

Abstract
Diabetes mellitus (DM) is a health condition characterized by hyperglycemia over a prolonged period. There are three main types of DM: DM type 1 (DM1), DM2 and gestational DM (GDM). Maternal diabetes, which includes the occurrence of DM1 and DM2 during pregnancy or GDM, increases the occurrence of gesttional complications and adverse fetal outcomes. The hyperglycemic intrauterine environment affects not only the fetus but also the placental development and function in humans and experimental rodents. The underlying mechanisms are still unclear, but some evidence indicates alterations in trophoblast proliferation, apoptosis and cell cycle control in diabetes. A proper coordination of trophoblast proliferation, differentiation and invasion is required for placental development. Initially, increased expression of proliferative markers in junctional and labyrinth zones of rat placentas and villous cytotrophoblast, syncytiotrophoblast, stromal cells and fetal endothelial cells in human placentas is reported among diabetics. Moreover, reduced apoptotic index and expression of some apoptotic genes are described in placentas of GDM women. In addition, cell cycle regulators including cyclins and cyclin-dependent kinase inhibitors seem to be affected by the hyperglycemic environment. More studies are necessary to check the balance between proliferation, apoptosis and differentiation in trophoblast cells during maternal diabetes.
Keywords: Diabetes, Placenta, Proliferation, Apoptosis, Differentiation, Trophoblast
Core tip: This review article focuses on current knowledge about the effects of diabetes on trophoblast function such as proliferation, apoptosis and cell cycle control during placental development in human and rodent animal models. It also briefly discusses some placental pathological findings as a consequence of altered metabolic environment during diabetes.
INTRODUCTION
Diabetes mellitus (DM) is a group of metabolic diseases characterized by hyperglycemia resulting from defects in insulin secretion, insulin action, or both[1]. DM is a public health problem worldwide which is increasing mainly because of the high prevalence of obesity and sedentarism. In 2000, a previous study including all age groups, estimated the global prevalence of DM at 2.8% expecting to rise up to 4.4% by 2030[2].
There are three main types of DM: DM type 1 (DM1), DM2 and gestational DM (GDM). About 10% of all diabetes cases are DM1: an autoimmune disease in which an absolute deficiency of insulin resulting from the pancreatic β cells destruction occurs. This destruction can be caused by an autoimmune process (type 1a) or it can be idiopathic (type 1b). The former represents the vast majority of DM1 cases and usually manifests itself before 30 years of age, being more common in individuals of European origin. On the other hand, the vast majority of cases of diabetes existing in the population is DM2, characterized by peripheral resistance to insulin action and relative insulin deficiency[3].
In general, the overall prevalence of DM has increased in recent years due to the aging population and lifestyle changes. Parallel to this trend, the number of pregnant women with pre-existing diabetes (DM1 and DM2) has been increasing worldwide, and in some countries, this numbers are even doubling[4,5]. This increase is closely related to the high number of diabetic patients in reproductive age as well as to advances in clinical care available for pregnant diabetic women. Until the mid-20th century, DM1 women either had not reached the child-bearing age or had had serious health problems that contraindicated pregnancy. The discovery and commercial availability of insulin changed this scenario and a better glycemic control for women with diabetes led to a considerable reduction in the rates of maternal and fetal complications[6].
GDM is the principal metabolic disorder that occurs during pregnancy and can affect 3% to 30% of pregnant women depending on the population studied and the diagnostic criteria used[7]. It is also defined as any degree of glucose intolerance of variable severity which arises or is diagnosed during pregnancy. Besides, it is characterized by the maternal pancreas inability to meet the growing demand of insulin as from the second trimester of gestation[8].
Maternal diabetes, which includes the occurrence of either DM1 or DM2 in pregnancy and GDM, creates an unfavorable environment for embryonic and fetoplacental development. Despite the several developmental and morphological differences between rodents and women placenta, the alterations induced by maternal diabetes are similar in diabetic patients and diabetic experimental models[9]. Several works have been published addressing the impact of diabetes on placental weight and growth and materno-placental oxygen supply[10,11].
As it is known, the placenta is a highly specialized organ in the interface between maternal and fetal circulation with fundamental functions for pregnancy. It permits the fetus anchorage to the uterus, O2/CO2 exchange, the nutrition and the waste products removal during embryonic and fetal development[12]. Also, it acts as a protective barrier against xenobiotics and releases a variety of steroids, hormones and cytokines[13]. Therefore, placental dysfunction has deleterious effects on adequate pregnancy support. Among placental cells, trophoblasts permit the embryo implantation and nutrition in the early pregnancy and thereafter they will contribute considerably to the development and function of the placenta. The underlying mechanisms of placental pathology during diabetes are still unclear, but some evidence indicates changes in trophoblast proliferation, apoptosis and cell cycle control.
MATERNAL DIABETES EFFECTS ON TROPHOBLAST PROLIFERATION
A proper coordination of trophoblast proliferation, differentiation and invasion is required for placental development. Initially, cell proliferation should be tightly controlled for proper tissue growth and differentiation. Throughout gestation, growth factors such as epidermal growth factor, vascular endothelial growth factor, platelet-derived growth factor, placental growth factor, colony stimulating factor 1, insulin-like growth factor I (IGF-I), or IGF-II are abundantly secreted from diverse cell types of the fetal-maternal interface and have been to promote proliferation, adhesion and/or invasion[14-17].
