

Abstract
Voltage gated calcium channel (VGCC) antibodies are generally associated with Lambert-Eaton myasthenic syndrome. However the presence of this antibody has been associated with paraneoplastic as well as non-paraneoplastic cerebellar degeneration. Most patients with VGCC-antibody-positivity have small cell lung cancer (SCLC). Lambert-Eaton myasthenic syndrome (LEMS) is an autoimmune disease of the presynaptic part of the neuromuscular junction. Its classical clinical triad is proximal muscle weakness, areflexia and autonomic dysfunction. Fifty to sixty percent of LEMS patients have a neoplasia, usually SCLC. The co-occurrence of SCLC and LEMS causes more severe and progressive disease and shorter survival than non-paraneoplastic LEMS. Treatment includes 3,4 diaminopyridine for symptomatic purposes and immunotherapy with prednisolone, azathioprine or intravenous immunoglobulin in patients unresponsive to 3,4 diaminopyridine. Paraneoplastic cerebellar degeneration (PCD) is a syndrome characterized with severe, subacute pancerebellar dysfunction. Serum is positive for VGCC antibody in 41%-44% of patients, usually with the co-occurrence of SCLC. Clinical and electrophysiological features of LEMS are also present in 20%-40% of these patients. Unfortunately, PCD symptoms do not improve with immunotherapy. The role of VGCC antibody in the immunopathogenesis of LEMS is well known whereas its role in PCD is still unclear. All patients presenting with LEMS or PCD must be investigated for SCLC.
Keywords: Voltage gated calcium channel antibody, Lambert-Eaton myasthenic syndrome, Paraneoplastic cerebellar degeneration, Onconeural antibodies, Small cell lung cancer
Core tip: Voltage gated calcium channel (VGCC) antibodies are generally associated with Lambert-Eaton myasthenic syndrome, but also with paraneoplastic or non-paraneoplastic cerebellar degeneration. The autoimmune nature of non-tumour Lambert-Eaton myasthenic syndrome is reflected in its association with various HLA subtypes and other autoimmune diseases such as vitiligo, myasthenia gravis and diabetes mellitus. The most common tumour associated with VGCC-antibody-positivity is small cell lung cancer. Knowledge on the relation between cerebellar degeneration and VGCC is limited, and treatment response is poor in this group of patients.
INTRODUCTION
Voltage gated calcium channels are immunologic targets for several disease. The calcium channels as a target of the pathogenic antibodies in Lambert–Eaton myasthenic syndrome (LEMS) was first suggested by Fukunaga et al[1] in 1983. Subsequent studies showed antibodies against P/Q type calcium channel as the most prominent in these patients[2].
Although voltage gated calcium channel (VGCC) antibodies are generally associated with LEMS, usually seen as a paraneoplastic syndrome with small cell lung cancer (SCLC), rarely non-paraneoplastic cerebellar degeneration may also occur in the presence of this antibody[3,4]. VGCC antibody positivity is observed in 85%-90% of LEMS patients whereas the ratio approaches 100% in LEMS patients with SCLC[5]. Approximately 40% of patients with subacute onset cerebellar degeneration, usually with SCLC, have VGCC antibody positivity[3,6]. Moreover these antibodies can also be detected in SCLC patients without neurological involvement[5].
VGCC
The VGCC is crucial in the depolarization of the cell membrane and cellular influx of calcium in response to action potential. It functions as a secondary messenger in electrical signalization and initiates several cellular mechanisms[7]. They are found in several cells, such as smooth and skeletal muscle fibers, endocrine cells, neurons[7]. The channel also locates on the presynaptic membrane of the axon terminal. VGCC opens by action potential and leads to the entry of calcium ions into the axon terminals. Calcium influx results in movement of acetylcholine vesicles towards the presynaptic membrane and acetylcholine is released into the synaptic cleft. In striated muscles, the VGCC on the membrane of transverse tubules directly activates ryanodine-sensitive calcium channels in the sarcoplasmic reticulum and initiates rapid contraction[7,8].
VGCC is divided into five types: L, P/Q, N, R, T depending on tissues and pharmacological properties[7]. The channel contains 4 or 5 subunits (α1, α2/δ, β and γ).The ion transition pore responsible for the biochemical and electrophysiological properties is the α1 subunit. This subunit contains six helical transmembrane segments (S1-S6) and 4 domains (I-IV)[9] (Figure 1). Ten different α1 subunits have been defined and CaV2.1 α1 subunit is found in P/Q type VGCC[7]. Voltage sensors are located in the S4 segment. The S5 and S6 segments are sensitive to calcium[9]. Antibodies against the S5-6 segments of α1 subunit are detected in 50% of LEMS patients[5]. Other antibodies detected in LEMS patients are against domain IV and β subunit[5,10]. However, the pathogenic role of β subunit antibodies is still controversial due to its intracellular location.
Figure 1.
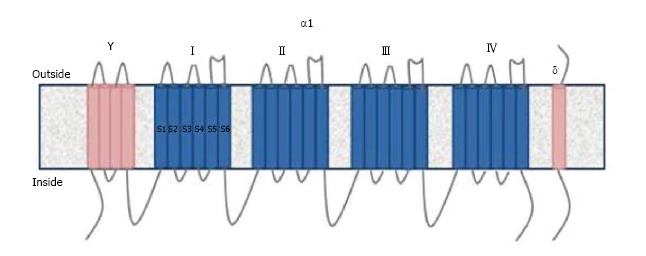
The structure of Voltage gated calcium channels.
