

Abstract
Vitiligo is a common pigmentary disorder. Many studies across decades and all over the world have attempted to illustrate the pathogenesis behind it; however, the pathogenesis of vitiligo remains elusive. This review article, we present the findings behind the most and updated theories behind this psychologically debilitating and disfiguring disease. The discussion begun with the role of genetic predisposition followed by neural theory first proposed in the 1950s. We highlight the autoimmune hypothesis, followed by the reactive oxygen species model, zinc-α2-glycoprotein deficiency hypothesis, viral theory, intrinsic theory and biochemical, molecular and cellular alterations accounting for loss of functioning melanocytes in vitiligo. Many theories were elaborated to clarify vitiligo pathogenesis. It is a multifactorial disease involving the interplay of several factors. Future research is needed to clarify the interaction of these factors for better understanding of vitiligo pathogenesis and subsequent successful treatment.
Keywords: Etiopathogenesis, Pigmentary disorder, Non-segmental vitiligo, Segmental vitiligo, Vitiligo
Core tip: The pathogenesis of vitiligo elaborated by several theory. Future research needed to clarify the interaction of these factors for better understanding of vitiligo pathogenesis and subsequent successful treatment.
INTRODUCTION
The pathogenesis is complex and involves the interplay of multiple factors; however, the exact pathogenesis is not well known. Lerner et al[1] in the 1950s firstly proposed the neural theory, and after that, model of reactive oxygen species (ROS), the autoimmune hypothesis and the melanocytorrhagy hypothesis have appeared.
DIFFERENT PATTERNS AND PHYSICAL DISTRIBUTION OF VITILIGO
Pruritus, elevated lesions, and erythematous margins present in inflammatory vitiligo. There are 2 main types: generalized vitiligo (GV) (widespread macules with a symmetrical distribution), whereas focal vitiligo (FV) (1 or few depigmented not elevated areas at a single site). After coalescing of vitiliginous areas in GV, or becomes extensive in the body with remaining of few normal areas, thus called vitiligo universalis. Non-segmental vitiligo enrolled FV and GV, but segmental vitiligo (SV) restricted to one unilateral region[2], (Figure 1). The lesions of vitiligo are asymptomatic except in inflammatory vitiligo, which is associated with pruritus and characterized by elevated lesions, and erythematous margins.
Figure 1.
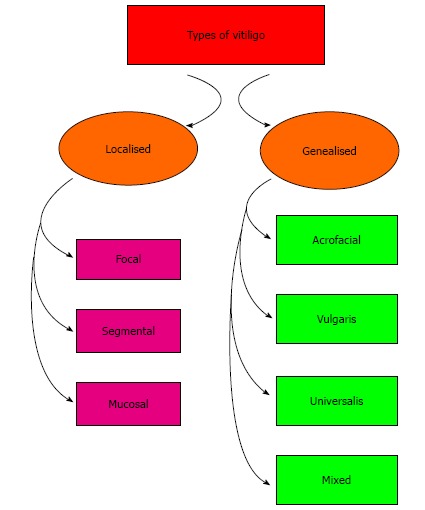
Types of vitiligo.
INHERITANCE OF VITILIGO
The inheritance is polygenic[3]. Family history exists in 6.25%-38% of patients with vitiligo[4]. Those who have recessive homozygosity at 3 epistatically interacting autosomal diallelic loci will be affected by vitiligo[5].
Molecular genetics-based studies
Spritz et al[6] (2004) revealed different loci or alleles for GV. Autoimmune susceptibility (AIS)-1, -2 (chromosome 7) and systemic lupus erythematosus vitiligo-related gene (SLEV1) (chromosome 17) both associated loci for GV and concomitant autoimmune diseases. Of 33 tested loci, only (XBP1, TSLP, and FOXP3) were primarily concomitant with GV. FOXP3 associated with X linked recessive multiple autoimmune disease syndrome. In addition, CTLA4 had association secondarily with GV, and the autoimmune diseases[7]. However, patients with GV also linked to AIS3 locus (chromosome 8)[6].
Methylation of deoxyribunucleic acid (DNA) conducted by DNA methyltransferases (DNMT1, -3a, -3b)[8]. Monocytes in vitiliginous patients and normal volunteers showed sensitivity to alterations in methylation and revealed association between IL-10 and reactivity of autoimmune system[9,10]. In comparison with controls, methylation was increased and hypermethylation of the methylation-sensitive region in IL-10 that could alter genes expression in autoimmunity[9]. In a similar way, the role of transforming growth factor beta-receptor II (TGFBR2), which inhibits the inflammatory pathways and lymphocyte activation was revealed[11,12].
The ultraviolet radiation resistance-associated gene (UVRAG), resists photo-damage, and plays a role in autophagy[13]. In 439 controls and 225 NSV patients, UVRAG has 2 SNPs which were significantly different[14].
Both diabetes mellitus type 1 and rheumatoid arthritis with SNPs found beside the insulin-dependent diabetes mellitus 8 locus (IDDM8)[15]. This region contains SMOC2, which enrolled in growth and development[16] and cell matrix interactions[17].
Also, melanocyte proliferating gene 1 (MYG1), is elevated in skin of both vitiliginous patients with activity, and without activity[18].
Human leukocyte antigen
Studies revealed that vitiligo associated with HLA-DRB1*07, HLA-A2, 11, 28, 31, 33, HLA-B17, 35, 40 and 44[19,20].
Susceptibility loci of vitiligo are on chromosome 6 and in the MHC[21]. A study genotyped 6623 patients with vitiligo and 10740 controls for 34 SNPs. At 6q27, 2 SNPS found with 3 unlinked genes. These gene include RNASET2, which responsible for ribonuclease (RNAse)[22]. The other two genes are the chemokine receptor 6 gene (CCR6)[21], and FGFR10P are imperative to progressing of the cycle of the cell that produce receptor of (FGF)[23]. Genes encode discoidin domain receptor 1 (DDR1)[24], and tyrosine kinase receptor play role in cell’s progression and function[25], both were involved in vitiligo.
THEORIES FOR VITILIGO PATHOGENESIS
The neural theory
Early theories: The “neural theory” supposed by Lerner’s (1959) was based on the fact that SV follows the course of the dermatome with exhibiting hyperhidrosis and emotional upset[1,26,27].
The sympathetic nervous system’s role: Dysfunction of sympathetic nervous system’s role (SNS) activity affect melanin production and lead to depigmentation. With iontophoresis and laser Doppler flowmetry level of microcirculation in lesions with vitiligo assessed to reveal SNS activity[28]. 10 subjects had facial SV, and 2 groups of controls were examined. 1 control group had 10 healthy, unaffected individuals, and the 2nd control group contained 10 non-segmental-type stable vitiligo patients. Patients were matched for gender and age. Approximately, the cutaneous blood flow was higher three times on the lesions vs normal skin in SV. The differences was not revealed in the non-SV.
Neuropeptide and neuronal markers: Neuropeptide Y (NPY), calcitonin gene-related peptide (CGRP), vasoactive intestinal polypeptide (VIP), and polyclonal general neuronal marker (PGP) tested for their immuno-reactivity in 12 patients with vitiligo and 7 unaffected control subjects[29]. NPY increased in the marginal areas of lesions in half of the patients vs normal, and associated with noradrenaline with exerting a local autonomic effect[29]. Lazarova et al[30] (2000) confirmed this finding; however, they found that CGRP was also non-significantly increased in vitiligo. Precipitating factor, as, stress, produce significant level of neuropeptides such as NPY that induct the disease[30,31]. A cohort study revealed increased levels of nerve growth factor (NGF) significantly in vitiligo[32]. Stress up regulates NGF expression in hair follicles, decreases the high affinity TrkA receptor, increases production of p75NTR NGF-receptor, and increases in dorsal root ganglia the substance P neurons[33].
Catecholamine metabolite levels [homovanillic acid (HVA), vanilmandelic acid (VMA), 3-methoxytyramine (MT), normetanephrine (NMN), metanephrine (MN), 3,4-dihydroxy mandelic acid (DOMAC), and 3,4-dihydroxy phenylacetic acid (DOPAC)] were measured in 1-d urinary samples of 150 vitiliginous subjects and 50 normal subjects. HVA and VMA levels corresponded to the activity of the disease[34]. Stressors result in catecholamines discharge, which bind α-R in the mucosa and skin arteriolar wall leading to vasoconstriction, hypoxia, and overproduction of oxygen radicals that destroy melanocytes[34]. Mental stress could stimulate the hypothalamic-pituitary-adrenal axis and then secretion of catecholamines[34,35].
