

Abstract
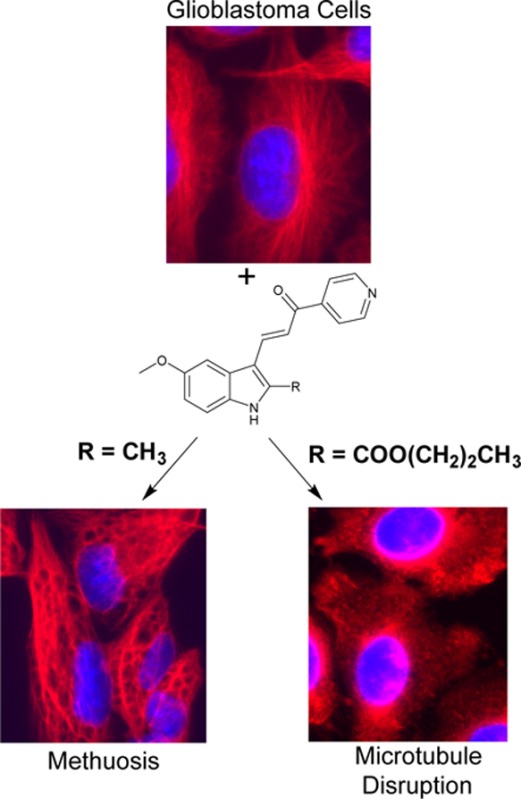
Methuosis is a form of nonapoptotic cell death characterized by an accumulation of macropinosome-derived vacuoles with eventual loss of membrane integrity. Small molecules inducing methuosis could offer significant advantages compared to more traditional anticancer drug therapies that typically rely on apoptosis. Herein we further define the effects of chemical substitutions at the 2- and 5-indolyl positions on our lead compound 3-(5-methoxy-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propene-1-one (MOMIPP). We have identified a number of compounds that induce methuosis at similar potencies, including an interesting analogue having a hydroxypropyl substituent at the 2-position. In addition, we have discovered that certain substitutions on the 2-indolyl position redirect the mode of cytotoxicity from methuosis to microtubule disruption. This switch in activity is associated with an increase in potency as large as 2 orders of magnitude. These compounds appear to represent a new class of potent microtubule-active anticancer agents.
Introduction
Conventional cancer chemotherapy relies heavily on drugs that trigger programmed cell death via activation of apoptosis pathways.1,2 However, many cancers harbor mutations in tumor suppressor genes that function in apoptosis signaling.3−5 Combined with increased capacity for drug efflux and DNA repair, these mutations attenuate apoptosis and contribute to the chemoresistance frequently encountered in recurrent tumors.6 Glioblastoma multiforme (GBM) is the most common primary brain tumor in adults, and it is a notorious example of cancer that is refractory to most forms of chemotherapy. Treatment for GBM typically includes surgical excision, followed by radiotherapy and administration of the DNA alkylating agent temozolomide.7 This standard-of-care therapy marginally increases patient survival time, but no curative treatment exists.7,8 Identification of molecules that can kill cancer cells via nonapoptotic cell death mechanisms may provide inroads to circumvent the insensitivity of GBM and other cancers to apoptosis. A number of nonapoptotic cell death pathways have been defined,9,10 and associated small molecules capable of inducing these pathways have been reported.11 Our group has focused on the development of compounds that can induce a novel form of cell death termed methuosis.12 First observed as a caspase-independent form of cell death triggered by overexpression of activated Ras in GBM cells,13,14 methuosis is characterized by the accumulation of large cytoplasmic vacuoles derived from macropinosomes and nonclathrin-coated endosomes. The vacuolated cells eventually undergo metabolic failure and lose membrane integrity. In pursuit of small molecules that might be used to induce methuosis in a therapeutic context, we initially identified indolyl-pyridinyl-propenones (also referred to as indole-based chalcones) that can trigger this form of cell death in GBM cells, including those resistant to temozolomide.15 Preliminary structure–activity relationship (SAR) studies defined key requirements for methuosis-inducing activity, resulting in a lead compound, 3-(5-methoxy-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propene-1-one, abbreviated MOMIPP (1a, Figure 1), with activity at low micromolar concentrations in cell-based assays.16 The following features contributed to optimal activity of the pharmacophore: (i) an indole-propenone-pyridine scaffold where the pyridine’s attachment is in the para (4′) position; (ii) a methoxy group at the 5-indolyl position; and, (iii) small alkyl substitutions at the 2-indolyl position, i.e., R = Me (1a) and Et (1b)16,17 (Figure 1). More recent studies of longer-chain aliphatic substitutions on the 2-indolyl position identified compounds that retained the ability to induce cellular vacuolization but lost the ability to kill GBM cells, as exemplified by compound 1c(17) (Figure 1). These surprising findings suggest that induction of methuotic cell death by specific indolyl-pyridinyl-propenones may involve pleiotropic effects on cellular pathways beyond those responsible for perturbation of macropinosome trafficking and vacuolization. Herein we evaluated a new series of compounds with additional modifications at the 2- and 5-indolyl positions. The results show that these compounds can either maintain good methuosis-inducing activity or uncouple vacuolization from cell death. An unexpected finding was that certain substitutions at the 2-indolyl position conferred a substantially altered biological profile, with disruption of microtubule polymerization becoming a prominent feature. This was accompanied by a dramatic increase in cytotoxicity. These compounds appear to represent a new class of potent microtubule-active anticancer agents.
Figure 1.

Structural requirements for optimal induction of methuotic cell death. Compound 1a is the current lead compound. The ethyl analogue, 1b, is of similar potency. The propyl derivative, 1c, induces cytoplasmic vacuolization, but is not cytotoxic.17
Results
Chemistry
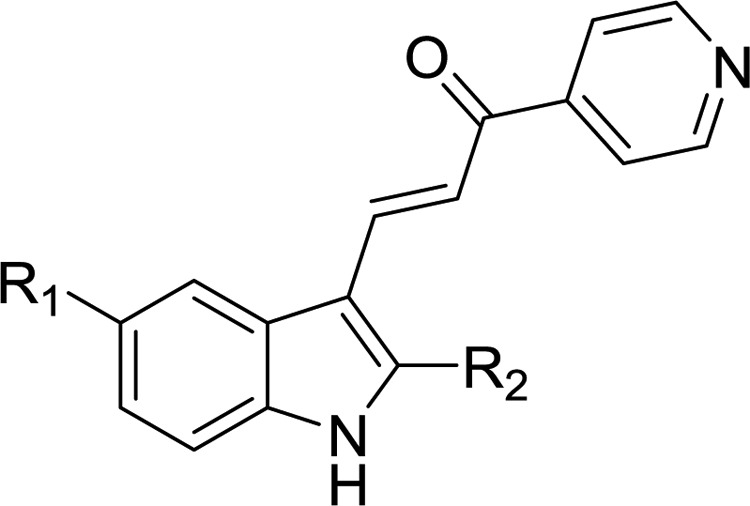
The 2- and 5-substituted indolyl-pyridinyl-propenones (Table 1, 2a–2q) were prepared by one of the following methodologies: (1) Regioselective acylation from 4-substituted N-BOC-protected 2-methylanilines (6a–6d, 18) and Weinreb amides18 (7, 19) followed by acid-catalyzed cyclization and deprotections (Schemes 1 and 4) to afford 1H-indole rings (8a–d, 20);19 or (2) Synthetic manipulation at specific positions on the formed indole ring to obtain 5-substituted amides (11, 12, 16 in Schemes 2 and 3), alkyl 2-indolylcarboxylates (23a–d in Scheme 5) or functionalized hydroxyl-based 2-indole intermediates (25, 30, 31 in Schemes 6 and 7). Synthesized indoles were then formylated under Vilsmeier–Haack conditions and subsequently condensed with 4-acetylpyridine producing final targets (2a–f, 2h, 2j–n, 2p, 2q) in a manner similar to our previous reports.16,17 Targets 2g, 2i, and 2o were prepared from their corresponding indolyl-pyridinyl-propenone scaffolds.
Table 1. Summary of Growth Inhibition Results for Compounds Generated By Schemes 1–7a.
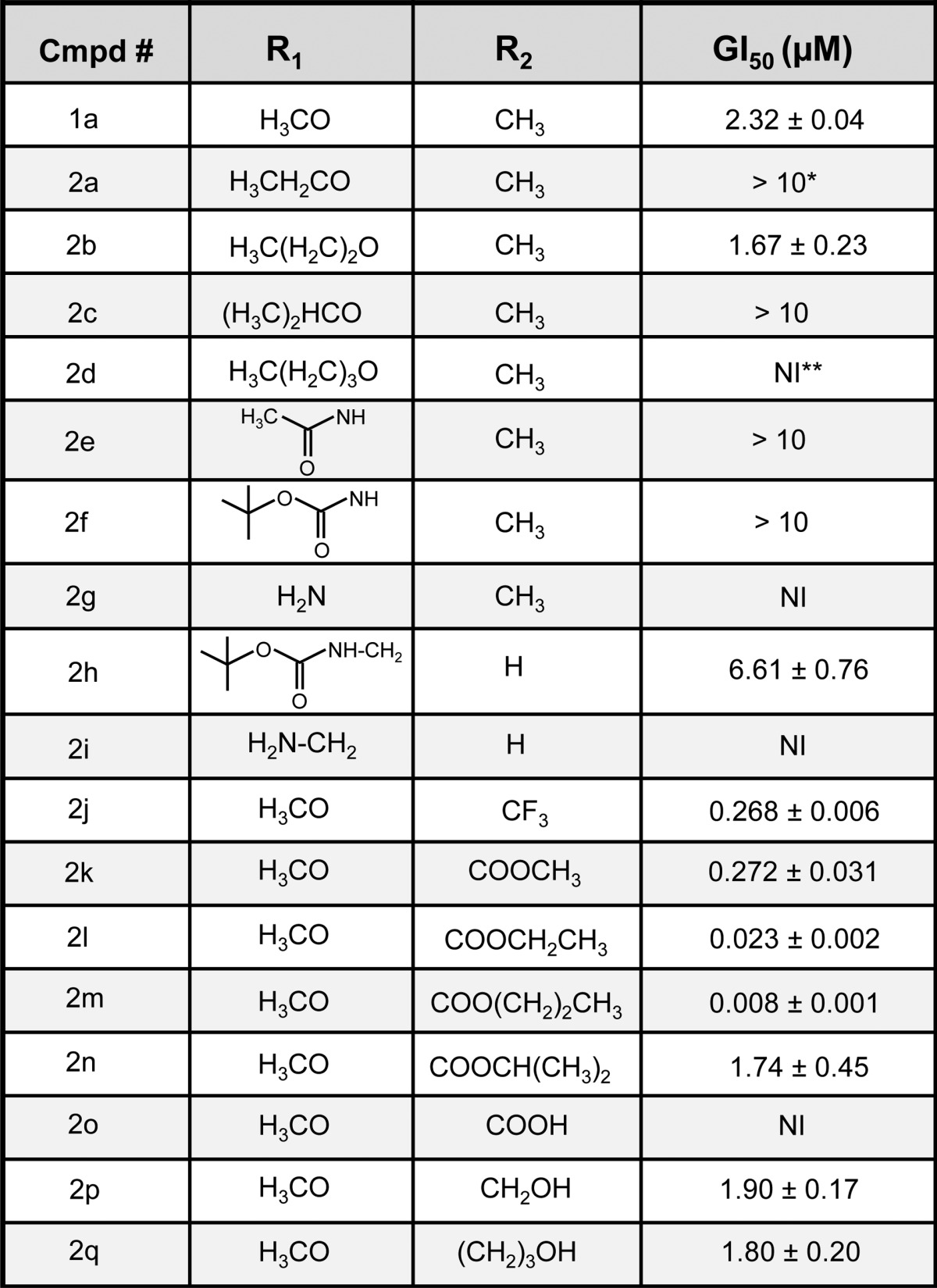
(*) >10: Growth inhibition relative to control did not reach 50% at the highest concentration tested (10 μM). (**) NI: No significant growth inhibition was detected relative to vehicle control.
Scheme 1. Synthesis of 5-Alkyloxy-2-methylindolyl Substituted Pyridinyl-Propenones 2a–d.

Reagents and conditions: (a) diethylsulfate in the case of 4a, or alkyl bromide, K2CO3, 2-butanone, reflux; (b) H2, Pd/C, EtOAc; (c) di-tert-butyl dicarbonate, THF, reflux; (d) 1. −40 to −50 °C, sec-butyllithium, THF; 2. −50 to −10 °C, 7; 3. TFA/DCM (e) 1. POCl3, DMF; 2. 1 N NaOH; (f) 4-acetylpyridine, piperidine, MeOH, reflux.
Scheme 4. Synthesis of 5-Methoxy-2-trifluoromethylindole-pyridinyl-propenone 2j.

Reagents and conditions: (a) 1. THF, sec-butyllithium, −40 °C to −50 °C; 2. Weinreb amide 19, −50 °C to −10 °C; 3. TFA/DCM; (b) 1. POCl3, DMF; 2. 1 N NaOH; (c) 4-acetylpyridine, piperidine, MeOH.
Scheme 2. Synthesis of 5-Amino-2-methylindolyl-pyridinyl-propenone (2g) and 5-Substituted Amides 2e and 2f.

Reagents and conditions: (a) acetic anhydride, reflux; (b) di-tert-butyldicarbonate, CH3CN, reflux; (c) 1. POCl3, DMF; 2. 1 N NaOH; (d) 4-acetylpyridine, MeOH, reflux; (e) 2f, TFA/MeOH.
Scheme 3. Synthesis of 5-Aminomethylindolyl-pyridinyl-propenones 2h and 2i.

Reagents and conditions: (a) di-tert-butyl dicarbonate, CH3CN, rt; (b) 1. POCl3, DMF; 2. 1 N NaOH; (c) 4-acetylpyridine, piperidine, MeOH, reflux; (d) TFA, MeOH, reflux.
Scheme 5. Synthesis of the Analogous Series of Substituted Alkyl 2-Indolylcarboxylate-pyridinyl-propenones 2k–2n, as well as the Free Acid Analogue 2o.

Reagents and conditions: (a) HCl (g), R-OH, reflux; (b) 1. POCl3, DMF; 2. 1 N NaOH; (c) 4-acetylpyridine, piperidine, R-OH, reflux; (d) 2k, MeOH, 1 N NaOH.
Scheme 6. Synthesis of 2-Hydroxymethyl-5-methoxyindole-pyridinyl-propenone 2p.

Reagents and conditions: (a) LiAlH4, THF, rt; (b) acetic anhydride, TEA, CH3CN, rt; (c) 1. POCl3, DMF; 2. 1 N NaOH; (d) 4-acetylpyridine, piperidine, MeOH, reflux.
Scheme 7. Synthesis of 2-Hydroxypropyl-5-methoxyindole-pyridinylpropenone 2q.

Reagents and conditions: (a) MnO2, EtOAc, reflux; (b) Ph3PCHCO2C2H5, THF, rt; (c) 1. LiAlH4, THF, 0 °C to rt; 2. H2, Pd/C, EtOAc/MeOH; (d) 30, acetic anhydride, TEA, CH3CN, rt; (e) 1. POCl3, DMF; 2. 1 N NaOH; (f) 4-acetylpyridine, piperidine, MeOH, reflux.
Schemes 1–3 depict the specific preparations of several 5-substituted analogues designed to probe the effects of increasing the hydrocarbon chain length in the ether linkage or replacing methoxy with nitrogen-containing functionalities. The 5-alkyloxyindole analogues (Scheme 1) were synthesized from commercially available 3-methyl-4-nitrophenol (3) by reaction with various alkyl-bromides, as in the case of 4b–d, or diethylsulfate for 4a. Hydrogenation reduced the nitro-group to amines 5a–5d, which were then protected with BOC (6a–6d). Regioselective acylation at the 2-methyl position was achieved using sec-butyllithium and Weinreb amide 7. The resulting ketone intermediate was cyclized and deprotected in a one-pot fashion utilizing excess TFA to yield the 5-alkyloxy-2-methylindoles 8a–8d. Reaction of these indoles with POCl3 and DMF, followed by treatment with 1 N NaOH, provided aldehydes 9a–9d which conveniently precipitated from the reaction media. The aldehydes were condensed with 4-acetylpyridine to provide the 5-substituted target compounds 2a–2d.
Analogues of 1a containing either amide or amine substitution at the 5-indolyl position were prepared as shown in Schemes 2 and 3. 5-Amino-2-methylindole (10, Scheme 2) was prepared by previously described methods16 and used to synthesize amide intermediates 11 and 12 by reaction with acetic anydride and di-tert-butyl dicarbonate, respectively. Interestingly, compound 11 was isolated as the diacetylated amide under conditions of reflux and excess reagent. The literature provides precedent for diacylation to occur when aniline is subjected to similar conditions.20 Conveniently, this readily isolable material could be deployed directly in the next step because a single acetyl-group was hydrolyzed during the NaOH-water workup leading to 13. Aldehydes 13 and 14 were condensed with 4-acetylpyridine to provide target compounds 2e and 2f. TFA deprotection of 2f in MeOH at rt yielded the free amine probe 2g. The des-methyl derivatives 2h and 2i (Scheme 3) were prepared from commercially available 15 in a fashion analogous to that for 2f and 2g but contain a methylene substitution between the amine and the indole ring.
Expanding previous SAR studies of 2-indolyl alkyl substitutions,17 additional functionalized probes were prepared as depicted in Schemes 4–7. Trifluoromethyl derivative 2j (Scheme 4), an isostere of 1a, was synthesized by acylating the 2-methyl substituent on the N-BOC-protected aniline with trifluoromethyl amide 19.18,21 Excellent regioselectivity was achieved by the dilithio species derived from 18 and sec-butyllithium in THF at −40 °C.19 Cyclization and deprotection occurred in a one-pot fashion with excess TFA affording 5-methoxy-2-trifluoromethylindole (20). Synthesis of aldehyde 21 and condensation to the final trifluoromethyl target 2j were accomplished by our standard methods. Alkyl carboxylates (23a–d) were synthesized from commercially available acid 22 by Fisher esterification conditions using their corresponding alcohols (Scheme 5). Formylation reactions to yield aldehydes 24a–24d, followed by Claisen-Schmidt condensation to provide 2k–2n, were conducted as described previously.16,17 Interestingly, the isopropyl ester 2n appeared to be the most sensitive to hydrolysis during workup and subsequent manipulation or storage, likely owing to its better leaving group among this series. Nevertheless, after saponification in aqueous base and MeOH, free acid 2o was also readily obtained from 2k which was available in greater supply.
Derivatives substituted with hydroxyl functionality were synthesized based upon the results obtained in our previous and current SAR studies for 2-substituted indole. Subtle chemical modifications at this position can alter activity and appear to be sensitive to both steric and electronic properties. Hydroxymethyl probe 2p was obtained according to Scheme 6. Previously prepared intermediate ethyl 5-methoxyindole-2-carboxylate (23b) was reduced by LAH to 25, which was then protected by acetylating the primary hydroxyl group (26).22 Conveniently, the acetyl group was then hydrolyzed during the alkaline conditions used to quench the acidic reagents generated during the Vilsmeier’s step that produce aldehyde 27. The aldehyde was condensed with 4-acetylpyridine to obtain 2p. This final target compound proved to be unstable when stored at rt in DMSO for 48 h (see associated note in the Experimental Section), presumably due to the presence of a benzylic alcohol conjugated to an α,β-unsaturated ketone. To circumvent the inherent instability of 2p, we also synthesized an analogue containing a hydroxypropyl functionality using the methodology in Scheme 7. Alcohol 25 was oxidized to 5-methoxyindole-2-carboxaldehyde (28) by MnO2 in EtOAc heated to reflux. The aldehyde was subjected to Wittig conditions using phosphonium ylide (Ph3PCHCO2C2H5) to produce the α,β-unsaturated ester 29, which was reduced to 2-hydroxypropylindole 30 using excess LAH. The primary hydroxyl group was protected as the O-acetate (31) under conditions of excess acetic anhydride, TEA, and CH3CN. Standard reactions of formylation (32) and condensation afforded hydroxypropyl analogue 2q. As anticipated, the O-acetyl group was hydrolyzed under these conditions.
Biological Activity
Growth Inhibition and Morphological Assays
Substitutions at the 5-Position of the Indole Ring
Cells experiencing methuosis initially undergo extreme cytoplasmic vacuolization which can be readily assessed by phase contrast microscopy. As viability is compromised (usually between 24 and 48 h), the cells detach from the culture surface and lose membrane integrity. Previous studies have established that the sulforhodamine B (SRB) colorimetric assay, which measures protein associated with adherent cells, is useful for evaluating the loss of viable cells and for ranking the relative potency of methuosis-inducing compounds.17 Although methuosis can occur in a broad spectrum of human tumor cell lines,12,14−16 our previous SAR studies were carried out with U251 glioblastoma cells. Therefore, we continued to use this cell line in the present studies. Growth inhibitory (GI) activities for all target compounds are recorded in Table 1 as the dose able to achieve 50% inhibition (GI50) compared to growth within control cultures treated with only vehicle.
When 1a’s methoxy-group is replaced by ethoxy (2a), isopropoxy (2c), or butoxy (2d) GI activity was markedly reduced. In contrast, the GI50 value for the 5-propoxy compound (2b) remained similar to that of 1a. As shown in Figure 2, the morphology of cells treated with 2.5 μM 1a was consistent with methuosis. Robust cytoplasmic vacuolization was observed within 4 h. The vacuoles persisted for the duration of the 48 h time-course with no increase in cell density. By 48 h, some nonviable cells could be observed detaching from the dish. Cytotoxicity was more evident at 10 μM, with very few viable cells remaining on the dish. The effects of 2b at 2.5 μM were essentially the same as those observed with 1a. In accord with its complete lack of GI activity, the morphological effect of the butoxy derivative 2d contrasted sharply with that of the propoxy 2b in that the former produced much fewer and smaller vacuoles and had little effect on cell density. Interestingly, the ethoxy (2a) and isopropoxy (2c) derivatives exhibited intermediate activity, with some induction of cellular vacuolization, but little or no inhibition of cell growth. This apparent dissociation between vacuolization and inhibition of cell growth/viability was similar to what we observed previously with some of the 2-indolyl-substituted pyridinyl-propenones upon lengthening the alkyl chain.17
Figure 2.