Antibodies to P/Q type channels are responsible for clinical symptoms of LEMS[5]. Thirty to forty percent of the patients with antibodies to P/Q type channels also have antibodies to N-type channels whereas in 25% patients also have antibodies to L-type channels[5]. Antibodies to N and P/Q type channels are detected in 40% of patients with cerebellar ataxia associated with SCLC[9]. Sry-like high-mobility group box (SOX-1), zic-4, anti-Hu are other antibodies detected in the sera of patients with PCD and SCLC and approximately70% of the patients have one of these antibodies[6].
LEMS
LEMS is the autoimmune disease of the presynaptic nerve terminals. It is a rare disease with a prevalence of 2.3 per million and an incidence of 0.5 per million[11]. It is associated with SCLC in 50%-60% of patients. As the clinical and laboratory features differ in patients with and without SCLC, the disease is divided into two groups as LEMS with SCLC (SCLC-LEMS) and non-tumour (NT-LEMS).The age of onset is 50 years or above and there is a male predominance in patients with SCLC-LEMS. On the other hand NT-LEMS can be seen in all age groups with a peak at the age of 35 and 60 and a female predominance[12].
LEMS hardly occurs in childhood; only 5% of LEMS patients are less than 18-year-old[13]. Our youngest LEMS patient was a eight year-old female.
Pathogenesis
LEMS is a disorder due to antibodies against P/Q type VGCC. VGCC antibodies interact with extracellular S5 and S6 segments of domain II, III and IV of α1 subunit and reduce the number of ion channels by cross binding[5,14-16]. The antibodies can also bind to other VGCC types without causing any dysfunction. Although VGCC antibodies usually generate an immune reaction, the response to epitopes varies in LEMS patients[17]. Antigenic modulation followed by clustering and reduction of VGCC leads to reduction in quantal release of acetylcholine in synapses and results in muscle weakness[18]. The down-regulation of the receptors of parasympathetic and sympathetic neurons that cause autonomic dysfunction is also associated with these antibodies.
VGCC antibodies can be detected by radioimmunoassay in 85%-90% of LEMS patients and 100% of patients with SCLC-LEMS[17,19,20]. P/Q type VGCC are expressed on SCLC cells and this expression results in production of antibodies and cross-reaction with presynaptic VGCC[21]. As the immune reaction is initiated early in tumour development, the diagnosis of LEMS frequently precedes the diagnosis of the SCLC[16]. Besides, VGCC antibodies may be detected in 3%-5% of patients with SCLC without muscle weakness or autonomic dysfunction[16].
Approximately 10%-15% of LEMS patients do not have serum antibodies. However, their serum reproduces LEMS-like symptoms when it transferred to mice. This finding suggests the presence of antibodies at very low concentrations or antibodies against different epitopes of the VGCC that are undetected by routine tests[5]. Antibodies against synaptotagmin, a synaptic vesicle protein, have been found in some seronegative and seropositive LEMS patients[14,22]. Although there is no evidence on their pathogenic role, the blocking of synaptotagmin, which is a Ca2+ sensor in the membrane of the pre-synaptic axon terminal may explain the muscle weakness in LEMS[23]. Another such antibody, whose pathogenic role is unclear, is against “sry-like high-mobility group box” (SOX-1) proteins. This antibody is positive in 67% of SCLC-LEMS patients, 22%-32% of patients with SCLC without LEMS and only in 5% of NT-LEMS patients[19].
The role of T lymphocytes in LEMS pathogenesis is unclear. Thymus and other lymphoid organs involved in myasthenia gravis (MG) do not present abnormalities in LEMS and the presynaptic terminals do not reveal T lymphocyte collection[16]. However regulatory T lymphocytes are down-regulated in SCLC-LEMS patients, but not in SCLC patients without LEMS, which suggests the dysfunction of regulatory T lymphocyte in SCLC-LEMS pathogenesis[24].
Unlike SCLC-LEMS patients, the trigger of the immune response is not defined in NT-LEMS.Various HLA associations, also reported with other autoimmune diseases, have been documented: Approximately two-third of patients have HLA-B8 (HLA class I) and HLA-DR3, -DQ2 (HLA class II) alleles[25,26]. Supporting this phenomenon, monozygous twins have been reported where one has LEMS and VGCC antibodies, and the other, myasthenia gravis and acetylcholine receptor antibodies[27]. On the other hand, HLA association was not found in SCLC-LEMS patients: because tumoral cells do not have strong HLA class I antigen expression, these molecules are possibly not involved in the immunopathogenesis[25]. These observations resemble the difference between MG with and without thymoma and reveal the existence of different autoimmune mechanisms in paraneoplastic disorders[16].
Clinical features
The classical triad of LEMS is proximal weakness, reduced tendon reflexes and autonomic dysfunction[16].
Proximal weakness, more prominent in lower extremities, is the first symptom in 80% of patients[16,28]. In the course of the disease, 80% of patients suffer from proximal weakness in both upper and lower extremities[28,29]. Facial, bulbar and distal weakness are also frequent[16]. The weakness is more severe and rapidly progressive in SCLC-LEMS patients[16,28].