The autoimmune hypothesis
The etio-pathogenesis of “generalized” or non-segmental vitiligo is better explained by autoimmune mechanisms as vitiligo often has autoimmune comorbidities and it often responds to immunosuppressive treatments[36]. The reaction of immunity are cell-mediated, humoral (antibody-mediated), or through the cytokines.
The role of humoral immunity: In 2010, tyrosine hydroxylase antibodies checked with radioimmunoassay (RIA) in sera were obtained from 79 non-SV patients, 8 patients with SV, 91 subjects with other autoimmune diseases and 28 healthy subjects. TH antibodies revealed significantly in non-SV. Also, in non-SV, antibodies against MCHR1 (melanin-concentrating hormone receptor 1), tyrosinase[37] and pigment cell-surface antigens[38] were noted.
In 80% of active vitiligo patients, immunoglobulin G (IgG) and immunoglobulin M (IgM) against melanocytes were found. Low levels IgA also found in the inactive and control groups[38].
Furthermore, anti-thyroglobulin antibodies, antithyroid antibodies, anti-thyroperoxidase, and antismooth muscle antibody are present. Those are typically related to thyroid disease and other autoimmune diseases[39,40].
Melanin concentrating hormone (MCH) binds MCHR1 thus increase calcium influx and acting as an antagonist of α-melanocyte-stimulating hormone (α-MSH)[41-43].
The role of cell-mediated immunity: Immunohisto-chemical examination of the inflammatory infiltrates in perilesional vitiligo skin using single and double immunostaining for melanocytes, Langerhans cells, T-cells, and macrophages revealed higher densities of melanocytes in normal skin, vs non-affected skin in subjects with vitiligo. These T cell had dramatic production of (IL-2R), and increased CD8:CD4 ratio. Thus, melanocytes destruction may be cytotoxic CD8 T-cell mediated. Perilesional HLA-DR production (MHC class II receptor) exhibited in all of the patients with vitiligo, especially along suprabasal and basal keratinocytes, due to local T cell reactivity. In addition, macrophages were numerous in vitiligo vs controls, whereas the CD36 subset of macrophages were higher in the later[44].
The role of cytokines in vitiligo: Beyond lymphocytes and antibodies, the immune system has a complex interplay of many cytokines. There are significantly increased expression of tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), and IL-10[45]. As IFN-γ and TNF-α are T helper cell-1 (Th1) cytokines, so vitiligo is mediated by the Th1 response[46].
IL-17 plays role with macrophages, keratinocytes, and fibroblasts. In addition, it activates the expression of others, as IL-1 and IL-6, and TNF-α[47,48]. Examination of sera and tissue of 30 vitiliginous subjects and 20 normal subjects showed significant higher levels of IL-17 toward vitiliginous subjects and disease duration[47].
The biochemical Theory- reactive oxygen species model
Oxidative stress hypothesis suggests that imbalanced redox (reduction-oxidation) state of the vitiliginous skin. This results in the dramatic production of reactive oxygen species (ROS), as H2O2. ROS oxidize components of the cell leading to melanocytes destruction and creating the depigmented macules[49].
The redox status of vitiligo patients: Sera of thirty-six patients with vitiligo (18 with inactive and 18 with unstable disease), and 40 normal subjects were examined for main factors of redox status involving selenium, malondialdehyde (MDA), vitamins A and E, and glutathione peroxidase (GPx) in the erythrocyte activities, catalase (CAT) and superoxide dismutase (SOD). Superoxide radicals are scavenged and their toxicity is reduced with SOD which transforms O2- to H2O2 and O2, and catalase transforms (H2O2) to (H2O) and (O2)[50]. As MDA results from lipid peroxidation, it is a marker of oxidative stress[51,52]. Selenium is required for GPx activity and vitamins A and E are important in antioxidant activity. Serum selenium, SOD and MDA are prominent in both unstable and inactive types. By enhancing SOD activity, the H2O2 accumulates. In addition, GPx detoxifies H2O2 (downstream enzyme). Therefore, GPx levels decreased in patients with vitiligo[50,53].
Increased SOD activity in patients with vitiligo is a response to oxidative stress; thus, H2O2 elevate as could not be eradicated by low level of CAT[53,54].
The role of tetrahydrobiopterin recycling in vitiligo: Another cellular pathway affected by accumulating H2O2 involves tetrahydrobiopterin. Tyrosinase is an imperative enzyme in formation of melanin[55]. L-tyrosine synthased from L-phenylalanine by the phenylalanine hydroxylase (PAH). The 5, 6, 7, 8-tetrahydrobiopterin or 6BH4 is essential cofactor for this process is. Defective recycling of 6BH4 lead to excess 7BH4 that is an inhibitor of PAH. Uncoupled PAH and 7BH4 found in suction blister material from the skin of vitiligo patients[56]. Kowlessur et al[57] (1996) also found that 7BH4 production yields H2O2. Haase et al[58] (2004) studied the enzyme dihydropteridine reductase (DHPR) (imperative to end the normal 6BH4 recycling). They assessed whole blood samples from 27 untreated vitiligo subjects and 8 normal subjects. The results showed that DHPR activity decrease with high concentrations of H2O2 and vice versa.
Effect of H2O2 on acetylcholinesterase (AchE) decrease in patients with vitiligo vs healthy controls[59,60]. Thus, AchE dependent on H2O2 concentration levels, i.e., low H2O2 concentrations (approximately 10-6M or mol/L) activate AchE whereas high concentrations (10-3M or mol/L) deactivate AchE[60]. Butyrylcholinesterase (BchE) mediates the hydrolysis of acetylcholine. The hydrolysis reaction is one of the rate-limiting steps in cholinergic signal transduction[61,62].
In 2008, showed that xanthine oxidase (XO) is a source of H2O2. High concentrations of H2O2 inhibit the activity of XO, and vice versa[63].
Briefly, there are at least 5 important pathways enrolled in H2O2 overproduction in vitiligo: (1) Defective recycling of 6BH4[56,64,65]; (2) Catecholamine formation increased as levels of monoamine oxidase A (MAO) increased[34,66,67]; (3) Inhibition of thioredoxin/thioredoxin reductase by calcium[68-70]; (4) NADPH oxidase activities increased by the cellular infiltrate[71]; and (5) Nitric oxide synthase (NOS) activities increased[71].
Oxidative stress affects calcium homeostasis at the cellular level[72] in melanocytes and keratinocytes in vitiliginous patients[69].
Zinc-α2-Glycoprotein deficiency hypothesis
For the first time, Bagherani et al[73] and Yaghoobi et al[74] pointed the probable association which might be present between ZAG and vitiligo[73,74]. It was suggested that the pathogenesis of vitiligo could be attributed to decrease in ZAG as follows: (1) Studies have demonstrated that ZAG is acting as a keratinocyte-derived factor influencing melanocyte proliferation and dendricity[75,76]. So, ZAG could be considered as a marker of cells differentiation and maturation[76]; (2) A chronic detachment of melanocytes is an imperative pillar in the pathogenesis of vitiligo[74,77,78]. Thus, melanocyte adhesions to the other cells in epidermis will be impaired in the lack of ZAG; (3) Topical steroids are the most safeand effective forms of treatment for vitiligo, especially for the localized one[74,79], because of their ability to increase ZAG expression[80,81]; (4) Some studies have shown that zinc can precipitate ZAG[74,82]. Thus, the effectivity of zinc in treating this disease is related to its ability to precipitate circulating ZAG at the site of vitiligo[73]; and (5) The linkage signals on chromosome 7 in patients with GV and associated autoimmune diseases have been reported[73,83]. Surprisingly, ZAG gene is located on the chromosome 7[76].
Viral theory
There is a strong association between vitiligo and chronic hepatitis C virus (HCV) infection and autoimmune hepatitis[84]. Akcan et al[85] in 2006 reported a low hepatitis B virus (HBV) sero-positivity in vitiliginous patients. Previous or concurrent cytomegalovirus (CMV) infections may induce the etio-pathogenesis or deterioration of vitiligo[85,86].
