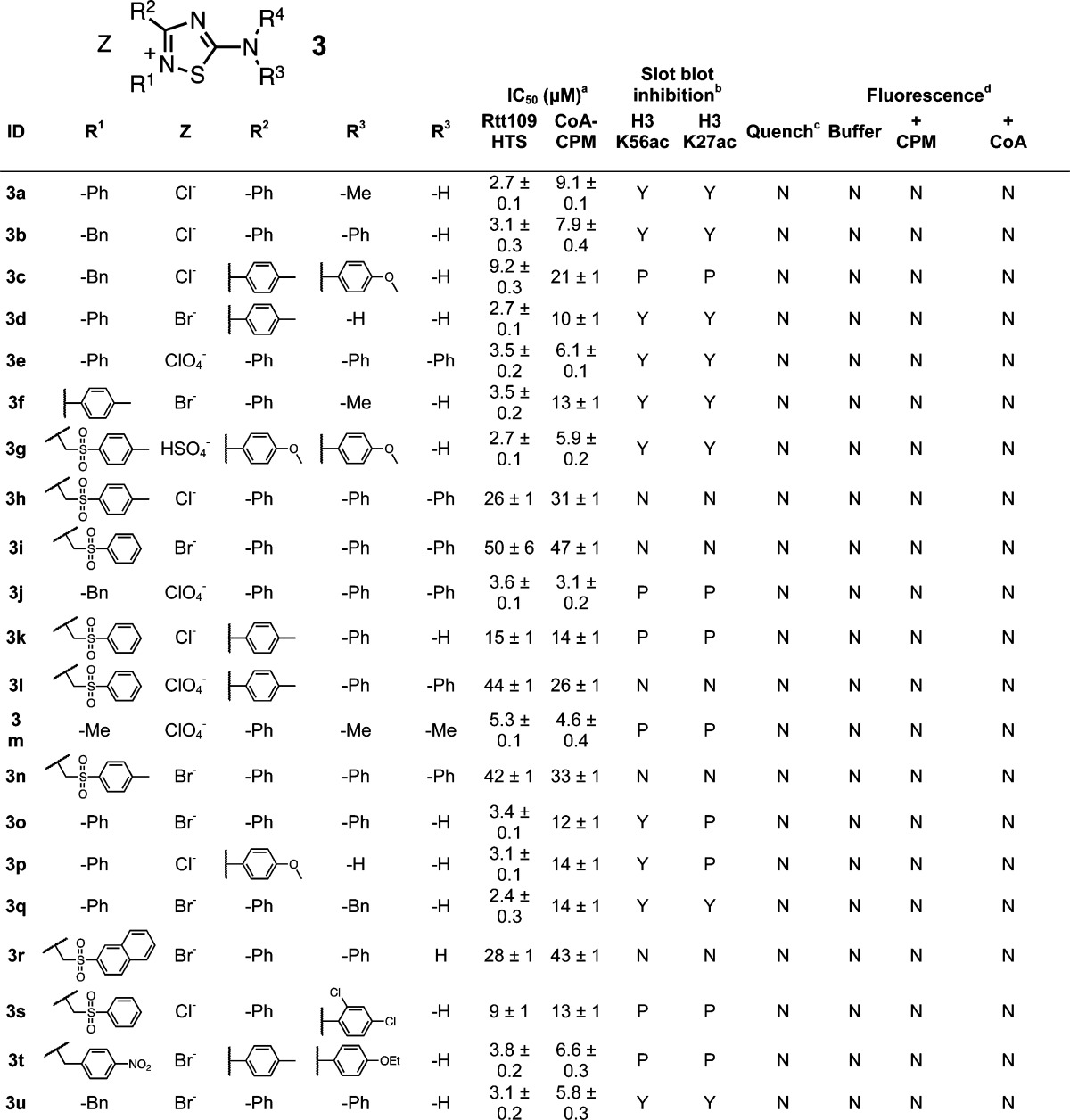
HTS refers to IC50 values calculated from the CPM-based Rtt109 HTS method, CoA-CPM refers to IC50 values calculated from the CoA-based HTS counter-screen.
HTS reaction aliquots from compounds tested at 125 μM final concentrations; Yes (“Y”), No (“N”) or Partial (“P”).
Compounds flagged as quenchers if greater than 20% assay signal reduction at 10 μM final concentrations.
Compounds flagged if fluorescence intensity greater than 20% assay signal at 125 μM final concentrations; compounds tested in either HTS buffer (“Buffer”), HTS buffer plus 20 μM CPM (“+ CPM”), or HTS buffer plus 7.5 μM CoA (“+ CoA”).
IC50 values shown are means ± SD for three replicates.
For the succinimide chemotype 4, our data is consistent with an elimination event followed by a Michael addition of a free thiol to the resulting maleimide. This is the same sulfhydryl-sensitive group present in the CPM probe used in our HTS. Elimination is likely, given the slightly alkaline pH of the assay buffer (pH 8.0). The thiol leaving groups (e.g., 4a″) were detected by UPLC–MS when the parent compound 4a was incubated in HTS buffer. This same proposed leaving group was not detected when 4a was incubated in neat MeOH (Figure 3A). Several trends also support this proposed mechanism. First, the proposed elimination product maleimides 5 showed nearly identical IC50 values in both the HTS and interference counter-screens compared to their parent succinimides (Table 4). Second, the apparent enzymatic inhibition and counter-screen IC50 values correlate well with the presumed leaving-group ability of the succinimide substituent. For instance, succinimides with S-linked aryl groups showed significant assay interference (Table 4), while nonaryl, S-linked leaving groups showed no significant activity and interference (Supporting Information, Figure S6). Third, succinimides with N-linked substituents did not appear to inhibit Rtt109 in our HTS or show interference in our counter-screens (Supporting Information, Figure S6). The substituents on the resulting maleimides did not have a noticeable effect on either the HTS or counter-screen IC50 values (Table 4). Most of the interfering succinimides 4 could only partially inhibit Rtt109 activity at higher compound concentrations in our HTS, which may be a reflection of the kinetics of the succinimide-to-maleimide conversion in buffer.
Table 4. Succinimide Seriese.
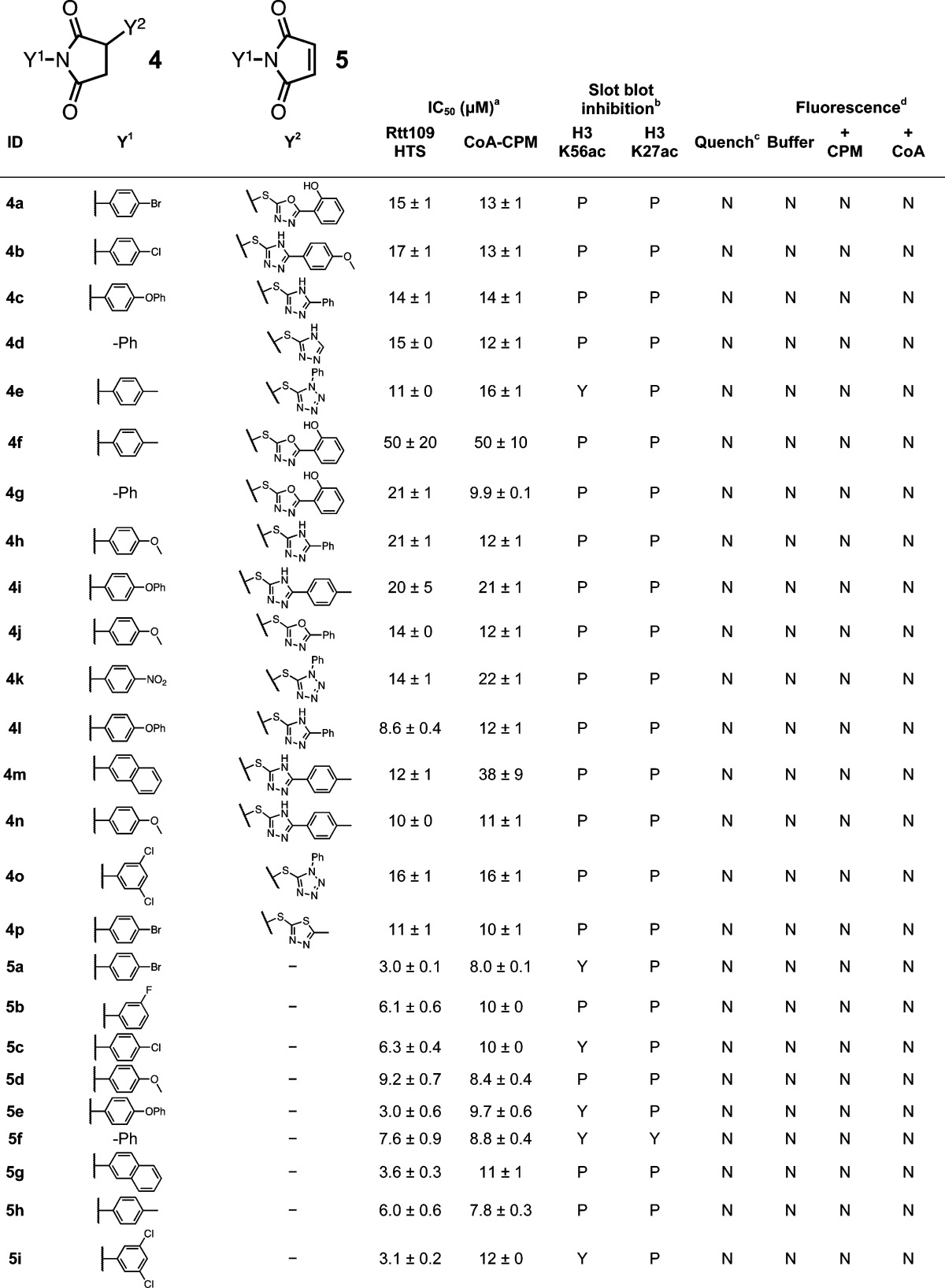
HTS refers to IC50 values calculated from the CPM-based Rtt109 HTS method, CoA-CPM refers to IC50 values calculated from the CoA-based HTS counter-screen
HTS reaction aliquots from compounds tested at 125 μM final concentrations; Yes (“Y”), No (“N”) or Partial (“P”).
Compounds flagged as quenchers if greater than 20% assay signal reduction at 10 μM final concentrations.
Compounds flagged if fluorescence intensity greater than 20% assay signal at 125 μM final concentrations; compounds tested in either HTS buffer (“Buffer”), HTS buffer plus 20 μM CPM (“+ CPM”), or HTS buffer plus 7.5 μM CoA (“+ CoA”).
IC50 values shown are means ± SD for three replicates.
The p-hydroxyarylsulfonamide chemotype 6 has been identified as a PAINS substructure.1 Others have shown this scaffold to be redox-active9 as well as subject to addition–elimination at the substituted C3-position.44 During our post-HTS triage, we also observed that many of these compounds produced H2O2 in our assay buffer, both in the presence and absence of the reducing agent DTT using a horseradish peroxidase–phenol red assay (HRP-PR; Supporting Information, Table S1, and data not shown). Several of these compounds did not produce detectable levels of H2O2 in our assay, however. This may be related to compound stability in assay buffer, as discussed below. Given these results, we suspected that H2O2 production might be another source of assay interference for this chemotype (by oxidizing the free thiol on CoA to sulfenic and sulfinic acids). However, we found that even relatively high levels of H2O2 (1 mM final concentrations) did not interfere with the assay readout in the CoA–CPM counter-screen when compared to control reactions (p = 0.61, n = 8). Even H2O2 present in levels greater than those observed in our HRP-PR redox assay did not appreciably inhibit the HAT activity of Rtt109–Vps75 or other HATs either in the presence or absence of DTT (Supporting Information, Table S2), suggesting these particular proteins are not overly susceptible to H2O2-mediated inactivation under our experimental conditions. Additionally, none of the other prototype compounds used in this report showed evidence of redox activity when tested (Supporting Information, Table S1). Together, these data suggest H2O2 release by these redox-active compounds is not the primary factor behind their compound-mediated reductions in HTS signal or enzymatic activity.
