

Abstract
Objective:
To determine whether myelin oligodendrocyte glycoprotein antibodies (MOG-Abs) were predictive of a demyelination phenotype in children presenting with acquired demyelinating syndrome (ADS).
Method:
Sixty-five children with a first episode of ADS (12 acute disseminated encephalomyelitis, 24 optic neuritis, 18 transverse myelitis, 11 other clinically isolated syndrome) were identified from 2 national demyelination programs in the United Kingdom and France. Acute serum samples were tested for MOG-Abs by cell-based assay. Antibodies were used to predict diagnosis of multiple sclerosis (MS) at 1 year.
Results:
Twenty-three of 65 (35%) children had MOG-Abs. Antibody-positive and antibody-negative patients were not clinically different at presentation, but identification of MOG-Abs predicted a non-MS course at 1-year follow-up: only 2/23 (9%) MOG-Ab–positive patients were diagnosed with MS compared to 16/42 (38%) MOG-Ab–negative patients (p = 0.019, Fisher exact test). Antibody positivity at outset was a useful predictor for a non-MS disease course, with a positive predictive value of 91% (95% confidence interval [CI] 72–99), negative predictive value of 38% (95% CI 24–54), positive likelihood ratio of 4.02 (CI 1.0–15.4), and odds ratio of 6.5 (CI 1.3–31.3).
Conclusions:
MOG-Abs are found at presentation in 35% of patients with childhood ADS, across a range of demyelinating disorders. Antibody positivity can be useful in predicting a non-MS disease course at onset.
Myelin oligodendrocyte glycoprotein (MOG) is exclusively expressed in the CNS. Although only a minor component (0.05%) of myelin, its location on the outermost lamellae of the myelin sheath1 makes it available for antibody binding and a potential target for autoantibody-mediated disease. MOG antibodies (MOG-Abs) have previously been shown to induce or contribute to demyelination in in vitro cultures and animal models.2,3 However, earlier ELISA and Western blot studies that identified antibodies to linear epitopes of the denatured MOG protein reported inconsistent results and positivity in healthy controls.1,4
More recent assays to detect antibodies that bind to conformational epitopes are more informative.3 MOG-Abs have been found in children with CNS demyelination, such as acute disseminated encephalomyelitis (ADEM), clinically isolated syndrome (CIS), multiple sclerosis (MS),5 and other recurrent forms of acquired demyelinating syndromes (ADS), more often than in adults.3,5 Techniques vary among laboratories, and there are conflicting reports of associations between various neurologic diseases and MOG-Abs. A cell-based assay (CBA) using only the extracellular and transmembrane domains of MOG identified binding to conformational MOG epitopes and seemed to be specific for non-MS demyelinating diseases,6 but when both we and others3 used the full-length protein, the sensitivity was higher but only specific when the serum was tested at a dilution of 1:160.3
Here we evaluated a pediatric cohort with a first episode of demyelination for the presence of MOG-Abs using the full-length MOG CBA. We reviewed the clinical and imaging phenotype of the patients and compared the antibody-positive and antibody-negative patients to determine whether MOG-Ab–positive children have a distinguishable phenotype, as has been reported in adults.6
METHODS
Patients.
Sixty-five consecutive children younger than 16 years with a first episode of ADS (12 ADEM, 24 optic neuritis [ON], 18 transverse myelitis [TM], 11 CIS) were identified from 2 established national demyelination programs in the UK7 (n = 49) and France (n = 16).8 Children presenting between September 2009 and October 2011 in whom a serum sample was available for testing were tested for MOG-Abs. Demographic and clinical data, including sex, age at onset, CSF analysis, and acute-onset first presentation MRI findings, were reviewed for each patient at presentation and at 1-year follow-up. MRI scans were reviewed blinded to clinical features. A standardized form was completed utilizing previously described nomenclature.8,9 Relapses, both clinical and radiologic, were defined by the reporting clinicians. Demyelinating phenotypes at onset and at 1-year follow-up were classified by a panel of pediatric neurologists within the respective programs based on International Pediatric Multiple Sclerosis Study Group criteria.10 Two groups were used as controls: adult patients with MS (n = 100) and aquaporin-4 (AQP4) antibody–positive adult patients (n = 100).
MOG-IgG cell-based immunofluorescence assay.