Autonomic dysfunction, observed in 80%-96% of patients, is the most common symptom although it is less disturbing than muscle weakness[12,28]. Erectile dysfunction and constipation are more frequent symptoms than urinary retention, dry eye and reduced sweating[5]. The rate and the nature of autonomic symptoms do not differ between SCLC-LEMS and NT-LEMS[16].
On neurological examination deep tendon reflexes are generally reduced or absent. However maximal isometric contraction of the muscle for 10 to 15 s may lead to the appearance of previously depressed or absent deep tendon reflexes and temporarily improve muscle strength, which is called “Post-exercise facilitation”. This phenomenon, a characteristic feature of LEMS, is not sensitive[5]. This phenomenon may also mask the reduction of deep tendon reflexes in 40% of patients so the neurological examination must be repeated after a resting period to verify the diagnosis[5].
Associated diseases
The most common co-existence of LEMS is with SCLC, reported in 50%-60% of LEMS patients[12]. Besides, 0.5%-3% of SCLC patients have LEMS[25]. This co-existence results in a more severe and rapidly progressive neurological disease with shortened survival compared to NT-LEMS which has a near normal survival[16]. Moreover SCLC-LEMS patients tend to be younger than other SCLC patients[19].
Older age, tobacco use, increased ESR support the probability of underlying SCLC. In a Dutch-English cohort study a scoring system called DELTA-P was implemented to predict SCLC in LEMS[12]. According to DELTA-P, dysarthria, dysphagia, chewing or neck weakness(D), erectile dysfunction (E), loss of weight (L), tobacco use (T), age of onset more than 50 (A) and Karnofsky performance less than 60 (P) have a predictive value: patients with a DELTA-P score three or more have higher than 94% risk of having SCLC.
The distinction of SCLC-LEMS from NT-LEMS is important, as treatment options and outcome are different. For this reason, new markers to diagnose SCLC-LEMS are still under investigation. Although SOX-1antibodies are detected in half of the SCLC-LEMS patients, the absence of a commercial kit, and the presence of these antibodies in NT-LEMS limit their use for differential diagnosis[30]. Another study revealed that VGCC antibody against domain IV positivity is more common in NT-LEMS patients than in SCLC-LEMS patients, but this is not used for clinical purposes yet[17].
The co-existence of LEMS with other malignancies such as non-small cell lung cancer, prostate carcinoma, orthymoma has been reported rarely[13] and a random association could not be excluded[5].
In children, the disease is generally non-paraneoplastic; lymphoproliferative malignancy and neuroblastoma are rare associations[13,31]. In accordance with the literature, our pediatric patient did not have any neoplasm.
Other autoimmune diseases such as thyroid disorders, alopecia, diabetes mellitus, MG can occur in NT-LEMS patients probably due to the presence of various HLA subtypes contributing to autoimmunity[32]. In our clinical experience, NT-LEMS patients may have associated vitiligo and myasthenia.
Diagnosis
The time lag between the onset of the symptoms and diagnosis is approximately 4 mo in SCLC-LEMS and 12 mo in NT-LEMS[12,29].
In patients with suspected LEMS, electrophysiological studies with repetitive nerve stimulation are among the most important diagnostic tests. The compound muscle action potential (CMAP) is low after the first stimulation and decreases further after repetitive stimulation at 2-5 Hz[33]. At least 10% reduction in CMAP after low frequency stimulation is considered abnormal and observed in 94%-98% of patients[34,35]. However, this finding may also be present in MG patients. To distinguish these two diseases, nerve stimulation at high frequency (50 Hz) or, as a less painful method for the patient, post-exercise measurement is employed, which increases CMAP by more than 100% in LEMS patients[34,35]. Optimum results will be obtained if treatment is interrupted 12 h before the study and the muscle temperature is above32 °C.
Although single fiber EMG is a sensitive test, it is used in combination with other tests as it cannot distinguish between MG and LEMS[33]. LEMS patients have increased jitter like MG patients. In case of severe neuromuscular junction dysfunction, the conduction defect in muscle fiber causes a decrease in amplitude and duration of motor unit potential as in myopathies[36].
VGCC antibodies are detected by RIA in 85%-90% of LEMS and in almost 100% of SCLC-LEMS patients[17,19,20]. Although the presence of VGCC antibodies supports the diagnosis of LEMS, the absence of antibodies in a patient with typical clinical features does not exclude the diagnosis.
In 50% of patients SCLC-LEMS, LEMS symptoms precede the diagnosis of SCLC. In addition to scoring systems such as DELTA-P, all patients with the diagnosis of LEMS must undergo computerized tomography of the thorax and positron emission tomography (PET). If the results are negative, the screening must be repeated every 3-6 mo until the second year of the disease[37].
The differential diagnosis of LEMS from seronegative and atypical myasthenia gravis can be challenging. Some clinical findings may be helpful: the progression of weakness is in the craniocaudal direction in MG and the reverse in LEMS; ptosis and facial weakness are less common and severe in LEMS. Electrophysiological studies described above and serological findings assist the clinician in the differential diagnosis.
LEMS with a subacute course can be misdiagnosed as Guillain Barré syndrome (GBS); the presence of sensorial symptoms, neuropathic pain, and elevated CSF protein favor the diagnosis of GBS. Amyotrophic lateral sclerosis may constitute another differential diagnosis, and can be distinguished by the asymmetrical weakness starting in the upper extremities and the presence of upper motor neuron signs.