UPLC–MS experiments provided important insights into the complex nature of this “triple-threat” chemotype. We first observed adducts 6a′–6e′, which are consistent with addition–elimination of a thiol on the parent compounds at the C3-position (Figure 4B,C).68 Importantly, these sulfonamides and the aforementioned adducts were not stable to our assay conditions. To our surprise, compounds 6a–6e (Table 5) and adducts 6a′–6e′ showed a time-dependent degradation in our HTS buffer when monitored by UPLC (Figure 4B,C and Supporting Information, Figure S7). These data are consistent with and extends a previous report examining a similar screening compound.69 On the basis of this data and plausible chemical mechanisms, we speculate that the degradation products are arylsulfonamides and naphthoquinones resulting from imine hydrolysis and perhaps other as yet unidentified intermediates. Evidence supporting the complex and subversive reactivity of this class of compounds includes the observation that treatment of 6a–6e with GSH led to a common compound adduct (6″) with an m/z of 446. We propose that this GSH adduct is formed by loss of the arylsulfonamide and water, perhaps by imine hydrolysis of 6a′–6e′ at the C–N bond (Figure 4C and Supporting Information, Figure S7). Further characterization of this degradation process is ongoing.
In the aqueous and slightly alkaline HTS conditions, it is likely chemotype 6 can also undergo imine hydrolysis to generate a reactive naphthoquinone in situ, although we were unable to observe this compound directly by our UPLC–MS setup. Naphthoquinone formation is consistent with the production of H2O2 and our observation of a common compound–glutathione adduct. It is likely the thiols could react with a resulting naphthoquinone via Michael addition–elimination. Interestingly, compounds with a quinone moiety 7 in place of a naphthoquinone generally showed less interference (Supporting Information, Figure S8) and none of these compounds were active in the slot blot or showed signs of redox activity (Supporting Information, Table S1). Taken together, these data suggest the napthoquinone moiety is an important structural factor for both redox activity and thiol reactivity, at least under our experimental conditions. Many compounds with the chemotype 6 inhibited Rtt109-catalyzed histone acetylation as determined by slot blot (Table 2), suggesting these compounds can inhibit enzymatic activity either by reacting with proteins and/or other nonspecific mechanism(s).
We also examined the ability of the presumed aromatic leaving groups formed from these substrates (e.g., 1a″) to interfere with the assay readout. Many of these leaving groups did not reduce the HTS or CoA-based counter-screen readouts, especially at the same low micromolar compound concentrations used for the prototype compounds (Supporting Information, Figure S9), and none inhibited enzymatic activity in the slot blot orthogonal assay. The severity of interference, however, appears to increase when they were allowed to incubate longer with CoA (unpublished observations). Despite containing a thiol group, none of these leaving groups formed fluorescent adducts with CPM, suggesting they are not sufficiently nucleophilic to react with the maleimide probe under the conditions tested. Interestingly, the only leaving groups that formed fluorescent adducts with CPM were some p-hydroxyarylsulfonamides (e.g., 6a) with thioglycolic or 3-mercaptopropionic acid substituents, as these compounds showed profiles consistent with false-negative enzymatic inhibition (Table 5).
Select Assay Artifacts Inhibit HAT-Catalyzed Histone Acetylation
Although compounds with chemotypes 1, 2, 3, 4, and 6 interfere with the assay readout by trapping CoA, several of these same compounds were shown to inhibit Rtt109-catalyzed histone acetylation at high compound concentrations by slot blot assay (Tables 1–5). We confirmed this inhibition for several compounds using a second, lower-throughput orthogonal HAT assay that utilized [3H]-acetyl-CoA. We found that most of these compounds inhibited Rtt109-catalyzed histone acetylation in the low micromolar range, particularly scaffolds 2, 3, and 6 (Table 6). This shows compounds with these chemotypes can inhibit Rtt109 enzymatic activity in vitro, but this is most likely via nonspecific protein reactivity, given the ability of these compounds to form thiol adducts. As expected for compounds with nonspecific thiol reactivity, these same compounds also inhibited the human HAT p300 and the yeast Gcn5–Ada2–Ada3 HAT complex at similar concentrations (Table 6). This inhibition was profoundly attenuated by the inclusion of DTT (Table 6), which is consistent with these chemotypes being thiol-reactive agents.
Table 6. Inhibition of HAT-Catalyzed Histone Acetylation by Select Compounds Using an Orthogonal [3H]-Acetyl-CoA HAT Assaya.
| ID | Rtt109 HTS IC50 (μM)b | slot blot activity (125 μM)b | Rtt109 IC50 (μM) (−) DTT | p300 IC50 (μM) (−) DTT | Gcn5 IC50 (μM) (−) DTT | (+) DTT IC50 effectc |
|---|---|---|---|---|---|---|
| 1a | 8.8 | P | 15 (11–22) | 7.5 (3.0–19) | 11 (4.6–26) | increase |
| 1i | 6 | P | 4.6 (2.9–7.2) | 6.2 (1.8–21) | 20 (9.3–44) | increase |
| 1j | 6.6 | P | 3.4 (1.9–6.2) | 6.2 (3.8–10) | 3.7 (2.8–4.8) | increase |
| 1k | 4.6 | P | 6.1 (4.1–12) | 11 (3.1–38) | 11 (5.3–22) | increase |
| 2a | 2.8 | P | 3.9 (2.7–5.6) | 2.2 (1.7–2.9) | 4.6 (3.4–6.3) | increase |
| 2e | 2.3 | Y | 0.77 (0.67–1.1) | 1.1 (0.7–1.6) | 1.6 (0.7–3.9) | increase |
| 3a | 2.7 | Y | 0.81 (0.59–1.1) | 1.9 (1.4–2.5) | 7.5 (5.3–11) | increase |
| 3d | 2.7 | Y | 0.64 (0.55–1.3) | 1.8 (1.4–2.4) | 5.3 (3.9–7.2) | increase |
| 3f | 3.5 | Y | 1.7 (1.5–2.2) | 2.0 (1.6–2.5) | 8.1 (5.8–11) | increase |
| 4a | 15 | P | 3.9 (3.1–5.0) | 3.7 (1.1–13) | 27 (12–61)) | increase |
| 4b | 17 | P | 1.2 (0.7–2.1) | 2.8 (1.5–5.2) | 6.7 (2.7–16) | increase |
| 4e | 11 | Y | 8.6 (4.6–16) | 9.8 (7.1–14) | 27 (10–70) | increase |
| 6a | NA | Y | 0.52 (0.45–1.7) | 1.0 (0.9–1.2) | 0.65 (0.3–1.2) | increase |
| 6b | 1.5 | Y | 0.67 (0.63–1.4) | 0.51 (0.40–1.4) | 2.5 (1.7–3.8) | increase |
| 6c | NA | Y | 0.15 (0.12–0.51) | 0.66 (0.57–1.3) | 1.3 (0.9–1.9) | increase |
| 6d | 3.9 | Y | 4.3 (3.1–6.0) | 3.1 (2.6–3.7) | 4.3 (2.3–7.7) | increase |
| 6e | 2.5 | Y | 1.6 (1.2–2.1) | 1.8 (1.3–2.5) | 1.2 (0.7–1.9) | increase |
| 6f | 0.91 | Y | 1.7 (1.4–1.9) | 1.1 (0.7–1.6) | 5.2 (4.0–6.7) | increase |
| 6y | NA | Y | 0.13 (0.12–0.64) | 0.33 (0.30–2.3) | 1.2 (0.9–1.7) | increase |
| CPM | Y | 0.26 (0.20–0.27) | 0.26 (0.23–0.35) | 1.1 (0.9–1.2) | increase | |
| garcinol | 13d | Y | 3.5 (3.3–3.8) | 1.7 (0.9–3.1) | 2.6 (1.4–4.9) | none |
| fluconazole | inactive | inactive | inactive | inactive | inactive | none |
In parentheses are the 95% confidence intervals for the IC50 values.
Compounds tested identically in the presence of 1 mM DTT; results similar versus Rtt109-Vps75, p300, and Gcn5; typically <20% inhibition was observed at 125 μM final compound concentrations.
Previously published value.42
Select Assay Artifacts Form Covalent Bonds with Protein Assay Components
To further examine the thiol reactivity of these problematic compounds, we performed protein mass spectrometry (LC–MS/MS) using tryptic digestions of samples containing select prototype compounds incubated with the protein components of the HTS assay. As expected for potent thiol-trapping compounds, we observed several ionized peptides with accurate mass measurements corresponding to covalently modified cysteine residues on Rtt109 (Figure 5 and Supporting Information, Table S3). Detectable adducts were also observed with select cysteine residues on Vps75 and Asf1 (Figure 5 and Supporting Information, Table S3). These compounds did not form detectable adducts with all the cysteines in the HTS proteins under our experimental conditions. We speculate this may be because some of the adducts were particularly labile under the experimental conditions or were not amenable to ionization or because sterically inaccessible and/or chemically inactivated cysteines (via sulfur oxidation) were not subject to reactivity. Further studies are needed to assess these possibilities.
Figure 5.

Selected spectra of compound–peptide adducts detected by peptide mass spectrometry. Prototype compounds were incubated with purified proteins from the Rtt109 HTS, and then samples were subjected to LC-MS/MS analyses after in-gel proteolysis. Shown are peptide MS/MS spectra with assigned y- and b-type fragments. (A) Compound 1a forms a detectable adduct with C94 on yeast Rtt109. (B) Compound 6a forms a detectable adduct with mono-oxidized C21 on yeast Vps75. Shown in each spectra are the sequences for the precursor peptide and a simplified reaction scheme for the adduct formation. See Supporting Information, Table S3, for additional examples of compound–peptide adducts detected by peptide mass spectrometry.
Overall, these data demonstrate the prototype in each of the chemical classes can covalently modify the protein components of our HAT assays in a promiscuous fashion. Given this data, and the strong attenuation of enzymatic inhibition by the inclusion of DTT in our radiolabeled HAT assays (Table 6), the most likely mechanism of enzymatic inhibition is nonspecific thiol reactivity. Because Rtt109 does not have a known catalytic cysteine residue,70 it is most likely the case that this thiol modification alters protein structure and dynamics rather than directly inhibiting the catalytic mechanism. The fact that several of the protein components included in the HTS method were modified (and that HAT inhibition can be significantly attenuated with DTT) further suggests these compounds react with thiols indiscriminately and may therefore show promiscuous bioactivity.
