

Abstract
Background
Signaling networks promoting cell growth and proliferation are frequently deregulated in cancer. Tumors often are highly dependent on such signaling pathways and may become hypersensitive to downregulation of key components within these signaling cascades. The classical mitogenic cascade transmits stimuli from growth factor receptors via Ras, Raf, MEK and ERK to the cell nucleus and provides attractive molecular targets for cancer treatment. For example, Ras and Raf kinase inhibitors are already in a number of ongoing phase II and phase III clinical trials. In this study the effect of the Raf kinase inhibitor BAY 43-9006 and of the MEK inhibitor CI-1040 (PD184352) on a Raf dependent lung tumor mouse model was analyzed in detail.
Methods
We have generated a lung cancer mouse model by targeting constitutively active C-Raf kinase to the lung. These mice develop adenomas within 4 months of life. At this time-point they received daily intraperitoneal injections of either 100 mg/kg BAY 43-9006 or CI-1040 for additional 21 days. Thereafter, lungs were isolated and the following parameters were analyzed using histology and immunohistochemistry: overall lung structure, frequency of adenoma foci, proliferation rate, ERK activity, caspase-3 activation, and lung differentiation.
Results
Both inhibitors were equally effective in vitro using a sensitive Raf/MEK/ERK ELISA. In vivo, the systemic administration of the MEK inhibitor CI-1040 reduced adenoma formation to a third and significantly restored lung structure. The proliferation rate of lung cells of mice treated with CL-1040 was decreased without any obvious effects on differentiation of pneumocytes. In contrast, the Raf inhibitor BAY 43-9006 did not influence adenoma formation in vivo.
Conclusion
The MEK inhibitor CI-1040 may be used for the treatment of Ras and/or Raf-dependent human malignancies.
Background
Oncogene-based therapeutics is a novel approach to inhibit proteins, which are essential for the initiation and maintenance of malignancies [1]. Agents such as Herceptin for the treatment of advanced breast cancer and Gleevec for chronic myelogenous leukemia have proven that cancer therapies targeting specific molecular alterations in signaling pathways are successful [2]. The Ras-MAP kinase pathway has a central role in regulating tumor cell growth and survival, differentiation and angiogenesis and has been targeted for therapeutic intervention in the past [3,4]. C-Raf kinase and MEK are downstream effectors of the Ras signaling cascade. Both kinases are essential for cellular homeostasis and induce both proliferation and survival by suppression of apoptosis [5]. Raf and Ras mutations found in human malignancies convey constitutive activity to these signaling molecules thereby converting them into an oncogenic state [6].
In this study the Raf inhibitor BAY 43-9006 [7] and the MEK inhibitor CI-1040 [8] were tested as potential drugs in a transgenic mouse lung cancer model [9]. We have previously established this mouse model by expressing mutated, constitutively active C-Raf kinase (C-Raf BxB) under the control of the human surfactant protein C (SP-C) promoter [10]. C-Raf BxB lacks the regulatory NH2-terminal sequences including the Ras interaction domain. Lung targeted expression of constitutively active C-Raf induced lung adenomas within 4 months of life [10]. Although these adenomas are stable for more than one year, deficiencies in other genes such as Bcl-2 or p53 were found to modulate adenoma growth or even switch the phenotype of tumor cells, respectively [11,12]. The effects of BAY 43-9006 and CI-1040 were assessed in this lung adenoma model by daily intraperitoneal injections of these drugs at concentrations of 100 mg per kg body weight over a period of three weeks to four months old C-Raf-BxB mice.
Methods
ELISA
Enzyme-linked immunosorbent assay for the MEK signaling cascade was done as previously described [13]. In brief, plates were coated with an anti ERK-antibody (Santa Cruz, sc-94). A kinase reaction was performed in the presence of ERK, Raf, MEK and the respective inhibitor. Phosphorylated ERK was detected by a sandwich technique using a secondary antibody (BioLabs, #9106L) and a peroxidase-linked species-specific tertiary antibody (Amersham Pharmacia Biotech, #NA 931). Similar results were obtained with constitutively active C-Raf BxB (data not shown).