Evaluation of the impact of increasing the indolyl 5-alkyloxy chain length on the morphological effects of 2-methylindolyl-pyridinyl-propenones in U251 glioblastoma cells. Phase contrast images of live cells were obtained as described in the Experimental Section. Compounds were added at the concentrations listed at the top of each panel. The control cells shown in the top panel received an equivalent volume of the DMSO vehicle.
The 5-amino derivative 2g had no GI activity in the concentration range tested (Table 1). It also failed to induce vacuolization at 2.5 μM, although it did cause vacuolization after 48 h at 10 μM (Figure 3). Likewise, neither the acetamide (2e), nor N-BOC (2f) analogues showed substantial growth inhibition below 10 μM (Table 1), although both compounds induced vacuolization when applied at 2.5 μM and 10 μM (Figure 3). Thus, the activities of 2e, 2f, and 2g were similar to 2a and 2c, insofar as all of these compounds triggered the accumulation of vacuoles without causing growth arrest or cytotoxicity. The 2-position des-methyl versions of the preceding N-BOC and free amine compounds, with a methylene spacer between the nitrogen and the indole ring at the 5-position (2h and 2i), gave mixed results. Primary amine 2i was completely inactive in both growth inhibition and vacuolization assays while the behavior of the N-BOC derivative 2h was somewhat anomalous. It did not induce vacuolization or cell death (Figure 3) but caused a modest reduction in the growth rate of the cells at concentrations above 2.5 μM (Table 1).
Figure 3.

Evaluation of the impact of amine, acetamide, and N-BOC substitutions at the 5-position of the indole ring on the morphological effects of 2-methylindolyl-pyridinyl-propenones. Phase contrast images of live cells were obtained as described in the Experimental Section. Compounds were added at the concentrations listed at the top of each panel.
Substitutions at the 2-Position of the Indole Ring
To further explore the influence of the 2-position on the biological activity of the pharmacophore, we substituted the methyl group of the prototype 1a with a lipophilic CF3 group. Surprisingly, the GI activity of this probe (2j) was increased by nearly an order of magnitude compared with 1a (Table 1). As shown in Figure 4, the majority of the cells treated with 2j at 2.5 μM rounded up and detached by 24–48 h, and most of the floating cells were determined to be nonviable by Trypan blue dye exclusion. Similar morphological effects were observed at concentrations as low as 0.6 μM. In cultures examined at 4 h, before the cells began to detach from the surface, we observed the formation of small cytoplasmic vacuoles and extensive blebbing of the plasma membrane (Figure 4). This phenotype is quite distinct from the extensive accumulation of large macropinocytotic vacuoles that we have come to associate with the onset of methuosis (e.g., Figure 2, 1a and 2b). It suggests that the increased cytotoxicity of the 2-trifluoromethyl derivative may be related to a different biological activity.
Figure 4.

Evaluation of the impact of different 2-indolyl substitutions on the morphological effects of 5-methoyxyindolyl-pyridinyl-propenones. Phase contrast images of live cells were obtained as described in the Experimental Section. Compounds were added at the concentrations listed at the top of each panel.
In light of the preceding findings, we expanded our compound library to include other lipophilic-linked arrangements such as derivatives containing alkyl 2-indolecarboxylates. As noted in Table 1, the methyl (2k), ethyl (2l), and n-propyl (2m) esters stood out as being much more potent inhibitors of cell growth and viability than any of the previously tested indolyl-pyridinyl-propenones, with the GI50 of 2m approaching 10 nM (Table 1). Morphologically, the cells subjected to these probes resembled those exposed to the trifluoromethyl derivative, 2j, with early membrane blebbing and cell rounding, followed by extensive loss of adherent cells by 24 h (Figure 4). In the cases of the most potent ethyl and propyl esters (2l and 2m), cytotoxic effects were observed as low as 0.1 μM, where the less potent methyl ester (2k) and CF3 (2j) analogues were no longer effective at this concentration. Interestingly, the GI activity and morphological effects of the isopropyl ester (2n) and the carboxylic acid (2o) were either markedly attenuated or completely eliminated (Table 1, Figure 4). The striking influence of the nature of the aliphatic chain on the biological activity of this series of alkyl carboxylates underscores the previously reported sensitivity of the indolyl-pyridinyl-propenones to synthetic manipulations at the 2-position of the indole ring,17 although this time within the context of lipophilic substituents that also appear to prompt a different mechanism.
A third type of 2-indolyl substitution is displayed by the hydroxymethyl (2p) and hydroxypropyl (2q) probes. Remarkably, reduction of the ester reverted the biological activity of these compounds to the methuosis phenotype, with GI values in the low micromolar range typical of the other methuosis-inducing compounds (e.g., 1a and 2b, Table 1). Likewise, cell morphology was characterized by extensive accumulation of large cytoplasmic vacuoles, with loss of viable adherent cells at concentrations ≥ 2.5 μM.
Cell Cycle Effects
The striking increase in cytotoxicity and the distinct morphological effects of the alkyl esters 2k–2m suggested that these compounds might be affecting cell growth and viability via a mechanism distinct from the methuosis-inducing compounds like 1a. Cell contraction, rounding, and detachment from the substratum are typically observed in cultured cells treated with mitotic inhibitors such as colchicine and vinblastine. Therefore, we utilized flow cytometry to generate DNA histograms from cells treated for 24 h with the two most potent agents, 2l and 2m. At a concentration of 3 μM, both compounds caused a substantial accumulation of cells in the G2/M phase of the cell cycle (Figure 5A). The apparent mitotic arrest was accompanied by a significant increase in the percentage of cells in the sub-G1/G0 compartment (Figure 5B), indicative of cell death by mitotic catastrophe. Similar cell cycle effects of 2l and 2m were observed at 0.1 μM (Figure 5C), which represents the lower end of the concentration range at which substantial morphological perturbations were observed in Figure 4. Interestingly, the percentage of cells in the G2/M phase actually was slightly higher in cultures treated with 2m at 0.1 μM compared with 3 μM (Figure 5B,C). This could be related to a higher percentage of dead cells derived from the G2/M-arrested population in the cultures treated with the higher concentration of 2m.
Figure 5.

Effects of selected indolyl-pyridinyl-propenones on cell cycle distribution in U251 cells. (A) DNA histograms of cells treated with the indicated compounds at 3 μM for 24 h were generated by flow cytometry as described in the Experimental Section. (B) Cells were treated with each compound for 24 h and the percentage of cells registering as having less than the G1/G0 DNA content (an indication of non-viable cells) is depicted in the left panel. In the same study the percentage of viable cells in each phase of the cell cycle was determined after gating out the sub-G1/G0 counts (right panel). (C) The assessment of cell death and cell cycle distribution was repeated in U251 cells treated with compounds at 0.1 μM instead of 3 μM. Values in B and C are means (± S.D.) derived from three separate cultures. The G2/M-phase and sub-G0/G1 cell populations in the cultures treated with 2l and 2m were significantly increased compared to the DMSO control at all concentrations (p ≤ 0.05).
In contrast to the results with 2l and 2m, the cell cycle distribution of cells treated with methuosis-inducing compounds such as 1a and 2b was similar to the DMSO-treated control (Figure 5). The latter finding is consistent with our previous studies of cells undergoing methuosis in response to overexpression of H-Ras (G12V),13 wherein we observed that the cell cycle distribution of the vacuolated cells did not change significantly prior to the loss of viability. It should also be noted that the presence of normal G1/G0 and S-phase populations in the cultures treated with 1a or 2b is not unexpected, since methuotic cell death triggered by 1a typically takes longer than 24 h at drug concentrations below 10 μM. In Figure 6, several additional compounds were tested for cell cycle effects at a single concentration (3 μM). Arrest of U251 cells in the G2/M phase, with a corresponding accumulation of sub-G1/G0 cells, was clearly evident in cultures treated with the 2-trifluoromethyl (2j) and methyl ester (2k) derivatives (Figure 6). The effects were very similar to those observed with colcemid, a known microtubule disruptor (Figure 6). In contrast, free acid 2o had no detectable effect on cell cycle distribution, reflecting its lack of activity in the SRB and morphology assays.
Figure 6.

Effects of additional indolyl-pyridinyl-propenones on cell cycle distribution in U251 cells. DNA histograms of cells treated with the indicated compounds at 3 μM for 24 h were generated by flow cytometry as described in the Experimental section. The results were analyzed to determine (A) the percentage of nonviable cells with sub-G1/G0 DNA content and (B) the percentage of viable cells in each phase of the cell cycle. Values are means (±S.D.) derived from three separate cultures. The G2/M-phase and sub-G0/G1 cell populations in the cultures treated with 2j, 2k, and colcemid were significantly increased compared to the DMSO control (p ≤ 0.05).
Effects on Microtubule Polymerization
The apparent mitotic arrest of cells treated with 2j–2m prompted us to examine the effects of this class of compounds on microtubule polymerization in intact cells. We began by assessing the integrity of the microtubule network by immunofluorescence microscopy, using an antibody against α-tubulin. As shown in Figure 7, the DMSO-treated control cells displayed a well-developed array of microtubules radiating from the juxtanuclear microtubule organizing center. In contrast, cells treated with 3 μM 2m had a tubulin staining pattern that was diffuse and disorganized. At a lower concentration (0.1 μM), 2m had a similar but somewhat less severe effect on microtubule organization. Cells treated with the methuosis-inducer 1a appeared to contain intact cable-like microtubules, although their organization was distorted by the accumulation of large vacuoles throughout the cytoplasm.
Figure 7.
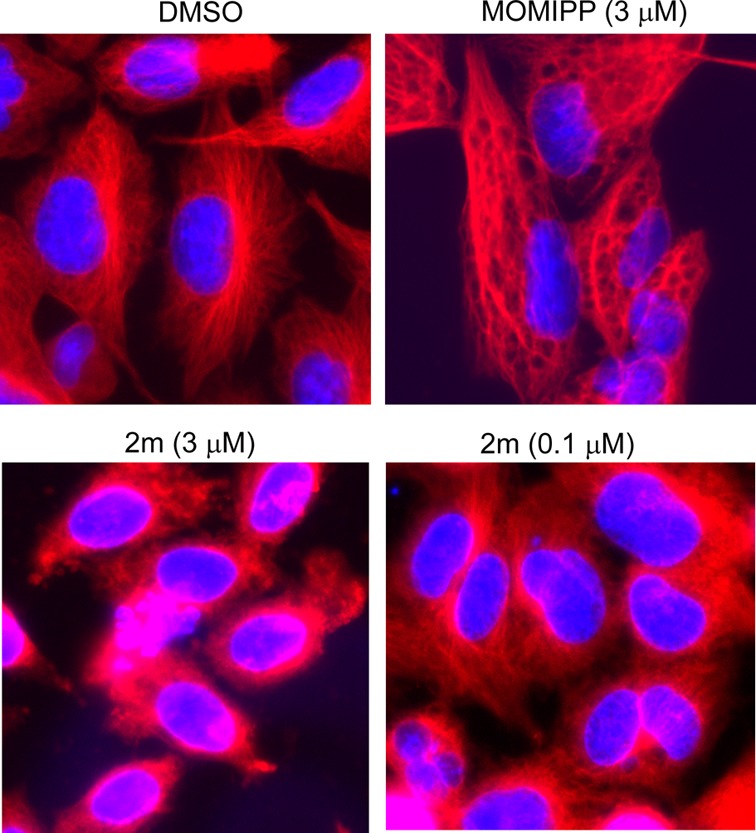
Immunofluorescence imaging of tubulin (red fluorescence) in cells treated for 24 h with the methuosis-inducing compound 1a and the 2-indolyl propyl ester 2m. The nuclei are visualized with DAPI (blue fluorescence).
To obtain a more direct indication of the effects of the compounds on tubulin polymerization, we utilized an established biochemical approach in which the drug-treated cells are lysed under conditions designed to preserve the native polymerization state of the microtubules. The polymerized tubulin is then separated from soluble tubulin by high-speed centrifugation and quantified by Western blot analysis. The results, depicted in Figure 8, demonstrate that the three most potent compounds, which caused mitotic arrest in Figures 5 and 6 (2j, 2l, and 2m), produced substantial declines in the relative amount of tubulin detected in the polymerized fraction. Lowering the concentration from 3 μM to 0.1 μM diminished but did not eliminate the effect on tubulin polymerization, consistent with the immunofluorescence observations in Figure 7. The results were very similar to those obtained with the positive control, colchicine, applied at the same concentrations. The proportion of polymerized tubulin in cells treated with the methuosis-inducing compounds, 1a and 2b, and the inactive derivative meta-MOMIPP,17 was similar to that detected in the DMSO control.
Figure 8.

Effects of selected indolyl-pyridinyl-propenones on tubulin polymerization in cultured U251 cells. Cells were treated for 4 h with the indicated compounds at a final concentration of 3 μM and then fractionated under conditions designed to preserve the native polymerization state of the microtubules. The percentage of polymerized versus soluble tubulin was determined by immunoblot analysis as described in the Experimental Section. For each compound the fraction of tubulin in the polymerized state was expressed as a percentage of the polymerized tubulin determined in a parallel control culture treated with DMSO alone. The results represent the mean (±S.D.) of determinations from three separate experiments. The decreases in the percentage of polymerized tubulin in cells treated with 2j, 2l, 2m and colchicine (at 3 μM) were significant at p ≤ 0.05.
Discussion
The indolyl-pyridinyl-propenones described in this report are related to naturally occurring flavonoid precursors termed chalcones, which consist of an α,β-unsaturated ketone linking two nonheterocyclic aromatic rings. Over the past two decades a large number of naturally occurring and synthetic chalcones and chalcone derivatives have been characterized as having anticancer activities.23−25 Our group has focused on a unique class of such molecules wherein the aromatic ring systems consist of indolyl- and pyridinyl-moieties. In particular, we have established that certain members of this family, namely, 1a and its des-methoxy analogue (MIPP), can induce methuosis and kill temozolomide-resistant glioma cells and doxorubicin-resistant breast cancer cells at low micromolar concentrations.15,16 The cytostatic and cytotoxic effects of 1a are moderately selective for cancer cells compared with normal fibroblasts16 (Supporting Information Figure S1). Recently, others have described unrelated small molecules (vacquinols) that also induce methuosis in cultured glioma cells and suppress the growth of tumor xenografts in mouse models.26 These studies have stimulated interest in evaluating the therapeutic potential of methuosis-inducing compounds.
Two fundamental questions about the methuosis-inducing compounds remain unresolved. The first relates to the identities of their protein targets. Previous studies suggested that induction of methuosis is due to drug interaction with specific proteins, rather than general covalent protein modification by Michael addition. In particular, the reversibility of methuosis upon drug wash-out during the early stages and the strict dependency of the biological effects on the para-configuration of the pyridinyl nitrogen support this concept.16,17 While reassuring in terms of highlighting specificity, the tendency of these compounds to be inactivated by many modifications on the pyridinyl or indolyl moieties has made the use of conventional affinity-based approaches for drug target identification quite challenging.
A second key question concerns the mechanism(s) leading to cell death, particularly the relationship between the formation of cytoplasmic vacuoles and the ultimate loss of cell viability. Our initial SAR studies with a directed library of compounds revealed that analogues that failed to induce vacuolization generally failed to cause cell death.15,16 Thus, we inferred that vacuolization was an important contributing factor in the cell death program. However, our recent studies with certain aliphatic 2-indolyl substitutions,17 coupled with the present observations with some of the substituents at the indolyl 5-position (e.g., 2a, 2c, 2e, 2f), indicate that vacuolization does not necessarily lead to growth arrest and cell death in every case. Thus, our current working hypothesis is that the cytotoxicity of certain indolyl-pyridinyl-propenones is due to pleiotropic effects, combining perturbations of macropinosome/endosome trafficking with alterations of cellular signaling or metabolic pathways that remain to be defined.
In the present study we have identified several new indolyl-pyridinyl-propenones that exhibit robust biological activities. For the purpose of discussion, we consider the active compounds as falling into three discernible classes based on the distinct cellular phenotypes that they induce in cultured cells (Figure 9). The compounds in Class 1 cause striking cellular vacuolization, but the cells continue to proliferate and remain viable. Examples of Class 1 compounds are drawn from this report (2a, 2c, 2e, 2f) and our previous publication.17 We envision that the Class 1 compounds target proteins involved in the formation of macropinosomes, the recycling of these structures, or the fusion of macropinosomes with lysosomes. The complexity of the phenotype suggests that there may be multiple Class 1 targets, but we cannot exclude the possibility that perturbation of a single enzyme, structural protein, or signaling molecule could be responsible. We also identified several new compounds with substantial methuosis-inducing activity (2b, 2p, 2q), comparable to that of our previously characterized lead compound 1a. In the context of these studies, we equate methuosis with the Class 2 phenotype. It is similar to the Class 1 phenotype in terms of massive cellular vacuolization, but in this case the cells detach and undergo metabolic failure, culminating with rupture of the cell membrane (i.e., methuosis). We speculate that in addition to interacting with Class 1 targets, the Class 2 compounds have affinity for a separate set of protein targets involved in metabolic and/or signaling pathways essential for cell viability. Through these combined effects, the Class 2 compounds trigger methuotic cell death. The most striking observation from the present study is that certain modifications at the 2-position of the indole ring increased the cytotoxic activity of the compounds by 1 or 2 orders of magnitude (e.g., 2j–2m). The increase in potency was accompanied by notable changes in the phenotype observed in glioma cells treated with these compounds, which we term Class 3. In contrast to the typical vacuolated morphology seen in cells undergoing methuosis, the cells treated with the 2-trifluoromethylindole and the alkyl 2-indolylcarboxylate esters underwent a rapid transition from early formation of small vacuoles to very active membrane blebbing and cell contraction. This was followed by microtubule depolymerization, mitotic arrest, and massive detachment of cells from the substratum. These changes lead us to conclude that compounds 2j–2m acquired the capacity to interact with one or more protein targets involved in microtubule assembly or maintenance (Class 3 targets). The early hint of vacuolization in cells treated with these compounds suggests that macropinocytosis may also have been affected, possibly via Class 1 or Class 2 targets. However, with the superimposition of potent microtubule-destabilizing activity, disruption of the cytoskeleton and consequent detachment of the cells occur before the Class 1 or Class 2 phenotypes can develop.
Figure 9.