Furthermore, other viruses as Epstein-Barr virus, hepatitis E virus, herpes virus and the human immunodeficiency virus (HIV) also have suspicious association with vitiligo[86,87].
Intrinsic theory
Melanocytes in vitiligo have an intrinsic defect leading to their death. They demonstrate different abnormalities, including abnormal rough endoplasmic reticulum or deficiency of unidentified melanocyte growth factors such as basic fibroblast growth factor (bFGF) and decrease in the number of melanocytes expressing the c-kit receptor in lesional skin[88,89].
Melanocytes require a constant keratinocyte-derived c-Kit stimulation for their maintenance[90], thus weak expression of keratinocyte-derived factors, as stem cell factor (SCF), may lead to passive melanocyte death and might explain the Koebner phenomenon[91].
Cellular, molecular and biochemical alterations and melanocytes loss of in vitiligo
Recently, malfunctioning melanocytes found in vitiliginous lesions[92]. Electron microscopy, reverse transcription PCR (polymerase chain reaction) and southern blotting experiments revealed sporadic survival of melanocyte in vitiliginous lesions[93,94]. Thus, this points to presence of immature melanocyte precursors/stem cells[95,96]. Now, 2 pathways have been supposed for melanocytes loss: highly programmed death by apoptosis[77,91,97,98] and accelerated cell senescence[99].
Apoptosis and accelerated cell senescence: Melanocytes from non-lesional skin of vitiligo patients have abnormalities as cytoplasm vacuolization, rough endoplasmic reticulum dilatation, DNA marginalization in the nucleus, loss of dendrites and detachment[88,99,100].
Regarding keratinocytes, apoptosis occurs at least in the traumatized vitiligo skin[101]. Thus, basal and suprabasal epidermal cells in the depigmented and normally pigmented skin show degeneration due to swelling of the membrane-bound organelles, formation of vacuoles and cytoplasm condensation[102].
As vitiligo could inducted by trauma (Koebner’s phenomenon). Lee et al[97] revealed the lower expression levels of the antiapoptotic Bcl-2 and FLIP proteins in vitiliginous skin vs the normally pigmented skin. On the other hand, there was dramatic levels of the proapoptoticBax and p53 proteins and of the active forms of caspase-3, 8 and 9[97].
Apoptosis triggered by normal developmental program, UV light, H2O2, staurosporine and other stimuli[91,103]. NALP1 gene that stimulates cellular apoptosis[102,104], is associated with vitiligo susceptibility[105,106].
Epidermal melanocytes from epidermal melanin unit produce growth factors (GF) for melanocytes[107]. Therefore, its damage have imperative effects on melanocyte survival[91]. Thus, low levels of GF as SCF, endothelin-1 (ET-1) or high levels of melanocyte inhibiting cytokines, as TNF-α and IL-6 may lead to keratinocyte apoptosis, and then apoptosis of melanocytes[108].
Life span of lesional keratinocytes is greatly shortened when compared to the life span of normal and non-lesional vitiligo keratinocytes. It also shows modification of proliferation and senescence marker expression (p16, p53, p21), when compared to keratinocytes from clinically noninvolved skin[98].
Although apoptosis and senescence of epidermal keratinocytes is a response to various stimuli, they also share some cellular mechanisms and controlled by similar molecular regulators[109]. Both apoptosis and aging induced by stress signals as ROS accumulation and DNA damage[110]. The paradigmatic proapoptotic factor p53[111], is also a guard keeper of DNA integrity which triggers cell cycle arrest in DNA damaged cells[109].
Melanocytorrhagy theory: Gauthier et al[78] in 2003 mentioned that NSV occurs due to “melanocytorrhagy”, or a chronic melanocytes detachment and loss caused by trauma and other stressors include catecholamines, ROS, or autoimmune elements. This theory combined the concepts from other theories mentioned before to elaborate a single integrated explanation of vitiligo pathogenesis[78].
A study done by Tobin et al[92] in 2000 proposed loss of melanocytes in vitiligo. They explained these findings because of oxidative stress caused by H2O2. Gauthier et al[78] (2003) also reported that impaired cell adhesion plays a role in vitiligo pathogenesis as the synthesis of extracellular matrix components by keratinocytes may be defective, the presence of focal gaps in the basement membrane and impaired formation of basement membrane. These abnormalities weaken the basal attachment of melanocytes. Trauma could aggravate this susceptibility with subsequent chronic melanocyte loss, known as melanocytorrhagy.
Le Poole et al[112] mentioned that the protein tenascin may play a role in decreasing melanocytes adhesion in vitiligo. This protein was highly expressed in patients with vitiligo than the controls[112]. This can explain the development of vitiligo by Koebner phenomenon, which represent “transepidermal migration”[78,113].
Integrated theory (Conversion theory)
Despite all the mentioned theories are attractive, it is likely that vitiligo is a result of the convergence of these pathological pathways. Most experts agree that vitiligo may be a syndrome with a multi-factorial etiology rather than a single entity[114] (Figure 2).
Figure 2.
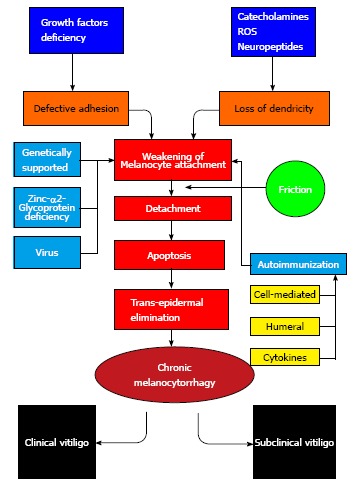
Pathogenesis of vitiligo.
TREATMENT
The disfigurement associated with vitiligo could cause serious emotional stress for the patient and affect his quality of life[115]. Sun protection of vitiliginous areas with sun blocks is imperative[115,116] to prevent sunburn, photodamage and occurrence of Koebner phenomenon. In addition, sun blocks decrease tanning of the uninvolved skin and thus lessen the contrast with vitiliginous lesions[117].
There is no treatment ensures complete cure of vitiligo. Therefore, there is a plethora of modalities, such as topical corticosteroids, vitamin-D derivatives, calcineurin inhibitors, photochemotherapy [psoralen plus UV-A (PUVA), psoralen with sunlight (PUVAsol)], phototherapy (UV-A, narrowband UV-B), surgical techniques[117-122], excimer laser[115,117,118-123], topical prostaglandin E (PGE2)[118], and combinations of topical therapies and light treatment[79]. Complementary and integrative therapies are also used, as ginkgo biloba[79], and levamisole[124], because of their immune-modulating properties[117].
Pseudocatalase cream with Dead Sea climatotherapy can also promote repigmentation[117]. Topical fluorouracil[125], topical melagenina I and II, minoxidil[118], oral L-phenylalanine[117,126-129], homeopathy, ayurvedic medicine, climtologic and balneologic therapies are other therapeutic options for vitiligo[118]. Patients with widespread disease (affecting more than half of body) seeking stable matching of skin color but for whom repigmentation is not expected will be more satisfied if normal pigmented areas are depigmented with 20% monobenzyl ether of hydroquinone, twice per day for almost one year. Another helpful modality for vitiligo universalis, is combined both the topical application of 4-methoxyphenol and the Q-switched (QS) ruby laser[115]. Q-Switched ruby laser destroy the melanosomes present in melanocytes and keratinocytes by selective photothermolysis[130].
According to the impact of oxidative stress on vitiligo, α-tochopherol can be used alone or with topical corticosteroids in combinedation with psoralen plus ultraviolet A (PUVA)[131,132]. Antioxidant pool (tochopherol acetate, ubiquinone, selenomethionine, methionine) could be used in vitiligo aiming to enhance the enzymatic and the non-enzymatic antioxidant activity[131,132]. Food additives, contaminants, preservatives and cosmetic products could exacerbate vitiligo due to oxidative stress in melanocytes[133].
High consumption of omega-6 and decreased omega-3 intake produce free radicals and pro-inflammatory cytokines. On the other hand, omega-3 intake exert protection by enhancing TGF-β mRNA levels and antioxidant enzymes[134] and inhibiting pro-inflammatory cytokines as TNF-α[135].