Demonstration of Compound–Protein Adducts by ALARM NMR
It is known that the reactivity of thiols in a proteinaceous microenvironment may be different than their reactivity with small-molecule thiols like GSH71 (and presumably CoA). To complete our study, we investigated whether these interfering compounds could react with protein cysteines from a completely unrelated protein, the La antigen, using ALARM NMR.18,72 Importantly, this assay utilizes a completely orthogonal detection method, that is, not based on fluorescence, mass spectrometry, antibodies, or radioactive substrates. We tested the prototype compounds (1a, 2a, 3a, 4a, 6a, and 6b) as well as positive and negative control compounds 2-chloro-1,4-naphthoquinone and fluconazole, respectively (Figure 6A). Consistent with the previous findings, all of these prototype compounds induced peak shifts in the regions of interest in the absence of DTT. These effects could be prevented by the inclusion of DTT in the assay buffer, the addition of which does not lead to peak shifts or signal attenuation (Figure 6A and Supporting Information, Figure S10). Together, these results indicate these prototype compounds (1a, 2a, 3a, 4a, 6a, and 6b) covalently modify cysteines located on the La antigen. In the case of the arylsulfonamides 6a and 6b, it appears the protein conformation is strongly perturbed (“denatured”) without the inclusion of DTT. Of possible relevance, related compounds have been recently reported as disrupters of protein–protein interactions.73
Figure 6.

Thiol reactivity of select screening compounds with the La protein as measured by ALARM NMR. (A) 2D 1H–13C HMQC spectra of selected 13C-labeled methyl groups for the selected compounds 1a, 2a, 3a, 4a, 6a, and 6b as tested by ALARM NMR for protein reactivity. These methyl groups have been shown to undergo peak shifts and intensity decreases in the presence of many compounds that covalently react with neighboring cysteine residues. Compounds were incubated with the La protein in either the presence or absence of 20 mM DTT. PC denotes the positive control compound, 2-chloro-1,4-naphthoquinone. Fluconazole is shown as a negative compound control. Shown are representative results from one of two independent experiments. (B) Summary of the additional compounds tested by ALARM NMR, including several negative compound controls that were inactive in the Rtt109 HTS and thiol-reactive counter-screen.
To further show the utility of this method and that the results were not exclusive to a select subset, we also tested several other analogues of these prototype compounds, including some negative controls comprised of structural analogues that did not show interference in our HTS counter-screens nor inhibition of Rtt109-catalyzed histone acetylation in the slot blot assay. As expected, all of the prototype analogues, but not the negative controls, were ALARM NMR-positive (Figure 6B). As before, including DTT in the sample buffer prevented the ALARM NMR reactivity. This demonstrates by a non-MS-based method that the interfering chemotypes are also susceptible to reactions with protein cysteines, a known source of nonspecific enzymatic inhibition and bioassay promiscuity.
Thiol-Reactive Chemotypes Show Promiscuous Behavior in Academic and Industrial Bioassays
As there is considerable chemical overlap in many academic screening libraries (unpublished observations), due in part to shared commercial vendors and the “combiphilic” nature (i.e., amenable to synthesis by combinatorial schemes) of many screening scaffolds, we examined the scientific literature and the PubChem database to gauge whether our findings may be more broadly applicable to other biological systems and assay formats.74−76 Not surprisingly, compounds with the interfering scaffolds and some closely related derivatives have been reported in the context of many biological systems with varying degrees of biological activity and claims of utility.77 Several compounds bearing the scaffolds described in this report also showed patterns of bioassay promiscuity in a simple search of PubChem bioassay records (Figure 7). On the basis of our findings, it is likely much of this bioassay promiscuity is due to nonspecific thiol reactivity.
Figure 7.

Select examples of compounds containing thiol-reactive chemotypes that demonstrate promiscuous PubChem bioassay profiles. Shown are conspicuous examples of compounds containing chemotypes 1, 2, 3, 4, and 6 that have promiscuous bioassay profiles according to a PubChem substructure search (accessed 1 March 2014). Accompanying each structure is the PubChem CID followed by the ratio (number of bioassays where the compound was classified as active/number of bioassays that the compound was tested).
Finally, we analyzed HTS records from a major pharmaceutical company for evidence of frequent-hitter behavior across the chemotypes that we have described above. It is commonly understood that academic and corporate libraries vary in size, composition, and chemical diversity, and therefore it is not immediately obvious that the trends seen in academic data would apply outside of this domain. For the purpose of comparison, we derived frequent-hitter scores for a large subset (>1 M compounds) of AstraZeneca’s corporate compound collection.78 The frequent-hitter scores are based on the body of historical HTS screening data for these compounds, typically, compounds in the corporate screening deck will have been tested in several tens to hundreds of HTS campaigns. The frequent-hitter score we derive takes into account the anticipated incidence of activity for an average compound, with high scores suggesting a higher-than-expected level of activity. A score cutoff is defined to identify those compounds with an unexpectedly high level of activity, thereby designating frequent hitters empirically. Details of the derivation of the frequent-hitter score (pBSF) have been described previously.78 It should be noted that the corporate data set used to identify frequent hitters covers a wide range of assay types, and does not solely encompass assays like those described earlier in this publication.
We examined the incidence of frequent hitters across various categories of nuisance chemotypes in the AstraZeneca collection (Table 7 and Supporting Information, Table S4). It is clear that some of the nuisance chemotypes derived from academic data display an elevated incidence of frequent-hitter behavior in the corporate data as well, although not all chemotypes showed the same degree of promiscuity. Chemotypes 1 (benzothiophene dioxides), 2 (benzothiadiazole/benzofurazans), and 6 (p-hydroxysulfonamides) exhibit high levels of promiscuous behavior in the AstraZeneca screening deck, suggesting their indiscriminate and deleterious influence is present in a wide range of assay technologies. For the p-hydoxysulfonamides 6, the observation that they may also cause protein denaturation in the ALARM NMR assay in this study suggests another mode of action along these lines (that is, in addition to their other liabilities of redox activity and thiol reactivity). The AstraZeneca corporate data showed high levels of assay promiscuity for chemotypes 6 and 7, which suggests the inactivity and weaker interference of chemotype 7 in our systems may be an assay-specific observation. That is, chemotype 7 may still be relatively promiscuous under other assays conditions,79 a speculation that may be pursued in future investigations. Nonsalt forms of the 1,2,4-thiadiazoles 3 show only slightly elevated levels of promiscuous behavior in the corporate data set (Supporting Information, Table S4), while the salt forms were relatively promiscuous (Table 7). The succinimide chemotype 4 did not exhibit high promiscuity in the AstraZeneca screening deck, but this may be an indication of problematic behavior under specific assay conditions such as alkaline assay buffers, which we expect would be needed to generate the reactive maleimides 5. We note that some of the assays used for generating the frequent-hitter scores have been stabilized with additions of DTT, which has the potential to mitigate the effects of reactive behavior depending on assay specifics. Therefore, the bioassay promiscuity emerging from this set of data may also be indicative of interference cause by mechanisms other than thiol reactivity. Overall, the observations derived from the larger set of corporate data corroborate the evidence derived from the academic data in this publication.
Table 7. Bioassay Promiscuity Analysis of Thiol-Reactive Chemotypes in an Industrial HTS Settingd.

Structure annotations: A, any atom; ns, number of substituents (e.g., “2s”); nr, number of connected ring bonds (e.g., “2r”); X, halogen.
Ndata designates the subset of compounds for which a pBSF score had been derived. This is dependent on the availability of HTS screening data.
Expected incidence of anomalous binders is 6% (averaged over all compounds).
Observed fractions of frequent hitters for structural classes (chemotypes). Note only biochemical assay data, and not cell-based assay data, were used to derive the frequent hitter score.
Discussion
In this article, we characterized the chemical basis of assay interference for five problematic chemotypes (1, 2, 3, 4, and 6) identified during the course of a recent triage of a sulfhydryl-scavenging HTS for inhibitors of Rtt109-catalyzed histone acetylation. These chemotypes were flagged as PAINS or have close chemical structural similarities to certain PAINS substructures. We first showed that while compounds containing any of these five scaffolds are capable of inhibiting Rtt109-catalyzed histone acetylation, this inhibition was confounded by the ability of these compounds to interfere with the HTS assay readout by reacting with free CoA in the Rtt109 HTS. We then demonstrated by UPLC–MS and LC–HRMS that these compounds can form adducts with other biological thiols such as GSH, and in another orthogonal enzymatic assay, can inhibit several different HATs in vitro only when DTT is absent in the reaction mixture. Protein mass spectrometry confirmed several of these compounds could covalently modify multiple cysteines in the HTS. Using ALARM NMR, yet another orthogonal detection method, we showed that the majority of these compounds can covalently modify cysteines on a completely unrelated protein system.
The findings described herein strongly suggest investigators (and reviewers) flag these problematic compounds and avoid their follow up. It is particularly troubling that many of these compound classes are still being reported in the patent literature and reputable scientific journals, some with dubious claims of biological utility (see Supporting Information). Perhaps not coincidentally, these compound types were active in other bioassays according to PubChem queries. The propagation of these nuisance compounds in reputable journals suggests that many academicians and reviewers alike are not fully aware of nuisance compounds such as PAINS and perhaps not appreciative of their potential to sidetrack early drug discovery projects.
On the basis of our studies, we highly recommend the problematic chemotypes described in this report be pursued as chemical leads with high levels of skepticism and that investigators currently working with these compounds carefully re-evaluate the interpretation of their results when there are claims of biological utility, including apparent enzymatic and cell-based selectivity, bioactivity, and mechanism-of-action studies. For instance, we posit that much of the selectivity observed for these chemotypes (a common defense for those publishing PAINS as bioactive compounds) is due to different susceptibilities of assay components (e.g., enzymes, cell lines) to thiol-reactive compounds or other nonspecific mechanisms and that observed bioactivity is likely attributable to off-target effects. With regards to mechanistic studies, it is likely some key component is missing from the experimental design (e.g., assessing the effect of DTT or rigorously testing for irreversibility). A recommended list of assays for evaluating the potential for compound–thiol reactivity is provided (Figure 8). We recommend that knowledge-based methods be supplemented by more than one of the experimental-based methods.
Figure 8.

Methods to help identify nonselective cysteine reactivity in compounds from HTS campaigns. Triage of active compounds from HTS (real or virtual) should always include knowledge-based methods to flag potential reactive entities. Flagged compounds should then either be removed from consideration or investigated more rigorously using two or more of the experimental-based methods described above. Notes: Several of these methods have been described in the text and elsewhere.45,105,106 The use of frontier molecular orbital (FMO) calculations has been reported as a gross method of flagging “frequent-hitters”.107 Certain cysteine proteases (e.g., caspase-1, -8) have been used as probes for reactivity including cysteines oxidation by redox-active compounds.108,109 MSTI = (E)-2-(4-mercaptostyryl)-1,3,3- trimethyl-3H-indol-1-ium;44 REOS, rapid elimination of swill.110
The emergence of epigenetic targets such as HATs has led to the development of several types of HTS assays to study epigenetic modifications like acetylation. HAT activity can be probed with antibody-based methods (e.g., Western blots, amplified luminescent proximity homogeneous assays, TR-FRET) or sulfhydryl-scavenging methods (e.g., fluorescent probes or coupled-enzyme reporters).32,46,80 Other methods such as radiolabeled substrates, mass spectrometry, and electrophoretic mobility, have been used to assay the status of protein acetylation, including cell-based adaptations.81−85 While each method has distinct advantages and disadvantages, each is still susceptible to false positives, assay artifacts, and identifying promiscuous “frequent hitters”.