Classification of biologically active idolyl-pyridinyl-propenones based on distinct cellular phenotypes elicited by modifications at R1 and R2. In the hypothetical model, Class 1 compounds are envisioned as interacting with one or more Class 1 protein targets to induce perturbations in macropinosome trafficking, resulting in cellular vacuolization without impairment of cell proliferation or viability. Class 2 compounds interact with the same Class 1 targets, but acquire the ability to bind additional Class 2 protein targets that function in metabolic or pro-survival signaling pathways. The combination of effects on Class 1 and Class 2 proteins results in a distinct phenotype characterized by extreme vacuolization and cell death via methuosis. The novel compounds in Class 3 gain the ability to inhibit proteins involved in microtubule assembly in a concentration range where Class 1 or 2 compounds have no effect on tubulin polymerization. Hence, mitotic arrest, cell rounding, and death (presumably by mitotic catastrophe) predominate as the main features of the Class 3 phenotype. It is unclear whether the Class 3 compounds retain the capacity to interact with Class 1 and 2 targets, although the early detection of vacuoles prior to microtubule disruption and cell rounding suggests that there may be some overlap.
In terms of summarizing specific SAR features that might be gleaned from the assembled compounds, several interesting points can be raised. First, it appears that the distinctive microtubule-related actions described above for the trifluoromethyl and ester analogues 2j–2m are prompted by the presence of an electron withdrawing substituent placed at the indolyl 2-position. In 1a the 2-methyl group is modestly electron donating and the biological profile is that of the Class 2 phenotype. Substitution with an electron withdrawing trifluoromethyl group (2j) redirects the profile to that of Class 3 and substitution with electron withdrawing carbonyl systems such as those present in esters 2l and 2m prompts Class 3 with even stronger potency. Alternatively, when the ester substituent is branched (2n) or is removed entirely so as to expose a carboxylic acid moiety (2o), activity is significantly reduced or lost completely. This is similar to our previous findings where exploration of this vicinity with various alkyl substituents suggested that there may be a pocket in 1a’s methuosis-related protein targets that prefers to accommodate a methyl or ethyl group.17 For that series, larger groups at the indolyl 2-position still caused vacuolization, but the cells did not proceed to methuotic death. Indeed, it was our suspicion about this pocket that led to the design of the more lipophilic 2j in order to further probe the importance of this feature for interaction with Class 2 targets. However, in addition to providing the desired increase in lipophilicity relative to 1a, the CF3 substitution also confers a significant electron withdrawing effect. Consistent with the results from the esters 2l and 2m, the latter appears to induce a shift in the biological activity of 2j from a Class 2 to Class 3 phenotype.
Contributing to construction of a topological map near the indolyl 2-position, relative to interactions with methuosis-related protein targets, are the unanticipated results for the two alcohols 2p and 2q. They are not complicated by having additional electron withdrawing effects and, as expected, do not evoke mitotic arrest and cell death by interaction with microtubules (Class 3). Instead, these substituents endow strong Class 2 effects even though they are less lipophilic than their corresponding alkyl systems. Furthermore, the high potency exhibited by the n-propyl alcohol 2q does not correspond to the previously limited size for simple alkyl arrangements wherein methyl or ethyl were preferred for methuosis activity.17 Indeed, the preference for a propyl in this case resembles the SAR for the Class 3 family of microtubule-interacting esters wherein the best alcohol adduct was also n-propyl. This could suggest that there are some commonalities among the protein targets engaged by the Class 2 and Class 3 indolyl-pyridinyl-propenones. At this point, for the specific methuosis-related proteins, we speculate that the topology of the putative binding pocket for the indolyl’s 2-position may approximate a narrow groove that can accommodate simple (nonbranched) alkyl groups that are lipophilic at the proximal end and capable of hydrogen bonding at the distal end, provided that ionizable moieties are not present anywhere along the alkyl chain.
Preferences for the topology around the indolyl 5-position appear to be more stringent, but the overall SAR is equally intriguing. Similar to our former study17 where the indolyl 2-position was probed with various alkyl groups, many of the substituents placed at the 5-position were able to retain Class 1 activity, but only a few demonstrated Class 2 methuosis activity. As shown previously,16 a phenolic substitution is not active, while a methoxy, as is present in 1a, prompts significant methuosis. Such activity is greatly reduced by ethoxy, is returned by n-propoxy, and then is greatly reduced again by branched alkyl or n-butoxy (2a, 2b, 2c, and 2d, respectively). Assuming that a model similar to that postulated for the indolyl 2-position also pertains to the indolyl 5-position, one might ask why a binding pocket that accommodates methoxy would optimally accept an n-propoxy substituent, but not an ethoxy or butoxy group. In this regard, it is worth noting that similar anomalies have been reported for well-known systems that have undergone extensive SAR studies. In one example, the “Goldilocks” nature27 of an ethyl group inserted between a bulky aryl-system and a simple methyl ester carbonyl moiety (thus constituting a 3-carbon system) was exploited as being “just right” to allow ready access for attack of β-adrenergic antagonists by esterases. While the 3-carbon system endowed an ultrashort duration of action, similar effects were not afforded by methyl (2-carbon system) or n-propyl (4-carbon system) spacers.28,29 In this case the author speculated that the additional conformational freedom afforded by the n-propyl group could cause the ester to become tucked back toward other bulky features present within the overall molecular framework. A second example of preferred conformational arrangements comes from the literature on dopamine receptor agonists. In this case, dopamine loses significant activity when its primary amine becomes substituted with a broad range of alkyl functionalities except for a distinct disubstitution situation wherein at least one of the substituents is an n-propyl group.30−32 In this heavily studied research arena, this remarkable effect is referred to as the “n-propyl phenomenon”, and it has prompted speculation that the dopamine receptor’s ligand-binding pocket maintains a “unique geometry to accommodate [just] an n-propyl group”. Our prior computational studies on substituted indolyl-pyridinyl-propenones suggest that their side-chain substitutions remain quite flexible, with only subtle differences in energy across several conformational possibilities for each increment of added carbon atoms to the 2-position.17 Thus, in our case, various arrangements being uniquely adopted by only certain homologues during interaction with a protein surface cannot be ruled out.
Further insight regarding the possible topography around the indolyl 5-position comes from our finding that an ionizable free amine (2g, 2i), a neutral amide (2e), and a pair of carboxamides having hydrogen bonding capabilities (2f, 2h) are all unable to induce methuosis. Although weak GI activity was observed in the specific case where a BOC group was synthesized on an amine spaced one methylene unit away from the indolyl moiety (2h), this activity did not match the Class 2 or 3 cytotoxic phenotypes. Thus, similar to the model for the indolyl 2-position, we speculate that the protein region interacting with the 5-position also may have a restrictive groove that can accommodate a methoxy group or the special case of a n-propoxy group, with the provisos that the latter must remain lipophilic throughout, and that additional substituents capable of hydrogen bonding or ionization are excluded anywhere along the alkyl chain.
In conclusion for our SAR, we find it quite remarkable that such profound changes in biological profiles can be unmasked around the indolyl-pyridinyl-propenone structural motif with subtle manipulations at either of just two positions. While we offer proposals for what the topography may look like in the relevant binding domains of the unknown Class 2 protein targets, the assumptions are based upon the present state of SAR with a limited spectrum of structural probes. Nevertheless, until specific proteins are identified, these early models can serve as useful conceptual tools for the design of new probes to map the structural determinants responsible for the distinct biological activities of the indolyl-pyridinyl-propenones.
Numerous chalcones and chalcone-derivatives displaying antimitotic activity have been characterized previously.24,33,34 While many of these compounds were identified by screening collections of synthetic chalcones in cell-based proliferation assays,35,36 others emerged from molecular modeling and design studies aimed at creating new structures that might compete for tubulin sites known to bind established microtubule inhibitors (e.g., colchicine, combretastatins).37 Most recently, efforts to develop curcumin analogues that might kill cancer cells by targeting the NFκB activation pathway serendipitously yielded a series of potent ortho-aryl substituted chalcones with the ability to bind to the colchicine site on tubulin and induce mitotic arrest and apoptotic cell death.38 Because of their distinct structural profiles and potencies, the trifluoromethyl and ester analogues described herein appear to represent a new class of potent microtubule-active anticancer compounds.
Among the various traditional chalcones investigated as potential anticancer agents, indole-based chalcones have received comparatively little attention until recently. In addition to the methuosis-inducing indolyl-pyridinyl-propenones described by our group,15−17 the other class of antiproliferative/cytotoxic indolyl-chalcones comprises the 1-(N-methylindolyl)-3-phenylpropenones.39−41 SAR studies of analogues with different substituents on the phenyl ring revealed that 3-(1-methyl-1H-indol-3-yl)-1-(2,4,6-trimethoxyphenyl)-2-propen-1-one, 33 (JAI-51)41−43 produced mitotic arrest at low micromolar concentrations. This was attributed to direct inhibition of tubulin polymerization based on the results of cell-free tubulin assembly assays.43 Although it is possible that our 2-trifluoromethylindole and ester derivatives might operate in a similar way to disrupt microtubules, we have not yet obtained definitive evidence to discriminate between direct tubulin binding and other indirect mechanisms, such as interference with microtubule-associated proteins. The possibility that the mechanism might differ from that described for 33 is suggested by the much higher potency and unique structural features of the indolyl-pyridinyl-propenones. In particular, our Class 3 microtubule-disrupting compounds lack the N-methyl group on the indole ring found in 33 and other antimitotic 1-(N-methylindolyl)-3-phenylpropenones. Another key difference between our current set of compounds and previously reported antimitotic chalcones relates to the pyridinyl moiety which is uniquely present in our scaffold and requisite for optimal activity. Specifically, cytotoxicity (via methuosis) was eliminated by switching the position of the pyridinyl nitrogen from para- to meta-.17 In a preliminary study, we have determined that altering the position of the pyridinyl nitrogen from para- to meta- in the context of 2l and 2m, has a similar negative influence on the antiproliferative activity of these microtubule-disrupting compounds, raising their GI50’s by approximately 2 orders of magnitude (Supporting Information Figure S2).
Drugs that affect microtubule dynamics are among the most widely employed anticancer agents.44,45 Distinct classes of antimitotic compounds include the Vinca alkaloids (e.g., vincristine, vinblastine) and colchicine analogues (e.g, combretastatins) both of which are considered microtubule destabilizers, and the taxanes (e.g., paclitaxel) which function as microtubule overstabilizing agents.45,46 Although they are effective against many types of cancer, these compounds are not without drawbacks in the clinic. Side effects, such as peripheral neuropathy and neutropenia, may limit the doses and treatment regiments that can be tolerated. Development of drug resistance is also a factor that limits efficacy in many cases. The latter may entail both induction of drug efflux pumps and alterations in the intrinsic properties of the microtubules (e.g., tubulin isotypes or posttranslational modifications).47,48 Finally, the Vinca alkaloids49 and taxanes50 exhibit low permeability through the blood–brain barrier, minimizing efficacy for treating primary or metastatic tumors in the central nervous system. For these reasons, the identification of new microtubule-directed agents with unique profiles continues to be of interest. Indolyl-pyridinyl-propenones are particularly intriguing candidates for further study because published findings with other indole-based chalcones suggest that they are poor substrates for drug efflux pumps and are able to cross the blood–brain barrier.43 Indeed, several functionalized chalcones have been shown to actively impair drug transporters like P-glycoprotein and breast cancer resistance protein.43,51,52 It remains to be determined if this property extends to the compounds described in this report. Our novel trifluoromethyl and ester analogues could be particularly attractive prototypes for future study, as they may operate through a combination of mechanisms to induce features of both methuosis (disruption of macropinosome trafficking) and microtubule destabilization. In this regard, it will be important to discern if these compounds trigger cell death through pathways similar to those promoted by conventional antitubulin compounds (i.e., apoptosis, mitotic catastrophe)53,54 or instead operate through a novel cell death program. Ultimately, the potential therapeutic utility of this class of compounds will depend on further evaluation of cytotoxicity and selectivity in a broad panel of transformed and nontransformed cell lines and determination of their pharmacokinetic properties in vivo.
Experimental Section
Chemistry
General Description
