

Abstract
The International Diabetes Federation estimates that 316 million people are currently affected by impaired glucose tolerance (IGT). Most importantly, recent forecasts anticipate a dramatic IGT increase with more that 470 million people affected by the year 2035. Impaired insulin sensitivity is major feature of obesity and diabetes and is strongly linked with adverse cardiometabolic phenotypes. However, the etiologic pathway linking impaired glucose tolerance and cardiovascular disease remains to be deciphered. Although insulin resistance has been attributed to inflammatory programs starting in adipose tissue, emerging evidence indicates that endothelial dysfunction may represent the upstream event preceding peripheral impairment of insulin sensitivity. Indeed, suppression of reactive oxygen species-dependent pathways in the endothelium has shown to restore insulin delivery to peripheral organs by preserving nitric oxide (NO) availability. Here we describe emerging theories concerning endothelial insulin resistance, with particular emphasis on the role oxidative stress. Complex molecular circuits including endothelial nitric oxide synthase, prostacyclin synthase, mitochondrial adaptor p66Shc, nicotinamide adenine dinucleotide phosphate-oxidase oxidase and nuclear factor kappa-B are discussed. Moreover, the review provides insights on the effectiveness of available compounds (i.e., ruboxistaurin, sildenafil, endothelin receptor antagonists, NO donors) in restoring endothelial insulin signalling. Taken together, these aspects may significantly contribute to design novel therapeutic approaches to restore glucose homeostasis in patients with obesity and diabetes.
Keywords: Endothelium, Insulin resistance, Oxidative stress, Obesity, Cardiometabolic risk, Vascular disease
Core tip: We present here the most recent advances in the understanding of endothelial insulin resistance, with a particular focus on the role of oxidative stress. The molecular pathways described may be instrumental for the development of mechanism-based therapeutic strategies to prevent maladaptive endothelial insulin signalling in patients with cardiometabolic disturbances.
PREVALENCE OF IMPAIRED GLUCOSE TOLERANCE
The most recent update of the International Diabetes Federation shows that 6.9% of the global population (316 million people) is currently affected by impaired glucose tolerance (IGT) and, most importantly, forecasts anticipate a dramatic IGT increase with more that 470 million people affected by the year 2035[1]. Such pandemic of metabolic syndromes and obesity-related disorders hints a proportional increase in the prevalence of type 2 diabetes (T2D), a major driver of morbidity and mortality worldwide[2]. Currently, 382 million people are affected by T2D, with a global age-adjusted prevalence of 10%. If these trends continue, 592 million people, or one adult in 10, will have diabetes by 2035[1]. The link between environmental factors (pollution, caloric intake, sedentary lifestyles), obesity and subsequent dysglycemia indicates that the progression to diabetes is not linear and involves different cellular mechanisms including alterations of insulin signalling, changes in glucose metabolism, free fatty acids oxidation as well as dysregulation of genes relevant to endothelial integrity[3,4]. The progression from obesity to T2D may take many years to occur, leading to different intermediate phenotypes with progressive changes in glucose parameters and shifts in glucose tolerance category. Yet, the etiologic pathway linking increased body weight, altered insulin signaling and subsequent hyperglycemia remained to be understood. Novel insights in this area may be instrumental to identify novel mechanism-based therapeutic strategies for the preservation of insulin signaling and, hence, diabetes development.