Treatment
The first choice for symptomatic treatment is 3,4 diaminopyridine[38]. This molecule blocks presynaptic voltage-gated potassium channels and provides a prolonged action potential which increases the quantal release of synaptic acetylcholine[39]. All randomized controlled studies of 3,4 diaminopyridine showed improvement in muscle strength and CMAP amplitudes. The drug is well tolerated although adverse effects like perioral and digital paresthesias and gastrointestinal symptoms are not uncommon. Seizures, which is the most frequent severe side effect, have been reported at high doses exceeding 100 mg/d[40,41]. In our experience, the drug is well tolerated and improves muscle strength at the dose of 40-60 mg/d.
Other treatments, which can increase the concentration of acetylcholine in synaptic cleft, are pyridostigmine and acetylcholine esterase inhibitors but they are not as effective as they are in MG patients[42].
In case of limited response to 3,4 diaminopyridine, immunosuppressive treatments must be considered. The combination of prednisolone and azathioprine is well studied and documented in LEMS patients[38,42]. Although there is not sufficient data, mycophenolate mofetil, cyclosporine and rituximab are also drugs employed in LEMS treatment[16,43]. Intravenous immunoglobulin (IVIg), another treatment option in paraneoplastic syndromes and MG, can also be used in LEMS. European Federation of Neurological Societies (EFNS) guidelines recommend IVIg in both SCLC-LEMS and NT-LEMS[44]. IVIg is also recommended in pregnant patients, as transplacental transmission of IgG antibodies may cause neonatal LEMS[45]. IVIg is generally preferred as its side effects are rare and it is easily used for the maintenance treatment, which is usually needed in LEMS patients. Plasma exchange whose effect is comparable to IVIg may carry technical difficulties and slightly higher rate of complications[46].
In patients with SCLC, the treatment of the tumour is crucial. The survival of SCLC-LEMS patients is better than in patients with SCLC alone, but there is no relation with VGCC or SOX-1 antibody positivity and survival[17,19]. The better prognosis in these patients may be correlated with the diagnosis time that is earlier in LEMS patients[12,29]. Moreover HLA-B8 positivity is related to prolonged survival in SCLC-LEMS patients[47].
Maintenance of optimal body weight, rehabilitation, frequent examinations for complications such as respiratory infections, and avoidance of drugs impairing neuromuscular transmission are other important aspects of the treatment.
CEREBELLAR DEGENERATION ASSOCIATED WITH VGCC ANTIBODY
Paraneoplastic cerebellar degeneration (PCD) is a syndrome characterized by subacute cerebellar dysfunction[48]. Clinical and pathological features of the syndrome were described by Brain and Wilkinson[49] in 1965 by the evaluation of 13 patients and 6 autopsy cases. Diffuse loss of Purkinje cells is the pathologic hallmark of the disease and usually accompanied by thinning of granular and molecular layers, degeneration of long tracts of spinal cord, dentate and olivary nuclei[4].
Most common neoplasms associated with cerebellar degeneration are lung, breast, ovarian cancers and Hodgkin lymphoma[48]. Onconeural antibodies such as anti-Hu, anti Yo, anti-Ri, anti-CV2, anti-Tr, anti-Ma, anti-Ta, anti zic 4, and anti-mGluR1 as well as VGCC antibody can be detected in PCD[50]. Clinical presentation, neuropathological findings and treatment responses of patients vary according to the type of the onconeural antibody, suggesting distinct immune mechanisms related to different antibodies[4].
Pathogenesis
Antibodies against VGCC of the P/Q type or N type are found in 41%-44% of PCD patients, generally associated with SCLC[3,6,9]. The P/Q type VGCC is highly expressed in cerebellar Purkinje cells and in the molecular layer of the cerebellum[9,51]. About 20%-40% of these patients also have clinical or electrophysiological diagnosis of LEMS[3,6]. Neuropathological findings of PCD with LEMS (PCD-LEMS) were reported in 1973 by Satoyoshi et al[51] for the first time. In a postmortem study of three PCD-LEMS patients with VGCC antibodies, 70%-80% of reduction in P/Q type VGCC of the molecular layer; loss of Purkinje cells and gliosis in the cerebellar cortex were observed[9,51]. The role of VGCC antibodies in the pathogenesis of PCD is still unclear. In a recent experimental study, antibodies of the IgG type purified from the serum of two VGCC antibody-positive patients with SCLC, one with PCD-LEMS and another patient with isolated LEMS were given to mice intrathecally, the antibodies associated with PCD-LEMS but not from isolated LEMS patients caused cerebellar ataxia in mice[52]. This finding suggests the presence of different epitopes of P/Q type VGCC antibodies which inhibit VGCC’s function in cerebellum, or of other additional, yet undiscovered pathogenic antibodies[52].
Clinical, laboratory and radiological features
Subacute and rapidly progressive gait unsteadiness is the presenting symptom of cerebellar degeneration[50]. Gait and limb ataxia, diplopia, dysarthria are the other prominent symptoms[50]. Sometimes these complaints may preceded by dizziness, nausea and viral infection-like symptoms[50]. Occasionally other signs and symptoms such as dysphagia, nystagmus and sensory deficits can be also seen during the course. Patients who had concomitant LEMS may also show proximal weakness and autonomic symptoms in addition to cerebellar symptoms[4].