The maleimide-based screens are subject to several mechanisms of chemical interference. We determined thiol-trapping compounds represented a significant source of assay artifacts in our CPM-based Rtt109 HTS, especially chemotypes 1, 2, 3, 4, and 6. These compounds interfered with our HTS readout by forming covalent adducts with the CoA produced by the HAT reaction, creating a convoluted readout of enzymatic inhibition. Nucleophilic screening compounds can also form adducts with CPM that can create either a false-positive or false-negative readout pattern, depending on the fluorescent characteristics of the adduct. Another artifact source is fluorescence quenching, although we did not encounter many examples of fluorescence quenchers in our post-HTS triage (data not shown). On the basis of the chemotypes in this report, along with the other triaged compounds, the majority of assay artifacts from our CPM-based HTS resulted from thiol-trapping rather than compound–CPM adducts. In our experience, a major (though not insurmountable) disadvantage of this screening method is the high levels of assay interference and the time and resources needed in the post-HTS phase to triage these artifacts. In fairness, it is worth noting that this method is capable of identifying compounds that inhibit enzymatic activity and has distinct advantages such as low cost and robustness.29,42
To prevent follow-up on bad chemical matter discovered by maleimide-based screens, we make several recommendations. First, we strongly recommend having a validated, robust orthogonal assay in place prior to conducting an HTS with this method. Relying solely on the CPM-based method could lead to the selection of thiol-trapping compounds, and if used for the basis of compound optimization could lead to the unfortunate case of optimizing for thiol reactivity rather than the desired enzymatic inhibition. This could conceivably happen if one were to view the apparent enzymatic inhibition data in Tables 1–5 as evidence of a preliminary SAR, when in fact it would be more appropriately called SIR (i.e., “structure–interference relationship”). As the nature of the CPM-based format contraindicates the use of DTT and other biological reducing agents, it would be advisible to have an orthogonal assay that can test candidate compounds in both the presence and absence of DTT or similar reducing agent to further rule out thiol reactivity (e.g., Table 6). Second, we recommend follow-up with the CoA–probe counter-screen, especially if no orthogonal assay is available. This assay can identify assay artifacts, and if used in parallel with an orthogonal assay, can identify potentially problematic thiol-reactive enzymatic inhibitors. Third, the ratio of acetyl-CoA to test compound should be kept as high as possible, although this must be balanced with other important factors such as the acetyl-CoA KM. If fluorescence quenching is a concern, we recommend the facile counter-screen used in this manuscript, as it should not be easily susceptible to interference from thiol-trapping compounds.
A deeper understanding and appreciation for the chemical mechanisms of assay interference and thiol reactivity can have important implications for early analogue selection and screening library design (see Figure 1 for the general chemotypes discussed here). For instance, benzothiophene 1,1-dioxides 1 are susceptible to certain addition–elimination reactions, and on the basis of our results, we recommend a leaving group analysis for such compounds. Therefore, selecting and/or testing analogues with weaker leaving groups or a reduction at the C2–C3 position (Supporting Information, Figure S2) may be a potential strategy to overcome thiol reactivity in this chemotype. The former strategy may be useful for certain succinimides 4 with good leaving groups (e.g., S-linked heteroaromatics; Supporting Information, Table S4). Certain 1,2,4-thiadiazoles 3 are susceptible to attack by thiol nucleophiles at the S1-position, specifically when the N2-position is positively charged. We note our findings with this chemotype are consistent with other previous mechanistic work on these compounds.56,57 Should investigators choose to pursue compounds bearing chemotype 3, it may be useful to assess the effect of switching to 1,2,4-oxadiazole analogues, as well as testing the nonsalt forms of 1,2,4-thiadiazoles. The benzothiadiazoles/benzofurazans 2 interfere by nucleophilic aromatic substitution, and this interference correlated with the apparent strength of the presumed leaving group. As with chemotypes 1, 4, and 6, a strategy for navigating away from this problematic chemotype would be to select analogues with weaker leaving groups or with less electron-withdrawing functional groups on the heteroaromatic core.86 These findings may be also useful for updating current PAINS filters. For instance, one example of updating PAINS filters would be to include a modified “diazox_B” substructure to include additional strong electron-withdrawing moieties such as nitro groups (Table 2).
The SAR/SIR of these chemotypes also raises important questions about the ability of certain PAINS to be converted to non-PAINS. It is interesting to note that even in the cases where incidence of anomalous behavior is high, presence of the offending substructure does not predispose all compounds to anomalous behavior. This suggests that it may be possible to “design out” such behavior if the activity seen in the assay is true after all. Nonetheless, presence of the nuisance chemotype does suggest that there is a very significant risk of failure in attempting such optimization, as chances are high the “active” is acting via a therapeutically uninteresting mechanism, thereby rendering such hits unattractive start points for HTS follow-up.
Both the succinimides 4 and the p-hydroxyarylsulfonamides 6 illustrate the susceptibility of screening compounds to undergo chemical transformations under certain assay conditions. The decomposition of chemotype 6 was unexpected, and it will be interesting to examine the conditions critical for this conversion as well as more detailed characterization of this decomposition process. For instance, it appears several of these problematic arylsulfonamides were also unstable in ALARM NMR buffer, suggesting this scaffold is likely unstable in many other biologically relevant aqueous buffers and not an isolated phenomenon (Supporting Information, Figure S11). We observed several quinone–protein adducts for both 6a and 6b by protein LC–MS/MS, further attesting to the instability of this scaffold in our assay conditions (Supporting Information, Table S3). Given the instability of scaffolds 4 and 6, we recommend assessing the stability of any promising compounds in assay buffer by analytical techniques to verify the structure of the active chemical entity in the biological context. This is a rather straightforward experiment, and in light of our findings, it may be an important confirmatory experiment to perform before proceeding to more extensive experiments, such as molecular modeling, that are based on correct structure identification and integrity.
There has been a recent resurgence of interest in covalent drugs (e.g., ibrutinib and dimethyl fumarate).87−92 This renewed interest has been used as a line of defense in the reporting of known reactive compounds, including PAINS, as viable drug leads.93,94 We have shown in this manuscript that all interference compounds are not created equal and that they can exhibit a distinct SIR. However, we suggest it is highly unlikely that compounds that show indiscriminate protein reactivity, as do PAINS, will ever be useful drug or probe leads. While there may be exceptions, we expect that most of the recently developed covalent drugs have either been purposefully designed as such or have undergone extensive mechanistic studies and medicinal chemistry optimization. They are usually not the outcome of the optimization of nonselective, reactive, and promiscuous compounds that sometimes are reported from HTS. Therefore, we recommend that the scientific community apply an extremely high standard of rigor to the review and publication of manuscripts that claim any drug- or probe-like potential for these types of compounds. Additionally, we caution researchers that commercially available “probes” that feature known thiol-reactive moieties, including but not limited to those chemotypes discussed herein, may be less selective versus the proteome than their “probe-like” status suggests (Supporting Information, Table S5).
Our findings highlight the importance of taking a chemocentric approach to HTS triage and hit prioritization and highlight the need for carefully planned counter-screens and orthogonal assays in a well-validated cascade of hit-triaging assays (Figure 8). We believe our investigation also demonstrates the importance of partnering with medicinal chemists in the post-HTS triage process and should serve as caution for lead selection based primarily on initial potency and SAR data without confirmation of activity by orthogonal methods. The continued growth of cheminformatics and the incorporation of PAINS filters into both commercial software suites (e.g., SYBYL, Schrodinger Canvas) and freeware is undoubtedly a positive advancement for the field. However, many compounds with chemotypes 1–4 were not flagged by our cheminformatics PAINS filters. This raises important concerns about the potential for overreliance on cheminformatics filters. For instance, unseasoned researchers (and reviewers) may fall into the trap that because PAINS were removed by substructure filters, that they no longer have to consider any related nuisance compounds. Strategies to mitigate this risk are to (1) take a chemocentric approach to HTS triage45 using a well-validated cascade of deconvoluting assays, (2) encourage more mechanistic studies of nuisance compounds to further the understanding of their behavior, (3) periodically update PAINS filters as more data is made available, and (4) mine the ever-increasing amount of HTS data for insights into PAINS substructures (e.g., along the lines shown in a recent report).78 Such undertakings also raise important follow-up questions for those in HTS triage about what exactly should constitute a PAINS and what criteria should form the basis for classifying a compound as “promiscuous” and/or “pan-assay”. Likely, these definitions will have to be dependent on the screening context, at least in part, and guided by those with sufficient expertise in HTS triage.
Materials and Methods
Molecular Libraries, Compounds, and Reagents
The chemical library has been described previously.42 The following reagents were obtained from Sigma-Aldrich: DMSO, CPM, CoA (sodium salt hydrate), acetyl-CoA (sodium salt), bovine serum albumin (BSA), H2O2, and Triton X-100. Compounds tested in post-HTS assays were repurchased as solid powders from standard chemical vendors (e.g., eMolecules). In a quality-control sampling of a random 5% of the chemical library samples used in this report, greater than 90% of the tested commercial samples had acceptable purities (>90%) by UPLC–MS analysis and 1H NMR and LRMS–ESI spectra consistent with their vendor-provided structures.
Rtt109 HTS and Dose–Response Experiments
The CPM-based Rtt109 assays have been detailed in a previous report with minor modifications.42 Briefly, all compounds studied in this report were rescreened in assay buffer containing freshly prepared 0.01% Triton X-100 (v/v) and enzyme concentrations of 50 nM Rtt109–Vps75 complex. For IC50 experiments, compounds were tested in triplicate at eight compound concentrations ranging from 200 nM to 125 μM final compound concentrations. Slot blots were performed on reaction aliquots using standard techniques with a Bio-Rad Bio-Dot SF microfiltration apparatus. Membranes were imaged with a LI-COR Odyssey and analyzed using Image Studio (LI-COR Biosciences). Equal protein loading was verified by Ponceau S staining of each membrane.
Data Analysis and Statistics
Z′ factors for each plate were calculated using eq 1:95
| 1 |
where σ and μ represent the standard deviation and mean of the positive (c+) and negative (c–) plate controls, respectively. All plates tested in these studies had Z′ factors ≥0.5. IC50 values were determined by fitting dose–response data to the sigmoidal dose–response variable slope four-parameter equation in GraphPad Prism 6.0. Other statistical analyses were also performed in Prism using standard procedures.
Assay Interference Counter-Screens
Compounds interfering with the CPM-based assay readout were identified as previously described with minor modifications.42 Compounds were tested in triplicate at eight concentrations ranging from 200 nM to 125 μM final compound concentrations using an adaption of the Rtt109 HTS assay format. Proteins and assay buffer were dispensed to assay plates analogously to the HTS procedure, then the acetyl-CoA substrate was replaced with CoA in concentrations titered to match the fluorescence intensity observed for the uninhibited enzyme reaction in the HTS assay (approximately 5 μM CoA). Compounds were incubated with CoA and allowed to react with CPM under conditions identical to the HTS procedure. Assay interference was quantified by comparing the background-corrected (compound + proteins + CoA + CPM) fluorescence intensities to the (DMSO + proteins + CoA + CPM) controls. To further investigate their fluorescence behavior under the HTS conditions, select compounds were also incubated with assay reagents (buffer-only, buffer + CoA, buffer + CPM) and their fluorescence intensity measured. The overall plate layout, controls, protocols, and assay readouts were unchanged from the aforementioned compound–CoA–CPM counter-screen.