All reactions were performed in oven-dried 2-neck round-bottom flasks under an atmosphere of either Ar or N2 and stirred with Teflon-coated magnetic bars. TLC (silica gel F254 plates, Baker-flex) was used to monitor progress of all reactions with visualization performed under 254 nm UV light. Reagent grade and anhydrous solvents were purchased from Sigma-Aldrich and used without further purification unless otherwise noted. Silica gel sorbent (230–400 mesh) was purchased from Fisher Scientific. Samples to be purified by column chromatography were dissolved in a minimal volume of solvent and then adsorbed onto silica gel (5–10× the amount of sample by weight) by evaporation under reduced pressure until the solid composite was free-flowing. The dry loaded sample was then applied to the top of the prepacked column bed allowing the solvent line of the column to be above the added adsorbent. Unless indicated otherwise, chromatography was conducted by flash column methods as described previously55 utilizing a gradient of increasingly polar eluent specifically indicated for each compound. Gradients were performed in a stepwise fashion in 200 mL increments. For EtOAc/hexanes systems, the column was charged with the defined starting eluent percentage. After the sample was applied, 200 mL of the initial eluent was used, followed by 200 mL at 10% increments until the product eluted into the final gradient. For MeOH/DCM systems, the column was charged with the defined starting eluent percentage. After the sample was applied, 200 mL of the initial eluent was used, followed by 200 mL at 2.5% increments until the product eluted into the final gradient. Isocratic separations are denoted on an individual basis. TLC was used to monitor product elution during flash column chromatography. Appropriate fractions were combined, and solvents were evaporated in vacuo (rotary evaporator under water aspirator vacuum) and then further dried by a vacuum pump (0.5 mm Hg) for 24 h unless described otherwise. Samples that were heated in a vacuum desiccator were equipped to a vacuum pump (0.5 mm Hg) and dried for a specified time and temperature denoted for the individual procedure. Solvent solutions dried with Na2SO4 were stored in a sealed flask and allowed to sit for at least 12 h. Upon completion, the drying agent was removed by vacuum filtration, and the solvents were evaporated in vacuo and then further dried by a vacuum pump (0.5 mm Hg) for 24 h. Samples reduced by hydrogenation utilized a PARR hydrogenator. The psi of the H2 is provided in the individual experimental detail. Melting points were performed in triplicate on an electrothermal digital melting point apparatus and are uncorrected. Proton (1H) and carbon (13C) NMR experiments were recorded on either a 600 MHz Bruker Avance, Inova 600 MHz, or an Inova 400 MHz instrument. Samples were referenced to TMS when present, or the solvent residual peak for 1H and 13C, respectively: (CDCl3; 7.27, 77.13; DMSO-d6; 2.50, 39.51; MeOH-d4; 3.31, 49.15). 1H NMR chemical shifts were given in ppm, and coupling constants (J values) were expressed in hertz (Hz) using the following designations: s (singlet), d (doublet), t (triplet), q (quartet), quin (quintet), sex (sextet), sep (septet), dd (doublet of doublets), m (multiplet). The 13C chemical shifts are reported for each compound in the Experimental Section and in all cases confirm structure. In a few cases 13C shifts were found to double-up in their peak locations. Fluorine (19F) NMR was recorded on an Inova 400 MHz instrument at 376 MHz. Samples were referenced externally to CFCl3. Purity for tested compounds (2a–2f, 2h, 2j–2q) was determined by combustion analysis (Atlantic Microlabs, Norcross GA), or in the cases of 2g and 2i by HPLC. All tested compounds possess ≥95% purity. Intermediate compounds were determined to possess ≥95% purity by combustion analysis except 4a and 6a. For these intermediates, the mp and spectral data matched literature values. Observed values for combustion analysis were considered acceptable within ±0.4% of calculated values. Synthetic derivatives reported as solvates are denoted in the text and were calculated by incorporating the minimal amount of appropriate solvent that was subjected to the compound during the purification process in order to be within the acceptable range (±0.4%). HPLC was performed on an Alliance instrument (#2659) equipped with a quaternary pump, an inline membrane degasser, autosampler, and photodiode array (PDA) detector (#2996) from Waters Corporation (Milford, MA). The column was a Nova-PakC18 column, 4 μm particle size (150 mm × 3.9 mm). Details for HPLC analysis are described in the individual procedures for 2g and 2i. High resolution mass spectrometry (HRMS) was performed by the University of Michigan’s Department of Chemistry as a technical service. Compounds 1a–1c, 10, and 18 were described previously.16,17 Compound 7 was prepared according to the literature.17,18 Compounds 3, 15, and 22 were purchased commercially from Sigma-Aldrich.
trans-3-(5-Ethoxy-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2a)
5-Ethoxy-2-methylindole-3-carboxaldehyde 9a (188 mg, 0.93 mmol) was dissolved in anhydrous methanol (12 mL). 4-Acetylpyridine (168 mg, 1.38 mmol) and piperidine (118 mg, 1.38 mmol) were added, and the solution was heated at reflux for 24 h. A precipitate slowly formed which was collected, washed with ice-cold MeOH (30 mL) and dried at 40 °C in a vacuum desiccator for 24 h to yield a bright yellow powder (244 mg, 86%): mp 255–257 °C. TLC Rf 0.30 (80% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 11.88 (s,1H), 8.80 (m, 2H), 8.09–8.06 (d, 1H, J = 15.3 Hz), 7.93–7.92 (m, 2H), 7.41 (d,1H, J = 2.22 Hz), 7.34–7.32 (d, 1H, J = 15.3 Hz), 7.30–7.29 (d, 1H, J = 8.64 Hz), 6.84–6.82 (dd, 1H, J1 = 8.7 Hz, J2 = 2.28 Hz), 4.14–4.11 (q, 2H, J = 6.96 Hz), 2.57 (s, 3H), 1.37–1.36 (t, 3H, J = 6.96 Hz). 13C NMR (150 MHz, DMSO-d6) δ 188.1, 154.4, 150.6, 145.8, 145.1, 139.6, 131.0, 126.6, 121.4, 112.82, 112.24, 111.3, 109.3, 104.4, 63.6, 14.9, 12.2. Elemental analysis calculated for C19H18N2O2: C, 74.49; H, 5.92; N, 9.14. Found: C, 74.30; H, 6.02; N, 9.04.
trans-3-(2-Methyl-5-propoxy-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2b)
2-Methyl-5-propoxyindole-3-carboxaldehyde 9b (200 mg, 0.920 mmol) was reacted in a similar manner to that for 2a to yield a bright yellow powder (204 mg, 69%): mp 228–229 °C. TLC Rf 0.40 (80% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 11.88 (s,1H), 8.81–8.80 (m, 2H), 8.08–8.06 (d, 1H, J = 15.3 Hz), 7.93–7.92 (m, 2H), 7.41 (d, 1H, J = 2.22 Hz), 7.34–7.31 (d, 1H, J = 15.24 Hz), 7.30–7.29 (d, 1H, J = 8.64 Hz), 6.85–6.83 (dd, 1H, J1 = 8.64 Hz, J2 = 2.28 Hz), 4.04–4.02 (t, 2H, 6.54 Hz), 2.56 (s, 3H), 1.80–1.74 (sex, 2H, J = 6.66 Hz), 1.03–1.01 (t, 3H, J = 7.38 Hz). 13C NMR (150 MHz, DMSO-d6) δ 188.2, 154.6, 150.6, 145.75, 145.18, 139.6, 131.0, 126.6, 121.4, 112.90, 112.25, 111.4, 109.3, 104.4, 69.6, 22.3, 12.2, 10.6. Elemental analysis calculated for C20H20N2O2: C, 74.98; H, 6.29; N, 8.74. Found: C, 75.00; H, 6.23; N, 8.69.
trans-3-(5-Isopropoxy-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2c)
5-Isopropyloxy-2-methylindole-3-carboxaldehyde 9c (225 mg, 1.04 mmol) was reacted in a similar manner to that for 2a to yield an orange powder (294 mg, 88%): mp 245–247 °C. TLC Rf 0.38 (80% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 11.88 (s,1H), 8.81–8.80 (m, 2H), 8.08–8.05 (d, 1H, J = 15.24 Hz), 7.92–7.91 (m, 2H), 7.42 (d,1H, J = 2.22 Hz), 7.33–7.30 (d, 1H, J = 15.3 Hz), 7.30–7.28 (d, 1H, J = 8.76 Hz), 6.84–6.82 (dd, 1H, J1 = 8.7 Hz, J2 = 2.28 Hz), 4.70–4.66 (quin, 1H, 6 Hz), 2.56 (s, 3H), 1.30–1.29 (d, 6H, J = 6 Hz). 13C NMR (150 MHz, DMSO-d6) δ 188.2, 153.1, 150.6, 145.88, 145.17, 139.6, 131.1, 126.7, 121.4, 112.82, 112.74, 112.23, 109.2, 106.8, 70.1, 22.0, 12.1. Elemental analysis calculated for C20H20N2O2: C, 74.98; H, 6.29; N, 8.74. Found: C, 74.86; H, 6.22; N, 8.62.
trans-3-(5-Butoxy-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2d)
5-Butoxy-2-methylindole-3-carboxaldehyde 9d (250 mg, 1.08 mmol) was reacted in a similar manner to that for 2a to yield a bright yellow powder (206 mg, 57%): mp 227–228 °C. TLC Rf 0.33 (80% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 11.39 (s, 1H), 8.81–8.80 (m, 2H), 8.08–8.06 (d, 1H, J = 15.3 Hz), 7.93–7.92 (m, 2H), 7.41 (d, 1H, J = 12.3 Hz), 7.34–7.31 (d, 1H, J = 15.24 Hz), 7.30–7.28 (d, 1H, J = 8.64 Hz), 6.84–6.82 (dd, 1H, J1 = 8.64 Hz, J2 = 2.34 Hz), 4.08–4.06 (t, 2H, J = 6.48 Hz), 2.57 (s, 3H), 1.76–1.71 (m, 2H), 1.52–1.45 (m, 2H), 0.97–0.94 (t, 3H, J = 7.38 Hz). 13C NMR (150 MHz, DMSO-d6) δ 188.2, 154.6, 150.6, 145.74, 145.19, 139.6, 131.0, 126.6, 121.4, 112.90, 112.25, 111.44, 109.3, 104.3, 64.8, 31.0, 18.9, 13.8, 12.2. Elemental analysis calculated for C21H22N2O2: C, 75.42; H, 6.63; N, 8.38. Found: C, 75.39; H, 6.55; N, 8.37
trans-3-(5-Acetamido-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2e)
5-Acetamido-2-methylindol-3-caboxaldehyde 13 (150 mg, 0.69 mmol) was dissolved in MeOH (6 mL) and stirred vigorously until dissolution was complete. Piperidine (91 mg, 1.07 mmol) and 4-acetylpyridine (208 mg, 1.72 mmol) were added, and the mixture was heated to reflux while stirring vigorously for 20 h. The resulting precipitate was filtered, washed with ice-cold MeOH (10 mL), and dried at 40 °C in a vacuum desiccator for 24 h to yield an orange solid (193 mg, 87%): mp >300 °C. TLC Rf 0.54 (80% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ11.94 (s, 1H), 9.89 (s, 1H), 8.84–8.83 (m, 2H), 8.26 (d, 1H, J = 1.9 Hz), 8.07–8.05 (d, 1H, J = 15 Hz), 7.87–7.86 (m, 1H), 7.48–7.46 (dd, 1H, J1 = 8.6 Hz, J2 = 1.9 Hz), 7.33–7.30 (m, 2H), 2.57 (s, 3H), 2.08 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 188.0, 167.8, 150.6, 145.82, 145.17, 139.6, 134.0, 132.4, 125.7, 121.1, 114.9, 112.8, 111.5, 110.03, 109.26, 24.0, 11.9. Elemental analysis calculated for C19H17N3O2·0.25 MeOH: C, 70.63; H, 5.54; N, 12.84. Found: C, 70.25; H, 5.29; N, 12.83. HRMS ESI+ calculated m/z for C19H17N3O2 (M + H)+ 320.1394. Found (M + H)+ 320.1392.
trans-3-(5-N-Boc-amino-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2f)
5-(N-Boc-amino)-2-methylindol-3-carboxaldehyde 14 (250 mg, 0.91 mmol) was reacted in a similar manner to that for 2e to yield an orange solid (332 mg, 96%): mp >300 °C (263 °C color change). TLC Rf 0.37 (75% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ11.92 (s, 1H), 9.28 (s, 1H), 8.85–8.84 (m, 2H), 8.08–8.05 (m, 2H), 7.92–7.91 (m, 2H), 7.50 (s, 1H), 7.39–7.37 (d, 1H, J = 15 Hz), 7.30–7.29 (d, 1H, J = 9 Hz), 2.57 (s, 3H), 1.52 (s, 9H); 13C NMR (150 MHz, DMSO-d6) δ 187.6, 153.0, 150.6, 145.8, 139.6, 134.1, 132.0, 125.9, 121.2, 114.0, 112.5, 111.4, 109.2, 78.8. Elemental analysis calculated for C22H23N3O3: C, 70.01; H, 6.14; N, 11.13. Found: C, 69.91; H, 6.10; N, 11.04.
trans-3-(5-Amino-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2g)
Compound 2f (35 mg, 0.0927 mmol) was suspended in MeOH (5 mL) and TFA (1 mL) and heated to reflux for 24 h. The volatiles were evaporated in vacuo, and the sample was redissolved in H2O (5 mL) and neutralized with 1 N NaOH (determined by pH paper). Purification by column chromatography (2.5% to 10% MeOH/DCM) and subsequent drying at 40 °C in a vacuum desiccator for 24 h yielded an orange solid (15 mg, 58%): mp 263–264 °C. TLC Rf 0.52 (10% MeOH/DCM). 1H NMR (600 MHz, d4-MeOH) δ 8.75 (m, 2H), 8.26–8.23 (d, 1H, J = 15.06 Hz), 7.96 (m, 2H), 7.40 (d, 1H, J = 1.8 Hz), 7.37–7.35 (d, 1H, J = 15 Hz), 7.16–7.14 (d, 1H, J = 8.4 Hz), 6.74–6.72 (dd, 1H, J1 = 8.4 Hz, J2 = 1.8 Hz), 2.56 (s, 3H). 13C NMR (150 MHz, MeOH-d4) δ 190.8, 151.2, 148.4, 147.6, 143.66, 143.21, 132.6, 128.7, 123.4, 114.4, 113.2, 111.6, 108.1, 12.1. HRMS ESI+ calculated m/z for C17H16N3O (M + H)+ 278.1288. Found (M + H)+ 278.1286. HPLC analysis: retention time =2.642 min; peak area, 99.48%; eluent A, 10 mM TEA solution with 0.1% formic acid (pH 3); eluent B, CH3CN; isocratic (15% eluent B) over 10 min with a flow rate of 1 mL min–1 and detection at 280 nm; injection of 10 μL of 59 μM 2g.
trans-3-(5-N-Boc-Aminomethyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2h)
5-N-Boc-(aminomethyl)-indole-3-carboxaldehyde 17 (152 mg, 0.554 mmol) was reacted in a similar manner to that for 2e to yield a yellow solid (150 mg, 72%): mp 227 °C. TLC Rf 0.31 (80% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 11.97 (s, 1H), 8.83–8.82 (m, 2H), 8.15–8.14 (d, 1H, J = 2.6 Hz), 8.08–8.06 (d, 1H, J = 15.6 Hz), 7.92–7.91 (m, 3H), 7.52–7.50 (d, 1H, J = 15.6 Hz), 7.44–7.42 (m, 2H), 7.17–7.16 (d,1H, J = 8.4 Hz), 4.30–4.29 (d, 2H, J = 6 Hz), 1.38 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 188.6, 155.8, 150.5, 144.8, 141.0, 136.5, 134.5, 133.4, 125.0, 122.5, 121.3, 118.6, 114.7, 112.55, 112.22, 77.6, 43.7, 28.2. Elemental analysis calculated for C22H23N3O3: C, 70.01; H, 6.14; N, 11.13. Found: C, 70.20; H, 5.98; N, 11.16.
trans-3-(5-Aminomethyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2i)
Compound 2h (100 mg, 0.265 mmol) was reacted in a similar manner to that for 2g except the reaction time was extended to 48 h, and the isocratic chromatography eluent was DCM/MeOH/TEA (90:9:1). An orange/yellow solid was obtained (69 mg, 94%): mp 170–172 °C (darkened 155–158 °C). TLC Rf 0.22 DCM/MeOH/TEA (90:9:1). 1H NMR (600 MHz, MeOH-d4) δ 8.78–8.77 (m, 2H), 8.19–8.17 (d, 1H, J = 15.6 Hz), 8.11 (d, J = 0.6 Hz, 1H), 7.99–7.98 (m, 2H), 7.96 (s, 1H), 7.60–7.57 (d, 1H, J = 15.6 Hz), 7.53–7.51 (d, 1H, J = 8.4 Hz), 7.34–7.32 (dd, 1H, J1 = 8.4 Hz, J2 = 1.8 Hz), 4.19 (s, 2H). 13C NMR (150 MHz, MeOH-d4) δ 191.4, 151.3, 147.7, 143.3, 139.5, 135.7, 130.9, 127.3, 125.1, 123.5, 121.8, 116.7, 115.1, 114.1, 45.8. HRMS ESI+ calculated m/z for C17H16N3O (M + H)+ 278.1288. Found (M+H)+ 278.1291. HPLC analysis: retention time = 2.869 min; peak area, 99.14%; eluent A, 10 mM TEA solution with 0.1% formic acid (pH 3); eluent B, CH3CN; isocratic (15% eluent B) over 10 min with a flow rate of 1 mL min–1 and detection at 280 nm; injection of 10 μL of 44 μM 2i.
trans-3-(5-Methoxy-2-trifluoromethyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2j)
5-Methoxy-2-trifluoromethylindole-3-carboxyaldehyde 21 (156 mg, 0.641 mmol) was dissolved in anhydrous MeOH (10 mL). 4-Acetylpyridine (157 mg, 1.30 mmol) and piperdine (111 mg, 1.30 mmol) were added, and the mixture was heated to reflux for 20 h. Upon completion, volatiles were evaporated in vacuo, and the sample was purified by column chromatography (30–70% EtOAc/hexanes). The resulting solid was dried at 40 °C in a vacuum desiccator for 36 h to yield a bright yellow solid (60 mg, 27%): mp 230–232 °C. TLC Rf 0.49 (75% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 13.03 (s, 1H), 8.86–8.85 (m 2H), 8.07–8.04 (dd, 1H, J1 = 15.7 Hz, J2 = 1.2 Hz), 7.99–7.98 (m, 2H), 7.75–7.72 (d, 1H, J = 15.6 Hz), 7.60 (d, 1H, J = 2.28 Hz), 7.52–7.51 (d, 1H, J = 8.94 Hz), 7.12–7.10 (dd,1H, J1 = 8.94 Hz, J2 = 2.34 Hz), 3.92 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 188.9, 155.8, 150.6, 143.8, 135.2, 131.0, 127.2 (q, 2JCF = 36.5 Hz) 125.0, 121.48, 121.23 (q, 1JCF = 268.8 Hz), 119.9, 115.8, 114.2, 111.4, 103.3, 55.6; 19F NMR (376 MHz, DMSO-d6) δ −51.4 (s, 3F). Elemental analysis calculated for C18H13F3N2O2: C, 62.43; H, 3.78; N, 8.09. Found: C, 62.59; H, 3.91; N, 8.04.
Methyl trans-3-(5-Methoxy-1H-indol-2-carboxylate-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2k)
Methyl 5-methoxyindole-3-carboxyaldehyde-2-carboxylate 24a (100 mg, 0.404 mmol), 4-acetylpyridine (73 mg, 0.607 mmol), and piperidine (52 mg, 0.607 mmol) were dissolved in anhydrous MeOH (10 mL) and heated to reflux for 20 h. The resulting yellow precipitate was filtered, washed with ice-cold MeOH (20 mL), and then dried at 40 °C in a vacuum desiccator for 36 h (110 mg, 81%): mp 272–273 °C. TLC Rf 0.36 (80% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 12.58 (s, 1H), 8.85–8.84 (d, 2H, J = 4.44 Hz), 8.74–8.71 (d, 2H, J = 16.02 Hz), 7.95–7.94 (d, 2H, J = 4.5 Hz), 7.71–7.68 (d, 1H, J = 16.0 Hz), 7.54 (s, 1H), 7.49–7.48 (d, 1H, J = 8.88 Hz), 7.10–7.09 (d, 1H, J = 8.94 Hz), 3.94 (s, 3H), 3.90 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 189.6, 161.0, 155.7, 150.6, 144.2, 139.1, 131.8, 128.4, 125.3, 121.5, 120.2, 116.6, 115.6, 114.4, 103.0, 55.5, 52.2. Elemental Analysis calculated for C19H16N2O4: C, 67.85; H, 4.80; N, 8.33. Found: C, 67.71; H, 4.76; N, 8.37.
Ethyl trans-3-(5-Methoxy-1H-indol-2-carboxylate-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2l)
Ethyl 5-methoxyindole-3-carboxyaldehyde-2-carboxylate 24b (250 mg, 1.01 mmol) was reacted in a manner similar to that for 2k except 2 equiv of 4-acetylpyridine and piperidine were added and cold EtOH (10 mL) was used to wash the precipitate to yield a bright yellow solid (303 mg, 86%): mp 245–247 °C. TLC Rf 0.42 (75% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 12.51 (s, 1H), 8.84–8.83 (m 2H), 8.75–8.72 (d, 1H, J = 16.02 Hz), 7.94–7.93 (m, 2H), 7.68–7.66 (d, 1H, J = 15.96 Hz), 7.54 (d, 1H, J = 2.22 Hz), 7.50–7.49 (d, 1H, J = 8.94 Hz), 7.10–7.08 (dd,1H, J1 = 8.94 Hz, J2 = 2.34 Hz), 4.41–4.38 (q, 2H, J = 7.14 Hz), 3.90 (s, 3H), 1.36–1.33 (t, 3H, J = 7.14 Hz). 13C NMR (150 MHz, DMSO-d6) δ 189.8, 160.6, 155.7, 150.6, 144.3, 139.4, 131.7, 128.6, 125.3, 121.5, 120.2, 116.6, 115.4, 114.3, 103.0, 61.1, 55.4, 14.0. Elemental analysis calculated for C20H18N2O4: C, 68.56; H, 5.18; N, 8.00. Found: C, 68.40; H, 5.07; N, 7.95.
Propyl trans-3-(5-Methoxy-1H-indol-2-carboxylate-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2m)
Propyl 5-methoxyindole-3-carboxyaldehyde-2-carboxylate 24c (223 mg, 0.853 mmol) was dissolved in n-PrOH (20 mL, dried over 3 Å molecular sieves). 4-Acetylpyridine (155 mg, 1.28 mmol) and piperidine (109 mg, 1.28 mmol) were added, and the was mixture heated to reflux for 24 h. An orange precipitate slowly began to form. Upon completion, the orange precipitate was collected, washed with ice-cold n-PrOH (20 mL), and dried at 40 °C in a vacuum desiccator for 36 h to yield an orange solid (245 mg, 79%): mp 221 °C. TLC Rf 0.48 (80% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 12.49 (s, 1H), 8.84–8.83 (m 2H), 8.74–8.71 (d, 1H, J = 16.02 Hz), 7.94–7.92 (m, 2H), 7.68–7.65 (d, 1H, J = 15.96 Hz), 7.54 (d, 1H, J = 2.28 Hz), 7.51–7.49 (d, 1H, J = 8.94 Hz), 7.10–7.08 (dd, 1H, J1 = 8.94 Hz, J2 = 2.34 Hz), 4.32–4.30 (t, 2H, J = 6.48 Hz), 3.90 (s, 3H), 1.76–1.71 (sex, 2H, J = 7.32 Hz), 1.00–0.98 (t, 3H, J = 7.44 Hz). 13C NMR (150 MHz, DMSO-d6) δ 189.9, 160.8, 155.8, 150.7, 144.4, 139.4, 131.9, 128.8, 125.4, 121.66, 120.41, 116.70, 115.50, 114.47, 103.1, 66.7, 55.6, 21.6, 10.4; Elemental analysis calculated for C21H20N2O4: C, 69.22; H, 5.53; N, 7.69. Found: C, 68.93; H, 5.66; N, 7.75.