In diabetes, the enlargement of the junctional zone (JZ) in the diabetic rat placenta is described as the increased number of glycogen and giant trophoblast cells[10]. Indeed, our previous stereological study confirmed a greater volume of spongiotrophoblast/glycogen cells in diabetic rats compared with controls[18]. Also, other works have shown changes in the size and organization of the spongiotrophoblast and glycogen cells in rat models of diabetes[10,19,20], suggesting the JZ as the placental compartment most sensitive to the diabetic condition[18,21].
Studies on the effects of maternal diabetes on placental development have solely reported increased expression of proliferative markers in the JZ and labyrinth zone (LZ) of rat placentas[22,23]. Zorn et al[23] showed that diabetes promotes an increased cell proliferation rate, detected by Ki67 immunostain, especially of spongiotrophoblast cells at gestational day 14 (gd 14), and of labyrinth cells, spongiotrophoblast and trophoblast giant cells at gd 17. Also, intense proliferating cell nuclear antigen (PCNA) immunostain in labyrinth and spongiotrophoblast cells on 17 d and in spongiotrophoblast and trophoblast giant cells on 21 d of pregnancy were noted in diabetic groups than in control groups, indicating deregulated cell proliferation in hyperglycemic condition which may explain the placentomegaly observed in diabetic animals at gd 20[22].
In humans, the placentas of GDM pregnancies are heavier than those of control patients[24,25] and the mechanism accounting for this increased placental mass is unknown. Enlargement of the capillary surface area with capillary proliferation and penetration of newly formed vessels have also been shown in DM[26]. Villous immaturity is present in 60% of diabetic placentas and is characterized by an increase in the number of mature and immature intermediate villi[27]. At the same time, a higher number of villous cytotrophoblast, villous stromal fibroblasts, macrophages, endothelial cells and syncytiotrophoblast nuclei in diabetes were noted[28-30]. As in diabetic animals, it was also reported, increased proliferative activity in villous cytotrophoblasts compared to normal placentas[27,30,31]. Leach et al[32] reported higher PCNA immunoreactivity in endothelial cells of diabetic placentas. In addition to PCNA staining, Ki67 and cyclin D3 (Figure 1) staining of villous cytotrophoblast, syncytiotrophoblast, villous stromal cells and fetal endothelial cells increased in diabetic placentas compared to controls[33].
Figure 1.
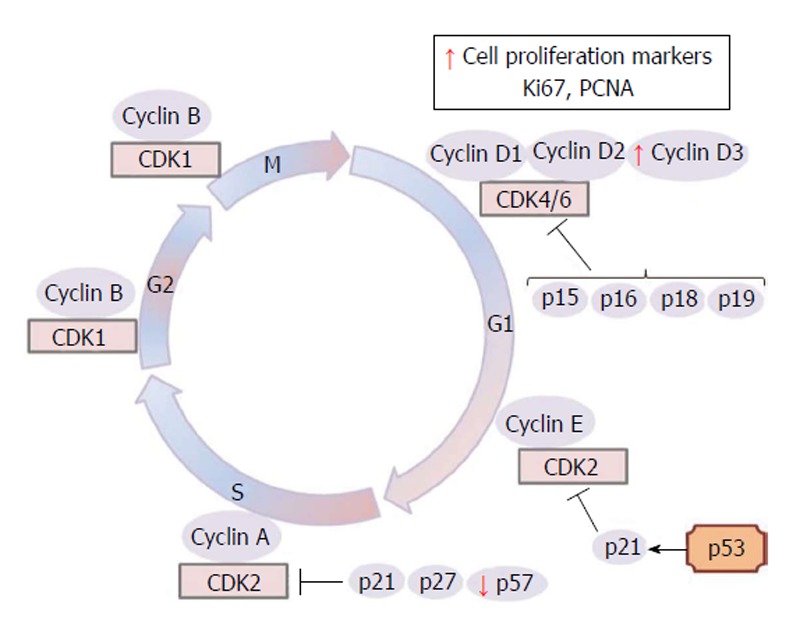
Schematic representation of eukaryotic cell cycle and key regulatory proteins which allow the transition from one cell cycle phase to another. CDKs inhibitors, such as p15, p16, p18, p19 (INK4 group), p21, p27 and p57 (CIP/KIP class) and proliferative markers are also showed. The arrows in red indicate increased or decreased expression of some trophoblast key regulatory proteins, CDKs inhibitors and proliferative markers in maternal diabetes. G1: Gap 1 phase; S: synthesis phase; G2: Gap 2 phase; M: Mitosis; CDKs: Cyclin-dependent kinases; PCNA: Proliferating cell nuclear antigen; p53: Tumor protein p53.