IMPACT OF INSULIN RESISTANCE ON CARDIOVASCULAR OUTCOME
Mortality from cardiovascular disease (CVD) is significantly higher in subjects with T2D than in those without[5]. Notably, the risk of macrovascular complications seems to be proportional to the impairment of glucose homeostasis[6]. Among different diabetes-related conditions, insulin resistance (IR) and hyperglycemia are major precursors of atherothrombotic events and poor CV outcome[7]. Waist circumference, an hallmark of IR, is a strong independent predictor of CVD[8]. Dagenais et al[9] showed that subjects in the upper tertile of waist circumference had an increased adjusted relative risk of 29% for CV death, 27% for myocardial infarction, and 35% for total mortality, suggesting a strong association between abdominal obesity and CV events (Figure 1). Along this line, elevated insulin and glucose concentrations are associated with increased CVD risk, regardless of diabetes[10,11]. Impaired insulin signalling is a key feature of the metabolic syndrome (MetS), defined by the presence of hyperglycemia, central obesity, low high density lipoprotein cholesterol level, high triglyceride level and elevated blood pressure or antihypertensive drugs use. MetS is highly represented in patients with type 1 diabetes (T1DM) (38% in men and 40% in women) and is an important predictor of CV events[12]. Indeed, MetS was associated with a 2.1-fold increased risk of CV events and a 2.5-fold increased risk of CV-related mortality after 5.5 years follow-up in 3783 patients with T1DM[13]. The main issue when it comes to insulin resistance is how to measure it in clinical practice. The Homeostasis Model Assessment IR (HOMA-IR) is emerging as well-established marker of IR with a high predictive value for incident coronary events and stroke[14-16]. This is likely due to the fact that HOMA-IR includes in its formula both fasting glucose and insulin levels thus showing a stronger association with cardiovascular disease than glucose or insulin alone[14]. Despite many investigations confirmed a potential predictive value of IR, the mechanisms underlying this phenomenon still remain poorly understood. First, it is not clear whether IR is an active process or rather the consequence of the inflammatory milieu observed in obese and diabetic patients. Second, it remains unknown if the impairment of insulin signaling occurs simultaneously in all insulin-sensitive organs or whether tissue-specific IR has a primary role in triggering maladaptive insulin responses in other tissues. In order to answer these complex questions, many researchers are now exploring the pathophysiology of IR in different organs as well as its impact on metabolic features and longevity.
Figure 1.
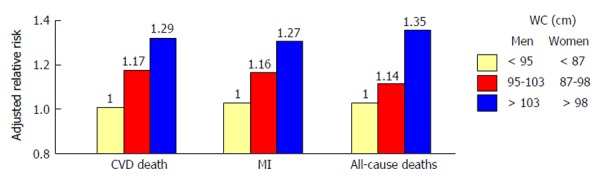
Association between abdominal obesity and cardiovascular disease. Subjects in the upper tertile of waist circumference had an increased adjusted relative risk of 29% for cardiovascular (CV) death, 27% for myocardial infarction (MI), and 35% for total mortality, suggesting a strong association between abdominal obesity and CV events. Adapted from Dagenais et al[9]. WC: Waist circumference; CVD: Cardiovascular disease death.
ENDOTHELIAL INSULIN RESISTANCE: AN EMERGING CONCEPT
Evidence accumulated over the last decade has shown that loss of insulin signaling in the endothelium accelerates atherosclerotic lesions and vascular dysfunction in mice[17-21]. Noteworthy, these effects occur regardless of concomitant CV risk factors, suggesting a central role of endothelial IR[4]. Although IR has been attributed to inflammation in adipocytes, recent work has overturned such “adipocentric paradigm”[22]. The novel concept that IR may primarily starts in the endothelium squares with the notion that endothelial cells are highly represented within the entire vascular system and, hence, within different organs[18,20,23]. Recent experimental work has demonstrated that the transcription factor nuclear factor kappa-B (NF-κB) is a key determinant of endothelial insulin resistance in mice[17]. NF-κB is a well known molecular complex involved in inflammatory programs enabling transcription of cytokines, activation of stress kinases and dysregulation of insulin-related pathways[24,25]. Of note, genetic disruption of IκB prevents inflammation and insulin resistance in obesity and T2D[26]. Hasegawa et al[17] have shown that mice with endothelium-specific suppression of NF-κB signaling (E-DNIκB) were protected against IR in adipose tissue and skeletal muscle. These mice displayed reduced oxidative stress markers, decreased macrophage infiltration