Acute samples taken within 3 months of presentation were tested (Y.H., P.W.) using CBAs in routine clinical use, as previously described (serum dilution 1:160).11 Here the binding of serum IgG to the surface of human embryonic kidney cells transfected with cDNA encoding the full-length MOG protein (courtesy of M. Reindl, Innsbruck, Austria) was visualized using a fluorescence-labeled secondary antibody. The observers were blinded to the clinical details. Positive serum samples were further diluted to determine their endpoint titers. A proportion of the UK children had been tested for MOG-Abs previously using the truncated MOG construct12 and were reanalyzed here using the full-length MOG construct alongside the remaining patients.
Statistical analysis.
Statistical analysis was performed using commercially available software (IBM SPSS, release 18.0 [IBM Corporation, Armonk, NY] or GraphPad Prism 6 [GraphPad Software Inc., La Jolla, CA]). Nonparametric statistical tests (Mann-Whitney tests) were used for continuous distributions, and χ2 or Fisher exact tests were used for nominal data. A regression decision tree analysis was used to create a tree-based classification model to aid in predicting risk of MS from first presentation. The classification tree χ2 Automatic Interaction Detector (CHAID) method builds segments and provides a final predictive model based on the best combination of predictor variables. The following predictor variables were used to grow the tree: MOG-Abs (positive/negative), age, intrathecal oligoclonal bands (OCBs) at onset (positive/negative), ADS phenotype at onset (ADEM, ON, TM, other CIS), anatomical distribution of radiologic features (cortical gray, periventricular, juxtacortical, deep white, intracallosal, basal ganglia/thalamus, brainstem, and cerebellar), and lesion characteristics (well-demarcated, diffuse, size, T1 hypointensity, and contrast enhancement).
Standard protocol approvals, registrations, and patient consents.
Ethical approval was obtained from the UK Multicentre Research Ethics Committee (09/H1202/92) and local research ethics committee in Paris (PP 12-024) for the respective national demyelination programs, and ethical approval for further antibody testing on referred samples was obtained from the Oxfordshire Regional Ethical Committee A (07/Q1604/28).
RESULTS
Of the 65 children with ADS tested, 23 were MOG-Ab positive (>1:160; 35%; median age 9.9 years, range 3.0–15.8 years, titers from 1:540 to 1:4,860) and 42 were MOG-Ab negative (65%; median age 13.1 years, range 1.3–15.5 years). None of the control patients were positive for MOG-Abs. All patients with ADS were confirmed to be AQP4-Ab negative.
MOG-Abs were identified across all ADS phenotypes (see the table for clinical data). MOG-Abs were detected in pediatric patients with ADEM (4/12; range 1:540–4,860, median 1:1,620), ON (12/24; range 1:540–4,860, median 1:1,620), TM (4/18; range 1:540–1,680, median 1:1,080), and other CIS (3/11; range 1:1,620–4,860. median 1:1,620) but not in AQP4-Ab–positive patients with neuromyelitis optica (NMO) or adults with MS (figure 1A). There was a trend to more MOG-Ab–positive patients with ON and fewer with TM, but the titers did not vary between the phenotypes (figure 1A) and there was no correlation between antibody titer and age at presentation (figure 1B). No significant differences between the antibody-positive and antibody-negative groups were observed in the presence of T2 abnormal brain and spine MRI, lesion characteristics (well-demarcated, diffuse, size, T1 hypointensity, and contrast enhancement), or both McDonald 2010 dissemination in space and dissemination in time criteria at onset for CIS cases. However, stratified to the anatomical involvement, MOG-Ab–positive patients were less likely to have intracallosal (1/23 vs 15/40, p = 0.006, Fisher exact test), deep white (5/23 vs 23/40, p = 0.008, Fisher exact test), periventricular (3/23 vs 17/40, p = 0.02, Fisher exact test), and cerebellar (3/23 vs 16/42, p = 0.05, Fisher exact test) lesions (see the table).
Table.
Demographics, clinical and paraclinical features, and outcome of MOG-Ab–positive and MOG-Ab–negative patients

Figure 1. Full-length MOG cell-based assay using a serum dilution of 1:160 as a cutoff for positivity (red line in both plots).