TROPHOBLAST APOPTOSIS IN MATERNAL DIABETES
The occurrence of apoptosis is shown during normal placental development and in morbid states[34-44]. In the normal human placenta, the presence of apoptotic cells could be associated with many events like trophoblast attachment and invasion[45,46], spiral artery remodelling[47,48], trophoblast differentiation[47-49] and labor[34,50]. However, the rates of placental apoptosis, even in normal human gestations, are still controversial. A predominance of apoptosis during early gestation, diminishing after the second trimester[41,51] and a significant increase in apoptosis as pregnancy progresses were reported[34,38,44].
Some works showed reduced apoptotic index, by TUNEL assay, in placentas from GDM[52,53] and DM1[36] patients compared to control placentas. Increased placental weight in GDM was associated with significantly reduced trophoblast apoptosis[52,53]. On the other hand, some authors reported increased apoptosis of villous cytotrophoblasts and syncytiotrophoblast nuclei in diabetic placentas in vivo[54] or in vitro[55]. Some technical differences such as the mode of delivery, the placental sampling or differences in gestational age could be responsible for study discrepancies[52]. Therefore, more studies are necessary to check the balance between proliferation and apoptosis in human diabetic placentas.
In the placenta, conditions like low oxygen and oxidative stress could induce to apoptosis that may be initiated by intrinsic or extrinsic pathways resulting in the activation of central apoptotic effectors, the caspases[56]. The extrinsic pathway involves members of the tumor necrosis factor (TNF) death receptor family, whose ligands include TNF-α, Fas ligand, Apo3 ligand (Apo3L) and Apo2L[44]. The activation of death receptors results in receptor aggregation and recruitment of adaptors molecules Fas-associated death domain or TNF-R-associated death domain[57]. As a consequence, procaspase-8 and procaspase-10 are recruited and become activated, initiating the cleavage of downstream effector caspases[58-60]. Caspase-8 could occasionally cleavage BH3-interacting domain death agonist that activates the intrinsic pathway[61].
The intrinsic pathway could be initiated by toxins, radiation, DNA damage and reactive oxygen species that lead to cellular stress and deficiency of growth factors[62]. This pathway induces mitochondrial membrane permeability modification by changes in the association of pro- and anti-apoptotic B-cell lymphoma 2 (BCL2) proteins[63]. The outer membrane permeabilization leads to the release of cytochrome c from the mitochondrial intermembrane space to the cytosol[64]. Them, the cytochrome c binds to the protease activating factor-1 forming the apoptosome[65]. The apoptosome cleaves procaspase-9, activating the terminal pathway of apoptosis. Additionally, the Smac (Second mitochondria-derived activator of caspase) is released from the mitochondria and eliminates the inhibitory effect of inhibitor of apoptosis proteins on caspases[66,67]. Both pathways culminate in a terminal pathway involving the cleavage and activation of caspase-3, -6, and -7, initiating cell destruction by activating DNAses and cleaving DNA repair enzymes such as poly (ADP-ribose) polymerase (PARP)[68,69].
Concerning proteins associated with cell death pathways reduced expression of BCL2 has been reported in placentas from diabetic patients compared to normoglycemic women[54]. Furthermore reduced gene expression of BCL2, BCL2L1, BCL2L2, myeloid cell leukemia 1 and X-linked inhibitor of apoptosis and reduced protein expression of the Fas receptor (FasR), FasL, caspase-3 and its PARP has been reported, indicating extrinsic and intrinsic pathways downregulation in placentas with GDM[50,53] (Figure 2).
Figure 2.
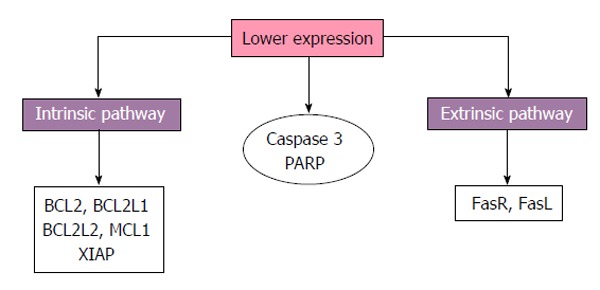
Schematic representation of trophoblast apoptosis findings in maternal diabetes. Reduced expression of apoptotic components from both intrinsic and extrinsic pathways, caspase-3 and poly (ADP-ribose) polymerase (PARP) are reported by some works. FasR: Fas receptor; FasL: Fas ligand; MCL1: Myeloid cell leukemia 1; BCL2: B-cell lymphoma 2; BCL2L1: BCL2-like 1; XIAP: X-linked inhibitor of apoptosis; BCL2L2: BCL2-like 2.
According to Rudge et al[70] (2012), severe diabetes in mice decrease placental TUNEL index from day 18 to 21 of pregnancy, at the same time that small for pregnancy age fetus and increased placental weight are also found. A GDM animal model (db/+ mice), when treated with TNF-α at gd 11.5, a pro-apoptotic peptide, there was an increased number of apoptotic cells, detected by cleaved caspase-3 immunostaining, in both labyrinth and trombospongium, at gd 18.5[71]. Unfortunately, little is known about the cleaved caspase-3 placental activity from other’s models of animal diabetes or even human diabetic pregnancies.