Cell membranes enriched with omega-3 polyunsa-turated fatty acids have elevated activity of glutathione peroxidase (GSH)[136]. In addition, omega-3 fatty acids contains indole-3-carbinol which activates CYP1A1 that is responsible for hydroxylation of estrogens to 2-hydroxyestrone[137]. Furthermore, omega-3 fatty acids have a vital role in the function of the central nervous system and affect the susceptibility and prognosis of depression[135]. Twenty percent of patients with vitiligo are found to have depression. This highlights the benefits of these lipids in vitiligo[137].
In conclusion, many theories were elaborated to clarify vitiligo pathogenesis. It is a multifactorial disease involving the interplay of several factors. Future research is needed to clarify the interaction of these factors for better understanding of vitiligo pathogenesis and subsequent successful treatment.
Footnotes
P- Reviewer: Kaliyadan F, Kostoglou-Athanassiou I S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
Conflict-of-interest: There are no conflicts of interest. There are no sponsors or fund for the research.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: September 28, 2014
First decision: October 28, 2014
Article in press: January 12, 2015
References
- 1.Lerner AB. Vitiligo. J Invest Dermatol. 1959;32:285–310. [PubMed] [Google Scholar]
- 2.Wolff K, Johnson RA, Suurmond D. Section 13: Pigmentary Disorders-VITILIGO. In: Fitzpatrick's Color Atlas & Synopsis of Clinical Dermatology, 6th ed., editors. New York: McGraw-Hill; 2009. Available from: http: //www.accessmedicine.com.login.ezproxy.library.ualberta.ca/content.aspx?aID=5187746. [Google Scholar]
- 3.Njoo MD, Westerhof W. Vitiligo. Pathogenesis and treatment. Am J Clin Dermatol. 2001;2:167–181. doi: 10.2165/00128071-200102030-00006. [DOI] [PubMed] [Google Scholar]
- 4.Nath SK, Majumder PP, Nordlund JJ. Genetic epidemiology of vitiligo: multilocus recessivity cross-validated. Am J Hum Genet. 1994;55:981–990. [PMC free article] [PubMed] [Google Scholar]
- 5.Kim SM, Chung HS, Hann SK. The genetics of vitiligo in Korean patients. Int J Dermatol. 1998;37:908–910. doi: 10.1046/j.1365-4362.1998.00549.x. [DOI] [PubMed] [Google Scholar]
- 6.Spritz RA, Gowan K, Bennett DC, Fain PR. Novel vitiligo susceptibility loci on chromosomes 7 (AIS2) and 8 (AIS3), confirmation of SLEV1 on chromosome 17, and their roles in an autoimmune diathesis. Am J Hum Genet. 2004;74:188–191. doi: 10.1086/381134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birlea SA, Jin Y, Bennett DC, Herbstman DM, Wallace MR, McCormack WT, Kemp EH, Gawkrodger DJ, Weetman AP, Picardo M, et al. Comprehensive association analysis of candidate genes for generalized vitiligo supports XBP1, FOXP3, and TSLP. J Invest Dermatol. 2011;131:371–381. doi: 10.1038/jid.2010.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet. 2002;3:662–673. doi: 10.1038/nrg887. [DOI] [PubMed] [Google Scholar]
- 9.Zhao M, Gao F, Wu X, Tang J, Lu Q. Abnormal DNA methylation in peripheral blood mononuclear cells from patients with vitiligo. Br J Dermatol. 2010;163:736–742. doi: 10.1111/j.1365-2133.2010.09919.x. [DOI] [PubMed] [Google Scholar]
- 10.Szalmás A, Bánáti F, Koroknai A, László B, Fehér E, Salamon D, Gergely L, Minárovits J, Kónya J. Lineage-specific silencing of human IL-10 gene expression by promoter methylation in cervical cancer cells. Eur J Cancer. 2008;44:1030–1038. doi: 10.1016/j.ejca.2008.02.046. [DOI] [PubMed] [Google Scholar]
- 11.Yun JY, Uhm YK, Kim HJ, Lim SH, Chung JH, Shin MK, Yim SV, Lee MH. Transforming growth factor beta receptor II (TGFBR2) polymorphisms and the association with nonsegmental vitiligo in the Korean population. Int J Immunogenet. 2010;37:289–291. doi: 10.1111/j.1744-313X.2010.00923.x. [DOI] [PubMed] [Google Scholar]
- 12.Basak PY, Adiloglu AK, Ceyhan AM, Tas T, Akkaya VB. The role of helper and regulatory T cells in the pathogenesis of vitiligo. J Am Acad Dermatol. 2009;60:256–260. doi: 10.1016/j.jaad.2008.09.048. [DOI] [PubMed] [Google Scholar]
- 13.Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–699. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 14.Jeong TJ, Shin MK, Uhm YK, Kim HJ, Chung JH, Lee MH. Association of UVRAG polymorphisms with susceptibility to non-segmental vitiligo in a Korean sample. Exp Dermatol. 2010;19:e323–e325. doi: 10.1111/j.1600-0625.2009.01039.x. [DOI] [PubMed] [Google Scholar]
- 15.Birlea SA, Gowan K, Fain PR, Spritz RA. Genome-wide association study of generalized vitiligo in an isolated European founder population identifies SMOC2, in close proximity to IDDM8. J Invest Dermatol. 2010;130:798–803. doi: 10.1038/jid.2009.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu P, Pazin DE, Merson RR, Albrecht KH, Vaziri C. The developmentally-regulated Smoc2 gene is repressed by Aryl-hydrocarbon receptor (Ahr) signaling. Gene. 2009;433:72–80. doi: 10.1016/j.gene.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maier S, Paulsson M, Hartmann U. The widely expressed extracellular matrix protein SMOC-2 promotes keratinocyte attachment and migration. Exp Cell Res. 2008;314:2477–2487. doi: 10.1016/j.yexcr.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 18.Kingo K, Philips MA, Aunin E, Luuk H, Karelson M, Rätsep R, Silm H, Vasar E, Kõks S. MYG1, novel melanocyte related gene, has elevated expression in vitiligo. J Dermatol Sci. 2006;44:119–122. doi: 10.1016/j.jdermsci.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 19.Hu DY, Ren YQ, Zhu KJ, Lv YM, Cheng H, Zhang Z, Li Y, He SM, Tang J, Liu JL, et al. Comparisons of clinical features of HLA-DRB1*07 positive and negative vitiligo patients in Chinese Han population. J Eur Acad Dermatol Venereol. 2011;25:1299–1303. doi: 10.1111/j.1468-3083.2010.03971.x. [DOI] [PubMed] [Google Scholar]
- 20.Misri R, Khopkar U, Shankarkumar U, Ghosh K. Comparative case control study of clinical features and human leukocyte antigen susceptibility between familial and nonfamilial vitiligo. Indian J Dermatol Venereol Leprol. 2009;75:583–587. doi: 10.4103/0378-6323.57719. [DOI] [PubMed] [Google Scholar]
- 21.Quan C, Ren YQ, Xiang LH, Sun LD, Xu AE, Gao XH, Chen HD, Pu XM, Wu RN, Liang CZ, et al. Genome-wide association study for vitiligo identifies susceptibility loci at 6q27 and the MHC. Nat Genet. 2010;42:614–618. doi: 10.1038/ng.603. [DOI] [PubMed] [Google Scholar]
- 22.Thompson DM, Parker R. The RNase Rny1p cleaves tRNAs and promotes cell death during oxidative stress in Saccharomyces cerevisiae. J Cell Biol. 2009;185:43–50. doi: 10.1083/jcb.200811119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Acquaviva C, Chevrier V, Chauvin JP, Fournier G, Birnbaum D, Rosnet O. The centrosomal FOP protein is required for cell cycle progression and survival. Cell Cycle. 2009;8:1217–1227. doi: 10.4161/cc.8.8.8248. [DOI] [PubMed] [Google Scholar]