Animals
Lung targeted expression of constitutively active C-Raf (SP-C C-Raf BxB), lacking the regulatory N-terminal sequences, which comprise the Ras interaction domain, induced lung adenomas within 4 months of life [10]. The adenomas were indistinguishable from those induced by expression of wild type C-Raf (data not shown). Litters of the transgenic mice were randomly assigned to the control groups (no treatment, n = 11, respectively placebo treatment, n = 12) and to the study group (BAY 43-9006 or CI-1040: daily intraperitoneal injections at a dose of 100 mg/kg from 4 months of age over a period of 21 days, n = 12 each group). At this dose both drugs were well tolerated (data not shown). Serum concentration of BAY 43-9006 was determined by liquid-liquid extraction and consecutive HPLC analysis [14]. Lungs were isolated and analyzed at the end of the treatment period. Animals were compared to non-treated or placebo-treated transgenic mice of the same litter. Placebo treatment did not cause any changes in comparison to untreated mice (data not shown).
Histology and immunohistochemistry
Lungs were fixed under 25 cm water pressure with formalin. Histology was done on formalin-fixed, paraffin-embedded lung specimen. 4 μm-cut sections were deparaffinized, rehydrated in graded alcohols and hematoxylin and eosin stained. Antigen retrieval was performed with citrate buffer, pH 6.0, and microwave treatment. Endogenous peroxidase activity was blocked by incubation with 3% methanol. Unspecific binding was blocked by normal goat serum. The primary antibody for p-ERK (Cell signaling 9101), PCNA (DAKO M 0879), for Ki-67 (DAKO M 7249) and Bmi-1 (Santa Cruz sc-10745) were detected by a peroxidase labelled secondary antibody (DAKO P0450). Corresponding secondary antibodies were used to detect activated caspase-3 primary antibody (Cell signaling 9664) and pro-SP-C antibody (generous gift of Dr. Jeffrey A. Whitsett, Cincinnati Children's Hospital Medical Center, OH/USA). Staining was performed using diaminobenzidine and hematoxylin counterstaining. Slides were analyzed in a blinded fashion. Random pictures were taken of each specimen. Adenoma foci were counted per mm2. Positive cells in adenoma foci were counted and expressed as percentage of all adenoma cells.
Results
BAY 43-9006 and CI-1040 are equally effective in vitro but differ in their in vivo properties
To determine whether the inhibitors used in our study are active we performed at first an in vitro assay. The activity of BAY 43-9006 and CI-1040 was measured using an ELISA method that detects phosphorylation of ERK that is dependent on Raf and MEK activity [13]. Both substances were equally effective in vitro with complete inhibition of ERK phosphorylation at concentrations of less than 1 μM (Fig. 1a). The IC50 values for inhibition of C-Raf by BAY 43-9006 and by CI-1040 in our assays were 16 nM and 12 nM, respectively. In addition, the effect of both inhibitors was also tested on cell lines. At a concentration of 0,01 μM the inhibitory effect of CI-1040 on ERK phosphorylation was more than 90 %, whereas BAY 43-9006 at 4 μM lead to an inhibition in the range between 60 – 80 % as determined by Western blotting and immunohistochemistry (data not shown).
Figure 1.
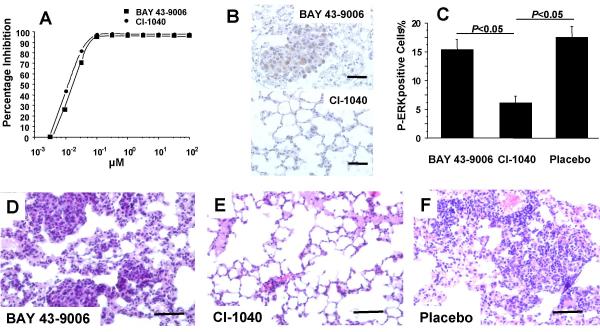
MEK inhibitor CI-1040 but not Raf inhibitor BAY 43-9006 reduces lung adenomas in vivo. (A) MEK activity was completely inhibited in vitro by BAY 43-9006 and CI-1040 as determined by ERK activation enzyme-linked immunosorbent assay. IC50 values for BAY 43-9006 and CI-1040 were 16 nM and 12 nM respectively. (B, C) The amount of P-ERK and the number of P-ERK positive cells were reduced in vivo by treatment with CI-1040 but not with BAY 43-9006. (D, E, F) Treatment with CI-1040 but not with BAY 43-9006 reduced adenomas and improved lung structure after 21d (hematoxylin and eosin staining; scale bars = 60 μm). Lung adenomas as well as the overall lung morphology was indistinguishable in placebo treated and untreated animals (data not shown).