CELL CYCLE CONTROL OF TROPHOBLAST IN MATERNAL DIABETES
The appropriate development of an organism depends on the balance between cell cycle exit and the differentiation process in all tissues. The cell cycle exit is required for terminal differentiation of many cell types and cell cycle progression is regulated by a series of cyclin-dependent kinases (CDKs) that consist of catalytic subunits, designated CDKs, and activating subunits, designated cyclins[72,73]. The activation and inactivation of different cyclin-CDKs at adequate moments is necessary for precise progression into the cell cycle[74,75].
Although placental growth is essentially a result of the coordination of trophoblast proliferation and differentiation, there is little information about the mitotic regulators that provide the synchronization of trophoblast proliferation and differentiation[76]. In the rat term placenta, cyclin D1 and cyclin D3 are expressed in placental fetal cells, whereas the G1/S cyclin E are present only in the spongiotrophoblast and labyrinthine trophoblast cells[77]. The D-type cyclins serve as growth factor sensors that integrate extracellular signals with the cell cycle machinery. Together with their partner kinases, CDK4 and CDK6, they operate in early-to-mid G1 to promote progression through the G1-S restriction point[73]. The nuclei expression of cyclins D1 and D3 in mesenchymal and labyrinthine trophoblast cells could infer a role in the differentiated state maintenance in late gestation[77]. In the human placenta, cyclins D1 and D3 have been observed in endothelial cells[78]. However, cyclin activity during diabetes has been little explored in placental cells. Only one work reported cyclin D3 staining intensities significantly increasing in villous parts, basal plates and chorionic plate of a diabetic group when compared to control placentas[33]; perhaps prominent cyclin D expression could contribute to the increased cell proliferation observed in diabetic placentas (Figure 1).
There are two families of CDK inhibitors that act to inhibit cell cycle progression. The INK4 family (p15ink4b, p16ink4a, p18ink4c, p19ink4d) inhibits the CDK4, CDK6 and cyclin D activities in the G1 phase and G1/S transition of cell cycle. In turn, the CIP/KIP family (p21waf/cip1, p27kip1 and p57kip2) inhibits the cell cycle at many checkpoints by acting on multiple cyclin-CDK complexes[74,75].
The altered metabolic environment in maternal diabetes could affect the expression of genes that control the cell cycle events as was observed for reduced p57 expression in diabetic rat placentas on days 17 and 21 of pregnancy[79] (Figure 1). In the normal rat placenta, immunostaining intensities of cell cycle inhibitors p27, and p57 were observed to be higher in the JZ compared to the LZ close to term[77]. Accordingly, since p57 is a cell cycle inhibitor and tumor suppressor, lack of p57 activity can lead to a loss of cell cycle control and hyperproliferation[33]. Therefore, less p57 expression may explain the reason why diabetic placentas are heavier and bigger[33].
In fact, abnormal placental development is present in p27 and p57 knockout mice[80]. The LZ is less vascularized and contains more trophoblasts than those from wild type placentas. Moreover, Takahashi et al[81] demonstrated that placentomegaly was observed in p57 deficient mice in which the numbers of placental cells in the LZ and spongiotrophoblasts were twice the number of those of the wild type. In addition to placentomegaly, deregulation of the cell cycle can result in the development or progression of some trophoblastic diseases like preeclampsia that can occur in diabetic women. In humans, p57 staining index of villous parts decreases significantly in diabetes[33]. Another CIP/KIP family member, p27, has no difference in staining intensity in the villous part between diabetic and control groups of human patients, but has different staining patterns in different placental cell types[33]. Some studies indicated that p27 and p57 have different functions in human placental development[82,83].
In summary, this article reviewed the current knowledge about the effects of hyperglycemia on trophoblast proliferation, apoptosis and cell cycle control during pregnancy. More detailed studies are required to check the balance between proliferation, apoptosis and differentiation in trophoblast cells during maternal diabetes.