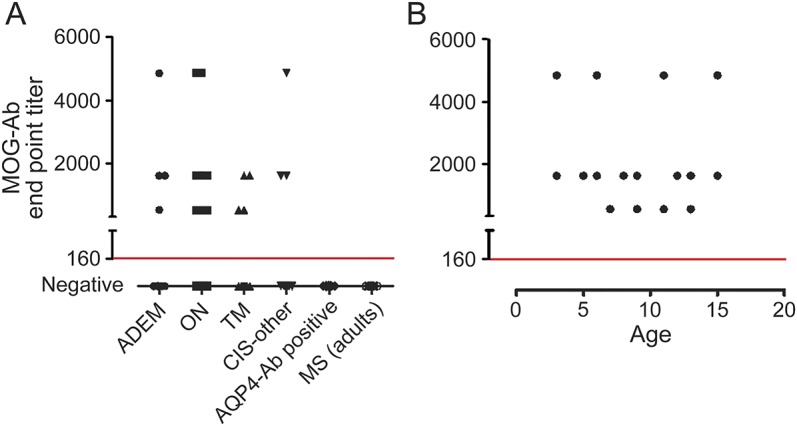
(A) Myelin olidgodendrocyte glycoprotein antibodies (MOG-Abs) were detected in a range of childhood demyelination syndromes but not in aquaporin-4 (AQP4)-Ab–positive neuromyelitis optica patients (0/100) or adults with multiple sclerosis (MS) (0/100). There was no correlation between MOG-Ab titer at onset and acquired demyelinating syndrome phenotype (A) or patient age (B). ADEM = acute disseminated encephalomyelitis; CIS = clinically isolated syndrome; ON = optic neuritis; TM = transverse myelitis.
CSF OCBs at disease onset were very rare in the MOG-Ab–positive group compared to the MOG-Ab–negative group (1/16 vs 14/35, p = 0.019, Fisher exact test). Considering the 51 patients who were tested for both OCBs and MOG-Abs, only 1/16 MOG-Ab–positive patients developed MS, compared with 14/35 MOG-Ab–negative patients. MOG-Ab status was additionally informative to OCB testing. Eleven of 15 OCB+ patients developed MS (73%), whereas 11 of 14 OCB+ and MOG-Ab− patients developed MS (79%; see figure 2).
Figure 2. Summary of the utility of MOG-Abs and OCB testing in predicting pediatric disease course at onset compared to clinical follow-up at 1 year.

Following testing with either myelin olidgodendrocyte glycoprotein antibody (MOG-Ab) or oligoclonal blands (OCBs), the additional testing of the respective other is represented by arrows to the respective outcomes. A MOG-Ab–positive test predicted a non–multiple sclerosis (MS) diagnosis, whereas OCB positivity was highly predictive of MS. Eleven of 15 OCB- positive patients developed MS (73%), whereas 11 of 14 OCB-positive and MOG-Abs–negative patients developed MS (79%). The one MOG-Ab–positive and OCB-positive patient did not have MS, and all MOG-Ab–positive and OCB-negative cases had a non-MS course, compared to 91% if only OCB was negative. Of the 14 patients not tested for intrathecal OCBs, 7 patients tested positive for MOG-Ab; 2 patients from the antibody-positive and 2 from the antibody-negative groups had a diagnosis of MS at 1-year follow-up. ADEM = acute disseminated encephalomyelitis; CIS = clinically isolated syndrome; N/D = not done; ON = optic neuritis; TM = transverse myelitis.
At 1-year follow-up, 16/42 MOG-Ab–negative patients were diagnosed with MS (15 clinical, 1 radiologic), compared to only 2/23 MOG-Ab–positive patients (p = 0.019, Fisher exact test; see the table and figure 3). MOG-Abs were found in 2/18 patients with MS compared to 21/47 in the non-MS group (p = 0.02, Fisher exact test).
Figure 3. Two of 23 MOG-Ab–positive patients were diagnosed with MS compared to 16/42 MOG-Ab–negative patients (p = 0.02, Fisher exact test).

As myelin oligodendrocyte glycoprotein antibodies (MOG-Abs) were only tested retrospectively, identification of the antibodies did not influence the final diagnosis. One hundred adult patients with multiple sclerosis (MS) and 100 adults with aquaporin-4 (AQP4)-Ab–positive neuromyelitis optica were all MOG-Ab negative. ADEM = acute disseminated encephalomyelitis; CIS = clinically isolated syndrome; ON = optic neuritis; TM = transverse myelitis.