of adipose tissue as well as increased blood flow and muscle mitochondrial content. Of note, capillary recruitment and subsequent insulin delivery were explained by restoration of nitric oxide (NO) levels in E-DNIκB animals[17]. This latter observation is important since endothelial nitric oxide synthase (eNOS) dysfunction may lead to a reduction in microcirculatory blood flow and, hence, reduced delivery of insulin within hormone-sensitive organs. Indeed, insulin-mediated glucose uptake is reduced in eNOS-/- as compared with WT mice[27]. In other words, microvascular dysfunction occurring in liver, adipose tissue and skeletal muscle explains the progressive decline of peripheral insulin distribution[28,29]. Of note, restoration of eNOS functionality due to suppression of endothelial NF-κB signaling was capable to rescue aging-associated insulin resistance and, most importantly, to prolong lifespan in mice[17]. In line with these experimental data, studies in humans demonstrated that insulin-dependent vasodilation may represent a significant contributor to insulin-stimulated glucose uptake[30-32]. Muris et al[33] proposed that approximately 40% of insulin-mediated glucose uptake by skeletal muscle can be attributed to capillary recruitment; according to this hypothesis, microvascular dysfunction not only precedes and predicts the development of T2D but also constitutes one of the links between IR and hypertension in MetS. Consistently, improvement of insulin sensitivity in patients with cardiometabolic disturbances is associated with restoration of flow-mediated vasodilation[34,35]. Another study showed that disruption of endothelial insulin signaling by genetic deletion of insulin receptor substrate-2 (IRS-2) alters insulin delivery in muscle thus affecting glucose tolerance in mice[18]. Of interest, endothelial IRS-2 and ApoE knockout mice showed a more severe atherosclerotic disease progression as compared to controls[18]. Further work demonstrated that knockout of three major FoxO isoforms in endothelial cells attenuates endothelial IR and atherosclerosis in low density lipoprotein receptor knockout mice, suggesting that FoxO inhibition may represent a potential therapeutic approach to prevent CVD and IR in patients with diabetes[36]. Accordingly, activity of protein kinase C (PKC)β2 and NF-κB in endothelial cells isolated from insulin resistant subjects was markedly enhanced and this finding was associated with blunted eNOS phosphorylation, reduced nitric oxide availability and impaired endothelial function[23].
Research discussed so far implies that reprogramming detrimental pathways in the vascular endothelium may be considered a novel approach to prevent metabolic disease.
ROLE OF OXIDATIVE STRESS IN MALADAPTIVE INSULIN SIGNALING
In obese subjects, exposure to environmental cues triggers many pathological processes including reprogramming of oxidant genes and subsequent redox changes in different tissues[37,38]. The importance of reactive oxygen species (ROS) has been claimed over the last 50 years since these mediators are likely the most pervasive precursors of maladaptive intracellular signalling[39]. Previous seminal work carried out in conditions of hyperglycemia has elegantly demonstrated how ROS accumulation can easily boost activation of detrimental downstream pathways such as advanced glycation end products (AGEs), polyol and hexosamine pathways as well as proinflammatory transcriptional programs initiated by NF-κB[40]. These ROS-sensitive molecular events are being translated to endothelial dysfunction and, hence, micro and macrovascular complications[7]. While the relation between hyperglycemia and oxidative stress has been clearly delineated, the etiologic path linking insulin resistance to ROS generation remains to be deciphered. Recent evidence suggests that oxidative stress may contribute to alter insulin sensitivity in the vascular endothelium. Du et al[19] have shown that enhanced oxidation of free fatty acids (FFAs) in aortic endothelial cells increases the production of superoxide by the mitochondrial electron transport chain thus triggering molecular pathways of maladaptive insulin signalling. Indeed, FFAs-induced overproduction of superoxide was able to activate an array of proinflammatory signals while hampering the activity of key anti-atherogenic enzymes such as prostacyclin synthase (PGIS) and eNOS. The importance of ROS in this setting was outlined by experiments in obese mice showing that inactivation of PGIS and eNOS was prevented by inhibition of FFAs release from the adipose tissue[19]. For the first time, this