Footnotes
P- Reviewer: Rajakumar A, Wadsack C S- Editor: Ji FF L- Editor: A E- Editor: Liu SQ
Conflict-of-interest: The authors declare that they have no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: August 14, 2014
First decision: November 3, 2014
Article in press: December 31, 2014
References
- 1.American Diabetes Association. Standards of medical care in diabetes--2009. Diabetes Care. 2009;32 Suppl 1:S13–S61. doi: 10.2337/dc09-S013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 3.American Diabetes Association. Standards of medical care in diabetes--2012. Diabetes Care. 2012;35 Suppl 1:S11–S63. doi: 10.2337/dc12-s011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hunt KJ, Schuller KL. The increasing prevalence of diabetes in pregnancy. Obstet Gynecol Clin North Am. 2007;34:173–199, vii. doi: 10.1016/j.ogc.2007.03.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet. 2014;383:69–82. doi: 10.1016/S0140-6736(13)60591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Negrato CA, Montenegro RM, Mattar R, Zajdenverg L, Francisco RP, Pereira BG, Sancovski M, Torloni MR, Dib SA, Viggiano CE, et al. Dysglycemias in pregnancy: from diagnosis to treatment. Brazilian consensus statement. Diabetol Metab Syndr. 2010;2:27. doi: 10.1186/1758-5996-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.American Diabetes Association (ADA) Standards of Medical Care in Diabetes. Diabetes Care. 2011;34 Suppl 1:S11–61. doi: 10.2337/dc11-S011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchanan TA, Xiang A, Kjos SL, Watanabe R. What is gestational diabetes? Diabetes Care. 2007;30 Suppl 2:S105–S111. doi: 10.2337/dc07-s201. [DOI] [PubMed] [Google Scholar]
- 9.Vambergue A, Fajardy I. Consequences of gestational and pregestational diabetes on placental function and birth weight. World J Diabetes. 2011;2:196–203. doi: 10.4239/wjd.v2.i11.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gewolb IH, Merdian W, Warshaw JB, Enders AC. Fine structural abnormalities of the placenta in diabetic rats. Diabetes. 1986;35:1254–1261. doi: 10.2337/diab.35.11.1254. [DOI] [PubMed] [Google Scholar]
- 11.Taricco E, Radaelli T, Nobile de Santis MS, Cetin I. Foetal and placental weights in relation to maternal characteristics in gestational diabetes. Placenta. 2003;24:343–347. doi: 10.1053/plac.2002.0913. [DOI] [PubMed] [Google Scholar]
- 12.Bauer MK, Harding JE, Bassett NS, Breier BH, Oliver MH, Gallaher BH, Evans PC, Woodall SM, Gluckman PD. Fetal growth and placental function. Mol Cell Endocrinol. 1998;140:115–120. doi: 10.1016/s0303-7207(98)00039-2. [DOI] [PubMed] [Google Scholar]
- 13.Furukawa S, Hayashi S, Usuda K, Abe M, Hagio S, Ogawa I. Toxicological pathology in the rat placenta. J Toxicol Pathol. 2011;24:95–111. doi: 10.1293/tox.24.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bischof P, Meisser A, Campana A. Paracrine and autocrine regulators of trophoblast invasion--a review. Placenta. 2000;21 Suppl A:S55–S60. doi: 10.1053/plac.2000.0521. [DOI] [PubMed] [Google Scholar]
- 15.Ferretti C, Bruni L, Dangles-Marie V, Pecking AP, Bellet D. Molecular circuits shared by placental and cancer cells, and their implications in the proliferative, invasive and migratory capacities of trophoblasts. Hum Reprod Update. 2007;13:121–141. doi: 10.1093/humupd/dml048. [DOI] [PubMed] [Google Scholar]
- 16.Lala PK, Hamilton GS. Growth factors, proteases and protease inhibitors in the maternal-fetal dialogue. Placenta. 1996;17:545–555. doi: 10.1016/s0143-4004(96)80071-3. [DOI] [PubMed] [Google Scholar]
- 17.Pollheimer J, Knöfler M. Signalling pathways regulating the invasive differentiation of human trophoblasts: a review. Placenta. 2005;26 Suppl A:S21–S30. doi: 10.1016/j.placenta.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 18.Farias PS, dos S Souza K, Fioretto ET, dos Santos MR, Aires MB. Maternal diabetes affects rat placental morphology and pregnancy. Endocrine. 2014;45:497–501. doi: 10.1007/s12020-014-0169-2. [DOI] [PubMed] [Google Scholar]
- 19.Prager R, Abramovici A, Liban E, Laron Z. Histopathological changes in the placenta of streptozotocin induced diabetic rats. Diabetologia. 1974;10:89–91. doi: 10.1007/BF00421419. [DOI] [PubMed] [Google Scholar]
- 20.Ne’eman Z, Barash V, Rosenmann E, Shafrir E. Localization of glycogen in the placenta of diabetic rats: a light and electron microscopic study. Placenta. 1987;8:201–208. doi: 10.1016/0143-4004(87)90023-3. [DOI] [PubMed] [Google Scholar]