Fluorescence Quenching Counter-Screen
Compounds were tested for evidence of fluorescence quenching using a modification of our published procedure.42 Briefly, CPM and CoA (20 and 5 μM final concentrations, respectively) were allowed to react to completion in assay buffer. Completion was defined as a stable signal plateau, usually after 5 min reaction time. The CPM–CoA adduct solution (20 μL per well) was then dispensed into assay plates preplated with DMSO and test compounds. Compounds were dispensed using an ECHO 550 contactless liquid dispenser (Labcyte). Microplates were shaken for 5 min and allowed to equilibrate for another 5 min at room temperature. Fluorescence intensity was measured, and the data was analyzed as percent signal reduction compared to DMSO controls.
Compound–Thiol Adduct Characterization
Selected compounds (1 equiv) and either CoA or reduced l-glutathione (2 equiv) were incubated under HTS-like conditions, except with 5% DMSO (v/v) and no detergent in the assay buffer.42 Compounds were also tested in HTS buffer or methanol minus the addition of biological thiols. Compounds were typically incubated at 0.5 mM final concentrations. Samples with visible precipitates were passed through 0.25 μm syringe filters to remove particulates. Sample injections were typically 1.0 μL in volume performed by an autosampler and were analyzed on a Waters UPLC system using a BEH C18 2.1 mm × 50 mm column. The flow rate was 0.250 mL/min with a standard gradient starting at 95% Solution A (950 mL H2O, 50 mL MeCN, 1 mL formic acid) and ending with 100% solution B (1000 mL MeCN plus 1 mL formic acid) over 6.5 min. The samples were monitored simultaneously using an ELS detector, a diode array detector (214, 220, 244, and 254 nm), and a ZQ mass spectrometer (ESI positive and negative modes).
Redox-Activity Assay
Selected compounds were assessed for redox activity using published protocols.9,42,96 Freshly prepared 100 μM H2O2 (Sigma) was included as a positive plate control, while NSC-663284 and 4-amino-1-naphthol were used as positive redox-active controls for DTT and DTT-free assay conditions, respectively. Fluconazole and DMSO were used as negative compound and plate controls, respectively. Compounds were tested in triplicate at eight final concentrations (200 nM to 125 μM via 2.5-fold dilutions) in either the presence or absence of 1 mM DTT final concentration. All active compounds did not interfere with the assay readout at A610 (data not shown).
[3H]-Acetyl-CoA HAT Assays
For selected compounds, inhibition of HAT activity was also checked with an orthogonal in vitro radiolabeled substrate assay. Rtt109 inhibition was tested at eight compound concentrations (200 nM to 125 μM final compound concentrations via 2.5-fold dilutions) in an adaptation of a previous procedure.42 Briefly, reactions were performed in standard volume 384-well microplates using 45 μL total reaction volumes containing the following in final concentrations: 50 mM Tris HCl, pH 8.0, 50 mM KCl, 0.1 mM EDTA, 1 mM DTT, 0.01% Triton X-100 (v/v), 50 ng/μL BSA, and 2.5 μM [3H]-acetyl-CoA (PerkinElmer). Purified recombinant yeast Rtt109–Vps75 was tested at approximately 5 nM final concentrations, while purified recombinant Asf1–dH3–H4 (approximately 250 nM) was used as acetylation substrate. Compounds and DMSO were plated with a multichannel pipet, followed by a similar addition of a solution containing enzyme and histone substrate (36 μL). Test compounds were allowed to equilibrate with enzyme and histone substrate for 10 min at 30 °C in an incubator. The HAT reaction was initiated by adding [3H]-acetyl-CoA solution (7.5 μL). DMSO content was kept constant across all reactions at 3% (v/v). After 5 min, the reactions were quenched by multichannel pipet transfer of reaction aliquots (35 μL) to adjacent microplate wells each containing 35 μL of 2-propanol. Aliquots (35 μL) of the quenched solutions were carefully spotted onto Whatman P-81 phosphocellulose paper filters (GE Healthcare) and air-dried. Filter papers were washed five times for 5 min per cycle with 50 mM NaHCO3, pH 9.0, then rinsed with acetone and then allowed to air-dry for 30 min. [3H]-Acetate incorporation was then measured by an LS6500 liquid scintillation counter (Beckman–Coulter). Percent inhibition was calculated as a percentage of DMSO control. Similar reactions minus Rtt109–Vps75 were used as background controls. Testing versus p300–BHC and the Gcn5–Ada2–Ada3 complex were performed similarly, except that the final enzyme concentrations were approximately 500 pM and the substrate was purified recombinant dH3–H4 tetramers.97−99
Protein Mass Spectrometry
Test compounds were incubated with purified Rtt109–Vps75 or Asf1 complexes. Compounds and proteins were incubated together at 30 °C for 60 min at 100 μM and 10 μM final concentrations, respectively. Reaction mixtures were denatured with gentle heating and then further resolved by SDS-PAGE. Protein bands were excised after staining with Coomassie blue. In-gel protease digestions were performed in an adaption of published procedures.100 Peptide extracts were dried in vacuo and reconstituted in 98:2:0.1 H2O:acetonitrile:TFA; approximately 0.2 μg of each gel band was analyzed by capillary LC–MS on a Velos Orbitrap mass spectrometer (Thermo Fisher) with higher energy collision induced dissociation activation.101 Peaks Studio 6.0 build 20120620 (Bioinformatics Solutions) software package was used for interpretation of tandem MS and protein inference.102 Search parameters for Rtt109, Vps75, and Asf1 proteins were UniProt database (Sacharomyces cerevisiae strain ATCC 204508/S288c, taxonomy ID 559292, accessed 19 May 2014) concatenated with the common lab contaminant proteins (www.thegpm.org); parent mass error tolerance = 20.0 ppm; fragment mass error tolerance = 0.1 Da; precursor mass search type = monoisotopic; enzyme trypsin with max missed cleavages = 2 and nonspecific trypsin cleavage; variable modifications = methionine oxidation and dioxidation, cysteine oxidation, and dioxidation, and suspected compound adducts; maximum variable modifications per peptide = 5; false discovery rate calculation = on; spectra merge options = 0.2 min within 10.0 ppm mass window; charge correction = on for charge states 2–8; spectral filter quality >0.65. Support for the detection of peptides plus adducts from each supporting tandem MS data was based on: (1) high confidence peaks peptide score (minimum −10 log P 35), (2) a minimum of five consecutive b- or y-type peptide fragment ions, (3) high precursor mass accuracy (<7 ppm), and (4) supporting signature ion peaks for the site localization of the pertinent cysteine modification on one or more peptide fragments.
Cheminformatics
Incidence of frequent-hitting behavior was checked in the AstraZeneca corporate screening deck by mining the historical screening data. We calculate a descriptor, pBSF, for each compound to determine whether it is more active than expected.78 The pBSF score is the negative logarithm of the probability that the observed pattern of activity and inactivity is observed by chance, given the known “average” behavior across all compounds in the screening deck and across the historical set of screening campaigns they have been measured in. If the likelihood of seeing the pattern at hand is high, the compound is likely not a frequent hitter and all is fine. However, if the probability of seeing the pattern is low, the resulting pBSF score will be high and the pattern should be regarded as anomalous. A cutoff of pBSF > 2 was used to designate compounds exhibiting suspicious binding behavior. To check the incidence of frequent-hitting behavior, we searched the corporate collection using substructures (with in-house tools), collated pBSF scores for the set, and counted the number of frequent-hitting compounds using the pBSF threshold stated in the above. For reference, the average fraction of compounds displaying frequent-hitting behavior across the collection of compounds with historical HTS data is 6%.78 The number of HTS data points is variable for each compound, as it depends on the number of times a compound has been screened. The median number of data points per compound in the data set is approximately 200, with only 10% of the compounds having less than 50 data points. Only biochemical assay data, and not cell-based assay data, were used to derive the frequent hitter score
ALARM NMR
ALARM NMR was performed as previously described with minor modifications.18,72 The gene encoding amino acids 100–324 of the human La antigen was cloned into pET-28b+ vector (Novagen) such that it contained both an N- and C-terminal His tag. The plasmid was freshly transformed into Escherichia coli Rosetta cells (Novagen) and cultured in M9 minimal media supplemented with 15NH4Cl (CIL) in an adaption of published procedures.103,104 The La antigen was enriched with 13C at the δ-methyl groups of leucine, the δ-methyl group of isoleucine, and the γ-methyl groups of valine by the addition [3-13C]-α-ketobutyrate and [3,3′-13C]-α-ketoisovalerate (sodium salts, CIL) to the culture medium 30 min before inducing in the presence of 1 mM IPTG for 8 h at 25 °C (OD600 was approximately 0.8 at time of induction). Harvested cells were lysed by French press in ice-cold lysis buffer consisting of 50 mM Tris, pH 7.6, 300 mM NaCl, 10% glycerol (v/v), 5 mM β-mercaptoethanol (BME), 5 mM imidazole, 2 mM MgCl2, benzonase (Sigma), and protease inhibitor cocktail. This solution containing the lysed cells was sonicated briefly (3 × 15 s pulse sequence) on ice, then loaded onto a prewashed Ni-bead column (GE Healthcare) kept at 4 °C. Proteins were eluted from the beads with an elution buffer consisting of 50 mM Tris, pH 7.6, 300 mM NaCl, 10% glycerol (v/v), 5 mM BME ,and an imidazole gradient ranging from 5 mM to 0.5 M. Pooled elution fractions containing the La antigen were dialyzed overnight (25 mM sodium phosphate, pH 7.0, 5 mM DTT), flash-frozen in liquid N2, and stored at −80 °C until further use. Prior to use, aliquots of 500 μM protein was incubated in the presence of 20 mM DTT at 37 °C for 1 h, then dialyzed versus 2 × 2 L of 25 mM sodium phosphate buffer, pH 7.0 (no DTT) at 4 °C with constant N2 bubbling. The 1H/13C-HMQC spectra were acquired in 25 mM sodium phosphate buffer, pH 7.0, 10% D2O (v/v; CIL) ± 200 μM test compounds delivered from 10 mM DMSO stock solutions, and ± 20 mM DTT. Compounds were incubated with proteins at 37 °C for 1 h and then 30 °C for 15 h prior to data collection. Data were recorded at 25 °C on a Bruker 700 MHz NMR spectrometer equipped with a cryoprobe (Bruker) and autosampler. Samples were loaded into Bruker 1.7 mM SampleJet tubes with 40 μL total sample volumes and stored at 4 °C while in queue. The ALARM NMR samples were tested at 50 μM protein concentrations using 16 scans, 2048 complex points in F2, and 80 points in F1 using standard protein HMQC and water suppression pulse sequences. Nonreactive compounds were identified by the absence of chemical shifts (13C-methyl) ± 20 mM DTT. Reactive compounds induced chemical shifts in certain diagnostic peaks in the absence of DTT, and this effect was significantly attenuated when 20 mM DTT was included in an otherwise identical sample.18 As an additional precaution against trace reactive contaminants, compounds tested by ALARM NMR were repurified in-house by standard HPLC procedures using mass-directed collection.
Chemical Synthesis and Characterization
Detailed adduct synthetic procedures and chemical characterization can be found in the Supporting Information.