study demonstrated that ROS may actively participate to impaired endothelial signalling thus favouring a pro-atherosclerotic phenotype in subjects with IR. Consistently, in ApoE-/- mice with endothelium-specific IR, generation of superoxide was strongly linked to hampered insulin sensitivity, vasorelaxation and atherosclerotic lesions[41]. A further study recently showed that nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase 2 (Nox2) may also be implicated in maladaptive insulin response by inducing a detrimental rearrangement of insulin receptors with subsequent deregulation of downstream kinase effectors, and eNOS dysfunction. Interestingly, obese mice with genetic disruption of Nox2 were protected against ROS accumulation and endothelial IR, suggesting that targeting Nox2 could represent a valuable therapeutic strategy in the context of prediabetes[42] (Figure 2). On such a background, we have recently explored the possibility that the mitochondrial adaptor p66Shc might participate to ROS-driven IR in the endothelium. The adaptor p66Shc is a pivotal modulator of mitochondrial ROS through oxidation of cytochrome c[43,44]. We have previously reported that genetic deletion of p66Shc protects against vascular dysfunction and oxidative stress in diabetic mice[45]. Moreover, p66Shc expression is increased in patients with T2D and correlates with plasma isoprostane levels, a reliable in vivo marker of oxidative stress[46]. We have recently found that in vivo gene silencing of p66Shc restored endothelial insulin response by affecting the IRS-1/Akt/eNOS pathway[47]. Furthermore, p66Shc knockdown in endothelial cells isolated from obese mice attenuated ROS production, FFAs oxidation and prevented dysregulation of redox-sensitive pathways such as NF-κB, AGE precursor methylglyoxal and PGI2 synthase. Collectively, our results show that p66Shc may contribute to the pathogenesis of IR and increased vascular risk in the context of obesity and T2D. Selective targeting of p66Shc may restore endothelial insulin sensitivity thus preventing adverse cardiometabolic phenotypes. In line with our findings, a recent work has shown that endothelium-specific overexpression of PKCβ2, a key molecular event eliciting ROS production, suppressed insulin-dependent pathways in APOE-/- mice[21]. Interestingly, expression of the potent vasoconstrictor endothelin-1 was highly increased in vessels isolated from APOE-/- animals with PKCβ2 overexpression (Figure 2). Taken together, these results indicate that p66Shc stands along a detrimental signalling cascade involved in ROS generation, microvascular dysfunction and, hence, peripheral insulin resistance. The clinical relevance of these experimental findings is supported by the notion that oxidative stress is significantly increased in cardiometabolic disorders. A cross-sectional study from the LIPGENE cohort revealed that levels of total nitrite, lipid peroxidation products, hydrogen peroxide (H2O2), superoxide dismutase and glutathione peroxidase activities were all strongly associated with metabolic syndrome traits[48]. Despite such evidence brings enthusiasms toward the possibility of targeting oxidative stress in humans, we are still far from having achieved satisfactory results in term of intermediate endpoints such as endothelial function and atherosclerotic lesions. Indeed, available antioxidants may not fully scavenge cellular ROS since they are unable to target intracellular enzymes involved in redox signalling. This notion is confirmed by the negative results of major trials with oral supplementation of high-dose vitamins[49].
Figure 2.
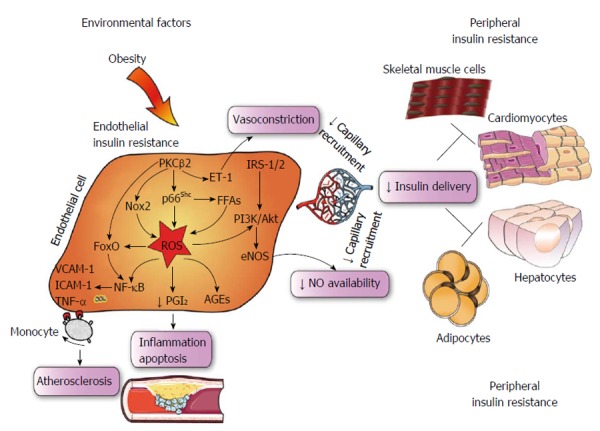
Central role of endothelial insulin resistance. Schematic representing intricate inflammatory and ROS-sensitive pathways responsible for maladaptive insulin signalling in the vascular endothelium. In obese subjects, environmental stimuli favour progressive impairment of endothelial cell function due to ROS accumulation and reduced NO bioavailability, leading to defective capillary recruitment and hampered insulin delivery to hormone sensitive organs. PKC: Protein kinase C; NF-κB: Nuclear factor kappa-B; IRS; Insulin receptor substrate; ROS; Reactive oxygen species; AGEs; Advanced glycation end products; PGI2; Prostacyclin; VCAM-1; Vascular cell adhesion molecule-1; ICAM-1; Intercellular cell adhesion molecule-1; NO; Nitric oxide; TNF-α: Tumor necrosis factor α; FFA: Free fatty acid; ET-1: Endothelin-1; Nox2: NADPH oxidase 2; FoxO: Forkhead box O; eNOS: Endothelial nitric oxide synthase. NADPH: Nicotinamide adenine dinucleotide phosphate.