Isopropyl trans-3-(5-Methoxy-1H-indol-2-carboxylate-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2n)
Isopropyl 5-methoxyindole-3-carboxyaldehyde-2-carboxylate 24d (210 mg, 0.804 mmol) was reacted in a manner similar to that for 2m except i-PrOH was used instead of n-PrOH to yield a bright yellow solid (238 mg, 81%): mp 233–236 °C. TLC Rf 0.46 (80% EtOAc/hexanes). 1H NMR (600 mHz, DMSO-d6) δ 12.44 (s, 1H), 8.84–8.83 (m 2H), 8.74–8.71 (d, 1H, J = 16.02 Hz), 7.92 (m, 2H), 7.65–7.62 (d, 1H, J = 16.02 Hz), 7.53 (d, 1H, J = 2.28 Hz), 7.50–7.49 (d, 1H, J = 9 Hz), 7.10–7.08 (dd,1H, J1 = 8.94 Hz, J2 = 2.34 Hz), 5.24–5.18 (sep, 1H, J = 6.24 Hz), 3.90 (s, 3H), 1.34–1.33 (d, 6H, J = 6.24 Hz). 13C NMR (150 MHz,DMSO-d6) δ 190.1, 160.3, 155.8, 150.7, 144.5, 139.7, 131.8, 129.2, 125.4, 121.7, 120.4, 116.61, 115.37, 114.47, 103.1, 69.0, 55.6, 21.7. Elemental analysis calculated for C21H20N2O4·0.25 C3H8O: C, 68.85; H, 5.84; N, 7.38. Found: C, 68.51; H, 5.61; N, 7.62.
Sodium trans-3-(5-Methoxy-1H-indol-2-carboxylate-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2o)
Compound 2k (52 mg, 0.18 mmol) was dissolved in methanol (3 mL) and NaOH (1 N, 2 mL). The reaction mixture was heated to reflux for 30 h. Upon completion, the final volume was reduced by approximately one-half of the original volume in vacuo, and the sample was stored at 4 °C overnight resulting in the formation of an orange precipitate. The orange precipitate was collected, washed dropwise with a 50% mixture of MeOH/H2O (10 mL, chilled at 4 °C for 1 h), and dried at 40 °C in a vacuum desiccator for 36 h to yield an orange salt (16 mg, 28%): mp >300 °C. TLC Rf 0.55 DCM/MeOH: Acetic acid (90:9:1). 1H NMR (600 MHz, DMSO-d6) δ 11.72 (s, 1H), 9.34–9.32 (d, 1H, J = 16.02 Hz), 8.79–8.78 (m, 2H), 7.88–7.87 (m, 2H), 7.43 (d, 1H, J = 1.8 Hz), 7.40–7.37 (m, 2H), 6.88–6.87 (dd, 1H, J1 = 8.76 Hz, J2 = 2.16 Hz), 3.86 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 189.2, 163.5, 155.0, 150.5, 145.6, 144.8, 130.2, 126.5, 121.6, 114.8, 113.6, 112.4, 110.8, 103.8, 55.5. Elemental analysis calculated for C18H13N2NaO4·2 H2O: C, 56.84; H, 4.51; N, 7.37. Found: C, 56.64; H, 4.60; N, 7.27. HRMS ESI– calculated m/z for C18H13N2O4 (M – H)− 321.0881. Found (M – H)− 321.0884.
3-(2-Hydroxymethyl-5-methoxy-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2p)
5-Methoxy-2-hydroxymethylindole-3-carboxaldehyde 27 (75 mg, 0.37 mmol), 4-acetylpyridine (118 mg, 0.37 mmol), and piperidine (82.6 mg, 0.37 mmol) were dissolved in methanol (1 mL) and heated to reflux. The reaction mixture was stirred for 45 min during which the solution became bright orange. The flask was then removed from heat and allowed to stir for 1 h. A precipitate formed which was collected, washed with ice-cold MeOH (5 mL), and dried in a vacuum desiccator for 24 h to yield an orange solid (28 mg, 24%). mp 229–230 °C. TLC Rf 0.65 (6% MeOH/DCM), 0.21 (80% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 11.96 (s, 1H), 8.82–8.81 (m, 2H), 8.17–8.14 (d, 1H, J = 15.36 Hz), 7.94–7.93 (m, 2H), 7.45–7.44 (d, 1H, J = 2.28 Hz), 7.38–7.34 (m, 2H), 6.88–6.86 (dd, 1H, J1 = 8.7 Hz, J2 = 2.34 Hz), 5.70–5.68 (t, 1H, J = 5.46 Hz), 4.85–4.84 (d, 2H, J = 5.46 Hz), 3.86 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 188.2, 155.2, 150.6, 148.0, 145.0, 139.3, 131.0, 126.7, 121.4, 113.8, 113.0, 111.6, 108.3, 103.4, 55.57, 55.32. Elemental Analysis calculated for C18H16N2O3·0.033 MeOH: C, 70.00; H, 5.26; N, 9.05. Found: C, 69.61; H, 5.09; N, 8.90. This compound showed significant degradation when stored in DMSO-d6 for 48 h as determined by 1H NMR. After 48 h, ∼50% of 2p was degraded, and the spectrum showed a number of unidentifiable compounds. All other derivatives (1a–c, 2a–o, 2q) were stable in prolonged exposure (up to 12 months) to DMSO. When stored in its isolated precipitate form, this compound is stable.
3-(2-Hydroxypropyl-5-methoxy-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2q)
2-Hydroxypropyl-5-methoxyindole-3-carboxaldehyde-O-acetate 32 (87 mg, 0.31 mmol) in MeOH (6 mL), piperidine (40 mg, 0.47 mmol), and 4-acetylpyridine (57 mg, 0.47 mmol) were heated to reflux and stirred for 20 h. The solvent was reduced by approximately one-half of the original volume in vacuo, and the flask was placed in a −20 °C freezer for 30 min. The resulting bright orange-red precipitate was collected, washed with ice-cold MeOH (5 mL) and dried in a vacuum desiccator for 24 h (16 mg, 15%): mp 221–223 °C. TLC Rf 0.23 (4% MeOH/DCM). 1H NMR (600 MHz, DMSO-d6) δ 11.87 (s, 1H), 8.81–8.80 (m, 2H), 8.12–8.10 (d, 1H, J = 15.24 Hz), 7.94–7.93 (m, 2H), 7.44 (d, 1H, J = 2.28 Hz), 7.40–7.37 (d, 1H, J = 15.24 Hz), 7.33–7.31 (d, 1H, J = 8.64 Hz), 6.87–6.85 (dd, 1H, J1 = 8.7 Hz, J2 = 2.4 Hz), 4.68–4.66 (t, 1H, J = 4.62 Hz), 3.86 (s, 3H), 3.48–3.45 (q, 2H, J = 6.0 Hz), 2.99–2.96 (t, 2H, J = 7.62 Hz), 1.86–1.81 (quin, 2H, J = 6.24 Hz). 13C NMR (150 MHz, DMSO-d6) δ 188.1, 155.2, 150.62, 149.80, 145.1, 139.5, 131.2, 126.4, 121.4, 113.01, 112.42, 111.1, 108.9, 103.7, 59.9, 55.6, 32.7, 22.7. Elemental Analysis for C20H20N2O3: C, 71.41; H, 5.99; N, 8.33. Found: C, 71.12; H, 6.05; N, 8.34.
4-Ethoxy-2-methyl-1-nitrobenzene (4a)
3-Methyl-4-nitrophenol 3 (500 mg, 3.0 mmol) was dissolved in 2-butanone (10 mL). Potassium carbonate (903 mg, 6.5 mmol) was added to the solution followed by diethylsulfate (0.45 mL, 3.2 mmol). The reaction mixture was heated at 70 °C for 2 h. Upon completion, the mixture was cooled to rt, and aqueous ammonia (30%, 1 mL) was added and stirred for 18 h at rt. The reaction mixture was filtered and washed with 2-butanone (30 mL). The filtrate was collected and evaporated in vacuo to yield a white solid (580 mg, 97%). mp 50–52 °C (lit.56 mp 51 °C). TLC Rf 0.65 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 8.11–8.09 (d, 1H, J = 8.88 Hz), 6.82–6.78 (m, 2H), 4.14–4.10 (q, 2H, J = 6.96 Hz), 2.65 (s, 3H), 1.48–1.46 (t, 3H, J = 6.95 Hz). 13C NMR (150 MHz, CDCl3) δ 162.5, 142.0, 137.1, 127.6, 117.9, 112.2, 64.2, 21.8, 14.6.
2-Methyl-4-propoxy-1-nitrobenzene (4b)
Compound 3 (1.5 g, 9.8 mmol) was dissolved in 2-butanone (40 mL). Potassium carbonate (3.4 g, 24.5 mmol) was added, and the solution was heated at reflux. After 5 min, 1-bromopropane (18.5 mmol, 2 mL) was added dropwise, and the reaction mixture continued to stir for 18 h. The solvent was evaporated in vacuo, and the crude residue was partitioned between EtOAc (150 mL) and saturated NaHCO3 (150 mL). The organic layer was separated, washed with brine (100 mL), and dried over Na2SO4. The resulting oil was purified by column chromatography (0% to 20% EtOAc/hexanes) to yield a yellow oil (1.89 g, 99%): TLC Rf 0.70 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 8.09–8.07 (d, 1H, J = 8.76 Hz), 6.79–6.77 (m, 2H), 3.99–3.97 (t, 2H, J = 6.54 Hz), 2.63 (s, 3H), 1.85–1.82 (sex, 2H, J = 7.44 Hz), 1.06–1.04 (t, 3H, J = 7.44 Hz). 13C NMR (150 MHz, CDCl3) δ 162.9, 142.2, 137.3, 127.8, 118.1, 112.4, 70.3, 22.60, 21.98, 10.6. Elemental analysis calculated for C10H13NO3·0.03 hexanes: C, 61.81; H, 6.84; N, 7.08. Found: C, 62.20; H, 6.78; N, 7.23.
4-Isopropoxy-2-methyl-1-nitrobenzene (4c)
This compound was prepared in a similar manner to that for 4b except that 2-bromopropane was deployed for the alkyl ether adduct and chromatography utilized a 0–15% gradient for elution to yield a yellow oil (1.87 g, 98%): TLC Rf 0.68 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 8.08–8.07 (d, 1H, J = 8.82 Hz), 6.77–6.75 (m, 2H), 4.66–4.62 (sep, 1H, J = 6.06 Hz), 2.62 (s, 3H), 1.38–1.37 (d, 6H, J = 6.12 Hz). 13C NMR (150 MHz, CDCl3) δ 162.6, 141.7, 137.1, 127.6, 118.9, 112.8, 70.6, 21.84, 21.78. Elemental analysis calculated for C10H13NO3·0.06 hexanes: C, 62.10; H, 6.96; N, 6.99. Found: C, 62.45; H, 6.69; N, 7.09.
4-Butoxy-2-methyl-1-nitrobenzene (4d)
This compound was prepared in a similar manner to that for 4b except 1-bromobutane was deployed for the alkyl ether adduct and the partition step during workup utilized DCM (150 mL) and water (150 mL) to provide a yellow oil (2.0 g, 99%): TLC Rf 0.70 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 8.09–8.07 (d, 1H, J = 8.88 Hz), 6.79–6.76 (m, 2H), 4.03–4.01 (t, 2H, J = 6.48 Hz), 2.63 (s, 3H), 1.80–1.78 (quin, 2H, J = 7.44 Hz), 1.53–1.47 (sex, 2H, J = 7.44 Hz), 1.0–0.97 (t, 3H, J = 7.38 Hz). 13C NMR (150 MHz, CDCl3) δ 162.7, 141.9, 137.1, 127.6, 117.9, 112.2, 68.3, 31.0, 21.8, 19.1, 13.8. Elemental analysis calculated for C11H15NO3: C, 63.14; H, 7.23; N, 6.69. Found: C, 63.25; H, 7.09; N, 6.69.
4-Ethoxy-2-methylaniline (5a)
Compound 4a (250 mg, 1.38 mmol) was dissolved in EtOAc (10 mL) and MeOH (10 mL) and transferred to a 250 mL hydrogenation flask. 10% Pd/C (25 mg, 10% w/w) was added, and the sample was hydrogenated for 4 h at 35 psi H2. Upon completion, the mixture was filtered over a bed of Celite and then concentrated in vacuo. The residue was dissolved in EtOAc (50 mL) and washed with NaHCO3 (50 mL × 3). The organic layer was dried over Na2SO4 to yield a dark-red oil (192 mg, 92%): TLC Rf 0.30 (20% EtOAc/hexanes). 1NMR (600 MHz, CDCl3) δ 6.67 (s, 1H), 6.62 (m, 2H), 3.97–3.93 (q, 2H, J = 6.96 Hz), 3.42 (s, 2H), 2.16 (s, 3H), 1.38–1.35 (t, 3H, J = 7.02 Hz). 13C NMR (150 MHz, CDCl3) δ 152.3, 138.1, 124.4, 117.45, 116.34, 113.1, 64.2, 17.9, 15.2. Elemental analysis calculated for C9H13NO: C, 71.49; H, 8.67; N, 9.26. Found: C, 71.42; H, 8.76; N, 9.04.
2-Methyl-4-propoxyaniline (5b)
Compound 4b (3.78 g, 19.4 mmol) was dissolved in EtOAc (20 mL) and MeOH (20 mL) and transferred to a 250 mL hydrogenation flask. 10% Pd/C (378 mg, 10% w/w) was added, and the sample was hydrogenated for 4 h at 35 psi H2. Upon completion, the mixture was filtered over a bed of Celite and then concentrated in vacuo. The residue was dissolved in EtOAc (100 mL) and washed with NaHCO3 (100 mL × 3). The organic layer was separated and then dried over Na2SO4 to provide a crude brown oil which was further purified by chromatography (20% to 50% EtOAc/hexanes) to yield an amber oil (2.72 g, 85%): TLC Rf 0.27 (20% EtOAc/hexanes). 1NMR (600 MHz, CDCl3) δ 6.67 (s, 1H), 6.62 (m, 2H), 3.85–3.38 (t, 2H, J = 6.66 Hz), 2.16 (s, 3H), 1.79–1.73 (sex, 2H, J = 7.44 Hz), 1.02–1.00 (t, 3H, J = 7.44 Hz). 13C NMR (150 MHz, CDCl3) δ 152.3, 137.8, 124.1, 117.2, 116.1, 112.9, 70.1, 22.7, 17.7, 10.5. Elemental analysis calculated for C10H15NO: C, 72.69; H, 9.15; N, 8.48. Found: C, 72.42; H, 9.14; N, 8.46.
4-Isopropoxy-2-methylaniline (5c)
This compound was prepared from 4c (3.64 g, 18.7 mmol) in a manner similar to that for 5b to yield an amber oil (2.5 g, 81%): TLC Rf 0.33 (20% EtOAc/hexanes). 1NMR (600 MHz, CDCl3) δ 6.67 (s, 1H), 6.62 (m, 2H), 4.39–4.35 (sep, 1H, J = 6.06 Hz), 2.15 (s, 3H), 1.29–1.28 (d, 6H, J = 6.06 Hz). 13C NMR (150 MHz, CDCl3) δ 150.8, 138.0, 124.1, 119.4, 116.10, 115.09, 71.0, 22.2, 17.7. Elemental analysis calculated for C10H15NO: C, 72.69; H, 9.15; N, 8.48. Found: C, 72.75; H, 9.11; N, 8.33.
4-Butoxy-2-methylaniline (5d)
This compound was prepared from 4d (1.92 g, 9.2 mmol) in a manner similar to that for 5b yielding an amber oil (1.48 g, 90%): TLC Rf 0.28 (20% EtOAc/hexanes). 1NMR (600 MHz, CDCl3) δ 6.67 (s, 1H), 6.62 (m, 2H), 3.89–3.87 (t, 2H, J = 6.54 Hz), 2.16 (s, 3H), 1.73–1.70 (quin, 2H, J = 6.6 Hz), 1.49–1.45 (sex, 2H, J = 7.5 Hz), 0.97–0.95 (t, 3H, J = 7.38 Hz). 13C NMR (150 MHz, CDCl3) δ 152.4, 137.8, 124.2, 117.24, 116.13, 112.9, 68.3, 31.5, 19.3, 17.7, 13.9. Elemental analysis calculated for C11H17NO: C, 73.70; H, 9.56; N, 7.81. Found: C, 73.55; H, 9.51; N, 7.96.
tert-Butyl (4-ethoxy-2-methylphenyl)carbamate (6a)
Compound 5a (210 mg, 1.39 mmol) and di-tert-butyl-dicarbonate (334 mg, 1.53 mmol) in THF (10 mL) were heated to reflux for 20 h. The reaction mixture was concentrated in vacuo and redissolved in DCM (30 mL). This mixture was washed with saturated NaHCO3 (30 mL) and brine (30 mL). The organic layer was separated and dried over Na2SO4 to produce a dark-red oil which was purified by chromatography (0% to 20% EtOAc/hexanes) to yield an orange solid (293 mg, 84%): mp 66–68 °C (lit.56 mp 66 °C). TLC Rf 0.50 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.51 (s, 1H), 6.72 (m, 2H), 6.06 (s, 1H), 4.01–3.97 (q, 2H, J = 6.96 Hz), 2.22 (s, 3H), 1.53 (s, 9H), 1.40–1.37 (t, 3H, J = 6.9 Hz). 13C NMR (150 MHz, CDCl3) δ 155.8, 153.7, 146.7, 129.0, 124.0, 116.6, 112.2, 80.1, 63.6, 28.4, 18.1, 14.9.
tert-Butyl (4-propoxy-2-methylphenyl)carbamate (6b)
Compound 5b (2.58 g, 15.6 mmol) and di-tert-butyl dicarbonate (5.11 g, 23.4 mmol) in THF (75 mL) were heated to reflux for 24 h. Volatiles were evaporated in vacuo, and the residue was purified by chromatography (0% to 20% EtOAc/hexanes) to yield a light-purple solid (4.06 g, 98%): mp 74–76 °C. TLC Rf 0.60 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.50 (s, 1H), 6.72–6.70 (m, 2H), 6.06 (s, 1H), 3.89–3.86 (t, 2H, J = 6.6 Hz), 2.22 (s, 3H), 1.81–1.75 (sex, 2H, J = 7.38 Hz), 1.50 (s, 9H), 1.03–1.00 (t, 3H, J = 7.38 Hz). 13C NMR (150 MHz, CDCl3) δ 156.1, 153.8, 131.0, 129.0, 124.0, 116.6, 112.2, 80.1, 69.7, 28.4, 22.6, 18.1, 10.5. Elemental analysis calculated for C15H23NO3: C, 67.90; H, 8.74; N, 5.28. Found: C, 67.78; H, 8.68; N, 5.24.
tert-Butyl (4-isopropoxy-2-methylphenyl)carbamate (6c)
This compound was prepared from 5c in a similar manner to that for 6b except that chromatography used a gradient of 0% to 15%. A white solid was obtained (3.78 g, 99%): mp 57–59 °C. TLC Rf 0.57 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.50 (s, 1H), 6.72–6.70 (m, 2H), 6.06 (s, 1H), 4.50–4.45 (sep, 1H, J = 6.06 Hz), 2.21 (s, 3H), 1.59 (s, 9H), 1.31–1.30 (d, 6H, J = 6.06 Hz). 13C NMR (150 MHz, CDCl3) δ 154.63, 153.72, 131.2, 129.0, 123.9, 118.2, 113.7, 80.1, 70.1, 28.4, 22.1, 18.0. Elemental analysis calculated for C15H23NO3: C, 67.90; H, 8.74; N, 5.28. Found: C, 67.97; H, 8.76; N, 5.24.
tert-Butyl (4-butoxy-2-methylphenyl)carbamate (6d)
Compound 5d (1.18 g, 6.6 mmol) and di-tert-butyl dicarbonate (1.58 g, 7.24 mmol) in THF (50 mL) were heated to reflux for 24 h. The solvent was evaporated in vacuo, and the residue was dissolved in EtOAc (50 mL) and washed with brine (50 mL × 3). The organic layer was separated and then dried over Na2SO4. The residue was purified by chromatography (0–20% EtOAc/hexanes) to provide a light-orange solid (4.06 g, 98%): mp 53–55 °C. TLC Rf 0.65 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.50 (s, 1H), 6.72–6.70 (m, 2H), 6.06 (s, 1H), 3.93–3.91 (t, 2H, J = 6.54 Hz), 2.22 (s, 3H), 1.76–1.71 (quin, 2H, J = 6.6 Hz), 1.50 (s, 9H), 1.49–1.44 (m, 2H), 0.98–0.95 (t, 3H, J = 7.38 Hz). 13C NMR (150 MHz, CDCl3) δ 156.0, 153.7, 131.1, 128.9, 124.0, 116.5, 112.2, 80.1, 67.8, 31.3, 28.4, 19.2, 18.0, 13.8. Elemental analysis calculated for C16H25NO3: C, 68.79; H, 9.02; N, 5.01. Found: C, 68.87; H, 8.94; N, 5.02.
5-Ethoxy-2-methylindole (8a)
Compound 6a (900 mg, 3.58 mmol) was dissolved in THF (15 mL) under an atmosphere of Argon. The solution was cooled to −40 °C over 10 min and sec-butyllithium (1.4 M, 5.37 mL) was added slowly as to maintain an internal temperature of < −25 °C. After reaching 1 equiv of sec-butyllithium (2.7 mL), the reaction mixture turned a bright yellow signifying deprotonation of the amide nitrogen. The reaction mixture was then cooled to −50 °C, and a solution of N-methoxy-N-methylacetylamide 7 (442.8 mg, 4.78 mmol) in THF (3 mL) was added over 5 min. The reaction mixture was warmed to −10 °C over 30 min. The mixture was partitioned between Et2O (75 mL) and 0.5 N HCl (75 mL). The aqueous layer was separated and extracted an additional two times with Et2O (50 mL). The Et2O phases were combined and washed with brine (75 mL) and then dried over Na2SO4 to yield a dark-brown oil. The crude intermediate was dissolved in DCM (20 mL). TFA (3 mL) was added to the mixture which was then stirred at rt for 48 h. Upon completion, the reaction mixture was added to a separatory funnel and washed with NaHCO3 (50 mL) followed by brine (50 mL). The organic layer was separated and then dried over Na2SO4 to provide a crude black oil (1 g) which was purified by chromatography (0–20% EtOAc/hexanes) to yield a brown solid (335 mg, 53%): mp 88–90 °C. TLC Rf 0.48 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.72 (s, 1H), 7.16–7.14 (d, 1H, J = 8.64 Hz), 6.99 (d, 1H, J = 2.4 Hz), 6.77–6.75 (dd, 1H, J1 = 8.7 Hz, J2 = 2.4 Hz), 6.13 (s, 1H), 4.07–4.04 (q, 2H, J = 6.96 Hz), 2.41 (s, 3H), 1.43–1.40 (t, 3H, J = 6.96 Hz). 13C NMR (150 MHz, CDCl3) δ 153.3, 135.8, 131.2, 129.5, 111.33, 110.75, 103.1, 100.3, 64.2, 15.1, 13.8. Elemental analysis calculated for C11H13NO: C, 75.40; H, 7.48; N, 7.99. Found: C, 75.11; H, 7.64; N, 7.46.
2-Methyl-5-propoxyindole (8b)