Acknowledgments
The authors acknowledge: Todd Rappe and Drs. Georges Mer, Chaohong Sun, and Youlin Xia for assistance with ALARM NMR; Drs. Sergei Gaidamakov and Richard Maraia for the plasmid containing the full-length human La antigen; Dr. Philip Cole for the p300-BHC plasmid; Dr. Rebecca J. Burgess for producing the Gcn5–Ada2–Ada3 complex; Sarah J. Sexton for assistance with compound procurement; Dr. Annamaria Szabolcs for assistance with chemical library preparation; Dr. Kathryn Nelson for careful review of this manuscript; Todd W. Markowski and Jacob Wragge for assistance with the protein mass spectrometry experiments; and the Minnesota NMR Center and University of Minnesota Center for Mass Spectrometry and Proteomics. This work was supported by the Minnesota Partnership for Biotechnology and Medical Genomics (73–01 to M.A.W. and Z.Z.), the NIH (GM72719 and GM81838 to Z.Z.), the Mayo Foundation for Medical Education and Research and the Minnesota Supercomputing Institute. J.L.D. was supported by an NIH predoctoral fellowship (F30 DK092026-01), a Pharmaceutical Research and Manufacturers of America Foundation predoctoral pharmacology/toxicology fellowship, and the Mayo Foundation. Funding for NMR instrumentation was provided by the Office of the Vice President for Research, the University of Minnesota Medical School, the University of Minnesota College of Biological Science, the NIH, the NSF, and the Minnesota Medical Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The opinions or assertions contained herein belong to the authors and are not necessarily the official views of the funders.
Glossary
Abbreviations Used
- Acetyl-CoA
acetyl coenzyme A
- ALARM NMR
a La assay to detect reactive molecules by nuclear magnetic resonance
- BME
β-mercaptoethanol
- BSA
bovine serum albumin
- CoA
coenzyme A
- CPM
N-[4-(7-diethylamino-4-methylcoumarin-3-yl)phenyl]maleimide
- DMSO
dimethyl sulfoxide
- DNA
Deoxyribonucleic acid
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- GSH
glutathione
- H3K27
histone H3 lysine 27
- H3K27ac
histone H3 lysine 27 acetylation
- H3K56
histone H3 lysine 56
- H3K56ac
histone H3 lysine 56 acetylation
- HAT
histone acetyltransferase
- HMQC
heteronuclear multiple quantum coherence
- HPLC
high-performance liquid chromatography
- HRMS
high-resolution mass spectrometry
- HRP-PR
horseradish peroxidase–phenol red
- HTS
high-throughput screen or high-throughput screening
- IC50
half-maximal inhibitory concentration
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- LC–HRMS
liquid chromatography–high-resolution mass spectrometry
- LC–MS/MS
liquid chromatography–tandem mass spectrometry
- log P
partition coefficient
- m/z
mass-to-charge ratio
- LRMS–ESI
low-resolution mass spectrometry–electrospray ionization
- MeCN
acetonitrile
- MeOH
methanol
- MS
mass spectrometry
- MTSI
(E)-2-(4-mercaptostyryl)-1,3,3-trimethyl-3H-indol-1-ium
- NMR
nuclear magnetic resonance
- PAINS
pan-assay interference compounds
- pBSF
negative log of binomial survivor function
- REOS
rapid elimination of swill
- SAR
structure–activity relationship
- SAR
structure–activity relationship
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TFA
trifluoroacetic acid
- TR-FRET
time-resolved fluorescence resonance energy transfer
- UPLC
ultraperformance liquid chromatography
Supporting Information Available
Detailed adduct synthetic procedures and chemical characterization (PDF and CSV). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
Designed the experiments: J.L.D., M.A.W., J.W.M.N., J.M.S. Performed the experiments: J.L.D., J.W.M.N., J.M.S., S.F., L.H., H.Z. Analyzed the data: J.L.D., M.A.W., J.W.M.N., J.M.S., S.F., L.H., Z.Z. Wrote the paper: J.L.D. Contributed with revisions: M.A.W., J.W.M.N., J.M.S., S.F., L.H., Z.Z.
The authors declare no competing financial interest.
Supplementary Material
References
- Baell J. B.; Holloway G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Ferrins L.; Falk H.; Nikolakopoulos G. PAINS: relevance to tool compound discovery and fragment-based screening. Aust. J. Chem. 2013, 66, 1483–1494. [Google Scholar]
- Baell J.; Walters M. A. Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [DOI] [PubMed] [Google Scholar]
- Feng B. Y.; Simeonov A.; Jadhav A.; Babaoglu K.; Inglese J.; Shoichet B. K.; Austin C. P. A high-throughput screen for aggregation-based inhibition in a large compound library. J. Med. Chem. 2007, 50, 2385–2390. [DOI] [PubMed] [Google Scholar]
- Schorpp K.; Rothenaigner I.; Salmina E.; Reinshagen J.; Low T.; Brenke J. K.; Gopalakrishnan J.; Tetko I. V.; Gul S.; Hadian K. Identification of small-molecule frequent hitters from AlphaScreen high-throughput screens. J. Biomol. Screening 2013, 19, 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai J. J.; Kirkup M. P.; Frank E. A.; Pachter J. A.; Bryant R. W. Compounds capable of generating singlet oxygen represent a source of artifactual data in scintillation proximity assays measuring phosphopeptide binding to SH2 domains. Anal. Biochem. 1999, 270, 33–40. [DOI] [PubMed] [Google Scholar]
- Simeonov A.; Jadhav A.; Thomas C. J.; Wang Y.; Huang R.; Southall N. T.; Shinn P.; Smith J.; Austin C. P.; Auld D. S.; Inglese J. Fluorescence spectroscopic profiling of compound libraries. J. Med. Chem. 2008, 51, 2363–2371. [DOI] [PubMed] [Google Scholar]
- Gul S.; Gribbon P. Exemplification of the challenges associated with utilising fluorescence intensity based assays in discovery. Expert Opin. Drug Discovery 2010, 5, 681–690. [DOI] [PubMed] [Google Scholar]
- Soares K. M.; Blackmon N.; Shun T. Y.; Shinde S. N.; Takyi H. K.; Wipf P.; Lazo J. S.; Johnston P. A. Profiling the NIH Small Molecule Repository for compounds that generate H2O2 by redox cycling in reducing environments. Assay Drug Dev. Technol. 2010, 8, 152–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaki B. U.; Frazblau S. G.; Chadwick L. R.; Lankin D. C.; Zhang F.; Wang Y.; Pauli G. F. Purity–activity relationships of natural products: the case of anti-TB active ursolic acid. J. Nat. Prod. 2008, 71, 1742–1748. [DOI] [PubMed] [Google Scholar]
- Huang Z. Impact of impurities on IC50 values of P450 inhibitors. Drug Metab. Lett. 2011, 5, 156–162. [DOI] [PubMed] [Google Scholar]
- Hermann J. C.; Chen Y.; Wartchow C.; Menke J.; Gao L.; Gleason S. K.; Haynes N.-E.; Scott N.; Petersen A.; Gabriel S.; Vu B.; George K. M.; Narayanan A.; Li S. H.; Qian H.; Beatini N.; Niu L.; Gan Q.-F. Metal impurities cause false positives in high-throughput screening campaigns. ACS Med. Chem. Lett. 2013, 4, 197–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbert P.-E.; Unterreiner V.; Siebert D.; Gubler H.; Parker C.; Gabriel D. Recommendations for the reduction of compound artifacts in time-resolved fluorescence resonance energy transfer assays. Assay Drug Dev. Technol. 2007, 5, 363–372. [DOI] [PubMed] [Google Scholar]
- Tarzia G.; Antonietti F.; Duranti A.; Tontini A.; Mor M.; Rivara S.; Traldi P.; Astarita G.; King A.; Clapper J. R.; Piomelli D. Identification of a bioactive impurity in a commercial sample of 6-methyl-2-p-tolylaminobenzo[d][1,3]oxazin-4-one (URB754). Ann. Chim. 2007, 97, 887–894. [DOI] [PubMed] [Google Scholar]
- Wipf P.; Arnold D.; Carter K.; Dong S.; Johnston P. A.; Sharlow E.; Lazo J. S.; Huryn D. A case study from the chemistry core of the Pittsburgh Molecular Library Screening Center: the Polo-like kinase polo-box domain (Plk1-PBD). Curr. Top. Med. Chem. 2009, 9, 1194–1205. [DOI] [PubMed] [Google Scholar]
- Ingólfsson H. I.; Thakur P.; Herold K. F.; Hobart E. A.; Ramsey N. B.; Periole X.; de Jong D. H.; Zwama M.; Yilmaz D.; Hall K.; Maretzky T.; Hugh C.; Hemmings J.; Blobel C.; Marrink S. J.; Koçer A.; Sack J. T.; Andersen O. S. Phytochemicals perturb membranes and promiscuously alter protein function. ACS Chem. Biol. 2014, 9, 1788–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe A.; James M. J.; Lee V. M. Y.; Smith A. B. III; Trojanowski J. Q.; Ballatore C.; Brunden K. R. Aminothienopyridazines and methylene blue affect tau fibrillization via cysteine oxidation. J. Biol. Chem. 2013, 288, 11024–11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huth J. R.; Mendoza R.; Olejniczak E. T.; Johnson R. W.; Cothron D. A.; Liu Y.; Lerner C. G.; Chen J.; Hajduk P. J. ALARM NMR: a rapid and robust experimental method to detect reactive false positives in biochemical screens. J. Am. Chem. Soc. 2005, 127, 217–224. [DOI] [PubMed] [Google Scholar]
- Thorne N.; Inglese J.; Auld D. S. Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem. Biol. 2010, 17, 646–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst K. J.; Allen M. D.; Zhang J. The cAMP-dependent protein kinase inhibitor H-89 attenuates the bioluminescence signal produced by Renilla luciferase. PLoS One 2009, 4, e5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitman L. H.; Veldhoven J. P. D. v.; Zweemer A. M.; Ye K.; Brussee J.; IJzerman A. P. False positives in a reporter gene assay: identification and synthesis of substituted N-pyridin-2-ylbenzamides as competitive inhibitors of firefly luciferase. J. Med. Chem. 2008, 51, 4724–4729. [DOI] [PubMed] [Google Scholar]
- Auld D. S.; Thorne N.; Maguire W. F.; Inglese J. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 3585–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld D. S.; Southall N. T.; Jadhav A.; Johnson R. L.; Diller D. J.; Simeonov A.; Austin C. P.; Inglese J. Characterization of chemical libraries for luciferase inhibitory activity. J. Med. Chem. 2008, 51, 2372–2386. [DOI] [PubMed] [Google Scholar]
- Auld D. S.; Lovell S.; Thorne N.; Lea W. A.; Maloney D. J.; Shen M.; Rai G.; Battaile K. P.; Thomas C. J.; Simeonov A.; Hanzlik R. P.; Inglese J. Molecular basis for the high-affinity binding and stabilization of firefly luciferase by PTC124. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 4878–4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranchak P.; MacArthur R.; Guha R.; Zuercher W. J.; Drewry D. H.; Auld D. S.; Inglese J. Profile of the GSK published protein kinase inhibitor set across ATP-dependent and-independent luciferases: implications for reporter-gene assays. PLoS One 2013, 8, e57888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanagel R.; Vengeliene V.; Jandeleit B.; Fischer W.; Grindstaff K.; Zhang X.; Gallop M.; Krstew E.; Lawrence A.; Kiefer F. Acamprosate produces its anti-relapse effects via calcium. Neuropsychopharmacology 2014, 39, 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole P. A. Chemical probes for histone-modifying enzymes. Nature Chem. Biol. 2008, 4, 590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtele H.; Tsao S.; Lépine G.; Mullick A.; Tremblay J.; Drogaris P.; Lee E.-H.; Thibault P.; Verreault A.; Raymond M. Modulation of histone H3 lysine 56 acetylation as an antifungal therapeutic strategy. Nature Med. 2010, 16, 774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes da Rosa J.; Bajaj V.; Spoonamore J.; Kaufman P. D. A small molecule inhibitor of fungal histone acetyltransferase Rtt109. Bioorg. Med. Chem. Lett. 2013, 23, 2853–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes da Rosa J.; Boyartchuk V. L.; Zhu L. J.; Kaufman P. D. Histone acetyltransferase Rtt109 is required for Candida albicans pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1594–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlin J. L.; Chen X.; Walters M. A.; Zhang Z. Histone-modifying enzymes, histone modifications and histone chaperones in nucleosome assembly: lessons learned from Rtt109 histone acetyltransferases. Crit. Rev. Biochem. Mol. Biol. 2014, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk H.; Connor T.; Yang H.; Loft K. J.; Alcindor J. L.; Nikolakopoulos G.; Surjadi R. N.; Bentley J. D.; Hattarki M. K.; Dolezal O.; Murphy J. M.; Monahan B. J.; Peat T. S.; Thomas T.; Baell J. B.; Parisot J. P.; Street I. P. An efficient high-throughput screening method for MYST family acetyltransferases, a new class of epigenetic drug targets. J. Biomol. Screening 2011, 16, 1196–1205. [DOI] [PubMed] [Google Scholar]