FUTURE PERSPECTIVES
The possibility to target specific machineries in the vascular endothelium may represent an attractive challenge to prevent or delay systemic features of IR favouring adiposity and related comorbidities. There are several examples suggesting that mechanism-based therapeutic approaches might be tested over the next decades. High doses of salicylates have been shown to ameliorate IR and improve glucose tolerance by suppressing NF-κB activity in patients with T2D[50]. Moreover, selective pharmacological inhibition of PKCβ with LY379196 in freshly isolated endothelial cells from T2D patients reduced basal eNOS phosphorylation and improved insulin-mediated eNOS activation[23]. Consistently, the Food and drug administration-approved PKC inhibitor ruboxistaurin ameliorates functional endothelial IR and smooth muscle cell hypersensitivity to insulin in experimental obesity and diabetes[51]. In conditions of IR also the phosphodiesterase 5 inhibitor sildenafil has shown to improve NOS activity in human endothelial cells, thus suggesting the potential therapeutic use of this compound to warrant glucose homeostasis[52]. Furthermore, preclinical work demonstrated that dual ET(A)/ET(B) receptor blockade enhanced endothelium-dependent vasodilatation in individuals with IR, thus restoring vascular recruitment and insulin delivery to peripheral organs[53]. Yet, strategies to drive compounds specifically to the vascular endothelium are still far to be applied in humans. The main problem when it comes to tissue-specific treatment is represented by drug delivery. It is clear that selective rearrangement of maladaptive pathways in the endothelium would provide invaluable to restore microvascular dysfunction and insulin distribution to the liver, adipose tissue and skeletal muscle. An alternative option may be represented by NO donors or administration of eNOS cofactors in order to improve tissue capillary recruitment. Unfortunately this approach has failed many times due to the high oxidative burden in patients with metabolic disease which rapidly inactivates NO, thus favouring accumulation of peroxinitrite (ONOO-), protein nitrosylation and cellular dysfunction. In this respect, an example is provided by a recent clinical trial where oral treatment with eNOS cofactor tetrahydrobiopterin (BH4) has shown limited effectiveness on endothelial function due to systemic oxidation and poor uptake into the vascular wall[54]. These latter results highlight the need for more mechanistic understanding and alternative strategies to counteract pathways triggering eNOS dysfunction in patients with IR. We have recently showed that in vivo RNA interference may represent a valid approach to target specific ROS-generating enzymes in the endothelium[55]. Distribution studies showed that in vivo delivery of small interfering RNA together with a cationic transfection reagent is able to target the vascular endothelium while sparing surrounding tissues. Indeed, we demonstrated that in vivo gene silencing of the adaptor p66Shc restores insulin-dependent vasorelaxation in obese mice, suggesting that blunting endothelial oxidant pathways may be efficient for the maintenance of glucose homeostasis[47]. This work will be instrumental to understand the efficacy and safety of such technology in humans, and whether other candidates may be considered for gene therapy in the setting of endothelial IR.
Footnotes
P- Reviewer: Armstrong EJ, Matei D S- Editor: Ji FF L- Editor: A E- Editor: Liu SQ
Conflict-of-interest: The authors do not have any conflict of interest with regard to the present article.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: September 23, 2014
First decision: November 3, 2014
Article in press: December 31, 2014
References
- 1.International Diabetes Federation. IDF Diabetes Atlas, 6th ed. Brussels, Belgium: International Diabetes Federation; 2013. Available from: http: //www.idf.org/diabetesatlas. [Google Scholar]
- 2.Hossain P, Kawar B, El Nahas M. Obesity and diabetes in the developing world--a growing challenge. N Engl J Med. 2007;356:213–215. doi: 10.1056/NEJMp068177. [DOI] [PubMed] [Google Scholar]