Presence of MOG-Abs at disease outset was a useful predictor for non-MS disease course, with a positive predictive value of 91% (95% confidence interval [CI] 72–99), negative predictive value of 38% (95% CI 24–54), positive likelihood ratio of 4.02 (CI 1.0–15.4), and odds ratio (OR) of 6.5 (CI 1.3–31.3). In MOG-positive patients, 1/6 with asymptomatic brain lesions had MS vs 1/13 without asymptomatic brain lesions (OR 2.4, 95% CI 0.12–46, p = 0.6). In MOG-negative patients, 14/22 with asymptomatic brain lesions had MS vs 2/12 without asymptomatic brain lesions (OR 8.8, 95% CI 1.5–50, p = 0.02). At follow-up, there was no difference in the disabilities of patients between both groups (see table).
A regression tree analysis generated with clinical and paraclinical features (including MOG-Ab testing) to predict probability of MS diagnosis at 1 year had an overall 94% accuracy (1 MS and 3 non-MS cases incorrectly predicted; figure e-1 at Neurology.org/nn). Neither MOG-Ab nor OCB had significant added utility over the established clinical (demyelinating phenotype) and imaging predictors of the subsequent demyelination course. In MOG-Ab–positive patients, however, the demyelinating syndrome (ADEM/ON/TM) had additional utility in predicting a non-MS course. In MOG-Ab–negative patients, lesion location (periventricular and deep gray), gadolinium enhancement, and demyelination syndrome contributed to the prediction model (detailed in figure e-1).
DISCUSSION
Defining biomarkers that will help identify children with MS at presentation is an important aim. We tested serum samples taken at presentation from 65 children with ADS for MOG-Ab 1 year after onset when a diagnosis had been made, and MOG-Ab positivity identified acutely at first presentation of ADS was predictive of patients not following an MS course. Furthermore, these children were less likely to have CSF OCB positivity and MRI lesion distributions characteristic of MS, parameters previously reported to be useful in distinguishing MS from other demyelinating syndromes at onset.9,13
Identification of differences in MRI lesion distributions between MOG-Ab–positive and MOG-Ab–negative patients reflects the higher rate of MS in the MOG-Ab–negative group, particularly in patients with abnormal MRI. Similar findings were recently reported in a large cohort of MOG-Ab patients with ADEM,14 but as patients with MS only rarely present with ADEM, the negative predictive value of the antibody cannot be determined from this study. Nevertheless, MOG-Ab–positive children were more likely to have radiologic lesion resolution and had a better outcome.14
The association between intrathecal OCBs and MS is well-established,15 and children with OCBs identified at presentation of a first demyelinating episode were significantly more likely to be diagnosed with MS,15 as also found here. On reviewing recent studies of pediatric patients with MOG-Ab, OCBs were found in only 4/17 (23%) children presenting with ON,11 in none of the 7 children presenting with ADEM followed by recurrent or monophasic ON,16 and in none of the 3 children with NMO spectrum disorders (NMOSD).17
Surprisingly, there were no differences in the age at onset or patient demographics between the antibody-positive and antibody-negative groups, and antibody titers did not correlate with age, as previously reported.18 Although the age at onset is a known predictor of disease course in children with an initial demyelinating event, it was not a useful marker in our predictive model either in isolation or in combination with other prognostic factors such as abnormal MRI.
MOG-Abs have been identified in 4/27 (15%) adults6 and in 3/6 (50%)17 children with AQP4-Ab–negative NMOSD, but none of the children, nor any in this cohort, currently fulfill the diagnostic criteria for NMO.19 None of our MOG-Ab–positive patients had received long-term immunosuppression, and only 3 had a clinical relapse (of which 2 have MS and are currently being evaluated for treatment). It remains unclear whether these patients should be treated similarly to patients with AQP4-positive NMO20 or whether MOG-positive patients with a restricted form of demyelination (recurrent ON and TM) should be considered as having NMOSD, a diagnosis currently unified by the detection of AQP4-Ab.20 This is important, as several conventional MS drugs such as interferon21 and natalizumab22 have been associated with disease worsening in patients with NMO (both AQP4-Ab positive and AQP4-Ab negative), so their use in other antibody-mediated demyelinating diseases needs to be cautiously evaluated.