The cerebrospinal fluid (CSF) may show mild lymphocytic pleocytosis with elevated protein and oligoclonal bands[48]. VGCC antibodies may also be detected in CSF and there is some evidence of intrathecalsynthesis of the antibodies, and detected in about 25% of the patients[3]. This low percentage may be explained by the absence of CSF analysis in some cases and further studies are needed to increase the rate of antibody presence in CSF.
Initial brain magnetic resonance images or tomography are normal in most patients[4] although in early stages of the disease fluorodeoxyglucose-PET scans may show cerebellar hypermetabolism[50,53], whereas cerebellar atrophy and cerebellar hypometabolism are seen in the advanced stage of the disease[50].
Treatment
Treatment of the underlying malignancy has the priority like other paraneoplastic syndromes[50]. Corticosteroids, plasma exchange, IVIG, tacrolimus and cyclophosphamide are the immunotherapeutic options to be used concurrently with tumour therapy, but most of the cases did not show sufficient improvement despite treatment[50]. Unlike in LEMS, immunotherapy does not result in symptomatic improvement in PCD: this suggests PCD may be associated with irreversible damage of Purkinje cells[3].
NON-PARANEOPLASTIC CEREBELLAR DEGENERATION WITH VGCC ANTIBODY
VGCC antibodies were also found in a few patients with non-paraneoplastic cerebellar degeneration. In a study of the antibody profile of 67 cases with sporadic, late-onset cerebellar ataxia of unknown etiology, VGCC antibodies were found in 12%[54]. Two cases with NT-LEMS who developed cerebellar ataxia during the course of their disease had VGCC antibodies in serum and CSF. Cerebellar symptoms of these patients showed no improvement with different immunotherapies, as in PCD[55]. However a few cases of non-paraneoplastic cerebellar degeneration showed favorable outcome under rituximab and IVIg treatment[56,57].
CONCLUSION
Diseases related to VGCC antibodies are usually associated with SCLC. Therefore, SCLC should be investigated in patients with LEMS and/or cerebellar degeneration. The role of VGCC antibodies in the immunopathogenesis of LEMS is clear, however their role in cerebellar degeneration is not known. Determination of the effect of VGCC antibodies on the pathogenesis of cerebellar degeneration may contribute to the design of more efficient treatment strategies. Therefore, experimental models and pathologic studies that investigate the effect of immune mechanisms at molecular level in the tissue are needed.
Footnotes
P- Reviewer: Tan XR, Zezos P S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
Conflict-of-interest: We certify that there is no actual or potential conflict of interest in relation to this article.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 27, 2014
First decision: November 27, 2014
Article in press: January 15, 2015
References
- 1.Fukunaga H, Engel AG, Lang B, Newsom-Davis J, Vincent A. Passive transfer of Lambert-Eaton myasthenic syndrome with IgG from man to mouse depletes the presynaptic membrane active zones. Proc Natl Acad Sci USA. 1983;80:7636–7640. doi: 10.1073/pnas.80.24.7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lennon VA, Kryzer TJ, Griesmann GE, O’Suilleabhain PE, Windebank AJ, Woppmann A, Miljanich GP, Lambert EH. Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med. 1995;332:1467–1474. doi: 10.1056/NEJM199506013322203. [DOI] [PubMed] [Google Scholar]
- 3.Graus F, Lang B, Pozo-Rosich P, Saiz A, Casamitjana R, Vincent A. P/Q type calcium-channel antibodies in paraneoplastic cerebellar degeneration with lung cancer. Neurology. 2002;59:764–766. doi: 10.1212/wnl.59.5.764. [DOI] [PubMed] [Google Scholar]
- 4.Mason WP, Graus F, Lang B, Honnorat J, Delattre JY, Valldeoriola F, Antoine JC, Rosenblum MK, Rosenfeld MR, Newsom-Davis J, et al. Small-cell lung cancer, paraneoplastic cerebellar degeneration and the Lambert-Eaton myasthenic syndrome. Brain. 1997;120(Pt 8):1279–1300. doi: 10.1093/brain/120.8.1279. [DOI] [PubMed] [Google Scholar]
- 5.Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011;10:1098–1107. doi: 10.1016/S1474-4422(11)70245-9. [DOI] [PubMed] [Google Scholar]