- 3.Eckel RH, Kahn SE, Ferrannini E, Goldfine AB, Nathan DM, Schwartz MW, Smith RJ, Smith SR. Obesity and type 2 diabetes: what can be unified and what needs to be individualized? J Clin Endocrinol Metab. 2011;96:1654–1663. doi: 10.1210/jc.2011-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paneni F, Costantino S, Cosentino F. Insulin resistance, diabetes, and cardiovascular risk. Curr Atheroscler Rep. 2014;16:419. doi: 10.1007/s11883-014-0419-z. [DOI] [PubMed] [Google Scholar]
- 5.Haffner SM, Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–234. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- 6.Fuller JH, Shipley MJ, Rose G, Jarrett RJ, Keen H. Coronary-heart-disease risk and impaired glucose tolerance. The Whitehall study. Lancet. 1980;1:1373–1376. doi: 10.1016/s0140-6736(80)92651-3. [DOI] [PubMed] [Google Scholar]
- 7.Paneni F, Beckman JA, Creager MA, Cosentino F. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Eur Heart J. 2013;34:2436–2443. doi: 10.1093/eurheartj/eht149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Després JP. Abdominal obesity and cardiovascular disease: is inflammation the missing link? Can J Cardiol. 2012;28:642–652. doi: 10.1016/j.cjca.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Dagenais GR, Yi Q, Mann JF, Bosch J, Pogue J, Yusuf S. Prognostic impact of body weight and abdominal obesity in women and men with cardiovascular disease. Am Heart J. 2005;149:54–60. doi: 10.1016/j.ahj.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 10.Purnell JQ, Zinman B, Brunzell JD. The effect of excess weight gain with intensive diabetes mellitus treatment on cardiovascular disease risk factors and atherosclerosis in type 1 diabetes mellitus: results from the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study (DCCT/EDIC) study. Circulation. 2013;127:180–187. doi: 10.1161/CIRCULATIONAHA.111.077487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kilpatrick ES, Rigby AS, Atkin SL. Insulin resistance, the metabolic syndrome, and complication risk in type 1 diabetes: “double diabetes” in the Diabetes Control and Complications Trial. Diabetes Care. 2007;30:707–712. doi: 10.2337/dc06-1982. [DOI] [PubMed] [Google Scholar]
- 12.Thorn LM, Forsblom C, Fagerudd J, Thomas MC, Pettersson-Fernholm K, Saraheimo M, Wadén J, Rönnback M, Rosengård-Bärlund M, Björkesten CG, et al. Metabolic syndrome in type 1 diabetes: association with diabetic nephropathy and glycemic control (the FinnDiane study) Diabetes Care. 2005;28:2019–2024. doi: 10.2337/diacare.28.8.2019. [DOI] [PubMed] [Google Scholar]
- 13.Thorn LM, Forsblom C, Wadén J, Saraheimo M, Tolonen N, Hietala K, Groop PH. Metabolic syndrome as a risk factor for cardiovascular disease, mortality, and progression of diabetic nephropathy in type 1 diabetes. Diabetes Care. 2009;32:950–952. doi: 10.2337/dc08-2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gast KB, Tjeerdema N, Stijnen T, Smit JW, Dekkers OM. Insulin resistance and risk of incident cardiovascular events in adults without diabetes: meta-analysis. PLoS One. 2012;7:e52036. doi: 10.1371/journal.pone.0052036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar R, Lee TT, Jeremias A, Ruisi CP, Sylvia B, Magallon J, Kirtane AJ, Bigelow B, Abrahamson M, Pinto DS, et al. Comparison of outcomes using sirolimus-eluting stenting in diabetic versus nondiabetic patients with comparison of insulin versus non-insulin therapy in the diabetic patients. Am J Cardiol. 2007;100:1187–1191. doi: 10.1016/j.amjcard.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 16.Uetani T, Amano T, Harada K, Kitagawa K, Kunimura A, Shimbo Y, Harada K, Yoshida T, Kato B, Kato M, et al. Impact of insulin resistance on post-procedural myocardial injury and clinical outcomes in patients who underwent elective coronary interventions with drug-eluting stents. JACC Cardiovasc Interv. 2012;5:1159–1167. doi: 10.1016/j.jcin.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 17.Hasegawa Y, Saito T, Ogihara T, Ishigaki Y, Yamada T, Imai J, Uno K, Gao J, Kaneko K, Shimosawa T, et al. Blockade of the nuclear factor-κB pathway in the endothelium prevents insulin resistance and prolongs life spans. Circulation. 2012;125:1122–1133. doi: 10.1161/CIRCULATIONAHA.111.054346. [DOI] [PubMed] [Google Scholar]