- Sippel T. O. New fluorochromes for thiols: maleimide and iodoacetamide derivatives of a 3-phenylcoumarin fluorophore. J. Histochem. Cytochem. 1981, 29, 314–316. [DOI] [PubMed] [Google Scholar]
- Sippel T. O. Microfluorometric analysis of protein thiol groups with a coumarinylphenylmaleimide. J. Histochem. Cytochem. 1981, 29, 1377–1381. [DOI] [PubMed] [Google Scholar]
- Trievel R. C.; Li F. Y.; Marmorstein R. Application of a fluorescent histone acetyltransferase assay to probe the substrate specificity of the human p300/CBP-associated factor. Anal. Biochem. 2000, 287, 319–328. [DOI] [PubMed] [Google Scholar]
- Palmer N. A.; Sattler S. E.; Saathoff A. J.; Sarath G. A continuous, quantitative fluorescent assay for plant caffeic acid O-methyltransferases. J. Agric. Food Chem. 2010, 58, 5220–5226. [DOI] [PubMed] [Google Scholar]
- Linsky T.; Fast W. A continuous, fluorescent, high-throughput assay for human dimethylarginine dimethylaminohydrolase-1. J. Biomol. Screening 2011, 16, 1089–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C. C.; Ohwaki K.; Schneeweis J. E.; Stec E.; Varnerin J. P.; Goudreau P. N.; Chang A.; Cassaday J.; Yang L.; Yamakawa T.; Kornienko O.; Hodder P.; Inglese J.; Ferrer M.; Strulovici B.; Kusunoki J.; Tota M. R.; Takagi T. A fluorescence-based thiol quantification assay for ultra-high-throughput screening for inhibitors of coenzyme A production. Assay Drug Dev. Technol. 2008, 6, 361–374. [DOI] [PubMed] [Google Scholar]
- Bulfer S.; McQuade T.; Larsen M. Application of a high-throughput fluorescent acetyltransferase assay to identify inhibitors of homocitrate synthase. Anal. Biochem. 2011, 410, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins R. J.; Dotson G. D. A continuous fluorescent enzyme assay for early steps of lipid A biosynthesis. Anal. Biochem. 2012, 425, 21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves V.; Brannigan J. A.; Thinon E.; Olaleye T. O.; Serwa R.; Lanzarone S.; Wilkinson A. J.; Tate E. W.; Leatherbarrow R. J. A fluorescence-based assay for N-myristoyltransferase activity. Anal. Biochem. 2012, 421, 342–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlin J. L.; Sinville R.; Solberg J.; Zhou H.; Francis S.; Strasser J.; John K.; Hook D. J.; Walters M. A.; Zhang Z. A cell-free fluorometric high-throughput screen for inhibitors of Rtt109-catalyzed histone acetylation. PLoS One 2013, 8, e78877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz J. T.; Huth J. R.; Hajduk P. J. Enhancement of chemical rules for predicting compound reactivity towards protein thiol groups. J. Comput.-Aided Mol. Des. 2007, 21, 139–144. [DOI] [PubMed] [Google Scholar]
- McCallum M. M.; Nandhikonda P.; Temmer J. J.; Eyermann C.; Simeonov A.; Jadhav A.; Yasgar A.; Maloney D.; Arnold L. A. High-throughput identification of promiscuous inhibitors from screening libraries with the use of a thiol-containing fluorescent probe. J. Biomol. Screening 2013, 18, 705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlin J. L.; Walters M. A. The essential roles of chemistry in high-throughput screening triage. Future Med. Chem. 2014, 6, 1265–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T.; Yang C.; Zheng Y. G. Comparative studies of thiol-sensitive fluorogenic probes for HAT assays. Anal. Bioanal. Chem. 2012, 421, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne N.; Auld D. S.; Inglese J. Apparent activity in high-throughput screening: origins of compound-dependent assay interference. Curr. Opin. Chem. Biol. 2010, 14, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoichet B. K. Interpreting steep dose–response curves in early inhibitor discovery. J. Med. Chem. 2006, 49, 7274–7277. [DOI] [PubMed] [Google Scholar]
- Prinz H. Hill coefficients, dose–response curves and allosteric mechanisms. J. Chem. Biol. 2010, 3, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanyam K.; Altaf M.; Varier R.; Swaminathan V.; Ravindran A.; Sadhale P.; Kundu T. Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J. Biol. Chem. 2004, 279, 33716–33726. [DOI] [PubMed] [Google Scholar]
- Federici L.; Sterzo C. L.; Pezzola S.; Matteo A. D.; Scaloni F.; Federici G.; Caccuri A. M. Structural basis for the binding of the anticancer compound 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol to human glutathione S-transferases. Cancer Res. 2009, 69, 8025–8034. [DOI] [PubMed] [Google Scholar]
- Montoya L. A.; Pearce T. F.; Hansen R. J. Development of selective colorimetric probes for hydrogen sulfide based on nucleophilic aromatic substitution. J. Org. Chem. 2013, 78, 6550–6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung-Toung R.; Wodzinska J.; Li W.; Lowrie J.; Kukreja R.; Desilets D.; Karimian K.; Tam T. F. 1,2,4-Thiadiazole: a novel Cathepsin B inhibitor. Bioorg. Med. Chem. 2003, 11, 5529–5537. [DOI] [PubMed] [Google Scholar]
- Leung-Toung R.; Tam T. F.; Wodzinska J. M.; Zhao Y.; Lowrie J.; Simpson C. D.; Karimian K.; Spino M. 3-Substituted imidazo[1,2-d][1,2,4]-thiadiazoles: a novel class of factor XIIIa inhibitors. J. Med. Chem. 2005, 48, 2266–2269. [DOI] [PubMed] [Google Scholar]
- Marrano C.; Macedo P. d.; Gagnon P.; Lapierre D.; Gravel C.; Keillor J. W. Synthesis and evaluation of novel dipeptide-bound 1,2,4-thiadiazoles as irreversible inhibitors of guinea pig liver transglutaminase. Bioorg. Med. Chem. 2001, 9, 3231–3241. [DOI] [PubMed] [Google Scholar]
- Lewandowicz A. M.; Vepsäläinen J.; Laitinen J. T. The ‘allosteric modulator’ SCH-202676 disrupts G protein-coupled receptor function via sulphydryl-sensitive mechanisms. Br. J. Pharmacol. 2006, 147, 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göblyös A.; Vries H. d.; Brussee J.; IJzerman A. P. Synthesis and biological evaluation of a new series of 2,3,5-substituted [1,2,4]-thiadiazoles as modulators of adenosine A1 receptors and their molecular mechanism of action. J. Med. Chem. 2005, 48, 1145–1151. [DOI] [PubMed] [Google Scholar]
- Wörffel U.; Behnisch R. 5-Sulfanilamido- und 3-sulfanilamido-1,2,4-thiodiazole. Arch. Pharm. 1962, 295, 811–816. [DOI] [PubMed] [Google Scholar]
- Kurzer F. 1,2,4-Thiadiazoles. Adv. Heterocycl. Chem. 1965, 5, 119–204. [Google Scholar]
- Wilkins D.; Bradley P.. 1,2,4-Thiadiazoles. In Comprehensive Heterocyclic Chemistry II; Katrizky A., Rees C., Scriven E., Eds.; Pergamon: New York, 1996; Vol. 4, pp 307–354. [Google Scholar]
- Clerici F. Thiazole and thiadiazole S-oxides. Adv. Heterocycl. Chem. 2002, 83, 71–115. [Google Scholar]
- Tam T.; Leung-Toung R.; Li W.; Spino M.; Karimian K. Medicinal chemistry and properties of 1,2,4-thiadiazoles. Mini-Rev. Med. Chem. 2005, 5, 367–379. [DOI] [PubMed] [Google Scholar]
- Zyabrev V. S.; Renskii M. A. Recyclization of 5-[methyl(phenyl)amino]-2-(4-nitrobenzyl)-3-p-tolyl-1,2,4-thiadiazolium perchlorate into 9-methyl-3a-(4-nitrophenyl)-2-p-tolyl-3a,9-dihydrobenzo[b]-imidazo[4,5-e][1,4]thiazine. Russ. J. Gen. Chem. 2011, 81, 2209–2211. [Google Scholar]
- Zhang F.; Estavillo C.; Mohler M.; Cai J. Non-enzymatic reduction of a 1,2,4-thiadiazolium derivative. Bioorg. Med. Chem. 2008, 18, 2172–2178. [DOI] [PubMed] [Google Scholar]
- Renskii M. A.; Zyabrev V. S. Recyclization of 2,3-disubstituted 5-arylamino-2,5-dihydro-1,2,4-thiadiazoles and related salts into benzothiazole derivatives. Russ. J. Gen. Chem. 2003, 73, 1324–1325. [Google Scholar]
- Zyabrev V. S.; Rensky M. A.; Rusanov E. B.; Drach B. S. Cycloaddition of N-(2,2,2-trichloroethylidene)-substituted carboxamides and carbamates to 1,2,4-thiadiazol-5(2H)-imines. Heteroat. Chem. 2003, 14, 474–480. [Google Scholar]
- Renskii M. A.; Zyabrev V. S.; Drach B. S. Heterocyclizations based on products of cycloaddition of malononitrile to substituted 1,2,4-thiadiazol-5-(2H)-imines. Russ. J. Gen. Chem. 2002, 72, 1826–1827. [Google Scholar]
- Li W.-W.; Heinze J.; Haehnel W. Site-specific binding of quinones to proteins through thiol addition and addition–elimination reactions. J. Am. Chem. Soc. 2005, 127, 6140–6141. [DOI] [PubMed] [Google Scholar]
- Reed D.; Shen Y.; Shelat A. A.; Arnold L. A.; Ferreira A. M.; Zhu F.; Mills N.; Smithson D. C.; Regni C. A.; Bashford D.; Cicero S. A.; Schulman B. A.; Jochemsen A. G.; Guy R. K.; Dyer M. A. Identification and characterization of the first small molecule inhibitor of MDMX. J. Biol. Chem. 2010, 287, 10786–10796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albaugh B. N.; Kolonko E. M.; Denu J. M. Kinetic mechanism of the Rtt109–Vps75 histone acetyltransferase–chaperone complex. Biochemistry 2010, 49, 6375–6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J. M.; Wu D.; Motiu-DeGrood R.; Hupe D. J. A spectrophotometric method for studying the rates of reaction of disulfides with protein thiol groups applied to bovine serum albumin. J. Am. Chem. Soc. 1980, 102, 359–363. [Google Scholar]
- Huth J. R.; Song D.; Mendoza R. R.; Black-Schaefer C. L.; Mack J. C.; Dorwin S. A.; Ladror U. S.; Severin J. M.; Walter K. A.; Bartley D. M.; Hajduk P. J. Toxicological evaluation of thiol-reactive compounds identified using a la assay to detect reactive molecules by nuclear magnetic resonance. Chem. Res. Toxicol. 2007, 20, 1752–1759. [DOI] [PubMed] [Google Scholar]
- Zhuang C.; Narayanapillai S.; Zhang W.; Sham Y. Y.; Xing C. Rapid identification of Keap1–Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J. Med. Chem. 2014, 57, 1121–1126. [DOI] [PubMed] [Google Scholar]
- Canny S. A.; Cruz Y.; Southern M. R.; Griffin P. R. PubChem promiscuity: a web resource for gathering compound promiscuity data from PubChem. Bioinformatics 2009, 28, 140–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L.; Wang Y.; Bryant S. H. A survey of across-target bioactivity results of small molecules in PubChem. Bioinformatics 2009, 25, 2251–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Bajorath J. What is the likelihood of an active compound to be promiscuous? Systematic assessment of compound promiscuity on the basis of PubChem confirmatory bioassay data. AAPS J. 2013, 15, 808–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See Supporting Information for references to papers containing examples of these problematic chemotypes.