- 24.Silva de Castro CC, do Nascimento LM, Walker G, Werneck RI, Nogoceke E, Mira MT. Genetic variants of the DDR1 gene are associated with vitiligo in two independent Brazilian population samples. J Invest Dermatol. 2010;130:1813–1818. doi: 10.1038/jid.2010.34. [DOI] [PubMed] [Google Scholar]
- 25.Yoshimura T, Matsuyama W, Kamohara H. Discoidin domain receptor 1: a new class of receptor regulating leukocyte-collagen interaction. Immunol Res. 2005;31:219–230. doi: 10.1385/IR:31:3:219. [DOI] [PubMed] [Google Scholar]
- 26.Koga M, Tango T. Clinical features and course of type A and type B vitiligo. Br J Dermatol. 1988;118:223–228. doi: 10.1111/j.1365-2133.1988.tb01778.x. [DOI] [PubMed] [Google Scholar]
- 27.Manolache L, Benea V. Stress in patients with alopecia areata and vitiligo. J Eur Acad Dermatol Venereol. 2007;21:921–928. doi: 10.1111/j.1468-3083.2006.02106.x. [DOI] [PubMed] [Google Scholar]
- 28.Wu CS, Yu HS, Chang HR, Yu CL, Yu CL, Wu BN. Cutaneous blood flow and adrenoceptor response increase in segmental-type vitiligo lesions. J Dermatol Sci. 2000;23:53–62. doi: 10.1016/s0923-1811(99)00090-0. [DOI] [PubMed] [Google Scholar]
- 29.Al’Abadie MS, Senior HJ, Bleehen SS, Gawkrodger DJ. Neuropeptide and neuronal marker studies in vitiligo. Br J Dermatol. 1994;131:160–165. doi: 10.1111/j.1365-2133.1994.tb08486.x. [DOI] [PubMed] [Google Scholar]
- 30.Lazarova R, Hristakieva E, Lazarov N, Shani J. Vitiligo-related neuropeptides in nerve fibers of the skin. Arch Physiol Biochem. 2000;108:262–267. doi: 10.1076/1381345520000710831zft262. [DOI] [PubMed] [Google Scholar]
- 31.Yehuda R, Brand S, Yang RK. Plasma neuropeptide Y concentrations in combat exposed veterans: relationship to trauma exposure, recovery from PTSD, and coping. Biol Psychiatry. 2006;59:660–663. doi: 10.1016/j.biopsych.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 32.Rateb AAH, Azzam OA, Rashed LA, El-Guindy NM, El-Din MS. The role of nerve growth factor in the pathogenesis of vitligo. JEWDS. 2005;1:18–24. Available from: http: //www.jewds.eg.net/pdf/2004/w1a1a3.PDF. [Google Scholar]
- 33.Peters EM, Handjiski B, Kuhlmei A, Hagen E, Bielas H, Braun A, Klapp BF, Paus R, Arck PC. Neurogenic inflammation in stress-induced termination of murine hair growth is promoted by nerve growth factor. Am J Pathol. 2004;165:259–271. doi: 10.1016/S0002-9440(10)63294-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morrone A, Picardo M, de Luca C, Terminali O, Passi S, Ippolito F. Catecholamines and vitiligo. Pigment Cell Res. 1992;5:65–69. doi: 10.1111/j.1600-0749.1992.tb00003.x. [DOI] [PubMed] [Google Scholar]
- 35.Stokes PE, Sikes CR. The hypothalamic-pituitary-adrenocortical axis in major depression. Neurol Clin. 1988;6:1–19. [PubMed] [Google Scholar]
- 36.Lepe V, Moncada B, Castanedo-Cazares JP, Torres-Alvarez MB, Ortiz CA, Torres-Rubalcava AB. A double-blind randomized trial of 0.1% tacrolimus vs 0.05% clobetasol for the treatment of childhood vitiligo. Arch Dermatol. 2003;139:581–585. doi: 10.1001/archderm.139.5.581. [DOI] [PubMed] [Google Scholar]
- 37.Kemp EH, Emhemad S, Akhtar S, Watson PF, Gawkrodger DJ, Weetman AP. Autoantibodies against tyrosine hydroxylase in patients with non-segmental (generalised) vitiligo. Exp Dermatol. 2011;20:35–40. doi: 10.1111/j.1600-0625.2010.01181.x. [DOI] [PubMed] [Google Scholar]
- 38.Harning R, Cui J, Bystryn JC. Relation between the incidence and level of pigment cell antibodies and disease activity in vitiligo. J Invest Dermatol. 1991;97:1078–1080. doi: 10.1111/1523-1747.ep12492607. [DOI] [PubMed] [Google Scholar]
- 39.Ingordo V, Gentile C, Iannazzone SS, Cusano F, Naldi L. Vitiligo and autoimmunity: an epidemiological study in a representative sample of young Italian males. J Eur Acad Dermatol Venereol. 2011;25:105–109. doi: 10.1111/j.1468-3083.2010.03696.x. [DOI] [PubMed] [Google Scholar]
- 40.Uncu S, Yaylı S, Bahadır S, Okten A, Alpay K. Relevance of autoimmune thyroiditis in children and adolescents with vitiligo. Int J Dermatol. 2011;50:175–179. doi: 10.1111/j.1365-4632.2010.04665.x. [DOI] [PubMed] [Google Scholar]
- 41.Chambers J, Ames RS, Bergsma D, Muir A, Fitzgerald LR, Hervieu G, Dytko GM, Foley JJ, Martin J, Liu WS, et al. Melanin-concentrating hormone is the cognate ligand for the orphan G-protein-coupled receptor SLC-1. Nature. 1999;400:261–265. doi: 10.1038/22313. [DOI] [PubMed] [Google Scholar]
- 42.Kemp EH, Waterman EA, Hawes BE, O’Neill K, Gottumukkala RV, Gawkrodger DJ, Weetman AP, Watson PF. The melanin-concentrating hormone receptor 1, a novel target of autoantibody responses in vitiligo. J Clin Invest. 2002;109:923–930. doi: 10.1172/JCI14643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ancans J, Hoogduijn MJ, Thody AJ. Melanosomal pH, pink locus protein and their roles in melanogenesis. J Invest Dermatol. 2001;117:158–159. doi: 10.1046/j.0022-202x.2001.01397.x. [DOI] [PubMed] [Google Scholar]
- 44.Le Poole IC, van den Wijngaard RM, Westerhof W, Das PK. Presence of T cells and macrophages in inflammatory vitiligo skin parallels melanocyte disappearance. Am J Pathol. 1996;148:1219–1228. [PMC free article] [PubMed] [Google Scholar]
- 45.Grimes PE, Morris R, Avaniss-Aghajani E, Soriano T, Meraz M, Metzger A. Topical tacrolimus therapy for vitiligo: therapeutic responses and skin messenger RNA expression of proinflammatory cytokines. J Am Acad Dermatol. 2004;51:52–61. doi: 10.1016/j.jaad.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 46.Taher ZA, Lauzon G, Maguiness S, Dytoc MT. Analysis of interleukin-10 levels in lesions of vitiligo following treatment with topical tacrolimus. Br J Dermatol. 2009;161:654–659. doi: 10.1111/j.1365-2133.2009.09217.x. [DOI] [PubMed] [Google Scholar]
- 47.Bassiouny DA, Shaker O. Role of interleukin-17 in the pathogenesis of vitiligo. Clin Exp Dermatol. 2011;36:292–297. doi: 10.1111/j.1365-2230.2010.03972.x. [DOI] [PubMed] [Google Scholar]
- 48.Kolls JK, Lindén A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 49.Khan R, Satyam A, Gupta S, Sharma VK, Sharma A. Circulatory levels of antioxidants and lipid peroxidation in Indian patients with generalized and localized vitiligo. Arch Dermatol Res. 2009;301:731–737. doi: 10.1007/s00403-009-0964-4. [DOI] [PubMed] [Google Scholar]
- 50.Ines D, Sonia B, Riadh BM, Amel el G, Slaheddine M, Hamida T, Hamadi A, Basma H. A comparative study of oxidant-antioxidant status in stable and active vitiligo patients. Arch Dermatol Res. 2006;298:147–152. doi: 10.1007/s00403-006-0680-2. [DOI] [PubMed] [Google Scholar]