- 18.Rask-Madsen C, Li Q, Freund B, Feather D, Abramov R, Wu IH, Chen K, Yamamoto-Hiraoka J, Goldenbogen J, Sotiropoulos KB, et al. Loss of insulin signaling in vascular endothelial cells accelerates atherosclerosis in apolipoprotein E null mice. Cell Metab. 2010;11:379–389. doi: 10.1016/j.cmet.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du X, Edelstein D, Obici S, Higham N, Zou MH, Brownlee M. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J Clin Invest. 2006;116:1071–1080. doi: 10.1172/JCI23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kubota T, Kubota N, Kumagai H, Yamaguchi S, Kozono H, Takahashi T, Inoue M, Itoh S, Takamoto I, Sasako T, et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 2011;13:294–307. doi: 10.1016/j.cmet.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 21.Li Q, Park K, Li C, Rask-Madsen C, Mima A, Qi W, Mizutani K, Huang P, King GL. Induction of vascular insulin resistance and endothelin-1 expression and acceleration of atherosclerosis by the overexpression of protein kinase C-β isoform in the endothelium. Circ Res. 2013;113:418–427. doi: 10.1161/CIRCRESAHA.113.301074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim JK. Endothelial nuclear factor κB in obesity and aging: is endothelial nuclear factor κB a master regulator of inflammation and insulin resistance? Circulation. 2012;125:1081–1083. doi: 10.1161/CIRCULATIONAHA.111.090134. [DOI] [PubMed] [Google Scholar]
- 23.Tabit CE, Shenouda SM, Holbrook M, Fetterman JL, Kiani S, Frame AA, Kluge MA, Held A, Dohadwala MM, Gokce N, et al. Protein kinase C-β contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation. 2013;127:86–95. doi: 10.1161/CIRCULATIONAHA.112.127514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 25.Semenkovich CF. Insulin resistance and atherosclerosis. J Clin Invest. 2006;116:1813–1822. doi: 10.1172/JCI29024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, Shoelson SE. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293:1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- 27.Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M, Vollenweider P, Pedrazzini T, Nicod P, Thorens B, et al. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation. 2001;104:342–345. doi: 10.1161/01.cir.104.3.342. [DOI] [PubMed] [Google Scholar]
- 28.Kim F, Pham M, Maloney E, Rizzo NO, Morton GJ, Wisse BE, Kirk EA, Chait A, Schwartz MW. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol. 2008;28:1982–1988. doi: 10.1161/ATVBAHA.108.169722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rask-Madsen C, King GL. Endothelium-dependent delivery of insulin to muscle interstitium. Cell Metab. 2011;13:236–238. doi: 10.1016/j.cmet.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laakso M, Edelman SV, Brechtel G, Baron AD. Decreased effect of insulin to stimulate skeletal muscle blood flow in obese man. A novel mechanism for insulin resistance. J Clin Invest. 1990;85:1844–1852. doi: 10.1172/JCI114644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clark MG, Wallis MG, Barrett EJ, Vincent MA, Richards SM, Clerk LH, Rattigan S. Blood flow and muscle metabolism: a focus on insulin action. Am J Physiol Endocrinol Metab. 2003;284:E241–E258. doi: 10.1152/ajpendo.00408.2002. [DOI] [PubMed] [Google Scholar]
- 32.Rask-Madsen C, Kahn CR. Tissue-specific insulin signaling, metabolic syndrome, and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2012;32:2052–2059. doi: 10.1161/ATVBAHA.111.241919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muris DM, Houben AJ, Schram MT, Stehouwer CD. Microvascular dysfunction: an emerging pathway in the pathogenesis of obesity-related insulin resistance. Rev Endocr Metab Disord. 2013;14:29–38. doi: 10.1007/s11154-012-9231-7. [DOI] [PubMed] [Google Scholar]
- 34.Vitale C, Mercuro G, Cornoldi A, Fini M, Volterrani M, Rosano GM. Metformin improves endothelial function in patients with metabolic syndrome. J Intern Med. 2005;258:250–256. doi: 10.1111/j.1365-2796.2005.01531.x. [DOI] [PubMed] [Google Scholar]
- 35.Naka KK, Papathanassiou K, Bechlioulis A, Pappas K, Kazakos N, Kanioglou C, Papafaklis MI, Kostoula A, Vezyraki P, Makriyiannis D, et al. Rosiglitazone improves endothelial function in patients with type 2 diabetes treated with insulin. Diab Vasc Dis Res. 2011;8:195–201. doi: 10.1177/1479164111408628. [DOI] [PubMed] [Google Scholar]