- 21.Giachini FR, Carriel V, Capelo LP, Tostes RC, Carvalho MH, Fortes ZB, Zorn TM, San Martin S. Maternal diabetes affects specific extracellular matrix components during placentation. J Anat. 2008;212:31–41. doi: 10.1111/j.1469-7580.2007.00839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Acar N, Korgun ET, Cayli S, Sahin Z, Demir R, Ustunel I. Is there a relationship between PCNA expression and diabetic placental development during pregnancy? Acta Histochem. 2008;110:408–417. doi: 10.1016/j.acthis.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 23.Zorn TM, Zúñiga M, Madrid E, Tostes R, Fortes Z, Giachini F, San Martín S. Maternal diabetes affects cell proliferation in developing rat placenta. Histol Histopathol. 2011;26:1049–1056. doi: 10.14670/HH-26.1049. [DOI] [PubMed] [Google Scholar]
- 24.Higgins M, Mc Auliffe F. A review of maternal and fetal growth factors in diabetic pregnancy. Curr Diabetes Rev. 2010;6:116–125. doi: 10.2174/157339910790909431. [DOI] [PubMed] [Google Scholar]
- 25.Ashfaq M, Janjua MZ, Channa MA. Effect of gestational diabetes and maternal hypertension on gross morphology of placenta. J Ayub Med Coll Abbottabad. 2005;17:44–47. [PubMed] [Google Scholar]
- 26.Sherer DM, Divon MY. Prenatal ultrasonographic assessment of the ductus arteriosus: a review. Obstet Gynecol. 1996;87:630–637. doi: 10.1016/0029-7844(95)00484-X. [DOI] [PubMed] [Google Scholar]
- 27.Benirschke K, Kaufmann P, Baergen RN. Pathology of the humanplacenta. 5th ed. New York: Springer; 2006. [Google Scholar]
- 28.Desoye G, Shafrir E. The human placenta in diabetic pregnancy. Diabet Rev. 1996;4:70–89. [Google Scholar]
- 29.Fox H. Pathology of the placenta in maternal diabetes mellitus. Obstet Gynecol. 1969;34:792–798. [PubMed] [Google Scholar]
- 30.Teasdale F. Histomorphometry of the placenta of the diabetic women: class A diabetes mellitus. Placenta. 1981;2:241–251. doi: 10.1016/s0143-4004(81)80007-0. [DOI] [PubMed] [Google Scholar]
- 31.Jones CJ, Fox H. An ultrastructural and ultrahistochemical study of the placenta of the diabetic woman. J Pathol. 1976;119:91–99. doi: 10.1002/path.1711190203. [DOI] [PubMed] [Google Scholar]
- 32.Leach L, Gray C, Staton S, Babawale MO, Gruchy A, Foster C, Mayhew TM, James DK. Vascular endothelial cadherin and beta-catenin in human fetoplacental vessels of pregnancies complicated by Type 1 diabetes: associations with angiogenesis and perturbed barrier function. Diabetologia. 2004;47:695–709. doi: 10.1007/s00125-004-1341-7. [DOI] [PubMed] [Google Scholar]
- 33.Unek G, Ozmen A, Mendilcioglu I, Simsek M, Korgun ET. Immunohistochemical distribution of cell cycle proteins p27, p57, cyclin D3, PCNA and Ki67 in normal and diabetic human placentas. J Mol Histol. 2014;45:21–34. doi: 10.1007/s10735-013-9534-3. [DOI] [PubMed] [Google Scholar]
- 34.Smith SC, Baker PN, Symonds EM. Placental apoptosis in normal human pregnancy. Am J Obstet Gynecol. 1997;177:57–65. doi: 10.1016/s0002-9378(97)70438-1. [DOI] [PubMed] [Google Scholar]
- 35.Chan CC, Lao TT, Cheung AN. Apoptotic and proliferative activities in first trimester placentae. Placenta. 1999;20:223–227. doi: 10.1053/plac.1998.0375. [DOI] [PubMed] [Google Scholar]
- 36.Burleigh DW, Stewart K, Grindle KM, Kay HH, Golos TG. Influence of maternal diabetes on placental fibroblast growth factor-2 expression, proliferation, and apoptosis. J Soc Gynecol Investig. 2004;11:36–41. doi: 10.1016/j.jsgi.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 37.Danihel L, Gomolcák P, Korbel M, Pruzinec J, Vojtassák J, Janík P, Babál P. Expression of proliferation and apoptotic markers in human placenta during pregnancy. Acta Histochem. 2002;104:335–338. doi: 10.1078/0065-1281-00683. [DOI] [PubMed] [Google Scholar]
- 38.Halperin R, Peller S, Rotschild M, Bukovsky I, Schneider D. Placental apoptosis in normal and abnormal pregnancies. Gynecol Obstet Invest. 2000;50:84–87. doi: 10.1159/000010287. [DOI] [PubMed] [Google Scholar]
- 39.Ishihara N, Matsuo H, Murakoshi H, Laoag-Fernandez J, Samoto T, Maruo T. Changes in proliferative potential, apoptosis and Bcl-2 protein expression in cytotrophoblasts and syncytiotrophoblast in human placenta over the course of pregnancy. Endocr J. 2000;47:317–327. doi: 10.1507/endocrj.47.317. [DOI] [PubMed] [Google Scholar]
- 40.Kokawa K, Shikone T, Nakano R. Apoptosis in human chorionic villi and decidua during normal embryonic development and spontaneous abortion in the first trimester. Placenta. 1998;19:21–26. doi: 10.1016/s0143-4004(98)90094-7. [DOI] [PubMed] [Google Scholar]
- 41.Yamada Z, Kitagawa M, Takemura T, Hirokawa K. Effect of maternal age on incidences of apoptotic and proliferative cells in trophoblasts of full-term human placenta. Mol Hum Reprod. 2001;7:1179–1185. doi: 10.1093/molehr/7.12.1179. [DOI] [PubMed] [Google Scholar]