- 51.Latha B, Babu M. The involvement of free radicals in burn injury: a review. Burns. 2001;27:309–317. doi: 10.1016/s0305-4179(00)00127-3. [DOI] [PubMed] [Google Scholar]
- 52.Yildirim M, Baysal V, Inaloz HS, Can M. The role of oxidants and antioxidants in generalized vitiligo at tissue level. J Eur Acad Dermatol Venereol. 2004;18:683–686. doi: 10.1111/j.1468-3083.2004.01080.x. [DOI] [PubMed] [Google Scholar]
- 53.Dammak I, Boudaya S, Ben Abdallah F, Turki H, Attia H, Hentati B. Antioxidant enzymes and lipid peroxidation at the tissue level in patients with stable and active vitiligo. Int J Dermatol. 2009;48:476–480. doi: 10.1111/j.1365-4632.2009.03998.x. [DOI] [PubMed] [Google Scholar]
- 54.Sravani PV, Babu NK, Gopal KV, Rao GR, Rao AR, Moorthy B, Rao TR. Determination of oxidative stress in vitiligo by measuring superoxide dismutase and catalase levels in vitiliginous and non-vitiliginous skin. Indian J Dermatol Venereol Leprol. 2009;75:268–271. doi: 10.4103/0378-6323.48427. [DOI] [PubMed] [Google Scholar]
- 55.Prota G. The role of peroxidase in melanogenesis revisited. Pigment Cell Res. 1992;Suppl 2:25–31. doi: 10.1111/j.1600-0749.1990.tb00344.x. [DOI] [PubMed] [Google Scholar]
- 56.Schallreuter KU, Wood JM, Pittelkow MR, Gütlich M, Lemke KR, Rödl W, Swanson NN, Hitzemann K, Ziegler I. Regulation of melanin biosynthesis in the human epidermis by tetrahydrobiopterin. Science. 1994;263:1444–1446. doi: 10.1126/science.8128228. [DOI] [PubMed] [Google Scholar]
- 57.Kowlessur D, Citron BA, Kaufman S. Recombinant human phenylalanine hydroxylase: novel regulatory and structural properties. Arch Biochem Biophys. 1996;333:85–95. doi: 10.1006/abbi.1996.0367. [DOI] [PubMed] [Google Scholar]
- 58.Hasse S, Gibbons NC, Rokos H, Marles LK, Schallreuter KU. Perturbed 6-tetrahydrobiopterin recycling via decreased dihydropteridine reductase in vitiligo: more evidence for H2O2 stress. J Invest Dermatol. 2004;122:307–313. doi: 10.1046/j.0022-202X.2004.22230.x. [DOI] [PubMed] [Google Scholar]
- 59.Iyengar B. Modulation of melanocytic activity by acetylcholine. Acta Anat (Basel) 1989;136:139–141. doi: 10.1159/000146813. [DOI] [PubMed] [Google Scholar]
- 60.Schallreuter KU, Elwary SM, Gibbons NC, Rokos H, Wood JM. Activation/deactivation of acetylcholinesterase by H2O2: more evidence for oxidative stress in vitiligo. Biochem Biophys Res Commun. 2004;315:502–508. doi: 10.1016/j.bbrc.2004.01.082. [DOI] [PubMed] [Google Scholar]
- 61.Rakonczay Z, Brimijoin S. Biochemistry and pathophysiology of the molecular forms of cholinesterases. Subcell Biochem. 1988;12:335–378. doi: 10.1007/978-1-4899-1681-5_10. [DOI] [PubMed] [Google Scholar]
- 62.Schallreuter KU, Gibbons NC, Zothner C, Elwary SM, Rokos H, Wood JM. Butyrylcholinesterase is present in the human epidermis and is regulated by H2O2: more evidence for oxidative stress in vitiligo. Biochem Biophys Res Commun. 2006;349:931–938. doi: 10.1016/j.bbrc.2006.08.138. [DOI] [PubMed] [Google Scholar]
- 63.Shalbaf M, Gibbons NC, Wood JM, Maitland DJ, Rokos H, Elwary SM, Marles LK, Schallreuter KU. Presence of epidermal allantoin further supports oxidative stress in vitiligo. Exp Dermatol. 2008;17:761–770. doi: 10.1111/j.1600-0625.2008.00697.x. [DOI] [PubMed] [Google Scholar]
- 64.Schallreuter KU, Büttner G, Pittelkow MR, Wood JM, Swanson NN, Körner C. Cytotoxicity of 6-biopterin to human melanocytes. Biochem Biophys Res Commun. 1994;204:43–48. doi: 10.1006/bbrc.1994.2423. [DOI] [PubMed] [Google Scholar]
- 65.Schallreuter KU, Wood JM, Ziegler I, Lemke KR, Pittelkow MR, Lindsey NJ, Gütlich M. Defective tetrahydrobiopterin and catecholamine biosynthesis in the depigmentation disorder vitiligo. Biochim Biophys Acta. 1994;1226:181–192. doi: 10.1016/0925-4439(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 66.Orecchia G, Frattini P, Cucchi ML, Santagostino G. Normal-range plasma catecholamines in patients with generalized and acrofacial vitiligo: preliminary report. Dermatology. 1994;189:350–353. doi: 10.1159/000246877. [DOI] [PubMed] [Google Scholar]
- 67.Schallreuter KU, Wood JM, Pittelkow MR, Buttner G, Swanson N, Korner C, Ehrke C. Increased monoamine oxidase A activity in the epidermis of patients with vitiligo. Arch Dermatol Res. 1996;288:14–18. doi: 10.1007/BF02505037. [DOI] [PubMed] [Google Scholar]
- 68.Schallreuter KU, Pittelkow MR, Wood JM. Free radical reduction by thioredoxin reductase at the surface of normal and vitiliginous human keratinocytes. J Invest Dermatol. 1986;87:728–732. doi: 10.1111/1523-1747.ep12456848. [DOI] [PubMed] [Google Scholar]
- 69.Schallreuter KU, Pittelkow MP. Defective calcium uptake in keratinocyte cell cultures from vitiliginous skin. Arch Dermatol Res. 1988;280:137–139. doi: 10.1007/BF00456842. [DOI] [PubMed] [Google Scholar]
- 70.Schallreuter KU, Wood JM. EF-hands calcium-binding regulates the thioredoxin reductase/thioredoxin electron transfer in human keratinocytes and melanocytes. In: Heizmann C, editor. Novel calcium-binding proteins. Berlin: Springer Verlag; 1991. pp. 339–360. [Google Scholar]
- 71.Hann SK, Chang JH, Lee HS, Kim SM. The classification of segmental vitiligo on the face. Yonsei Med J. 2000;41:209–212. doi: 10.3349/ymj.2000.41.2.209. [DOI] [PubMed] [Google Scholar]
- 72.Marks DB, Marks AD, Smith CM. Oxygen metabolism and oxygen toxicity. In: Basic Medical Biochemistry a clinical Approach. Williams and Wilkins, Baltimore, Manyland; 1996. pp. 327–340. [Google Scholar]
- 73.Bagherani N, Yaghoobi R, Omidian M. Hypothesis: zinc can be effective in treatment of vitiligo. Indian J Dermatol. 2011;56:480–484. doi: 10.4103/0019-5154.87116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yaghoobi R, Omidian M, Bagherani N. Vitiligo: a review of the published work. J Dermatol. 2011;38:419–431. doi: 10.1111/j.1346-8138.2010.01139.x. [DOI] [PubMed] [Google Scholar]
- 75.Gordon PR, Mansur CP, Gilchrest BA. Regulation of human melanocyte growth, dendricity, and melanization by keratinocyte derived factors. J Invest Dermatol. 1989;92:565–572. doi: 10.1111/1523-1747.ep12709595. [DOI] [PubMed] [Google Scholar]
- 76.Hassan MI, Waheed A, Yadav S, Singh TP, Ahmad F. Zinc alpha 2-glycoprotein: a multidisciplinary protein. Mol Cancer Res. 2008;6:892–906. doi: 10.1158/1541-7786.MCR-07-2195. [DOI] [PubMed] [Google Scholar]
- 77.Gauthier Y, Cario Andre M, Taïeb A. A critical appraisal of vitiligo etiologic theories. Is melanocyte loss a melanocytorrhagy? Pigment Cell Res. 2003;16:322–332. doi: 10.1034/j.1600-0749.2003.00070.x. [DOI] [PubMed] [Google Scholar]
- 78.Gauthier Y, Cario-Andre M, Lepreux S, Pain C, Taïeb A. Melanocyte detachment after skin friction in non lesional skin of patients with generalized vitiligo. Br J Dermatol. 2003;148:95–101. doi: 10.1046/j.1365-2133.2003.05024.x. [DOI] [PubMed] [Google Scholar]