At next, the in vivo effects of a systemic administration of the inhibitors were analyzed using our Raf dependent lung tumor mice. Comparable serum concentrations of both inhibitors were reached after intraperitoneal injection as determined by liquid-liquid extraction of mouse serum and consecutive HPLC analysis [14] and Sirrenberg unpublished data). Lung sections of transgenic mice that have been treated for three weeks with either CI-1040 or BAY 43-9006 were examined for the amount of phosphorylated ERK. After treatment with CI-1040 the amount of phosphorylated ERK was clearly reduced as determined by immunohistochemistry (Fig. 1b) or Western blotting (data not shown). The number of cells that were stained by the Phospho-ERK antibody was reduced almost threefold (Fig. 1c). In contrast, phosphorylation of ERK in lung adenomas in vivo was virtually not affected by systemic treatment of mice with BAY 43-9006 (Fig. 1b,1c) or in controls (data not shown). Moreover, treatment with CI-1040 did not only reduce adenomas but also improve the overall lung structure with thin alveolar walls, whereas BAY 43-9006 again has no detectable effect (Fig. 1d,1e,1f).
The number of adenoma foci per mm2 was determined and found to be reduced more than 75 % after CI-1040 treatment in comparison to BAY 43-9006 treated or placebo treated animals (Fig. 2a). This treatment results in a reduced ratio of lung to body weight of lungs obtained from mice after CI-1040 treatment (7.5 ± 0.3 mg/g) in comparison to lungs isolated from mice after BAY 43-9006 treatment (10.4 ± 0.6 mg/g, P < 0.05).
Figure 2.
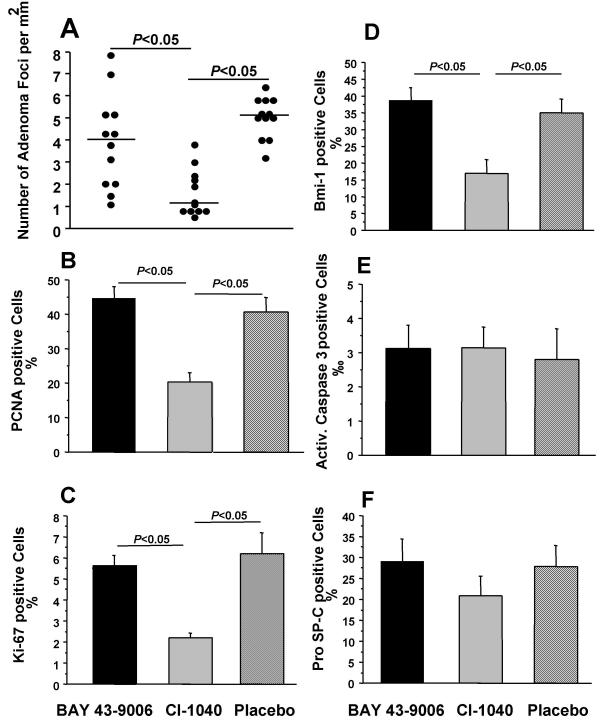
MEK inhibitor CI-1040 reduces proliferation in lung adenomas but has no influence on apoptosis or lung specific differentiation. (A) The number of adenoma foci per mm2 was reduced after treatment with CI-1040 but not with BAY 43-9006. (B) CI-1040 reduced the PCNA positive cells in lung adenomas by half but not BAY 43-9006. (C, D) Ki-67 and Bmi-1 positive cells were reduced to a third in lung adenomas after CI-1040 treatment. (E) Neither BAY 43-9006 nor CI-1040 activated the caspase-3 mediated apoptotic pathway in lung adenomas. (F) BAY 43-9006 or CI-1040 treatment did not change cell differentiation in lung adenomas, which was assessed by expression of pro-SP-C, a marker for alveolar type II cells.