- 6.Sabater L, Höftberger R, Boronat A, Saiz A, Dalmau J, Graus F. Antibody repertoire in paraneoplastic cerebellar degeneration and small cell lung cancer. PLoS One. 2013;8:e60438. doi: 10.1371/journal.pone.0060438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011;3:a003947. doi: 10.1101/cshperspect.a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsien RW. Calcium channels in excitable cell membranes. Annu Rev Physiol. 1983;45:341–358. doi: 10.1146/annurev.ph.45.030183.002013. [DOI] [PubMed] [Google Scholar]
- 9.van Coevorden-Hameete MH, de Graaff E, Titulaer MJ, Hoogenraad CC, Sillevis Smitt PA. Molecular and cellular mechanisms underlying anti-neuronal antibody mediated disorders of the central nervous system. Autoimmun Rev. 2014;13:299–312. doi: 10.1016/j.autrev.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 10.Verschuuren JJ, Dalmau J, Tunkel R, Lang B, Graus F, Schramm L, Posner JB, Newsom-Davis J, Rosenfeld MR. Antibodies against the calcium channel beta-subunit in Lambert-Eaton myasthenic syndrome. Neurology. 1998;50:475–479. doi: 10.1212/wnl.50.2.475. [DOI] [PubMed] [Google Scholar]
- 11.Wirtz PW, Nijnuis MG, Sotodeh M, Willems LN, Brahim JJ, Putter H, Wintzen AR, Verschuuren JJ. The epidemiology of myasthenia gravis, Lambert-Eaton myasthenic syndrome and their associated tumours in the northern part of the province of South Holland. J Neurol. 2003;250:698–701. doi: 10.1007/s00415-003-1063-7. [DOI] [PubMed] [Google Scholar]
- 12.Titulaer MJ, Maddison P, Sont JK, Wirtz PW, Hilton-Jones D, Klooster R, Willcox N, Potman M, Sillevis Smitt PA, Kuks JB, et al. Clinical Dutch-English Lambert-Eaton Myasthenic syndrome (LEMS) tumor association prediction score accurately predicts small-cell lung cancer in the LEMS. J Clin Oncol. 2011;29:902–908. doi: 10.1200/JCO.2010.32.0440. [DOI] [PubMed] [Google Scholar]
- 13.Wirtz PW, Smallegange TM, Wintzen AR, Verschuuren JJ. Differences in clinical features between the Lambert-Eaton myasthenic syndrome with and without cancer: an analysis of 227 published cases. Clin Neurol Neurosurg. 2002;104:359–363. doi: 10.1016/s0303-8467(02)00054-9. [DOI] [PubMed] [Google Scholar]
- 14.Takamori M, Takahashi M, Yasukawa Y, Iwasa K, Nemoto Y, Suenaga A, Nagataki S, Nakamura T. Antibodies to recombinant synaptotagmin and calcium channel subtypes in Lambert-Eaton myasthenic syndrome. J Neurol Sci. 1995;133:95–101. doi: 10.1016/0022-510x(95)00162-u. [DOI] [PubMed] [Google Scholar]
- 15.Iwasa K, Takamori M, Komai K, Mori Y. Recombinant calcium channel is recognized by Lambert-Eaton myasthenic syndrome antibodies. Neurology. 2000;54:757–759. doi: 10.1212/wnl.54.3.757. [DOI] [PubMed] [Google Scholar]
- 16.Gilhus NE. Lambert-eaton myasthenic syndrome; pathogenesis, diagnosis, and therapy. Autoimmune Dis. 2011;2011:973808. doi: 10.4061/2011/973808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pellkofer HL, Armbruster L, Krumbholz M, Titulaer MJ, Verschuuren JJ, Schumm F, Voltz R. Lambert-eaton myasthenic syndrome differential reactivity of tumor versus non-tumor patients to subunits of the voltage-gated calcium channel. J Neuroimmunol. 2008;204:136–139. doi: 10.1016/j.jneuroim.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 18.Quartel A, Turbeville S, Lounsbury D. Current therapy for Lambert-Eaton myasthenic syndrome: development of 3,4-diaminopyridine phosphate salt as first-line symptomatic treatment. Curr Med Res Opin. 2010;26:1363–1375. doi: 10.1185/03007991003745209. [DOI] [PubMed] [Google Scholar]
- 19.Titulaer MJ, Klooster R, Potman M, Sabater L, Graus F, Hegeman IM, Thijssen PE, Wirtz PW, Twijnstra A, Smitt PA, et al. SOX antibodies in small-cell lung cancer and Lambert-Eaton myasthenic syndrome: frequency and relation with survival. J Clin Oncol. 2009;27:4260–4267. doi: 10.1200/JCO.2008.20.6169. [DOI] [PubMed] [Google Scholar]
- 20.Motomura M, Lang B, Johnston I, Palace J, Vincent A, Newsom-Davis J. Incidence of serum anti-P/O-type and anti-N-type calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. J Neurol Sci. 1997;147:35–42. doi: 10.1016/s0022-510x(96)05303-8. [DOI] [PubMed] [Google Scholar]
- 21.Roberts A, Perera S, Lang B, Vincent A, Newsom-Davis J. Paraneoplastic myasthenic syndrome IgG inhibits 45Ca2+ flux in a human small cell carcinoma line. Nature. 1985;317:737–739. doi: 10.1038/317737a0. [DOI] [PubMed] [Google Scholar]
- 22.Takamori M, Hamada T, Komai K, Takahashi M, Yoshida A. Synaptotagmin can cause an immune-mediated model of Lambert-Eaton myasthenic syndrome in rats. Ann Neurol. 1994;35:74–80. doi: 10.1002/ana.410350112. [DOI] [PubMed] [Google Scholar]