- 79.Whitton ME, Ashcroft DM, González U. Therapeutic interventions for vitiligo. J Am Acad Dermatol. 2008;59:713–717. doi: 10.1016/j.jaad.2008.06.023. [DOI] [PubMed] [Google Scholar]
- 80.Bing C, Bao Y, Jenkins J, Sanders P, Manieri M, Cinti S, Tisdale MJ, Trayhurn P. Zinc-alpha2-glycoprotein, a lipid mobilizing factor, is expressed in adipocytes and is up-regulated in mice with cancer cachexia. Proc Natl Acad Sci USA. 2004;101:2500–2505. doi: 10.1073/pnas.0308647100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Russell ST, Tisdale MJ. The role of glucocorticoids in the induction of zinc-alpha2-glycoprotein expression in adipose tissue in cancer cachexia. Br J Cancer. 2005;92:876–881. doi: 10.1038/sj.bjc.6602404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hale LP. Zinc alpha-2-glycoprotein regulates melanin production by normal and malignant melanocytes. J Invest Dermatol. 2002;119:464–470. doi: 10.1046/j.1523-1747.2002.01813.x. [DOI] [PubMed] [Google Scholar]
- 83.Boissy RE, Spritz RA. Frontiers and controversies in the pathobiology of vitiligo: separating the wheat from the chaff. Exp Dermatol. 2009;18:583–585. doi: 10.1111/j.1600-0625.2008.00826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Akbayir N, Gökdemir G, Mansur T, Sökmen M, Gündüz S, Alkim C, Barutcuoglu B, Erdem L. Is there any relationship between hepatitis C virus and vitiligo? J Clin Gastroenterol. 2004;38:815–817. doi: 10.1097/01.mcg.0000139052.74801.98. [DOI] [PubMed] [Google Scholar]
- 85.Akcan Y, Kavak A, Sertbas Y, Olut AI, Korkut E, Bicik Z, Kisacik B. The low seropositivity of hepatitis B virus in vitiligo patients. J Eur Acad Dermatol Venereol. 2006;20:110–111. doi: 10.1111/j.1468-3083.2005.01335.x. [DOI] [PubMed] [Google Scholar]
- 86.Toker SC, Sarycaoglu H, Karadogan SK, Mistik R, Baskan EB, Tunaly S. Is there any relation between vitiligo and cytomegalovirus? J Eur Acad Dermatol Venereol. 2007;21:141–142. doi: 10.1111/j.1468-3083.2006.01822.x. [DOI] [PubMed] [Google Scholar]
- 87.Niamba P, Traoré A, Taïeb A. [Vitiligo in a black patient associated with HIV infection and repigmentation under antiretroviral therapy] Ann Dermatol Venereol. 2007;134:272–273. doi: 10.1016/s0151-9638(07)91512-4. [DOI] [PubMed] [Google Scholar]
- 88.Boissy RE, Liu YY, Medrano EE, Nordlund JJ. Structural aberration of the rough endoplasmic reticulum and melanosome compartmentalization in long-term cultures of melanocytes from vitiligo patients. J Invest Dermatol. 1991;97:395–404. doi: 10.1111/1523-1747.ep12480976. [DOI] [PubMed] [Google Scholar]
- 89.Norris A, Todd C, Graham A, Quinn AG, Thody AJ. The expression of the c-kit receptor by epidermal melanocytes may be reduced in vitiligo. Br J Dermatol. 1996;134:299–306. [PubMed] [Google Scholar]
- 90.Wehrle-Haller B. The role of Kit-ligand in melanocyte development and epidermal homeostasis. Pigment Cell Res. 2003;16:287–296. doi: 10.1034/j.1600-0749.2003.00055.x. [DOI] [PubMed] [Google Scholar]
- 91.Lee AY, Kim NH, Choi WI, Youm YH. Less keratinocyte-derived factors related to more keratinocyte apoptosis in depigmented than normally pigmented suction-blistered epidermis may cause passive melanocyte death in vitiligo. J Invest Dermatol. 2005;124:976–983. doi: 10.1111/j.0022-202X.2005.23667.x. [DOI] [PubMed] [Google Scholar]
- 92.Tobin DJ, Swanson NN, Pittelkow MR, Peters EM, Schallreuter KU. Melanocytes are not absent in lesional skin of long duration vitiligo. J Pathol. 2000;191:407–416. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH659>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 93.Kim YC, Kim YJ, Kang HY, Sohn S, Lee ES. Histopathologic features in vitiligo. Am J Dermatopathol. 2008;30:112–116. doi: 10.1097/DAD.0b013e3181651511. [DOI] [PubMed] [Google Scholar]
- 94.Gottschalk GM, Kidson SH. Molecular analysis of vitiligo lesions reveals sporadic melanocyte survival. Int J Dermatol. 2007;46:268–272. doi: 10.1111/j.1365-4632.2006.03000.x. [DOI] [PubMed] [Google Scholar]
- 95.Falabella R. Vitiligo and the melanocyte reservoir. Indian J Dermatol. 2009;54:313–318. doi: 10.4103/0019-5154.57604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Davids LM, du Toit E, Kidson SH, Todd G. A rare repigmentation pattern in a vitiligo patient: a clue to an epidermal stem-cell reservoir of melanocytes? Clin Exp Dermatol. 2009;34:246–248. doi: 10.1111/j.1365-2230.2008.02793.x. [DOI] [PubMed] [Google Scholar]
- 97.Lee AY, Youm YH, Kim NH, Yang H, Choi WI. Keratinocytes in the depigmented epidermis of vitiligo are more vulnerable to trauma (suction) than keratinocytes in the normally pigmented epidermis, resulting in their apoptosis. Br J Dermatol. 2004;151:995–1003. doi: 10.1111/j.1365-2133.2004.06136.x. [DOI] [PubMed] [Google Scholar]
- 98.Bondanza S, Maurelli R, Paterna P, Migliore E, Giacomo FD, Primavera G, Paionni E, Dellambra E, Guerra L. Keratinocyte cultures from involved skin in vitiligo patients show an impaired in vitro behaviour. Pigment Cell Res. 2007;20:288–300. doi: 10.1111/j.1600-0749.2007.00385.x. [DOI] [PubMed] [Google Scholar]
- 99.van den Wijngaard R, Wankowicz-Kalinska A, Le Poole C, Tigges B, Westerhof W, Das P. Local immune response in skin of generalized vitiligo patients. Destruction of melanocytes is associated with the prominent presence of CLA+ T cells at the perilesional site. Lab Invest. 2000;80:1299–1309. doi: 10.1038/labinvest.3780138. [DOI] [PubMed] [Google Scholar]
- 100.Boissy RE. Histology of vitiliginous skin. Hann S, Nordlund JJ, eds.Vitiligo, 1st ed. Oxford: Blackwell Science Ltd; 2008. pp. 23–34. [Google Scholar]
- 101.Moretti S, Fabbri P, Baroni G, Berti S, Bani D, Berti E, Nassini R, Lotti T, Massi D. Keratinocyte dysfunction in vitiligo epidermis: cytokine microenvironment and correlation to keratinocyte apoptosis. Histol Histopathol. 2009;24:849–857. doi: 10.14670/HH-24.849. [DOI] [PubMed] [Google Scholar]
- 102.Tschopp J, Martinon F, Burns K. NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol. 2003;4:95–104. doi: 10.1038/nrm1019. [DOI] [PubMed] [Google Scholar]
- 103.Raj D, Brash DE, Grossman D. Keratinocyte apoptosis in epidermal development and disease. J Invest Dermatol. 2006;126:243–257. doi: 10.1038/sj.jid.5700008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Church LD, Cook GP, McDermott MF. Primer: inflammasomes and interleukin 1beta in inflammatory disorders. Nat Clin Pract Rheumatol. 2008;4:34–42. doi: 10.1038/ncprheum0681. [DOI] [PubMed] [Google Scholar]