This compound was prepared from 6b (1.00 g, 3.77 mmol) in a manner similar to that for 8a except: after the addition of TFA the mixture was stirred for 24 h at rt then heated at reflux for an additional 2 h to ensure complete BOC indole deprotection, and the gradient for chromatography was 0–15%. A yellow solid was obtained (0.28 g, 39%): mp 66–67 °C. TLC Rf 0.50 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.72 (s, 1H), 7.16–7.14 (d, 1H, J = 8.7 Hz), 6.99 (d, 1H, J = 2.34 Hz), 6.77–6.76 (dd, 1H, J1 = 8.7 Hz, J2 = 2.4 Hz), 6.13 (s, 1H), 3.96–3.93 (t, 2H, J = 6.66 Hz), 2.41 (s, 3H), 1.84–1.78 (sex, 2H, J = 7.32 Hz), 1.06–1.03 (t, 3H, J = 7.44 Hz). 13C NMR (150 MHz, CDCl3) δ 153.5, 135.8, 131.1, 129.5, 111.3, 110.7, 103.1, 100.3, 70.4, 22.8, 13.8, 10.6. Elemental analysis calculated for C12H15NO: C, 76.16; H, 7.99; N, 7.40. Found: C, 76.13; H, 7.90; N, 7.36.
5-Isopropoxy-2-methylindole (8c)
This compound was prepared from 6c (1.00 g, 3.77 mmol) in a manner similar to that for 8b to yield a yellow solid (0.43 g, 60%): mp 64–65 °C. TLC Rf 0.52 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.69 (s, 1H), 7.14–7.13 (d, 1H, J = 8.7 Hz), 7.01 (d, 1H, J = 2.28 Hz), 6.75–6.74 (dd, 1H, J1 = 8.64 Hz, J2 = 2.34 Hz), 6.12 (s, 1H), 4.50–4.46 (sep, 1H, J = 6.06 Hz), 2.40 (s, 3H), 1.33–1.32 (d, 6H, J = 6.06 Hz). 13C NMR (150 MHz, CDCl3) δ 152.2, 135.8, 131.6, 129.7, 113.1, 110.6, 106.2, 100.3, 71.5, 22.3, 13.8. Elemental analysis calculated for C12H15NO: C, 76.16; H, 7.99; N, 7.40. Found: C, 76.12; H, 7.86; N, 7.29.
5-Butoxy-2-methylindole (8d)
This compound was prepared from 6d in a manner similar to that for 8a to yield a brown solid (0.356 g, 49%): mp 58–60 °C. TLC Rf 0.50 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.72 (s, 1H), 7.16–7.14 (d, 1H, J = 8.7 Hz), 6.99 (d, 1H, J = 2.4 Hz), 6.77–6.75 (dd, 1H, J1 = 8.7 Hz, J2 = 2.4 Hz), 6.13 (s, 1H), 4.00–3.98 (t, 2H, J = 6.6 Hz), 2.41 (s, 3H), 1.80–1.75 (quin, 2H, J = 6.6 Hz), 1.54–1.49 (sex, 2H, J = 7.5 Hz), 0.99–0.96 (t, 3H, J = 7.44 Hz). 13C NMR (150 MHz, CDCl3) δ 153.6, 135.8, 131.1, 129.5, 111.3, 110.7, 103.0, 100.3, 68.5, 31.6, 19.3, 13.94, 13.81. Elemental analysis calculated for C13H17NO: C, 76.81; H, 8.43; N, 6.89. Found: C, 76.84; H, 8.31; N, 6.78.
5-Ethoxy-2-methylindole-3-carboxaldehyde (9a)
DMF (2 mL) was cooled to 0 °C. POCl3 (0.5 mL) was added and the reaction mixture was stirred for 10 min. A solution of compound 8a (250 mg, 1.43 mmol) in DMF (3 mL) was added dropwise over 10 min and then stirred for an additional 40 min while warming to rt. The reaction mixture was added to ice-cold 1 N NaOH (35 mL), and the solution was stirred for 30 min in an ice-bath. The resulting precipitate was collected, washed with ice-cold H2O (20 mL), and dried at 40 °C for 24 h in a vacuum desiccator to yield a light-brown solid (238 mg, 82%): mp 199–200 °C. TLC Rf 0.51 (80% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 11.85 (s, H) 10.00 (s, 1H), 7.55 (d, 1H, J = 2.34 Hz), 7.27–7.25 (d, 1H, J = 8.7 Hz), 6.78–6.76 (dd, 1H, J1 = 8.7 Hz J2 = 2.4 Hz), 4.03–3.99 (q, 2H, J = 6.96 Hz), 2.64 (s, 3H), 1.35–1.33 (t, 3H, J = 6.96 Hz). 13C NMR (150 MHz, DMSO-d6) δ 184.5, 155.2, 149.0, 130.5, 126.8, 114.1, 112.71, 112.52, 103.6, 63.7, 15.3, 12.0. Elemental analysis calculated for C12H13NO2: C, 70.92; H, 6.45; N, 6.89. Found: C, 70.66; H, 6.57; N, 6.97.
2-Methyl-5-propoxyindole-3-carboxaldehyde (9b)
This compound was prepared from 8b (263 mg, 1.39 mmol) in a manner similar to that for 9a except for after the dropwise addition of substituted indole, the solution was stirred for 1 h. A cream-colored powder was obtained (228 mg, 75%): mp 156–158 °C. TLC Rf 0.52 (75% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 11.85 (s, 1H), 10.00 (s, 1H), 7.55 (d, 1H, J = 2.46 Hz), 7.27–7.25 (d, 1H, J = 8.7 Hz), 6.79–6.77 (dd, 1H, J1 = 8.7 Hz, J2 = 2.52 Hz), 3.92–3.90 (t, 2H, J = 6.54 Hz), 2.64 (s, 3H), 1.77–1.71 (sex, 2H, J = 7.38 Hz), 1.00–0.98 (t, 3H, J = 7.38 Hz). 13C NMR (150 MHz, DMSO-d6) δ 184.0, 154.9, 148.5, 130.0, 126.4, 113.6, 112.26, 112.04, 103.2, 69.3, 22.2, 11.5, 10.5. Elemental analysis calculated for C13H15NO2: C, 71.87; H, 6.96; N, 6.45. Found: C, 71.74; H, 6.81; N, 6.42.
5-Isopropoxy-2-methylindole-3-carboxaldehyde (9c)
This compound was prepared from 8c (347 mg, 1.83 mmol) in a manner similar to that for 9a except for after the dropwise addition of substituted indole, the solution was stirred for 90 min. A cream-colored powder was obtained (313 mg, 78%): mp 179–181 °C. TLC Rf 0.43 (75% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 11.84 (s, 1H), 10.00 (s, 1H), 7.56 (d, 1H, J = 2.4 Hz), 7.26–7.25 (d, 1H, J = 8.64 Hz), 6.77–6.76 (dd, 1H, J1 = 8.7 Hz, J2 = 2.46 Hz), 4.56–4.50 (sep, 1H, J = 6 Hz), 2.64 (s, 3H), 1.27–1.26 (d, 6H, J = 6 Hz). 13C NMR (150 MHz, DMSO-d6) δ 184.0, 153.4, 148.6, 130.1, 126.4, 113.58, 113.56, 112.0, 105.5, 69.9, 22.0, 11.5. Elemental analysis calculated for C13H15NO2: C, 71.87; H, 6.96; N, 6.45. Found: C, 71.70; H, 6.83; N, 6.60.
5-Butoxy-2-methylindole-3-carboxaldehyde (9d)
This compound was prepared from 8d (300 mg, 1.48 mmol) in a manner similar to that for 9b to yield a cream-colored powder (320 mg, 94%): mp 160–161 °C. TLC Rf 0.45 (3:1 EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 11.85 (s, 1H), 10.00 (s, 1H), 7.55 (d, 1H, J = 2.46 Hz), 7.26–7.25 (d, 1H, J = 8.7 Hz), 6.79–6.77 (dd, 1H, J1 = 8.7 Hz, J2 = 2.46 Hz), 3.96–3.94 (t, 2H, J = 6.48 Hz), 2.64 (s, 3H), 1.73–1.68 (quin, 2H, J = 6.48 Hz), 1.49–1.42 (sex, 2H, J = 7.38 Hz), 0.95–0.93 (t, 3H, J = 7.38 Hz). 13C NMR (150 MHz, DMSO-d6) δ 185.0, 154.9, 148.5, 130.0, 126.4, 113.6, 112.26, 112.04, 103.2, 67.4, 31.0, 18.8, 13.8, 11.5. Elemental analysis calculated for C14H17NO2: C, 72.70; H, 7.41; N, 6.06. Found: C, 72.87; H, 7.47; N, 6.13.
5-(N,N-Diacetyl-amino)-2-methylindole (11)
5-Amino-2-methylindole 10 (200 mg, 1.37 mmol) was dissolved in acetic anhydride (25 mL, 2.65 mmol), and the solution was heated at 60 °C for 20 h. Upon completion, the volatiles were evaporated and the oil partitioned between DCM (15 mL) and water (10 mL). The organic layer was separated, dried over Na2SO4, and purified by chromatography (isocratic 50% EtOAc/hexanes) to yield an orange-brown solid (200 mg, 64%): mp 130–134 °C. TLC Rf 0.33 (1:1 EtOAc/hexanes). 1H NMR (400 mHz, CDCl3) δ 8.12 (s, 1H), 7.26 (s, 1H), 7.25–7.24 (d, 1H, J = 0.8 Hz), 6.82–6.80 (dd, 1H, J1 = 8.4 Hz, J2 = 2 Hz), 6.21 (s, 1H), 2.42 (s, 3H), 2.31 (s, 6H); 13C NMR (150 MHz, CDCl3) δ 174.2, 137.3, 135.8, 130.9, 129.6, 120.3, 119.0, 111.6, 100.2, 27.1, 13.5. Elemental analysis calculated for C13H14N2O2: C, 67.81; H, 6.13; N, 12.17. Found: C, 67.73; H, 6.16; N 11.99.
5-(N-Boc-Amino)-2-methylindole (12)
5-Amino-2-methylindole 10 (400 mg, 1.62 mmol) and di-tert-butyl-dicarbonate (712 mg, 3.26 mmol) were dissolved in CH3CN (12 mL) and stirred at rt for 20 h. The volatiles were evaporated in vacuo, the residue was purified by chromatography (isocratic 50% EtOAc/hexanes), and the product was dried in a vacuum desiccator for 20 h to yield a light-brown solid (615 mg, 91%): mp 134–137 °C. TLC Rf 0.66 (1:1 EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.85 (s, 1H), 7.54 (s, 1H), 7.14–7.13 (d, 1H, J = 8.4 Hz), 7.01–6.99 (d, 1H, J = 7.8 Hz), 6.44 (s, 1H), 6.13 (s, 1H), 2.39 (s, 3H), 1.53 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 153.9, 136.4, 133.2, 130.5, 129.3, 114.5, 110.6, 100.2, 80.0, 28.6, 13.7. Elemental analysis calculated for C14H18N2O2: C, 68.27; H, 7.37; N, 11.37. Found: C, 68.30; H, 7.39; N 11.21.
5-Acetamido-2-methylindole-3-carboxaldehyde (13)
This compound was prepared from 11 (320 mg, 1.39 mmol) in a manner similar to that for 9a except for after dropwise addition of substituted indole the solution was stirred for 2 h to yield an orange precipitate. The sample was further purified by chromatography (0% to 10% MeOH/DCM) to yield a white solid (222 mg, 74%): Mp 263 °C (darkening). TLC Rf 0.46 (10% MeOH/DCM). 1H NMR (600 MHz, CD3OD) δ 9.99 (s, 1H), 8.18 (d, 1H, J = 1.8 Hz), 7.47–7.45 (dd, 1H, J1 = 8.4 Hz J2 = 1.8 Hz), 7.30–7.29 (d, 1H, J = 9 Hz), 2.70 (s, 3H), 2.14 (s, 3H). 13C NMR (150 MHz, CD3OD) δ 186.2, 171.6, 151.4, 135.03, 134.29, 127.4, 118.6, 115.6, 113.8, 112.3, 23.7, 11.8. Elemental analysis calculated for C12H12N2O2·0.15 H2O: C, 65.83; H, 5.66; N, 12.80. Found: C, 65.75; H, 5.59; N, 12.42.
5-(N-Boc-Amino)-2-methylindole-3-carboxaldehyde (14)
This compound was prepared from 12 (615 mg, 2.24 mmol) in a manner similar to that for 9a except for after the dropwise addition of substituted indole the solution was stirred for 4 h to provide an orange precipitate. The precipitate was further purified by chromatography (isocratic 5% MeOH/DCM) to yield a white solid (250 mg, 36%): mp 201–203 °C. TLC Rf 0.33 (75% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 9.70 (s, 1H), 8.08 (s, 1H), 7.26–7.24 (m, 2H), 2.69 (s, 3H), 1.52 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 184.4, 153.4, 149.4, 134.5, 131.6, 125.9, 115.6, 113.9, 111.4, 110.3, 79.0, 28.5, 11.7. Elemental analysis calculated for C15H18N2O3·0.125 H2O: C, 65.14; H, 6.65; N, 10.13. Found: C, 64.82; H, 6.81; N, 10.14. HRMS ESI+ calculated for C15H18N2O3 (M + H)+ 275.1390; (M + Na)+ 297.1210. Found (M + H)+ 275.1393; (M + Na)+ 297.1210.
N-Boc-5-(aminomethyl)indole (16)
Compound 15 (200 mg, 1.37 mmol) and di-tert-butyl dicarbonate (358 mg, 1.64 mmol) in CH3CN (10 mL) were stirred at rt for 48 h. Upon completion, solvents were evaporated in vacuo. The residue was purified by chromatography (10–50% EtOAc/hexanes) and dried at 40 °C in a vacuum desiccator for 24 h to yield a colorless oil (297 mg, 88%): TLC Rf 0.69 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 8.51 (s, 1H), 7.53 (s, 1H), 7.32–7.31 (d, 1H, J = 8.4 Hz), 7.18 (s, 1H), 7.12–7.11 (d, 1H, J = 7.8 Hz), 6.50 (s, 1H), 4.89 (s, 1H), 4.41–4.40 (d, 2H, J = 4.2 Hz), 1.48 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 156.2, 135.4, 130.2, 128.2, 125.0, 122.2, 120.0, 111.5, 102.6, 79.5, 45.5, 28.7. Elemental analysis calculated for C14H18N2O2: C, 68.27; H, 7.37; N, 11.37. Found: C, 68.11; H, 7.24; N, 11.17. Although we obtained a colorless oil, a previous report notes a light yellow solid with a mp 86–89 °C.57
N-Boc-5-(aminomethyl)indole-3-carboxaldehyde (17)
This compound was prepared from 16 (297 mg, 1.21 mmol) in a manner similar to that for 9a to yield a cream-colored solid (243 mg, 73%): mp 126–128 °C. TLC Rf 0.50 (80% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 12.10 (s, 1H), 9.91 (s, 1H), 8.27 (m, 1H), 8.00 (s, 1H), 7.45–7.43 (m, 2H), 7.17–7.15 (d, 1H, J = 8.4 Hz), 4.22–4.20 (d, 2H, J = 6 Hz), 1.40 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 184.7, 155.7, 138.6, 136.0, 134.0, 124.0, 123.1, 119.2, 118.0, 112.0, 77.6, 43.7, 28.2. Elemental analysis calculated for C15H18N2O3: C, 65.68; H, 6.61; N, 10.21. Found: C, 65.75; H, 6.61; N, 10.20.
N-Methoxy-N-methyltrifluoroacetamide (19)
O,N-Dimethylhydroxylamine hydrochloride (0.77g, 7.8 mmol) and trifluoroacetic anhydride (1.5 g, 1.0 mL, 7.1 mmol) in anhydrous DCM (25 mL) were stirred for 15 min at 0 °C. Pyridine (1.27 mL, 15.7 mmol) was added dropwise over 15 min. The reaction was then removed from the ice bath and stirred for 3.5 h at rt. The reaction mixture was washed with 0.5 N HCl (50 mL × 2), saturated NaHCO3 (50 mL) and brine (50 mL). The organic layer was separated and dried over Na2SO4. Bulb-to-bulb distillation was performed on the sample under water aspirator vacuum to obtain a colorless oil (578 mg, 52%): 1H NMR (600 MHz, CDCl3) δ 3.78 (s, 3H), 3.30 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 156.9 (q, 2JFC = 39 Hz), 116.1(q, 1JFC = 284 Hz), 62.2, 32.9. 19F NMR (376 MHz, CDCl3) δ −72.2 (s, 3F). Elemental analysis calculated for C4H6F3NO2: C, 30.58; H, 3.85; N, 8.92. Found: C, 30.65; H, 4.04; N, 8.83.
5-Methoxy-2-trifluoromethylindole (20)
This compound was prepared from 18 (1.0 g, 4.2 mmol) in a manner similar to that for 8a except Weinreb amide 19 (726 mg, 4.6 mmol) was utilized as the acylating agent to yield a yellow powder (0.390 g, 43%): mp 55 °C. TLC Rf 0.52 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 8.30 (s, 1H), 7.32–7.31 (d, 1H, J = 8.9 Hz), 7.10 (d, 1H, J = 2.4 Hz), 7.00–6.98 (dd, 1H, J1 = 8.9 Hz, J2 = 2.4 Hz), 6.85 (s, 1H), 3.85 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 154.9, 131.2, 127.09, 126.15 (q, 2JFC = 38 Hz), 121.2 (q, 1JFC = 266 Hz), 115.8, 112.6, 104.0 (q, 3JFC = 3 Hz), 102.7, 55.8. 19F NMR (376 MHz, CDCl3) δ −60.9 (s, 3F). Elemental analysis calculated for C10H8F3NO· 0.04 hexanes: C, 56.26; H, 3.95; N, 6.41. Found: C, 56.63; H, 3.95; N, 6.37.
5-Methoxy-2-trifluoromethylindole-3-carboxaldehyde (21)
DMF (1 mL) was cooled to 0 °C and POCl3 (0.25 mL) was added and stirred for 10 min. Compound 20 (336 mg, 1.69 mmol) in DMF (2 mL) was added dropwise over 10 min. The solution was stirred for an additional 1 h warming to rt and then heated to 85 °C and allowed to react for an additional 4 h. The solution was poured into ice-cold 1 N NaOH (40 mL) and stirred for 30 min in an ice-bath resulting in the formation of a precipitate. The precipitate was collected, washed with cold H2O, and dried overnight at 40 °C in a vacuum desiccator to yield a cream-colored solid. The sample was further purified by chromatography (7–20% EtOAc/hexanes) to obtain a white solid (178 mg, 43%): mp 236–239 °C. TLC Rf 0.51 (30% EtOAc/hexanes). 1H NMR (600 MHz, MeOH-d4) δ 10.22 (s, 1H), 7.79 (d, 1H, J = 2.46 Hz), 7.45–7.43 (d, 1H, J = 9 Hz), 7.06–7.04 (dd, 1H, J1 = 8.94 Hz, J2 = 2.52 Hz), 3.86 (s, 3H). 13C NMR (150 MHz, MeOH-d4) δ 186.2, 159.0, 132.0, 127.1, 123.4, 121.6, 118.2, 117.2, 114.8, 104.1, 56.2. 19F NMR (376 MHz, CDCl3) δ −58.7 (s, 3F). Elemental analysis calculated for C11H8F3NO2: C, 54.33; H, 3.32; N, 5.76. Found: C, 54.53; H, 3.32; N, 5.76.
Methyl 5-Methoxyindole-2-carboxylate (23a)
Methanolic HCl was prepared by bubbling anhydrous HCl (g) into anhydrous methanol (50 mL) for 5 min. 5-Methoxyindole-2-carboxylic acid 22 (2.0 g, 10.5 mmol) was added to the solution, and the reaction was heated to reflux for 24 h. The mixture was concentrated in vacuo and partitioned between 1 N NaOH (15 mL) /saturated NaHCO3 solution (85 mL) and EtOAc (100 mL). The organic layer was separated, washed with brine (50 mL), and then dried over Na2SO4. The resulting crude brown solid was purified by chromatography (20% to 75% EtOAc/hexane) to produce a light-brown solid (1.69 g, 78%). mp 183–186 °C (lit.58 177–178 °C). TLC Rf 0.67 in 50% EtOAc/Hex. 1H NMR (600 MHz, MeOH-d4) δ 7.33–7.31 (d, 1H, J = 9 Hz), 7.08 (m, 2H), 6.93–6.91 (dd, 1 H, J1 = 9 Hz, J2 = 2.5 Hz), 3.90 (s, 3H), 3.80 (s, 3 H). 13C NMR (150 MHz, MeOH-d4) δ 164.1, 156.1, 134.6, 129.07, 128.85, 117.9, 114.2, 109.1, 103.2, 56.1, 52.3. Elemental Analysis calculated for C11H11NO3: C, 64.38; H, 5.40; N, 6.83. Found: C, 64.55; H, 5.45; N; 6.95.
Ethyl 5-Methoxyindole-2-carboxylate (23b)
This compound was prepared in a manner similar to that for 23a except ethanolic HCl was deployed as the reactive solvent to produce a light-brown solid (1.74 g, 76%): mp 158–160 °C (lit.22 156–159 °C). TLC Rf 0.45 (25% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 8.88 (s, 1H), 7.32–7.31 (d, 1H, J = 8.9 Hz), 7.14 (m, 1H), 7.08 (d, 1H, J = 2.3 Hz), 7.01–6.99 (dd, 1 H, J1 = 8.9 Hz, J2 = 2.5 Hz), 4.42–4.39 (q, 2H, J = 7.1 Hz), 3.85 (s, 3 H), 1.42–1.40 (t, 3H, J = 7.1 Hz). 13C NMR (150 MHz, CDCl3) δ 161.9, 154.7, 132.1, 127.87, 127.84, 117.0, 112.8, 108.2, 102.5, 61.0, 55.7, 14.4. Elemental Analysis calculated for C12H13NO3: C, 65.74; H, 5.98; N, 6.39. Found: C, 66.02; H, 6.09; N; 6.45.
Propyl 5-Methoxyindole-2-carboxylate (23c)
To a suspension of 22 (670 mg, 3.50 mmol) in anhydrous n-PrOH (20 mL) was added HCl (4 N in dioxane, 2.5 mL). The reaction mixture was heated at reflux for 36 h. Upon completion, volatiles were evaporated in vacuo, and the resulting oil was purified by chromatography (0% to 20% EtOAc/hexanes) to produce a white powder (721 mg, 88%): mp 104–107 °C. TLC Rf 0.51 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 8.82 (s, 1H), 7.32–7.31 (d, 1H, J = 8.94 Hz), 7.15 (m, 1H), 7.08 (d, 1H, J = 2.4 Hz), 7.01–6.99 (dd, 1 H, J1 = 8.94 Hz, J2 = 2.46 Hz), 4.32–4.29 (t, 2H, J = 6.72 Hz), 3.85 (s, 3H), 1.84–1.78 (sex, 2H, J = 7.38 Hz), 1.05–1.03 (t, 3H, J = 7.38 Hz). 13C NMR (150 MHz, CDCl3) δ 162.0, 154.7, 132.1, 127.88, 127.85, 117.0, 112.7, 108.2, 102.5, 66.5, 55.7, 22.2, 10.5. Elemental Analysis calculated for C13H15NO3: C, 66.94; H, 6.48; N, 6.00. Found: C, 66.87; H, 6.44; N; 6.00.
Isopropyl 5-Methoxyindole-2-carboxylate (23d)