- 42.Weiss U, Cervar M, Puerstner P, Schmut O, Haas J, Mauschitz R, Arikan G, Desoye G. Hyperglycaemia in vitro alters the proliferation and mitochondrial activity of the choriocarcinoma cell lines BeWo, JAR and JEG-3 as models for human first-trimester trophoblast. Diabetologia. 2001;44:209–219. doi: 10.1007/s001250051601. [DOI] [PubMed] [Google Scholar]
- 43.Moley KH, Chi MM, Knudson CM, Korsmeyer SJ, Mueckler MM. Hyperglycemia induces apoptosis in pre-implantation embryos through cell death effector pathways. Nat Med. 1998;4:1421–1424. doi: 10.1038/4013. [DOI] [PubMed] [Google Scholar]
- 44.Straszewski-Chavez SL, Abrahams VM, Mor G. The role of apoptosis in the regulation of trophoblast survival and differentiation during pregnancy. Endocr Rev. 2005;26:877–897. doi: 10.1210/er.2005-0003. [DOI] [PubMed] [Google Scholar]
- 45.Uckan D, Steele A, Cherry BY, Chamizo W, Koutsonikolis A, Gilbert-Barness E, Good RA. Trophoblasts express Fas ligand: a proposed mechanism for immune privilege in placenta and maternal invasion. Mol Hum Reprod. 1997;3:655–662. doi: 10.1093/molehr/3.8.655. [DOI] [PubMed] [Google Scholar]
- 46.Galán A, O’Connor JE, Valbuena D, Herrer R, Remohí J, Pampfer S, Pellicer A, Simón C. The human blastocyst regulates endometrial epithelial apoptosis in embryonic adhesion. Biol Reprod. 2000;63:430–439. doi: 10.1093/biolreprod/63.2.430. [DOI] [PubMed] [Google Scholar]
- 47.Huppertz B, Frank HG, Kingdom JC, Reister F, Kaufmann P. Villous cytotrophoblast regulation of the syncytial apoptotic cascade in the human placenta. Histochem Cell Biol. 1998;110:495–508. doi: 10.1007/s004180050311. [DOI] [PubMed] [Google Scholar]
- 48.Mor G, Straszewski S, Kamsteeg M. Role of the Fas/Fas ligand system in female reproductive organs: survival and apoptosis. Biochem Pharmacol. 2002;64:1305–1315. doi: 10.1016/s0006-2952(02)01267-4. [DOI] [PubMed] [Google Scholar]
- 49.Mayhew TM, Leach L, McGee R, Ismail WW, Myklebust R, Lammiman MJ. Proliferation, differentiation and apoptosis in villous trophoblast at 13-41 weeks of gestation (including observations on annulate lamellae and nuclear pore complexes) Placenta. 1999;20:407–422. doi: 10.1053/plac.1999.0399. [DOI] [PubMed] [Google Scholar]
- 50.Smith SC, Leung TN, To KF, Baker PN. Apoptosis is a rare event in first-trimester placental tissue. Am J Obstet Gynecol. 2000;183:697–699. doi: 10.1067/mob.2000.106555. [DOI] [PubMed] [Google Scholar]
- 51.Sakuragi N, Luo , MN , Furata I, Watari H, Tsumura N, Nishiya M, Hirahatake K, Takeda N, Takeda T, et al. BCL-2 expression and apoptosis in human trophoblast. Troph Res. 1997;9:63–74. [Google Scholar]
- 52.Magee TR, Ross MG, Wedekind L, Desai M, Kjos S, Belkacemi L. Gestational diabetes mellitus alters apoptotic and inflammatory gene expression of trophobasts from human term placenta. J Diabetes Complications. 2014;28:448–459. doi: 10.1016/j.jdiacomp.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Belkacemi L, Kjos S, Nelson DM, Desai M, Ross MG. Reduced apoptosis in term placentas from gestational diabetic pregnancies. J Dev Orig Health Dis. 2013;4:256–265. doi: 10.1017/S2040174413000068. [DOI] [PubMed] [Google Scholar]
- 54.Sgarbosa F, Barbisan LF, Brasil MA, Costa E, Calderon IM, Gonçalves CR, Bevilacqua E, Rudge MV. Changes in apoptosis and Bcl-2 expression in human hyperglycemic, term placental trophoblast. Diabetes Res Clin Pract. 2006;73:143–149. doi: 10.1016/j.diabres.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 55.Araújo JR, Correia-Branco A, Ramalho C, Keating E, Martel F. Gestational diabetes mellitus decreases placental uptake of long-chain polyunsaturated fatty acids: involvement of long-chain acyl-CoA synthetase. J Nutr Biochem. 2013;24:1741–1750. doi: 10.1016/j.jnutbio.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 56.Sharp AN, Heazell AE, Crocker IP, Mor G. Placental apoptosis in health and disease. Am J Reprod Immunol. 2010;64:159–169. doi: 10.1111/j.1600-0897.2010.00837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chinnaiyan AM, Tepper CG, Seldin MF, O’Rourke K, Kischkel FC, Hellbardt S, Krammer PH, Peter ME, Dixit VM. FADD/MORT1 is a common mediator of CD95 (Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. J Biol Chem. 1996;271:4961–4965. doi: 10.1074/jbc.271.9.4961. [DOI] [PubMed] [Google Scholar]