CI-1040 reduces proliferation in lung adenomas but has no influence on apoptosis or lung specific differentiation
The adenoma foci were further analyzed for proliferation, apoptosis and differentiation to decipher the mechanism of action by which CI-1040 reduced adenomas. We assessed proliferation by immunohistochemistry for proliferating cellular antigen (PCNA), Ki-67 and Bmi-1. The percentage of PCNA positive cells was clearly reduced by half after CI-1040 treatment (Fig. 2b). The percentage of Ki-67 and Bmi-1 positive cells was reduced to a third after treatment with CI-1040 (Fig. 2c,2d). Apoptotic cells were detected by immunohistochemistry for activated caspase-3 and no difference was detected after BAY 43-9006 or CI-1040 treatment (Fig. 2e). Adenoma cells were analyzed for the expression of pro-SP-C as a marker of alveolar type II cell differentiation. The percentage of pro-SP-C positive cells did not change after BAY 43-9006 or CI-1040 treatment (Fig. 2f).
Discussion
We were able to show inhibition of ERK phosphorylation in vitro by both, the MEK inhibitor CI-1040 and the Raf inhibitor BAY 43-9600. In addition, both drugs inhibited ERK phosphorylation to a comparable level when tested on cell lines in tissue culture (data not shown). Although both inhibitors reached comparable serum concentrations after intraperitoneal administration (data not shown), the MEK inhibitor was active in mice whereas the Raf inhibitor was not. Why BAY 43-9006 is not active in our Raf dependent lung tumor mouse model and whether Raf and MEK might fulfill different functional roles with respect to lung adenoma formation needs to be further addressed in detail in future.
One possible explanation for the observed effects may relate to differences in the in vivo accessibility of these inhibitors to sub-cellular sites where C-Raf is localized. Alternatively, inhibition of Raf kinases might be more difficult to achieve due to multiple feedback mechanisms [15], whereas the regulation of MEK is less complex and inhibition of MEK is easier achieved in the living organism. Along this line it was already discussed previously that inhibitors targeting the kinase activity of Raf might not be useful as anticancer drugs because inhibition of Raf is always counterbalanced by reactivation [16]. Finally, it was recently suggested that C-Raf has signaling properties that are independent of its protein kinase activity [17]. Therefore, it has also to be considered that BAY 43-9006-bound kinase-inactive C-Raf has still transforming activity in pneumocyte type II cells. But this possibility appears unlikely in the light of earlier results demonstrating the loss of transforming activity and concomitant gain of dominant-negative function of c-Raf genes with mutations that inactivate the protein kinase activity of C-Raf [18-20]. Moreover, there is a direct correlation between reversal of a transformed phenotype by BAY43-9006 and Phospho-ERK suppression (Sirrenberg and Rapp, unpublished data).
Previous work on CI-1040 has already established that this drug has a cytostatic effect on subcutaneously implanted colon tumors of human and mouse origin harboring RAS mutations [21]. In addition, in a mouse model of metastatic melanoma that was dependent on a B-Raf mutation CI-1040 was preventing the formation of new pulmonary metastases and caused rapid regression of already established pulmonary metastases [22]. Both publications as well as our report indicate that CI-1040 has a broad spectrum of antitumor activity and will be useful for the treatment of cancers that are dependent on mutations or overexpression of members of the mitogenic cascade upstream of ERK.
We also determined the effects of both inhibitors on expression of Bmi-1. The number of Bmi-1 positive cells in lung sections was reduced more than twofold by CI-1040, whereas BAY 43-9006 had no effect. The observed effects of CI-1040 may be limited to proliferation as neither apoptosis nor alveolar differentiation was directly affected. Bmi-1 was originally cloned as c-Myc cooperating oncogene in murine lymphomas [23,24]. A role for Bmi-1 in human tumorigenesis was recently suggested by the discovery of Bmi-1 amplification in mantle cell lymphomas [25] and of Bmi-1 overexpression in a variety of tumors including non-small cell lung cancer [26]. Since CI-1040 might be more effective in inhibiting the proliferation of Bmi-1 positive adenoma cells, we want to address in future whether Bmi-1 is a self-renewal factor of lung stem cells or lung adenoma stem cells.