- Nissink J. W. M.; Blackburn S. Quantification of frequent-hitter behavior based on historical high-throughput screening data. Future Med. Chem. 2014, 6, 1113–1126. [DOI] [PubMed] [Google Scholar]
- Vuorinen A.; Engeli R.; Meyer A.; Bachmann F.; Griesser U. J.; Schuster D.; Odermatt A. Ligand-based pharmacophore modeling and virtual screening for the discovery of novel 17β-hydroxysteroid dehydrogenase 2 inhibitors. J. Med. Chem. 2014, 57, 5995–6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Tanner K. G.; Denu J. M. A continuous, nonradioactive assay for histone acetyltransferases. Anal. Biochem. 2000, 280, 308–314. [DOI] [PubMed] [Google Scholar]
- Fanslau C.; Pedicord D.; Nagulapalli S.; Gray H.; Pang S.; Jayaraman L.; Lippy J.; Blat Y. An electrophoretic mobility shift assay for the identification and kinetic analysis of acetyl transferase inhibitors. Anal. Biochem. 2010, 402, 65–68. [DOI] [PubMed] [Google Scholar]
- Ait-Si-Ali S.; Ramirez S.; Robin P.; Trouche D.; Harel-Bellan A. A rapid and sensitive assay for histone acetyl-transferase activity. Nucleic Acids Res. 1998, 26, 3869–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turlais F.; Hardcastle A.; Rowlands M.; Newbatt Y.; Bannister A.; Kouzarides T.; Workman P.; Aherne G. High-throughput screening for identification of small molecule inhibitors of histone acetyltransferases using scintillating microplates (FlashPlate). Anal. Biochem. 2001, 298, 62–68. [DOI] [PubMed] [Google Scholar]
- Wu H.-Y.; Huang F.-Y.; Chang Y.-C.; Hsieh M.-C.; Liao P.-C. Strategy for determination of in vitro protein acetylation sites by using isotope-labeled acetyl coenzyme A and liquid chromatography–mass spectrometry. Anal. Chem. 2008, 80, 6178–6189. [DOI] [PubMed] [Google Scholar]
- Robers M. B.; Loh C.; Carlson C. B.; Yang H.; Frey E. A.; Hermanson S. B.; Bi K. Measurement of the cellular deacetylase activity of SIRT1 on p53 via LanthaScreen technology. Mol. BioSyst. 2011, 7, 59–66. [DOI] [PubMed] [Google Scholar]
- Xie F.; Li B. X.; Broussard C.; Xiao X. Identification, synthesis and evaluation of substituted benzofurazans as inhibitors of CREB-mediated gene transcription. Bioorg. Med. Chem. Lett. 2013, 23, 5371–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan M. E.; Abramite J. A.; Anderson D. P.; Aulabaugh A.; Dahal U. P.; Gilbert A. M.; Li C.; Montgomery J.; Oppenheimer S. R.; Ryder T.; Schuff B. P.; Uccello D. P.; Walker G. S.; Wu Y.; Brown M. F.; Chen J. M.; Hayward M. M.; Noe M. C.; Obach R. S.; Philippe L.; Shanmugasundaram V.; Shapiro M. J.; Starr J.; Stroh J.; Che Y. Chemical and computational methods for the characterization of covalent reactive groups for the prospective design of irreversible inhibitors. J. Med. Chem. 2014, 57, 10072–10079. [DOI] [PubMed] [Google Scholar]
- Toledo Warshaviak D.; Golan G.; Borrelli K. W.; Zhu K.; Kalid O. Structure-based virtual screening approach for discovery of covalently bound ligands. J. Chem. Inf. Model. 2014, 54, 1941–1950. [DOI] [PubMed] [Google Scholar]
- Davids M. S.; Brown J. R. Ibrutinib: a first in class covalent inhibitor of Bruton’s tyrosine kinase. Future Oncol. 2014, 10, 957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalgutkar A. S.; Dalvie D. K. Drug discovery for a new generation of covalent drugs. Expert Opin. Drug Discovery 2012, 7, 561–581. [DOI] [PubMed] [Google Scholar]
- Venci J. V.; Gandhi M. A. Dimethyl fumarate (Tecfidera): a new oral agent for multiple sclerosis. Ann. Pharmacother. 2013, 47, 1697–1702. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Sabnis Y.; Zhao Z.; Zhang T.; Buhrlage S. J.; Jones L. H.; Gray N. S. Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. (Oxford, U. K.) 2013, 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinko W.; Wang Y.; Zhu W.; Zhang Y.; Feixas F.; Cox C. L.; Mitchell D. A.; Oldfield E.; McCammon J. A. Undecaprenyl diphosphate synthase inhibitors: antibacterial drug leads. J. Med. Chem. 2014, 57, 5693–5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancos I.; Bida J. P.; Tian D.; Bundrick M.; John K.; Holte M. N.; Her Y. F.; Evans D.; Saenz D. T.; Poeschla E. M.; Hook D.; Georg G.; Maher L. J. High-throughput screening for growth inhibitors using a yeast model of familial paraganglioma. PLoS One 2013, 8, e56827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Chung T.; Oldenburg K. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screening 1999, 4, 67–73. [DOI] [PubMed] [Google Scholar]
- Johnston P. A.; Soares K. M.; Shinde S. N.; Foster C. A.; Shun T. Y.; Takyi H. K.; Wipf P.; Lazo J. S. Development of a 384-well colorimetric assay to quantify hydrogen peroxide generated by the redox cycling of compounds in the presence of reducing agents. Assay Drug Dev. Technol. 2008, 6, 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancy B. M.; Crump N. T.; Peterson D. J.; Mukherjee C.; Bowers E. M.; Ahn Y.-H.; Yoshida M.; Zhang J.; Mahadevan L. C.; Meyers D. J.; Boeke J. D.; Cole P. A. Live-cell studies of p300/CBP histone acetyltransferase activity and inhibition. ChemBioChem 2012, 13, 2113–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Han J.; Kang B.; Burgess R.; Zhang Z. Human histone acetyltransferase 1 protein preferentially acetylates H4 histone molecules in H3.1-H4 over H3.3-H4. J. Biol. Chem. 2012, 287, 6573–6581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrios A.; Selleck W.; Hnatkovich B.; Kramer R. Expression and purification of recombinant yeast Ada2/Ada3/Gcn5 and Piccolo NuA4 histone acetyltransferase complexes. Methods 2007, 41, 271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A.; Wilm M.; Vorm O.; Mann M. Mass spectrometric sequencing of proteins from silver-stained polyacrylaminde gels. Anal. Chem. 1996, 68, 850–858. [DOI] [PubMed] [Google Scholar]
- Lin-Moshier Y.; Sebastian P.; Higgins L.; Sampson N.; Hewitt J.; Marchant J. Re-evaluation of the role of calcium homeostasis endoplasmic reticulum protein (CHERP) in cellular calcium signaling. J. Biol. Chem. 2013, 288, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B.; Zhang K.; Hendrie C.; Liang C.; Li M.; Doherty-Kirby A.; Lajoie G. PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 2337–2342. [DOI] [PubMed] [Google Scholar]
- Gardner K. H.; Zhang X.; Gehring K.; Kay L. E. Solution NMR studies of a 42 kDa Escherichia coli maltose binding protein/β-cyclodextrin complex: chemical shift assignments and analysis. J. Am. Chem. Soc. 1998, 120, 11738–11748. [Google Scholar]
- Hajduk P. J.; Augeri D. J.; Mack J.; Mendoza R.; Yang J.; Betz S. F.; Fesik S. W. NMR-based screening of proteins containing 13C-labeled methyl groups. J. Am. Chem. Soc. 2000, 122, 7898–7904. [Google Scholar]
- Copeland R. A.Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists. 1st ed.; Wiley-Interscience: New York, 2005. [PubMed] [Google Scholar]
- Lor L. A.; Schneck J.; McNulty D. E.; Diaz E.; Brandt M.; Thrall S. H.; Schwartz B. A simple assay for detection of small-molecule redox activity. J. Biomol. Screening 2007, 12, 881–890. [DOI] [PubMed] [Google Scholar]
- Curpan R.; Avram S.; Vianello R.; Bologa C. Exploring the biological promiscuity of high-throughput screening hits through DFT calculations. Bioorg. Med. Chem. 2014, 22, 2461–2468. [DOI] [PubMed] [Google Scholar]
- Smith G.; Barrett D.; Blackburn K.; Cory M. Expression, preparation, and high-throughput screening of caspase-8: discovery of redox-based and steroid diacid inhibition. Arch. Biochem. Biophys. 2002, 399, 195–205. [DOI] [PubMed] [Google Scholar]
- Crowe A.; Huang W.; Ballatore C.; Johnson R. L.; Hogan A.-M. L.; Huang R.; Wichterman J.; McCoy J.; Huryn D.; Auld D. S.; Smith A. B.; Inglese J.; Trojanowski J. Q.; Austin C. P.; Brunden K. R.; Lee V. M. Y. Identification of aminothienopyridazine inhibitors of tau assembly by quantitative high-throughput screening. Biochemistry 2009, 48, 7732–7745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters W. P.; Namchuk M. A guide to drug discovery: designing screens: how to make your hits a hit. Nature Rev. Drug Discovery 2003, 2, 259–266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.