- 23.Fukuda M, Moreira JE, Liu V, Sugimori M, Mikoshiba K, Llinás RR. Role of the conserved WHXL motif in the C terminus of synaptotagmin in synaptic vesicle docking. Proc Natl Acad Sci USA. 2000;97:14715–14719. doi: 10.1073/pnas.260491197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tani T, Tanaka K, Idezuka J, Nishizawa M. Regulatory T cells in paraneoplastic neurological syndromes. J Neuroimmunol. 2008;196:166–169. doi: 10.1016/j.jneuroim.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 25.Titulaer MJ, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: tumor versus nontumor forms. Ann N Y Acad Sci. 2008;1132:129–134. doi: 10.1196/annals.1405.030. [DOI] [PubMed] [Google Scholar]
- 26.Wirtz PW, Roep BO, Schreuder GM, van Doorn PA, van Engelen BG, Kuks JB, Twijnstra A, de Visser M, Visser LH, Wokke JH, et al. HLA class I and II in Lambert-Eaton myasthenic syndrome without associated tumor. Hum Immunol. 2001;62:809–813. doi: 10.1016/s0198-8859(01)00270-1. [DOI] [PubMed] [Google Scholar]
- 27.Punga AR, Nygren I, Askmark H, Stålberg EV. Monozygous twins with neuromuscular transmission defects at opposite sides of the motor endplate. Acta Neurol Scand. 2009;119:207–211. doi: 10.1111/j.1600-0404.2008.01082.x. [DOI] [PubMed] [Google Scholar]
- 28.Titulaer MJ, Wirtz PW, Kuks JB, Schelhaas HJ, van der Kooi AJ, Faber CG, van der Pol WL, de Visser M, Sillevis Smitt PA, Verschuuren JJ. The Lambert-Eaton myasthenic syndrome 1988-2008: a clinical picture in 97 patients. J Neuroimmunol. 2008;201-202:153–158. doi: 10.1016/j.jneuroim.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 29.Pellkofer HL, Armbruster L, Linke R, Schumm F, Voltz R. Managing non-paraneoplastic Lambert-Eaton myasthenic syndrome: clinical characteristics in 25 German patients. J Neuroimmunol. 2009;217:90–94. doi: 10.1016/j.jneuroim.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 30.Sabater L, Titulaer M, Saiz A, Verschuuren J, Güre AO, Graus F. SOX1 antibodies are markers of paraneoplastic Lambert-Eaton myasthenic syndrome. Neurology. 2008;70:924–928. doi: 10.1212/01.wnl.0000281663.81079.24. [DOI] [PubMed] [Google Scholar]
- 31.Bosdure E, Attarian S, Mancini J, Mikaeloff Y, Chabrol B. [Lambert-Eaton myastenic syndrome revealing neuroblastoma in 2 children] Arch Pediatr. 2006;13:1121–1124. doi: 10.1016/j.arcped.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 32.Wirtz PW, Bradshaw J, Wintzen AR, Verschuuren JJ. Associated autoimmune diseases in patients with the Lambert-Eaton myasthenic syndrome and their families. J Neurol. 2004;251:1255–1259. doi: 10.1007/s00415-004-0528-7. [DOI] [PubMed] [Google Scholar]
- 33.Medicine AQACAAoE. Practice parameter for repetitive nerve stimulation and single fiber EMG evaluation of adults with suspected myasthenia gravis or Lambert-Eaton myasthenic syndrome: summary statement. Muscle Nerve. 2001;24:1236–1238. doi: 10.1002/mus.1139. [DOI] [PubMed] [Google Scholar]
- 34.Oh SJ, Kurokawa K, Claussen GC, Ryan HF. Electrophysiological diagnostic criteria of Lambert-Eaton myasthenic syndrome. Muscle Nerve. 2005;32:515–520. doi: 10.1002/mus.20389. [DOI] [PubMed] [Google Scholar]
- 35.Tim RW, Massey JM, Sanders DB. Lambert-Eaton myasthenic syndrome (LEMS). Clinical and electrodiagnostic features and response to therapy in 59 patients. Ann N Y Acad Sci. 1998;841:823–826. doi: 10.1111/j.1749-6632.1998.tb11024.x. [DOI] [PubMed] [Google Scholar]
- 36.O’Neill JH, Murray NM, Newsom-Davis J. The Lambert-Eaton myasthenic syndrome. A review of 50 cases. Brain. 1988;111(Pt 3):577–596. doi: 10.1093/brain/111.3.577. [DOI] [PubMed] [Google Scholar]
- 37.Titulaer MJ, Soffietti R, Dalmau J, Gilhus NE, Giometto B, Graus F, Grisold W, Honnorat J, Sillevis Smitt PA, Tanasescu R, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. 2011;18:19–e3. doi: 10.1111/j.1468-1331.2010.03220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keogh M, Sedehizadeh S, Maddison P. Treatment for Lambert-Eaton myasthenic syndrome. Cochrane Database Syst Rev. 2011:CD003279. doi: 10.1002/14651858.CD003279.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Molgó J, Lundh H, Thesleff S. Potency of 3,4-diaminopyridine and 4-aminopyridine on mammalian neuromuscular transmission and the effect of pH changes. Eur J Pharmacol. 1980;61:25–34. doi: 10.1016/0014-2999(80)90378-7. [DOI] [PubMed] [Google Scholar]