Conclusions
These experiments demonstrate an essential role of MEK/ERK signaling for the maintenance of Raf induced transformation in vivo. Effects on the ERK pathway in vivo may be predictive for effective tumor drug design and therapy of Raf driven tumors. CI-1040 is currently evaluated in phase II trials and may be a promising anti-proliferative drug for Ras and/or Raf-dependent human malignancies.
Competing interests
None declared.
Authors' contributions
BWK was primarily involved and responsible for work related to histology and immunohistochemistry. RG carried out or supervised all work related to the ELISA and the drug administration. URR conceived the study and participated in its design and coordination. All authors were involved in drafting the manuscript and all approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
We thank Ralf Schreck for assistance in preparation of the manuscript, Tamara Potapenko for expert help with the mouse work and Ludmilla Wixler and Renate Metz for Phospho-ERK assays. Furthermore, we thank Christian Sirrenberg (Merck KGaA, Darmstadt) for the collaboration and his support. Work was funded by the DFG (SFB487) and the Mildred Scheel Foundation.
Contributor Information
Boris W Kramer, Email: Kramer_b@kinderklinik.uni-wuerzburg.de.
Rudolf Götz, Email: goetz@mail.uni-wuerzburg.de.
Ulf R Rapp, Email: rappur@mail.uni-wuerzburg.de.
References
- Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- Shawver LK, Slamon D, Ullrich A. Smart drugs: tyrosine kinase inhibitors in cancer therapy. Cancer Cell. 2002;1:117–123. doi: 10.1016/S1535-6108(02)00039-9. [DOI] [PubMed] [Google Scholar]
- Herrera R, Sebolt-Leopold JS. Unraveling the complexities of the Raf/MAP kinase pathway for pharmacological intervention. Trends Mol Med. 2002;8:S27–31. doi: 10.1016/S1471-4914(02)02307-9. [DOI] [PubMed] [Google Scholar]
- Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL, Franklin RA, McCubrey JA. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. 2003;17:1263–1293. doi: 10.1038/sj.leu.2402945. [DOI] [PubMed] [Google Scholar]
- Kerkhoff E, Rapp UR. The Ras-Raf relationship: an unfinished puzzle. Adv Enzyme Regul. 2001;41:261–267. doi: 10.1016/S0065-2571(00)00023-6. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Lyons JF, Wilhelm S, Hibner B, Bollag G. Discovery of a novel Raf kinase inhibitor. Endocr Relat Cancer. 2001;8:219–225. doi: 10.1677/erc.0.0080219. [DOI] [PubMed] [Google Scholar]
- Allen LF, Sebolt-Leopold J, Meyer MB. CI-1040 (PD184352), a targeted signal transduction inhibitor of MEK (MAPKK) Semin Oncol. 2003;30:105–116. doi: 10.1053/j.seminoncol.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Rapp UR, Fensterle J, Albert S, Gotz R. Raf kinases in lung tumor development. Adv Enzyme Regul. 2003;43:183–195. doi: 10.1016/S0065-2571(03)00002-5. [DOI] [PubMed] [Google Scholar]
- Kerkhoff E, Fedorov LM, Siefken R, Walter AO, Papadopoulos T, Rapp UR. Lung-targeted expression of the c-Raf-1 kinase in transgenic mice exposes a novel oncogenic character of the wild-type protein. Cell Growth Differ. 2000;11:185–190. [PubMed] [Google Scholar]
- Fedorov LM, Tyrsin OY, Papadopoulos T, Camarero G, Gotz R, Rapp UR. Bcl-2 determines susceptibility to induction of lung cancer by oncogenic CRaf. Cancer Res. 2002;62:6297–6303. [PubMed] [Google Scholar]
- Fedorov LM, Papadopoulos T, Tyrsin OY, Twardzik T, Gotz R, Rapp UR. Loss of p53 in craf-induced transgenic lung adenoma leads to tumor acceleration and phenotypic switch. Cancer Res. 2003;63:2268–2277. [PubMed] [Google Scholar]