- 36.Tsuchiya K, Tanaka J, Shuiqing Y, Welch CL, DePinho RA, Tabas I, Tall AR, Goldberg IJ, Accili D. FoxOs integrate pleiotropic actions of insulin in vascular endothelium to protect mice from atherosclerosis. Cell Metab. 2012;15:372–381. doi: 10.1016/j.cmet.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes. 2009;58:2718–2725. doi: 10.2337/db09-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paneni F, Costantino S, Volpe M, Lüscher TF, Cosentino F. Epigenetic signatures and vascular risk in type 2 diabetes: a clinical perspective. Atherosclerosis. 2013;230:191–197. doi: 10.1016/j.atherosclerosis.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 39.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 41.Gage MC, Yuldasheva NY, Viswambharan H, Sukumar P, Cubbon RM, Galloway S, Imrie H, Skromna A, Smith J, Jackson CL, et al. Endothelium-specific insulin resistance leads to accelerated atherosclerosis in areas with disturbed flow patterns: a role for reactive oxygen species. Atherosclerosis. 2013;230:131–139. doi: 10.1016/j.atherosclerosis.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 42.Du J, Fan LM, Mai A, Li JM. Crucial roles of Nox2-derived oxidative stress in deteriorating the function of insulin receptors and endothelium in dietary obesity of middle-aged mice. Br J Pharmacol. 2013;170:1064–1077. doi: 10.1111/bph.12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- 44.Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 45.Camici GG, Schiavoni M, Francia P, Bachschmid M, Martin-Padura I, Hersberger M, Tanner FC, Pelicci P, Volpe M, Anversa P, et al. Genetic deletion of p66(Shc) adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress. Proc Natl Acad Sci USA. 2007;104:5217–5222. doi: 10.1073/pnas.0609656104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pagnin E, Fadini G, de Toni R, Tiengo A, Calò L, Avogaro A. Diabetes induces p66shc gene expression in human peripheral blood mononuclear cells: relationship to oxidative stress. J Clin Endocrinol Metab. 2005;90:1130–1136. doi: 10.1210/jc.2004-1283. [DOI] [PubMed] [Google Scholar]
- 47.Paneni F, Costantino S, Cosentino F. p66(Shc)-induced redox changes drive endothelial insulin resistance. Atherosclerosis. 2014;236:426–429. doi: 10.1016/j.atherosclerosis.2014.07.027. [DOI] [PubMed] [Google Scholar]
- 48.Yubero-Serrano EM, Delgado-Lista J, Peña-Orihuela P, Perez-Martinez P, Fuentes F, Marin C, Tunez I, Tinahones FJ, Perez-Jimenez F, Roche HM, et al. Oxidative stress is associated with the number of components of metabolic syndrome: LIPGENE study. Exp Mol Med. 2013;45:e28. doi: 10.1038/emm.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Münzel T, Gori T, Bruno RM, Taddei S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur Heart J. 2010;31:2741–2748. doi: 10.1093/eurheartj/ehq396. [DOI] [PubMed] [Google Scholar]
- 50.Hundal RS, Petersen KF, Mayerson AB, Randhawa PS, Inzucchi S, Shoelson SE, Shulman GI. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest. 2002;109:1321–1326. doi: 10.1172/JCI14955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu X, Bean JS, Kassab GS, Rekhter MD. Protein kinase C inhibition ameliorates functional endothelial insulin resistance and vascular smooth muscle cell hypersensitivity to insulin in diabetic hypertensive rats. Cardiovasc Diabetol. 2011;10:48. doi: 10.1186/1475-2840-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mammi C, Pastore D, Lombardo MF, Ferrelli F, Caprio M, Consoli C, Tesauro M, Gatta L, Fini M, Federici M, et al. Sildenafil reduces insulin-resistance in human endothelial cells. PLoS One. 2011;6:e14542. doi: 10.1371/journal.pone.0014542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shemyakin A, Salehzadeh F, Esteves Duque-Guimaraes D, Böhm F, Rullman E, Gustafsson T, Pernow J, Krook A. Endothelin-1 reduces glucose uptake in human skeletal muscle in vivo and in vitro. Diabetes. 2011;60:2061–2067. doi: 10.2337/db10-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cunnington C, Van Assche T, Shirodaria C, Kylintireas I, Lindsay AC, Lee JM, Antoniades C, Margaritis M, Lee R, Cerrato R, et al. Systemic and vascular oxidation limits the efficacy of oral tetrahydrobiopterin treatment in patients with coronary artery disease. Circulation. 2012;125:1356–1366. doi: 10.1161/CIRCULATIONAHA.111.038919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paneni F, Mocharla P, Akhmedov A, Costantino S, Osto E, Volpe M, Lüscher TF, Cosentino F. Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circ Res. 2012;111:278–289. doi: 10.1161/CIRCRESAHA.112.266593. [DOI] [PubMed] [Google Scholar]