This compound was prepared in a manner similar to that for 23c except anhydrous i-PrOH was deployed as the reactive solvent, and the mixture was reacted under conditions of reflux for 4 d to produce a white powder (415 mg, 53%): mp 140–144 °C. TLC Rf 0.52 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 8.78 (s, 1H), 7.32–7.30 (d, 1H, J = 8.94 Hz), 7.13 (m, 1H), 7.07 (d, 1H, J = 2.34 Hz), 7.00–6.98 (dd, 1 H, J1= 8.94 Hz, J2= 2.4 Hz), 5.30–5.25 (sep, 1H, J = 6.3 Hz), 3.85 (s, 3 H), 1.39–1.38 (d, 6H, J = 6.3 Hz). 13C NMR (150 MHz, CDCl3) δ 161.4, 154.7, 132.0, 128.31, 127.86, 116.8, 112.7, 108.0, 102.5, 68.5, 55.7, 22.0. Elemental Analysis calculated for C13H15NO3: C, 66.94; H, 6.48; N, 6.00. Found: C, 66.80; H, 6.42; N; 6.16.
Methyl 5-Methoxyindole-3-carboxaldehyde-2-carboxylate (24a)
POCl3 (0.75 mL, 8 mmol) and DMF (2 mL) were stirred at 0 °C for 10 min. To this solution was added 23a (516 mg, 2.5 mmol) in DMF (2 mL) dropwise over 10 min. The reaction was stirred for an additional 30 min, removed from the ice bath, and warmed to rt. The reaction mixture was heated to 90 °C for 1.5 h and then poured into ice-cold 1 N NaOH (40 mL). The crude precipitate was filtered, washed with ice-cold water (25 mL), and dried at 40 °C in a vacuum desiccator for 24 h. The sample was purified by chromatography (40–70% EtOAc/hexanes) to provide a yellow solid (265 mg, 45%): mp 243–246 °C. TLC Rf 0.43 (50% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 12.82 (s, 1H), 10.58 (s, 1H), 7.69–7.68 (d, 1H, J = 2.4 Hz), 7.47–7.45 (d, 1H, J = 9 Hz), 7.05–7.03 (dd, 1H, J1 = 9.6 Hz, J2 = 2.5 Hz, 1H), 3.98 (s, 3H), 3.80 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 187.6, 160.6, 156.6, 132.1, 130.9, 125.6, 118.18, 117.28, 114.2, 102.3, 55.3, 52.7. Elemental analysis calculated for C12H11NO4: C, 61.80; H, 4.75, N, 6.01. Found: C, 61.83; H, 4.85; N, 6.18.
Ethyl 5-Methoxyindole-3-carboxaldehyde-2-carboxylate (24b)
This compound was prepared from 23b (553 mg, 2.52 mmol) in a manner similar to that for 24a except no chromatographic purification was required for the precipitate formed upon quenching with 1 N NaOH. A cream-colored solid was obtained (580 mg, 93%): mp 233–237 °C (lit.59 231 °C). TLC Rf 0.50 (50% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 12.77 (s, 1H), 10.60 (s, 1H), 7.70 (d, 1H, J = 2.46 Hz), 7.48–7.47 (d, 1H, J = 8.94 Hz), 7.05–7.04 (dd, 1H, J1 = 8.94 Hz, J2 = 2.52 Hz), 4.46–4.43 (q, 2H, J = 7.08 Hz), 3.81 (s, 3H), 1.41–1.38 (t, 3H, J = 7.08 Hz). 13C NMR (150 MHz, DMSO-d6) δ 187.4, 160.0, 156.5, 132.3, 130.7, 125.5, 118.0, 117.1, 114.1, 102.2, 61.6, 55.2, 14.0. Elemental analysis calculated for C13H13NO4: C, 63.15; H, 5.30, N, 5.67. Found: C, 62.93; H, 5.40; N, 5.76.
Propyl 5-Methoxyindole-3-carboxaldehyde-2-carboxylate (24c)
This compound was prepared from 23c (310 mg, 1.33 mmol) in a manner similar to that for 24b except after warming to rt the solution was stirred for 3 h to yield a cream-colored solid (322 mg, 92%): mp 198–201 °C. TLC Rf 0.72 (50% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 12.74 (s, 1H), 10.59 (s, 1H), 7.70 (d, 1H, J = 2.46 Hz), 7.49–7.47 (d, 1H, J = 8.94 Hz), 7.05–7.03 (dd, 1H, J1 = 8.94 Hz, J2 = 2.52 Hz), 4.37–4.35 (t, 2H, J = 6.6 Hz), 3.80 (s, 3H), 1.82–1.76 (sex, 2H, J = 7.38 Hz), 1.02–0.99 (t, 3H, J = 7.38 Hz). 13C NMR (150 MHz, DMSO-d6) δ 187.9, 160.7, 157.1, 132.80, 131.33, 126.1, 118.59, 117.72, 114.7, 102.8, 67.6, 55.7, 22.0, 10.9. Elemental analysis calculated for C14H15NO4: C, 64.36; H, 5.79, N, 5.36. Found: C, 64.24; H, 5.84; N, 5.40.
Isopropyl 5-Methoxyindole-3-carboxaldehyde-2-carboxylate (24d)
This compound was prepared from 23d (248 mg, 1.06 mmol) in a manner similar to that for 24c to yield a light yellow powder (261 mg, 93%): mp 201–203 °C. TLC Rf 0.63 (50% EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 12.70 (s, 1H), 10.59 (s, 1H), 7.69 (d, 1H, J = 2.46 Hz), 7.48–7.47 (d, 1H, J = 8.94 Hz), 7.05–7.03 (dd, 1H, J1 = 8.94 Hz, J2 = 2.52 Hz), 5.28–5.24 (sep, 1H, J = 6.3 Hz), 3.80 (s, 3H), 1.40–1.39 (d, 6H, J = 6.3 Hz). 13C NMR (150 MHz, DMSO-d6) δ 187.5, 159.7, 156.6, 132.7, 130.8, 125.6, 118.04, 117.20, 114.2, 102.3, 69.7, 55.3, 21.6. Elemental analysis calculated for C14H15NO4·0.2 H2O: C, 63.48; H, 5.86, N, 5.29. Found: C, 63.24; H, 5.73; N, 5.22.
2-Hydroxymethyl-5-methoxyindole (25).22,60
Compound 23b (2.12g, 9.67 mmol) in THF (40 mL) was stirred under Ar at 0 °C for 15 min. 2 M LAH in THF (3 eq, 15 mL) was added to the solution dropwise. The reaction mixture was stirred at rt for 1 h before the solvent was evaporated in vacuo. The residue was carefully quenched with ice-cold 0.5 N HCl (200 mL) and then extracted with EtOAc (200 mL). The organic layer was separated and dried over Na2SO4. The resulting crude residue was purified by chromatography (30% to 70% EtOAc/hexanes) to yield a yellow oil. Drying at 40 °C in a vacuum desiccator for 24 h produced a yellow solid (1.35 g, 79%): mp 85–88 °C (lit.22 80–83 °C). TLC Rf 0.63 (EtOAc). 1H NMR (600 MHz, DMSO-d6) δ 10.81 (s, 1H), 7.20–7.18 (d, 1H, J = 8.7 Hz), 6.95 (d, 1H, J = 2.4 Hz), 6.67–6.65 (dd, 1H, J1 = 8.7 Hz, J2 = 2.46 Hz), 6.18 (t, 1H, J = 1.2 Hz), 5.20–5.18 (t, 1H, J = 5.64 Hz), 4.56- 4.55 (d, 2H, J = 5.58 Hz), 3.72 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 153.0, 140.6, 131.1, 128.1, 111.5, 110.3, 101.4, 98.3, 56.8, 55.1; Elemental analysis calculated for C10H11NO2: C, 67.78; H, 6.26; N, 7.90. Found: C, 67.56; H, 6.23; N, 7.89.
2-Hydroxymethyl-5-methoxyindole-O-Acetate (26)
Compound 25 (3.05 mmol, 540 mg), triethylamine (3.36 mmol, 0.47 mL), and acetic anhydride (3.66 mmol, 0.35 mL) in CH3CN (20 mL) was stirred at rt for 3 h. The solvent was evaporated in vacuo and partitioned between EtOAc (50 mL) and saturated NaHCO3 (75 mL). The organic layer was separated, washed with brine (75 mL), and dried over Na2SO4. The residue was purified by chromatography (10–40% EtOAc/hexanes) to provide a light-yellow solid (600 mg, 90%): mp 87–88 °C. TLC Rf 0.28 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 8.47 (s, 1H), 7.24–7.23 (d, 1H, J = 8.8 Hz), 7.05–7.04 (d, 1H, J = 2.4 Hz), 6.88–6.86 (dd, 1H, J1 = 8.82 Hz, J2 = 2.5 Hz), 6.46–6.45 (d, 1H, J = 1.6 Hz), 5.20 (s, 2H), 3.84 (s, 3H), 2.10 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 172.3, 154.3, 133.6, 131.7, 127.9, 113.3, 111.9, 103.7, 102.3, 59.8, 55.8, 21.0. Elemental Analysis calculated for C12H13NO3: C, 65.74; H, 5.98; N, 6.39. Found: C, 65.91; H, 5.86; N, 6.37.
5-Methoxy-2-hydroxymethylindole-3-carboxaldehyde (27)
POCl3 (0.6 mL, 2.28 mmol) in DMF (2 mL) was stirred for 10 min at 0 °C. Compound 26 (470 mg, 2.14 mmol) in DMF (2 mL) was added dropwise over 10 min. The solution continued to stir at 0 °C for an additional 10 min and then warmed to rt, after which it was stirred for an additional 45 min. The mixture was poured into an ice-cold solution of 1 N NaOH (60 mL). The aqueous mixture was extracted with EtOAc (75 mL × 2) and the latter dried over Na2SO4 to provide a crude brown solid which was purified by chromatography (0–5% MeOH/DCM) to yield an off-white solid (348 mg, 66%): mp 195–197 °C. TLC Rf 0.57 (3% MeOH/DCM). 1H NMR (600 MHz, DMSO-d6) δ 11.98 (s, 1H), 10.07 (s, 1H), 7.57 (d, 1H, J = 2.64 Hz), 7.35–7.33 (d, 1H, J = 8.7 Hz), 6.83–6.81 (dd, 1H, J1 = 8.76 Hz, J2 = 2.52 Hz), 5.73–5.71 (t, 1H, J = 5.58 Hz), 4.96–4.95 (d, 2H, J = 5.52 Hz), 3.77 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 184.1, 155.6, 151.2, 130.2, 126.4, 112.80, 112.52, 112.34, 102.3, 55.25, 55.16. Elemental analysis calculated for C11H11NO3: C, 64.38; H, 5.40; N, 6.83. Found: C, 64.18; H, 5.25; N, 6.73.
5-Methoxyindole-2-carboxaldehyde (28)
Compound 25 (100 mg, 0.56 mmol) and MnO2 (490 mg, 5.6 mmol) in EtOAc (5 mL) were heated to reflux for 24 h. The mixture was filtered over Celite, and the filtrate was concentrated in vacuo and purified by chromatography (0% to 10% EtOAc/hexanes) to yield a pale-yellow powder (75 mg, 76%): mp 143–144 °C (lit.61 140–141 °C). TLC Rf 0.28 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 9.81 (s, 1H), 9.05 (s, 1H), 7.36–7.34 (d, 1H, J = 12 Hz), 7.20–7.19 (m, 1H), 7.12–7.11 (d, 1H, J = 6 Hz), 7.09–7.07 (dd, 1H, J1 = 9 Hz, J2 = 2.5 Hz), 3.86 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 181.8, 155.0, 136.3, 133.4, 127.7, 119.4, 114.15, 113.36, 102.8, 55.7. Elemental Analysis calculated for C10H9NO2: C, 68.56; H, 5.18; N, 8.00. Found: C, 68.42; H, 5.34; N, 7.85.
Ethyl 3-(5-Methoxyindol-2-yl)-2-propenolate (29)
To compound 28 (603 mg, 3.44 mmol) in THF (25 mL) was added (carbethoxymethylene)triphenylphosphorane (1.8 g, 5.16 mmol). The solution was stirred at rt for 20 h. The solvent was evaporated in vacuo, and the residue was dissolved in DCM (50 mL), washed with brine (50 mL) and then dried over Na2SO4. The resulting material was purified by chromatography (10% to 40% EtOAc/hexanes) to provide a pale-yellow powder (715 mg, 85%): mp 138–142 °C (lit.22 136–139 °C). TLC Rf 0.43 (30% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 8.35 (s, 1H), 7.67–7.64 (d, 1H, J = 16.02 Hz), 7.24 (s, 1H), 7.04–7.03 (d, 1H, J = 2.34 Hz), 6.94–6.92 (dd, 1H, J1 = 8.82 Hz, J2 = 2.4 Hz), 6.74 (d, 1H, J = 1.62 Hz), 6.22–6.20 (d, 1H, J = 16.02 Hz), 4.30–4.26 (q, 2H, J = 7.14 Hz), 3.84 (s, 3H), 1.36–1.33 (t, 3H, J = 7.08 Hz). 13C NMR (150 MHz, CDCl3) δ 167.3, 154.9, 134.69, 134.23, 133.33, 129.2, 115.97, 115.53, 112.3, 108.8, 102.5, 60.9, 56.0, 14.7. Elemental Analysis calculated for C14H15NO3•0.15 CH2Cl2: C, 65.87; H, 5.98; N, 5.43. Found: C, 66.07; H, 6.04; N, 5.45.
2-Hydroxypropyl-5-methoxyindole (30)
Compound 29 (631 mg, 2.57 mmol) was dissolved in THF (20 mL) and stirred at 0 °C under Argon. 2 M LAH in THF (3 eq, 15 mL) was added dropwise. The resulting mixture was warmed to rt for 1 h, and volatiles were evaporated in vacuo. The residue was carefully quenched with ice-cold 0.5 N HCl (200 mL) and then extracted with EtOAc (200 mL). The organic layer was separated and dried over Na2SO4. The resulting crude oil was purified by chromatography (30% to 70% EtOAc/hexanes) to provide a crude yellow solid, which demonstrated ∼85% purity based on 1H NMR. The impurity was determined to be a partially reduced alkene intermediate. The yellow solid was further reacted by dissolving in EtOAc (10 mL) and MeOH (10 mL) in a glass hydrogenation flask. 10% Pd/C (58 mg, 10% w/w) was added and the sample was hydrogenated for 4 h at 35 psi of H2 to ensure complete reduction. The mixture was filtered over Celite and purified by chromatography (20% to 70% EtOAc/hexanes) to yield a light-yellow solid (216 mg, 41%): mp 85–88 °C (lit.22 62–65 °C). TLC Rf 0.22 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 8.07 (s, 1H), 7.18–7.17 (d, 1H, J = 8.7 Hz), 7.01 (d, 1H, J = 2.4 Hz), 6.78–6.77 (dd, 1H, J1 = 8.7 Hz, J2 = 2.46 Hz), 6.19–6.18 (d, 1H, J = 1.98 Hz), 3.84 (s, 3H), 3.75–3.73 (t, 2H, J = 6.06 Hz), 2.87–2.85 (t, 2H, J = 7.2 Hz), 1.97–1.94 (quin, 2H, J = 6.12 Hz). 13C NMR (150 MHz, CDCl3) δ 154.2, 140.0, 131.2, 129.3, 111.16, 111.04, 102.1, 99.7, 62.3, 56.0, 31.9, 24.9. Elemental analysis for C12H15NO2: C, 70.22; H, 7.37; N, 6.82. Found: C, 70.10; H, 7.23; N, 6.74.
2-Hydroxypropyl-5-methoxyindole-O-acetate (31)
To compound 30 (300 mg, 1.46 mmol) in CH3CN (10 mL) was added TEA (0.22 mL, 1.61 mmol) and acetic anhydride (0.18 mL, 1.75 mmol). The solution was stirred at rt for 20 h. Volatiles were evaporated in vacuo, and the residue was dissolved in EtOAc (50 mL), washed with saturated NaHCO3 (75 mL) and brine (50 mL). The organic layer was separated, dried over Na2SO4, and purified by chromatography (0% to 50% EtOAc/hexanes to yield a pale-yellow solid (190 mg, 53%): TLC Rf 0.18 (17% EtOAc/hexanes). mp 86–90 °C. 1H NMR (600 MHz, CDCl3) δ 7.98 (s, 1H), 7.20–7.19 (d, 1H, J = 8.7 Hz), 7.01 (d, 1H, J = 2.46 Hz), 6.79–6.77 (dd, 1H, J1 = 8.7 Hz, J2 = 2.4 Hz), 6.19 (d, 1H, J = 2.1 Hz), 4.18–4.16 (t, 2H, J = 6.36 Hz), 3.84 (s, 3H), 2.82–2.80 (t, 2H, J = 7.38 Hz), 2.07 (s, 3H), 2.06–2.02 (quin, 2H, J = 6.36 Hz). 13C NMR (150 MHz, CDCl3) δ 171.6, 154.4, 139.4, 131.2, 129.4, 111.29, 111.25, 102.2, 100.0, 63.8, 56.1, 28.7, 24.9, 21.2. Elemental analysis calculated for C14H17NO3: C, 68.00; H, 6.93; N, 5.66. Found: C, 67.95; H, 6.91; N, 5.65.
2-Hydroxypropyl-5-methoxyindole-3-carboxaldehyde-O-acetate (32)
POCl3 (0.17 mL, 1.82 mmol) and DMF (1.5 mL) stirred at 0 °C for 10 min. Compound 31 (150 mg, 0.61 mmol) in DMF (2 mL) was added dropwise at 0 °C for an additional 10 min. The sample was removed from the ice-bath and reacted for 2 h at rt. The reaction mixture was poured into ice-cold H2O (15 mL) and 1 N NaOH was added dropwise until a pH of 10 was determined by pH paper (10 mL). The flask was refrigerated for 20 min to facilitate the formation of a precipitate. The precipitate was collected, washed with ice-cold H2O (20 mL) and dried at 40 °C in a vacuum desiccator for 24 h to provide a cream-colored solid (108 mg, 64%): mp 170–174 °C. TLC Rf 0.45 (4:1 EtOAc/hexanes). 1H NMR (600 MHz, DMSO-d6) δ 11.91 (s, 1H), 10.03 (s, 1H), 7.58 (d, J = 2.52 Hz, 1H), 7.30–7.29 (d, J = 8.76 Hz, 1H), 6.82–6.80 (dd, J1 = 8.7 Hz, J2 = 2.52 Hz, 1H), 4.04–4.02 (t, J = 6.42 Hz, 2H), 3.77 (s, 3H), 3.13–3.10 (t, J = 7.5 Hz, 2H), 2.06–2.01 (quin, J = 6.42 Hz, 2H), 1.97 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 184.0, 170.4, 155.5, 151.3, 130.2, 126.3, 113.5, 112.26, 112.15, 102.3, 63.0, 55.3, 28.5, 22.2, 20.7. Elemental Analysis calculated for C15H17NO4·0.19H2O: C, 64.64; H, 6.28; N, 5.03. Found: C, 64.25; H, 6.49; N, 5.04.
Cell Proliferation
The human U251 glioblastoma cell line was obtained from the DCT Tumor Repository (National Cancer Institute, Fredrick, MD). Cells were maintained in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum, as described previously.16 The effects of compounds on cell growth were assessed using the sulphorhodamine B (SRB) colorimetric assay,62 as described previously.17 Cells were seeded at an initial density of 2000 cells per well in 96-well plates, with four replicate wells for each drug concentration. All test compounds were dissolved in DMSO and serially diluted in DMSO so that the desired final drug concentrations could be achieved by making a 1/1000 dilution into the culture medium. One day after plating, four wells were assayed to establish a predrug (time-0) baseline. At the same time, fresh medium containing test compounds was added to the remaining wells. Control wells received DMSO alone. SRB assays were performed at a 48 h end point. To minimize the impact of potential variations in stability among the compounds, medium with test compound was replenished after the first 24 h. The concentration of each compound producing 50% growth inhibition (GI50) relative to the control without drug was calculated as described in the NCI-60 human cell line screening protocol (http://dtp.nci.nih.gov/branches/btb/ivclsp.html).
Cell Morphology
For acquisition of phase-contrast images of live cells, U251 cells were seeded on 35 mm diameter plastic culture dishes at 105 cells per dish. After 1 day, fresh medium with test compound was added. Images were obtained at the indicated time intervals after addition of drug, using an Olympus IX70 inverted microscope equipped with a DP-80 digital camera and CellSens 1.9 imaging software (Olympus America, Center Valley, PA).
Cell Cycle Analysis
Guava Cell Cycle Reagent was purchased from EMD Millipore Corporation, Hayward, CA. U251 cells were seeded at 200 000 cells per 35 mm dish 1 day prior to addition of test compounds. Cell cycle analyses were performed 24 h after addition of test compounds, using a Guava Personal Cytometer (EMD Millipore) according to the manufacturer’s protocol for the Guava Cell Cycle Reagent. CytoSoft 2.1.4 software was used to analyze the resulting DNA histograms and calculate the percentage of cells in each phase of the cell cycle, based on a total of 5000 events per sample. Statistical significance of differences between cells treated with specific compounds and control (DMSO-treated) cells was assessed using Student’s two tailed t test.
Immunofluorescence Microscopy
U251 cells were seeded on glass coverslips in 60 mm dishes at 350 000 cells per dish. One day after plating, fresh medium was added with compounds at the indicated concentration. Cells were fixed with ice-cold methanol, and α-tubulin was detected by immunofluorescence microscopy, using a primary mouse monoclonal antibody (Sigma Chemical Co., St. Louis, MO) followed by Alexa Fluor 568-labeled goat antimouse IgG (Life Technologies, Grand Island, NY). After washing away excess antibodies, nuclear DNA was stained for 5 min with 300 nM 4′,6-diamidino-2-phenyl-indole (DAPI) (Sigma Chemical Co.). Fluorescent images were obtained on an Olympus IX70 inverted microscope.
Tubulin Fractionation Assay