- Mallon R, Feldberg LR, Kim SC, Collins K, Wojciechowicz D, Hollander I, Kovacs ED, Kohler C. An enzyme-linked immunosorbent assay for the Raf/MEK1/MAPK signaling cascade. Anal Biochem. 2001;294:48–54. doi: 10.1006/abio.2001.5151. [DOI] [PubMed] [Google Scholar]
- Afify S, Rapp UR, Högger P. Validation of a simple chromatography assay for the quantification of the Raf kinase inhibitor BAY 43-9006 in small samples of serum. J Chromatogr B Biomed Appl. [DOI] [PubMed]
- Cohen PT, Browne GJ, Delibegovic M, Munro S. Assay of protein phosphatase 1 complexes. Methods Enzymol. 2003;366:135–144. doi: 10.1016/s0076-6879(03)66012-x. [DOI] [PubMed] [Google Scholar]
- Hall-Jackson CA, Eyers PA, Cohen P, Goedert M, Boyle FT, Hewitt N, Plant H, Hedge P. Paradoxical activation of Raf by a novel Raf inhibitor. Chem Biol. 1999;6:559–568. doi: 10.1016/S1074-5521(99)80088-X. [DOI] [PubMed] [Google Scholar]
- Huser M, Luckett J, Chiloeches A, Mercer K, Iwobi M, Giblett S, Sun XM, Brown J, Marais R, Pritchard C. MEK kinase activity is not necessary for Raf-1 function. Embo J. 2001;20:1940–1951. doi: 10.1093/emboj/20.8.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolch W, Heidecker G, Lloyd P, Rapp UR. Raf-1 protein kinase is required for growth of induced NIH/3T3 cells. Nature. 1991;349:426–428. doi: 10.1038/349426a0. [DOI] [PubMed] [Google Scholar]
- Bruder JT, Heidecker G, Rapp UR. Serum-, TPA-, and Ras-induced expression from Ap-1/Ets-driven promoters requires Raf-1 kinase. Genes Dev. 1992;6:545–556. doi: 10.1101/gad.6.4.545. [DOI] [PubMed] [Google Scholar]
- Troppmair J, Bruder JT, Munoz H, Lloyd PA, Kyriakis J, Banerjee P, Avruch J, Rapp UR. Mitogen-activated protein kinase/extracellular signal-regulated protein kinase activation by oncogenes, serum, and 12-O-tetradecanoylphorbol-13-acetate requires Raf and is necessary for transformation. J Biol Chem. 1994;269:7030–7035. [PubMed] [Google Scholar]
- Sebolt-Leopold JS, Dudley DT, Herrera R, Van Becelaere K, Wiland A, Gowan RC, Tecle H, Barrett SD, Bridges A, Przybranowski S, Leopold WR, Saltiel AR. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med. 1999;5:810–816. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- Collisson EA, De A, Suzuki H, Gambhir SS, Kolodney MS. Treatment of metastatic melanoma with an orally available inhibitor of the Ras-Raf-MAPK cascade. Cancer Res. 2003;63:5669–5673. [PubMed] [Google Scholar]
- Haupt Y, Alexander WS, Barri G, Klinken SP, Adams JM. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell. 1991;65:753–763. doi: 10.1016/0092-8674(91)90383-a. [DOI] [PubMed] [Google Scholar]
- van Lohuizen M, Verbeek S, Scheijen B, Wientjens E, van der Gulden H, Berns A. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell. 1991;65:737–752. doi: 10.1016/0092-8674(91)90382-9. [DOI] [PubMed] [Google Scholar]
- Bea S, Tort F, Pinyol M, Puig X, Hernandez L, Hernandez S, Fernandez PL, van Lohuizen M, Colomer D, Campo E. BMI-1 gene amplification and overexpression in hematological malignancies occur mainly in mantle cell lymphomas. Cancer Res. 2001;61:2409–2412. [PubMed] [Google Scholar]
- Vonlanthen S, Heighway J, Altermatt HJ, Gugger M, Kappeler A, Borner MM, van Lohuizen M, Betticher DC. The bmi-1 oncoprotein is differentially expressed in non-small cell lung cancer and correlates with INK4A-ARF locus expression. Br J Cancer. 2001;84:1372–1376. doi: 10.1054/bjoc.2001.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]