- 40.Lindquist S, Stangel M. Update on treatment options for Lambert-Eaton myasthenic syndrome: focus on use of amifampridine. Neuropsychiatr Dis Treat. 2011;7:341–349. doi: 10.2147/NDT.S10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanders DB, Massey JM, Sanders LL, Edwards LJ. A randomized trial of 3,4-diaminopyridine in Lambert-Eaton myasthenic syndrome. Neurology. 2000;54:603–607. doi: 10.1212/wnl.54.3.603. [DOI] [PubMed] [Google Scholar]
- 42.Skeie GO, Apostolski S, Evoli A, Gilhus NE, Illa I, Harms L, Hilton-Jones D, Melms A, Verschuuren J, Horge HW. Guidelines for treatment of autoimmune neuromuscular transmission disorders. Eur J Neurol. 2010;17:893–902. doi: 10.1111/j.1468-1331.2010.03019.x. [DOI] [PubMed] [Google Scholar]
- 43.Maddison P, McConville J, Farrugia ME, Davies N, Rose M, Norwood F, Jungbluth H, Robb S, Hilton-Jones D. The use of rituximab in myasthenia gravis and Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 2011;82:671–673. doi: 10.1136/jnnp.2009.197632. [DOI] [PubMed] [Google Scholar]
- 44.Elovaara I, Apostolski S, van Doorn P, Gilhus NE, Hietaharju A, Honkaniemi J, van Schaik IN, Scolding N, Soelberg Sørensen P, Udd B. EFNS guidelines for the use of intravenous immunoglobulin in treatment of neurological diseases: EFNS task force on the use of intravenous immunoglobulin in treatment of neurological diseases. Eur J Neurol. 2008;15:893–908. doi: 10.1111/j.1468-1331.2008.02246.x. [DOI] [PubMed] [Google Scholar]
- 45.Reuner U, Kamin G, Ramantani G, Reichmann H, Dinger J. Transient neonatal Lambert-Eaton syndrome. J Neurol. 2008;255:1827–1828. doi: 10.1007/s00415-008-0988-2. [DOI] [PubMed] [Google Scholar]
- 46.Weimer MB, Wong J. Lambert-eaton myasthenic syndrome. Curr Treat Options Neurol. 2009;11:77–84. doi: 10.1007/s11940-009-0010-z. [DOI] [PubMed] [Google Scholar]
- 47.Wirtz PW, Willcox N, van der Slik AR, Lang B, Maddison P, Koeleman BP, Giphart MJ, Wintzen AR, Roep BO, Verschuuren JJ. HLA and smoking in prediction and prognosis of small cell lung cancer in autoimmune Lambert-Eaton myasthenic syndrome. J Neuroimmunol. 2005;159:230–237. doi: 10.1016/j.jneuroim.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 48.Ko MW, Dalmau J, Galetta SL. Neuro-ophthalmologic manifestations of paraneoplastic syndromes. J Neuroophthalmol. 2008;28:58–68. doi: 10.1097/WNO.0b013e3181677fcc. [DOI] [PubMed] [Google Scholar]
- 49.Brain L, Wilkinson M. Subacute cerebellar degeneration associated with neoplasms. Brain. 1965;88:465–478. doi: 10.1093/brain/88.3.465. [DOI] [PubMed] [Google Scholar]
- 50.Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol. 2008;7:327–340. doi: 10.1016/S1474-4422(08)70060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fukuda T, Motomura M, Nakao Y, Shiraishi H, Yoshimura T, Iwanaga K, Tsujihata M, Eguchi K. Reduction of P/Q-type calcium channels in the postmortem cerebellum of paraneoplastic cerebellar degeneration with Lambert-Eaton myasthenic syndrome. Ann Neurol. 2003;53:21–28. doi: 10.1002/ana.10392. [DOI] [PubMed] [Google Scholar]
- 52.Martín-García E, Mannara F, Gutiérrez-Cuesta J, Sabater L, Dalmau J, Maldonado R, Graus F. Intrathecal injection of P/Q type voltage-gated calcium channel antibodies from paraneoplastic cerebellar degeneration cause ataxia in mice. J Neuroimmunol. 2013;261:53–59. doi: 10.1016/j.jneuroim.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 53.Choi KD, Kim JS, Park SH, Kim YK, Kim SE, Smitt PS. Cerebellar hypermetabolism in paraneoplastic cerebellar degeneration. J Neurol Neurosurg Psychiatry. 2006;77:525–528. doi: 10.1136/jnnp.2005.075325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bürk K, Wick M, Roth G, Decker P, Voltz R. Antineuronal antibodies in sporadic late-onset cerebellar ataxia. J Neurol. 2010;257:59–62. doi: 10.1007/s00415-009-5262-8. [DOI] [PubMed] [Google Scholar]
- 55.Lorenzoni PJ, Scola RH, Lang B, Kay CS, Teive HA, Kowacs PA, Werneck LC. Cerebellar ataxia in non-paraneoplastic Lambert-Eaton myasthenic syndrome. J Neurol Sci. 2008;270:194–196. doi: 10.1016/j.jns.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 56.Pellkofer HL, Voltz R, Kuempfel T. Favorable response to rituximab in a patient with anti-VGCC-positive Lambert-Eaton myasthenic syndrome and cerebellar dysfunction. Muscle Nerve. 2009;40:305–308. doi: 10.1002/mus.21315. [DOI] [PubMed] [Google Scholar]
- 57.Rigamonti A, Lauria G, Stanzani L, Mantero V, Andreetta F, Salmaggi A. Non-paraneoplastic voltage-gated calcium channels antibody-mediated cerebellar ataxia responsive to IVIG treatment. J Neurol Sci. 2014;336:169–170. doi: 10.1016/j.jns.2013.10.031. [DOI] [PubMed] [Google Scholar]