- 105.Jin Y, Bennett DC, Amadi-Myers A, Holland P, Riccardi SL, Gowan K, Fain PR, Spritz RA. Vitiligo-associated multiple autoimmune disease is not associated with genetic variation in AIRE. Pigment Cell Res. 2007;20:402–404. doi: 10.1111/j.1600-0749.2007.00398.x. [DOI] [PubMed] [Google Scholar]
- 106.Jin Y, Mailloux CM, Gowan K, Riccardi SL, LaBerge G, Bennett DC, Fain PR, Spritz RA. NALP1 in vitiligo-associated multiple autoimmune disease. N Engl J Med. 2007;356:1216–1225. doi: 10.1056/NEJMoa061592. [DOI] [PubMed] [Google Scholar]
- 107.Imokawa G. Autocrine and paracrine regulation of melanocytes in human skin and in pigmentary disorders. Pigment Cell Res. 2004;17:96–110. doi: 10.1111/j.1600-0749.2003.00126.x. [DOI] [PubMed] [Google Scholar]
- 108.Moretti S, Spallanzani A, Amato L, Hautmann G, Gallerani I, Fabiani M, Fabbri P. New insights into the pathogenesis of vitiligo: imbalance of epidermal cytokines at sites of lesions. Pigment Cell Res. 2002;15:87–92. doi: 10.1034/j.1600-0749.2002.1o049.x. [DOI] [PubMed] [Google Scholar]
- 109.Gandarillas A. Epidermal differentiation, apoptosis, and senescence: common pathways? Exp Gerontol. 2000;35:53–62. doi: 10.1016/s0531-5565(99)00088-1. [DOI] [PubMed] [Google Scholar]
- 110.Cordisco S, Maurelli R, Bondanza S, Stefanini M, Zambruno G, Guerra L, Dellambra E. Bmi-1 reduction plays a key role in physiological and premature aging of primary human keratinocytes. J Invest Dermatol. 2010;130:1048–1062. doi: 10.1038/jid.2009.355. [DOI] [PubMed] [Google Scholar]
- 111.Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 112.Le Poole IC, van den Wijngaard RM, Westerhof W, Das PK. Tenascin is overexpressed in vitiligo lesional skin and inhibits melanocyte adhesion. Br J Dermatol. 1997;137:171–178. doi: 10.1046/j.1365-2133.1997.18011894.x. [DOI] [PubMed] [Google Scholar]
- 113.Cario-André M, Pain C, Gauthier Y, Taïeb A. The melanocytorrhagic hypothesis of vitiligo tested on pigmented, stressed, reconstructed epidermis. Pigment Cell Res. 2007;20:385–393. doi: 10.1111/j.1600-0749.2007.00396.x. [DOI] [PubMed] [Google Scholar]
- 114.Halder RM, Chappell JL. Vitiligo update. Semin Cutan Med Surg. 2009;28:86–92. doi: 10.1016/j.sder.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 115.Lebwohl MG, Heymann WR, Berth-Jones J, Coulson I. Treatment of Skin Disease: Comprehensive Therapeutic Strategies. 2nd ed. Philadelphia, USA: Mosby Elsevier; 2006. pp. 683–687. [Google Scholar]
- 116.Burns T, Breathnach S, Cox N, Griffiths C. Rook’s Textbook of Dermatology. 7th ed, Vol. II. Oxford: Blackwell Science; 2004. pp. 52–57. [Google Scholar]
- 117.Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ. Fitzpatrick's Dermatology in General Medicine. 7th ed, Vol. I. USA: Mac Graw Hill; 2007. pp. 616–621. [Google Scholar]
- 118.Lotti T, Gori A, Zanieri F, Colucci R, Moretti S. Vitiligo: new and emerging treatments. Dermatol Ther. 2008;21:110–117. doi: 10.1111/j.1529-8019.2008.00178.x. [DOI] [PubMed] [Google Scholar]
- 119.Szczurko O, Boon HS. A systematic review of natural health product treatment for vitiligo. BMC Dermatol. 2008;8:2. doi: 10.1186/1471-5945-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bolognia JL, Jorizzo JL, Rapini R. Dermatology. 2nd edn, Vol. I. USA: Mosby Elsivier; 2008. pp. 913–920. [Google Scholar]
- 121.Forschner T, Buchholtz S, Stockfleth E. Current state of vitiligo therapy--evidence-based analysis of the literature. J Dtsch Dermatol Ges. 2007;5:467–475. doi: 10.1111/j.1610-0387.2007.06280.x. [DOI] [PubMed] [Google Scholar]
- 122.Grimes PE. New insights and new therapies in vitiligo. JAMA. 2005;293:730–735. doi: 10.1001/jama.293.6.730. [DOI] [PubMed] [Google Scholar]
- 123.Ostovari N, Passeron T, Zakaria W, Fontas E, Larouy JC, Blot JF, Lacour JP, Ortonne JP. Treatment of vitiligo by 308-nm excimer laser: an evaluation of variables affecting treatment response. Lasers Surg Med. 2004;35:152–156. doi: 10.1002/lsm.20057. [DOI] [PubMed] [Google Scholar]
- 124.Pasricha JS, Khera V. Effect of prolonged treatment with levamisole on vitiligo with limited and slow-spreading disease. Int J Dermatol. 1994;33:584–587. doi: 10.1111/j.1365-4362.1994.tb02903.x. [DOI] [PubMed] [Google Scholar]
- 125.Tsuji T, Hamada T. Topically administered fluorouracil in vitiligo. Arch Dermatol. 1983;119:722–727. [PubMed] [Google Scholar]
- 126.van den Wijngaard R, Wankowicz-Kalinska A, Pals S, Weening J, Das P. Autoimmune melanocyte destruction in vitiligo. Lab Invest. 2001;81:1061–1067. doi: 10.1038/labinvest.3780318. [DOI] [PubMed] [Google Scholar]
- 127.Michaëlsson G, Juhlin L, Vahlquist A. Effects of oral zinc and vitamin A in acne. Arch Dermatol. 1977;113:31–36. doi: 10.1001/archderm.1977.01640010033003. [DOI] [PubMed] [Google Scholar]
- 128.Hillström L, Pettersson L, Hellbe L, Kjellin A, Leczinsky CG, Nordwall C. Comparison of oral treatment with zinc sulphate and placebo in acne vulgaris. Br J Dermatol. 1977;97:681–684. doi: 10.1111/j.1365-2133.1977.tb14277.x. [DOI] [PubMed] [Google Scholar]
- 129.Burrows NP, Turnbull AJ, Punchard NA, Thompson RP, Jones RR. A trial of oral zinc supplementation in psoriasis. Cutis. 1994;54:117–118. [PubMed] [Google Scholar]
- 130.Kim YJ, Chung BS, Choi KC. Depigmentation therapy with Q-switched ruby laser after tanning in vitiligo universalis. Dermatol Surg. 2001;27:969–970. doi: 10.1046/j.1524-4725.2001.01101.x. [DOI] [PubMed] [Google Scholar]
- 131.Koshevenko IuN. [alpha-Tocopherol in the combined treatment of vitiligo] Vestn Dermatol Venerol. 1989;10:70–72. [PubMed] [Google Scholar]
- 132.Mandel AS, Haberman HF, Pawlowski D, Goldstein E. Non PUVA nonsurgical therapies for vitiligo. Clin Dermatol. 1997;15:907–919. doi: 10.1016/s0738-081x(97)00132-6. [DOI] [PubMed] [Google Scholar]
- 133.Bickers DR, Athar M. Oxidative stress in the pathogenesis of skin disease. J Invest Dermatol. 2006;126:2565–2575. doi: 10.1038/sj.jid.5700340. [DOI] [PubMed] [Google Scholar]
- 134.Fernandes G. Dietary lipids and risk of autoimmune disease. Clin Immunol Immunopathol. 1994;72:193–197. doi: 10.1006/clin.1994.1129. [DOI] [PubMed] [Google Scholar]
- 135.Logan AC. Omega-3 fatty acids and major depression: a primer for the mental health professional. Lipids Health Dis. 2004;3:25. doi: 10.1186/1476-511X-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Joulain C, Prigent AF, Némoz G, Lagarde M. Increased glutathione peroxidase activity in human blood mononuclear cells upon in vitro incubation with n-3 fatty acids. Biochem Pharmacol. 1994;47:1315–1323. doi: 10.1016/0006-2952(94)90329-8. [DOI] [PubMed] [Google Scholar]
- 137.Namazi MR. Prescribing cyclic antidepressants for vitiligo patients: which agents are superior, which are not? Psychother Psychosom. 2003;72:361–362. doi: 10.1159/000073036. [DOI] [PubMed] [Google Scholar]