- 58.Varfolomeev EE, Boldin MP, Goncharov TM, Wallach D. A potential mechanism of “cross-talk” between the p55 tumor necrosis factor receptor and Fas/APO1: proteins binding to the death domains of the two receptors also bind to each other. J Exp Med. 1996;183:1271–1275. doi: 10.1084/jem.183.3.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muzio M, Chinnaiyan AM, Kischkel FC, O’Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death--inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 60.Wang J, Chun HJ, Wong W, Spencer DM, Lenardo MJ. Caspase-10 is an initiator caspase in death receptor signaling. Proc Natl Acad Sci USA. 2001;98:13884–13888. doi: 10.1073/pnas.241358198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 63.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 64.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 65.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G, Vandenabeele P. Toxic proteins released from mitochondria in cell death. Oncogene. 2004;23:2861–2874. doi: 10.1038/sj.onc.1207523. [DOI] [PubMed] [Google Scholar]
- 67.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 68.Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 69.Koh DW, Dawson TM, Dawson VL. Mediation of cell death by poly(ADP-ribose) polymerase-1. Pharmacol Res. 2005;52:5–14. doi: 10.1016/j.phrs.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 70.Rudge MVC, Costa E, Barbisan LF, Damasceno DC, Bueno A, Saito FH, Calderon IMP, Rodrigues MMP. Evaluation of cell proliferation and apoptosis in placentas of rats with severe diabetes. Braz Arch Biol Technol. 2012;55:243–250. [Google Scholar]
- 71.Bobadilla RA, van Bree R, Vercruysse L, Pijnenborg R, Verhaeghe J. Placental effects of systemic tumour necrosis factor-α in an animal model of gestational diabetes mellitus. Placenta. 2010;31:1057–1063. doi: 10.1016/j.placenta.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 72.Lew DJ, Dulić V, Reed SI. Isolation of three novel human cyclins by rescue of G1 cyclin (Cln) function in yeast. Cell. 1991;66:1197–1206. doi: 10.1016/0092-8674(91)90042-w. [DOI] [PubMed] [Google Scholar]
- 73.Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 74.Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 75.Zavitz KH, Zipursky SL. Controlling cell proliferation in differentiating tissues: genetic analysis of negative regulators of G1--> S-phase progression. Curr Opin Cell Biol. 1997;9:773–781. doi: 10.1016/s0955-0674(97)80077-4. [DOI] [PubMed] [Google Scholar]
- 76.Levy R, Smith SD, Yusuf K, Huettner PC, Kraus FT, Sadovsky Y, Nelson DM. Trophoblast apoptosis from pregnancies complicated by fetal growth restriction is associated with enhanced p53 expression. Am J Obstet Gynecol. 2002;186:1056–1061. doi: 10.1067/mob.2002.122250. [DOI] [PubMed] [Google Scholar]
- 77.Korgun ET, Unek G, Herrera E, Jones CJ, Wadsack C, Kipmen-Korgun D, Desoye G. Mapping of CIP/KIP inhibitors, G1 cyclins D1, D3, E and p53 proteins in the rat term placenta. Histochem Cell Biol. 2011;136:267–278. doi: 10.1007/s00418-011-0841-z. [DOI] [PubMed] [Google Scholar]
- 78.De Falco M, Fedele V, Cobellis L, Mastrogiacomo A, Giraldi D, Leone S, De Luca L, Laforgia V, De Luca A. Pattern of expression of cyclin D1/CDK4 complex in human placenta during gestation. Cell Tissue Res. 2004;317:187–194. doi: 10.1007/s00441-004-0880-z. [DOI] [PubMed] [Google Scholar]
- 79.Acar N, Korgun ET, Ustunel I. Cell cycle inhibitor p57 expression in normal and diabetic rat placentas during some stages of pregnancy. Histol Histopathol. 2012;27:59–68. doi: 10.14670/HH-27.59. [DOI] [PubMed] [Google Scholar]
- 80.Zhang P, Wong C, DePinho RA, Harper JW, Elledge SJ. Cooperation between the Cdk inhibitors p27(KIP1) and p57(KIP2) in the control of tissue growth and development. Genes Dev. 1998;12:3162–3167. doi: 10.1101/gad.12.20.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Takahashi K, Kobayashi T, Kanayama N. p57(Kip2) regulates the proper development of labyrinthine and spongiotrophoblasts. Mol Hum Reprod. 2000;6:1019–1025. doi: 10.1093/molehr/6.11.1019. [DOI] [PubMed] [Google Scholar]
- 82.Genbacev O, McMaster MT, Fisher SJ. A repertoire of cell cycle regulators whose expression is coordinated with human cytotrophoblast differentiation. Am J Pathol. 2000;157:1337–1351. doi: 10.1016/S0002-9440(10)64648-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Korgun ET, Celik-Ozenci C, Acar N, Cayli S, Desoye G, Demir R. Location of cell cycle regulators cyclin B1, cyclin A, PCNA, Ki67 and cell cycle inhibitors p21, p27 and p57 in human first trimester placenta and deciduas. Histochem Cell Biol. 2006;125:615–624. doi: 10.1007/s00418-006-0160-y. [DOI] [PubMed] [Google Scholar]