One day after seeding U251 cells at 1.4 × 106 cells per 10 cm dish, cells were treated for 4 h with the indicated concentration of drugs. At the end of the incubation, the medium was aspirated, and the cells were quickly scraped into 700 μL of lysis buffer consisting of 100 mM PIPES, pH 6.9, 5 mM MgCl2, 1 mM EGTA, 30% glycerol, 0.1% NP-40, 0.1% Triton X-100, 0.1% Tween-20, 0.1% 2-mercaptoethanol, 2 mM GTP, and protease inhibitor cocktail (Cystoskeleton Inc., Denver, CO). The lysis buffer was briefly prewarmed to 37 °C prior to addition to the cells. 600 μL of the lysate was transferred to a prewarmed tube and centrifuged at 100000g for 1 h at 37 °C. The resulting supernatant solution contained the nonpolymerized tubulin and the pellet contained the polymerized tubulin. 0.24% of each fraction was separated on a 10% SDS-polyacrylamide gel, and tubulin was detected by immunoblot analysis using a mouse monoclonal antibody against α-tubulin (Sigma) using established methods.14 Chemiluminescent signals from the tubulin immunoblots were quantified using an Alpha Innotech FluorChem HD2 imaging system (San Leandro, CA). Statistical significance of differences between cells treated with specific compounds and control (DMSO-treated) cells was assessed using Student’s two tailed t test.
Acknowledgments
We thank Sage George for technical assistance with some of the SRB assays and Franz Patrick Corrales for providing the supplementary data in Figure S2. We thank Ms. Zhaoqi Zhang and Dr. Jeff Sarver for HPLC analysis. We also thank Dr. Yong Wah Kim for maintenance of the UT NMR facility. The work was supported by Grant R01 CA115495 (W.A.M.) from the National Institutes of Health, and by the Helen and Harold McMaster Endowment for Biochemistry and Molecular Biology.
Glossary
Abbreviations
- CH3CN
acetonitrile
- EtOAc
ethyl acetate
- GBM
gliobastoma multiforme
- GI
growth inhibitory
- MeOH
methanol
- MOMIPP
3-(5-methoxy-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propene-1-one
- TEA
triethylamine
- SRB
sulforhodamine B
Supporting Information Available
Figure S1 (comparison of the effects of 1a on the growth of U251 GBM cells versus normal human skin fibroblasts), Figure S2 (illustration of the GI effects of changing the pyridine nitrogen from para- to meta- for compounds 2l, 2m) and 1H NMR spectra for each described compound. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ (For E.M.A.) Department of Medicinal Chemistry, University of Minnesota College of Pharmacy, 8-101 Weaver Densford Hall, 308 Harvard St. S.E., Minneapolis, MN 55455.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. E.M.A., E.J.C., and H.M.K. contributed equally as third author.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hurley L. H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200. [DOI] [PubMed] [Google Scholar]
- Andon F. T.; Fadeel B. Programmed cell death: molecular mechanisms and implications for safety assessment of nanomaterials. Acc. Chem. Res. 2013, 46, 733–742. [DOI] [PubMed] [Google Scholar]
- Delbridge A. R.; Valente L. J.; Strasser A. The role of the apoptotic machinery in tumor suppression. Cold Spring Harbor Perspect. Biol. 2012, 4, a008789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10, 194–204. [DOI] [PubMed] [Google Scholar]
- Holohan C.; Van Schaeybroeck S.; Longley D. B.; Johnston P. G. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [DOI] [PubMed] [Google Scholar]
- Gatti L.; Zunino F. Overview of tumor cell chemoresistance mechanisms. Methods Mol. Med. 2005, 111, 127–148. [DOI] [PubMed] [Google Scholar]
- Stupp R.; Mason W. P.; van den Bent M. J.; Weller M.; Fisher B.; Taphoorn M. J.; Belanger K.; Brandes A. A.; Marosi C.; Bogdahn U.; Curschmann J.; Janzer R. C.; Ludwin S. K.; Gorlia T.; Allgeier A.; Lacombe D.; Cairncross J. G.; Eisenhauer E.; Mirimanoff R. O. Radiotherapy plus concomitant and adjuvant Temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [DOI] [PubMed] [Google Scholar]
- Grossman S. A.; Ye X.; Piantadosi S.; Desideri S.; Nabors L. B.; Rosenfeld M.; Fisher J.; Consortium N. C. Survival of patients with newly diagnosed glioblastoma treated with radiation and Temozolomide in research studies in the United States. Clin. Cancer Res. 2010, 16, 2443–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L.; Vitale I.; Abrams J. M.; Alnemri E. S.; Baehrecke E. H.; Blagosklonny M. V.; Dawson T. M.; Dawson V. L.; El-Deiry W. S.; Fulda S.; Gottlieb E.; Green D. R.; Hengartner M. O.; Kepp O.; Knight R. A.; Kumar S.; Lipton S. A.; Lu X.; Madeo F.; Malorni W.; Mehlen P.; Nunez G.; Peter M. E.; Piacentini M.; Rubinsztein D. C.; Shi Y.; Simon H. U.; Vandenabeele P.; White E.; Yuan J.; Zhivotovsky B.; Melino G.; Kroemer G. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzaler P.; Watson C. J. Killing a cancer: what are the alternatives?. Nat. Rev. Cancer 2012, 12, 411–424. [DOI] [PubMed] [Google Scholar]
- Kornienko A.; Mathieu V.; Rastogi S. K.; Lefranc F.; Kiss R. Therapeutic agents triggering nonapoptotic cancer cell death. J. Med. Chem. 2013, 56, 4823–4839. [DOI] [PubMed] [Google Scholar]
- Maltese W. A.; Overmeyer J. H. Methuosis: nonapoptotic cell death associated with vacuolization of macropinosome and endosome compartments. Am. J. Pathol. 2014, 184, 1630–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overmeyer J. H.; Kaul A.; Johnson E. E.; Maltese W. A. Active ras triggers death in glioblastoma cells through hyperstimulation of macropinocytosis. Mol. Cancer Res. 2008, 6, 965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhanot H.; Young A. M.; Overmeyer J. H.; Maltese W. A. Induction of nonapoptotic cell death by activated Ras requires inverse regulation of Rac1 and Arf6. Mol. Cancer Res. 2010, 8, 1358–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overmeyer J. H.; Young A. M.; Bhanot H.; Maltese W. A. A chalcone-related small molecule that induces methuosis, a novel form of non-apoptotic cell death, in glioblastoma cells. Mol. Cancer 2011, 10, 69–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. W.; Overmeyer J. H.; Young A. M.; Erhardt P. W.; Maltese W. A. Synthesis and evaluation of indole-based chalcones as inducers of methuosis, a novel type of nonapoptotic cell death. J. Med. Chem. 2012, 55, 1940–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabbic C. J.; Dietsch H. M.; Alexander E. M.; Nagy P. I.; Robinson M. W.; Overmeyer J. H.; Maltese W. A.; Erhardt P. W. Differential induction of cytoplasmic vacuolization and methuosis by novel 2-indolyl-substituted pyridinylpropenones. ACS Med. Chem. Lett. 2014, 5, 73–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahm S.; Weinreb S. M. N-Methoxy-N-methylamides as effective acylating agents. Tetrahedron Lett. 1981, 22, 3815–3818. [Google Scholar]
- Clark R. D.; Muchowski J. M.; Fisher L. E.; Flippin L. A.; Repke D. B.; Souchet M. Preparation of indoles and oxindoles from N-(tert-butoxycarbonyl)-2-alkylanilines. Synthesis 1991, 10, 871–878. [Google Scholar]
- Ayyangar N. R.; Srinivasan K. V. Effect of substituents in the formation of diacetanilides. Can. J. Chem. 1984, 62, 1292–1296. [Google Scholar]
- Graham S. L.; Scholz T. H. Preparation and utility of dianions from N-tert-butylthiophene-2-sulfonamide. J. Org. Chem. 1991, 56, 4260–4263. [Google Scholar]
- Tsotinis A.; Afroudakis P. A.; Davidson K.; Prashar A.; Sugden D. Design, synthesis, and melatoninergic activity of new azido- and isothiocyanato-substituted indoles. J. Med. Chem. 2007, 50, 6436–6440. [DOI] [PubMed] [Google Scholar]
- Go M. L.; Wu X.; Liu X. L. Chalcones: an update on cytotoxic and chemoprotective properties. Curr. Med. Chem. 2005, 12, 481–499. [DOI] [PubMed] [Google Scholar]
- Orlikova B.; Tasdemir D.; Golais F.; Dicato M.; Diederich M. Dietary chalcones with chemopreventive and chemotherapeutic potential. Genes Nutr. 2011, 6, 125–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jandial D. D.; Blair C. A.; Zhang S.; Krill L. S.; Yan-Bing Z.; Zi X. Molecular targeted approaches to cancer therapy and prevention using chalcones. Curr. Cancer Drug Targets 2014, 14, 181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitambi S. S.; Toledo E. M.; Usoskin D.; Wee S.; Harisankar A.; Svensson R.; Sigmundsson K.; Kalderen C.; Niklasson M.; Kundu S.; Aranda S.; Westermark B.; Uhrbom L.; Andang M.; Damberg P.; Nelander S.; Arenas E.; Artursson P.; Walfridsson J.; Forsberg Nilsson K.; Hammarstrom L. G.; Ernfors P. Vulnerability of glioblastoma cells to catastrophic vacuolization and death induced by a small molecule. Cell 2014, 157, 313–328. [DOI] [PubMed] [Google Scholar]
- Erhardt P. W.Case Study: “Esmolol Stat”. In Analogue-based Drug Discovery; Fisher J., Ganellin C. R., Eds.; Wiley-VCH: Weinheim, 2006; pp 233–246. [Google Scholar]
- Erhardt P. W.Esmolol. In Chronicles of Drug Discovery, Lednicer D., Eds.; American Chemical Society: Washington D.C., 1993; Vol. 3, pp 191–206. [Google Scholar]
- Erhardt P. W.; Woo C. M.; Anderson W. G.; Gorczynski R. J. Ultra-short-acting beta-adrenergic receptor blocking agents. 2. (Aryloxy)propanolamines containing esters on the aryl function. J. Med. Chem. 1982, 25, 1408–1412. [DOI] [PubMed] [Google Scholar]
- Ginos J. Z.; Cotzias G. C.; Tolosa E.; Tang L. C.; LoMonte A. Cholinergic effects of molecular segments of apomorphine and dopaminergic effects of N,N-dialkylated dopamines. J. Med. Chem. 1975, 18, 1194–1200. [DOI] [PubMed] [Google Scholar]
- Kohli J. D.; Weder A. B.; Goldberg L. I.; Ginos J. Z. Structure activity relationships of N-substituted dopamine derivatives as agonists of the dopamine vascular and other cardiovascular receptors. J. Pharmacol. Exp. Ther. 1980, 213, 370–374. [PubMed] [Google Scholar]
- Kaiser C.Structure-Activity Relationships of Dopamine Receptor Agonists. In Dopamine Receptor Agonists, Poste G., Crooke S. T., Eds.; Plenum Press: New York, 1984; pp 87–137. [Google Scholar]
- Edwards M. L.; Stemerick D. M.; Sunkara P. S. Chalcones: a new class of antimitotic agents. J. Med. Chem. 1990, 33, 1948–1954. [DOI] [PubMed] [Google Scholar]
- Ducki S. Antimitotic chalcones and related compounds as inhibitors of tubulin assembly. Anti-Cancer Agents Med. Chem. 2009, 9, 336–347. [DOI] [PubMed] [Google Scholar]
- Boumendjel A.; Boccard J.; Carrupt P. A.; Nicolle E.; Blanc M.; Geze A.; Choisnard L.; Wouessidjewe D.; Matera E. L.; Dumontet C. Antimitotic and antiproliferative activities of chalcones: forward structure-activity relationship. J. Med. Chem. 2008, 51, 2307–2310. [DOI] [PubMed] [Google Scholar]
- Dyrager C.; Wickstrom M.; Friden-Saxin M.; Friberg A.; Dahlen K.; Wallen E. A.; Gullbo J.; Grotli M.; Luthman K. Inhibitors and promoters of tubulin polymerization: synthesis and biological evaluation of chalcones and related dienones as potential anticancer agents. Bioorg. Med. Chem. 2011, 19, 2659–2665. [DOI] [PubMed] [Google Scholar]
- Kim do Y.; Kim K. H.; Kim N. D.; Lee K. Y.; Han C. K.; Yoon J. H.; Moon S. K.; Lee S. S.; Seong B. L. Design and biological evaluation of novel tubulin inhibitors as antimitotic agents using a pharmacophore binding model with tubulin. J. Med. Chem. 2006, 49, 5664–5670. [DOI] [PubMed] [Google Scholar]
- Zhu C.; Zuo Y.; Wang R.; Liang B.; Yue X.; Wen G.; Shang N.; Huang L.; Chen Y.; Du J.; Bu X. Discovery of potent cytotoxic ortho-aryl chalcones as new scaffold targeting tubulin and mitosis with affinity-based fluorescence. J. Med. Chem. 2014, 57, 6364–6382. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Kumar N. M.; Akamatsu K.; Kusaka E.; Harada H.; Ito T. Synthesis and biological evaluation of indolyl chalcones as antitumor agents. Bioorg. Med. Chem. Lett. 2010, 20, 3916–3919. [DOI] [PubMed] [Google Scholar]
- Champelovier P.; Chauchet X.; Hazane-Puch F.; Vergnaud S.; Garrel C.; Laporte F.; Boutonnat J.; Boumendjel A. Cellular and molecular mechanisms activating the cell death processes by chalcones: Critical structural effects. Toxicol. In Vitro 2013, 27, 2305–2315. [DOI] [PubMed] [Google Scholar]
- Martel-Frachet V.; Kadri M.; Boumendjel A.; Ronot X. Structural requirement of arylindolylpropenones as anti-bladder carcinoma cells agents. Bioorg. Med. Chem. 2011, 19, 6143–6148. [DOI] [PubMed] [Google Scholar]
- Champelovier P.; Mininno M.; Duchamp E.; Nicolle E.; Curri V.; Boumendjel A.; Boutonnat J. Cytotoxicity of chalcone derivatives towards glioblastoma. Anticancer Res. 2011, 31, 3213–3218. [PubMed] [Google Scholar]
- Boumendjel A.; McLeer-Florin A.; Champelovier P.; Allegro D.; Muhammad D.; Souard F.; Derouazi M.; Peyrot V.; Toussaint B.; Boutonnat J. A novel chalcone derivative which acts as a microtubule depolymerising agent and an inhibitor of P-gp and BCRP in in-vitro and in-vivo glioblastoma models. BMC Cancer 2009, 9, 242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmann K. H. The cytoskeleton and its interactions with small molecules. Bioorg. Med. Chem. 2014, 22, 5038–5039. [DOI] [PubMed] [Google Scholar]
- Jordan M. A.; Wilson L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [DOI] [PubMed] [Google Scholar]
- Mukhtar E.; Adhami V. M.; Mukhtar H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavallaris M.; Tait A. S.; Walsh B. J.; He L.; Horwitz S. B.; Norris M. D.; Haber M. Multiple microtubule alterations are associated with Vinca alkaloid resistance in human leukemia cells. Cancer Res. 2001, 61, 5803–5809. [PubMed] [Google Scholar]
- Dumontet C.; Sikic B. I. Mechanisms of action of and resistance to antitubulin agents: microtubule dynamics, drug transport, and cell death. J. Clin. Oncol. 1999, 17, 1061–1070. [DOI] [PubMed] [Google Scholar]
- Boyle F. M.; Eller S. L.; Grossman S. A. Penetration of intra-arterially administered vincristine in experimental brain tumor. Neuro-oncol. 2004, 6, 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellner S.; Bauer B.; Miller D. S.; Schaffrik M.; Fankhanel M.; Spruss T.; Bernhardt G.; Graeff C.; Farber L.; Gschaidmeier H.; Buschauer A.; Fricker G. Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo. J. Clin. Invest. 2002, 110, 1309–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. L.; Tee H. W.; Go M. L. Functionalized chalcones as selective inhibitors of P-glycoprotein and breast cancer resistance protein. Bioorg. Med. Chem. 2008, 16, 171–180. [DOI] [PubMed] [Google Scholar]
- Valdameri G.; Gauthier C.; Terreux R.; Kachadourian R.; Day B. J.; Winnischofer S. M.; Rocha M. E.; Frachet V.; Ronot X.; Di Pietro A.; Boumendjel A. Investigation of chalcones as selective inhibitors of the breast cancer resistance protein: critical role of methoxylation in both inhibition potency and cytotoxicity. J. Med. Chem. 2012, 55, 3193–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakifahmetoglu H.; Olsson M.; Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ. 2008, 15, 1153–1162. [DOI] [PubMed] [Google Scholar]
- Galan-Malo P.; Vela L.; Gonzalo O.; Calvo-Sanjuan R.; Gracia-Fleta L.; Naval J.; Marzo I. Cell fate after mitotic arrest in different tumor cells is determined by the balance between slippage and apoptotic threshold. Toxicol. Appl. Pharmacol. 2012, 258, 384–393. [DOI] [PubMed] [Google Scholar]
- Still W. C.; Kahn M.; Mitra A. Rapid chromatographic technique for preparative separations with moderate resolution. J. Org. Chem. 1978, 43, 2923–2925. [Google Scholar]
- Peters R.; Waldmeier P.; Joncour A. S. Efficient synthesis of a 5-HT2C receptor agonist precursor. Org. Process Res. Dev. 2005, 9, 508–512. [Google Scholar]
- Molander G. A.; Shin I. Synthesis and Suzuki-Miyaura cross-coupling reactions of potassium boc-protected aminomethyltrifluoroborate with aryl and hetaryl halides. Org. Lett. 2011, 13, 3956–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coowar D.; Bouissac J.; Hanbali M.; Paschaki M.; Mohier E.; Luu B. Effects of indole fatty alcohols on the differentiation of neural stem cell derived neurospheres. J. Med. Chem. 2004, 47, 6270–6282. [DOI] [PubMed] [Google Scholar]
- Skibo E. B.; Xing C.; Dorr R. T. Aziridinyl quinone antitumor agents based on indoles and cyclopent[b]indoles: structure-activity relationships for cytotoxicity and antitumor activity. J. Med. Chem. 2001, 44, 3545–3562. [DOI] [PubMed] [Google Scholar]
- Spadoni G.; Balsamini C.; Bedini A.; Diamantini G.; Di Giacomo B.; Tontini A.; Tarzia G.; Mor M.; Plazzi P. V.; Rivara S.; Nonno R.; Pannacci M.; Lucini V.; Fraschini F.; Stankov B. M. 2-[N-Acylamino(C1-C3)alkyl]indoles as MT1 melatonin receptor partial agonists, antagonists, and putative inverse agonists. J. Med. Chem. 1998, 41, 3624–3634. [DOI] [PubMed] [Google Scholar]
- Bhat G. A.; Siddappa S. Synthesis of indole-2-carbaldehydes, 2-(2-aminoethyl)-indoles and 2-(2-amino-propyl)-indoles. J. Chem. Soc. C 1971, 1, 178–181. [Google Scholar]
- Skehan P.; Storeng R.; Scudiero D.; Monks A.; McMahon J.; Vistica D.; Warren J. T.; Bokesch H.; Kenney S.; Boyd M. R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.