A limitation of our study is the short follow-up period. A longer follow-up would allow for more certainty of the course of the demyelination syndrome23; however, in a large Canadian cohort, the median time to a second clinical event or change on brain MRI for children with MS was 127 days (interquartile range 91–222).13 As all our patients were treated empirically with corticosteroids and/or IV immunoglobulin (IVIg) (none received plasma exchange), some may have had MOG-Ab testing following treatment, potentially confounding the results. Reports of low antibody positivity (typically for multiple antigens) following treatment with IVIg are unlikely to be the cause of the very high titers seen in our antibody-positive patients. Another limitation of the study is that MOG-Ab–positive patients, including the 3 relapsing ones, did not have a repeat sample taken (neither interval nor at the time of relapse), so we cannot say whether the persistence of MOG-Abs predicted a relapsing course, as has been previously reported.17,18,24 Recently, in a study of adult patients with NMO and suspected limited forms of the disease, MOG-Ab titers could not aid in differentiating between the different clinical phenotypes, including monophasic and relapsing diseases. Moreover, decrease of MOG-Ab titers was not associated with a monophasic course or better outcome, and persistence of antibodies for several years was reported in patients with clinical symptom resurgence.25
The phenotypic similarities between the different demyelinating syndromes at onset remain a significant clinical challenge when trying to identify children with MS. Here we identified MOG-Ab as an early predictor of the subsequent course of demyelination, a finding that requires replication in other and preferably larger cohorts.
Detecting MOG-Abs and determining their significance in various demyelinating syndromes remains an evolving field of investigation. The absence of MRI and CSF features typically seen in both adults and children with MS further supports the idea that MOG-Ab positivity indicates a different disease phenotype from MS. Ultimately, the presence of MOG-Abs cannot replace the other previously validated factors (clinical, OCB, and MRI) that predict MS risk but may prove useful either as an adjunct when the previously validated factors are already strongly predictive of MS (CIS, OCB-positive, and MRI changes) or as a conjunct when some of these factors are less striking or have not been performed.
Supplementary Material
GLOSSARY
- Ab
antibody
- ADEM
acute disseminated encephalomyelitis
- ADS
acquired demyelinating syndrome
- AQP4
aquaporin-4
- CBA
cell-based assay
- CI
confidence interval
- CIS
clinically isolated syndrome
- IVIg
IV immunoglobulin
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- NMO
neuromyelitis optica
- NMOSD
NMO spectrum disorder
- OCB
oligoclonal band
- OR
odds ratio
Footnotes
Supplemental data at Neurology.org/nn
NOTE ADDED IN PROOF
We have recently improved the sensitivity and specificity for MOG-Ab detection by using a more IgG specific secondary antibody for detection26 that does not detect IgM binding to MOG. Subsequent studies on all the sera reported here confirmed that they were IgG, not IgM.
AUTHOR CONTRIBUTIONS
The study was designed by Y.H., M.L., M.A., and P.W., with suggestions from A.V. P.W. and A.V. were responsible for supervising the laboratory studies performed by Y.H. and M.W. Y.H., M.A., K.D., C.H., J.P., P.N., M.T., E.W., and M.L. contributed to the evaluation and recording of clinical data. M.L. and P.W. supervised the study as a whole and contributed to the key message of the manuscript. Y.H. and M.A. prepared early drafts of the manuscript and all authors contributed to the revisions of the manuscript.
STUDY FUNDING
This work was supported by Oxford University Clinical Academic Graduate School, Oxford (Y.H.), the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre based at Oxford University Hospitals NHS Trust and the University of Oxford (Y.H., P.W., and A.V.), and by the NHS specialized services for rare diseases (neuromyelitis optica; P.W., M.W., J.P., and A.V.).
DISCLOSURE
Y. Hacohen has received research support from NIHR. M. Absoud is on the data safety monitoring board for a study sponsored by Neurim Pharmaceuticals; is on the editorial advisory board for the International Journal of Language & Communication Disorders; and has received research support from MS Society and Action Medical Research Charities. K. Deiva has received travel funding from Biogen and Merck Serono. C. Hemingway has received speaker honoraria from Biogen Idec, Terumo Products, Merck Serono, and Bayer and has received research support from Multiple Sclerosis Action Research. P. Nytrova has received speaker honoraria from Biogen and Novartis and has received research support from Czech Ministries of Education and Health. M. Woodhall reports no disclosures. J. Palace has been a UK advisory board participant for Merck Serono, Bayer Schering Pharma, Biogen Idec, Teva Pharmaceuticals Industries Ltd, Novartis Pharmaceuticals UK Ltd, and Sanofi-Aventis; has received travel support from Merck Serono, Novartis, and Biogen Idec; has a patent application for the use of metabolomics as a method to diagnose and stage disease in multiple sclerosis (University of Oxford); has consulted for Ono Pharmaceuticals Ltd, Chugai Pharma Ltd, CI Consulting, Biogen Idec, and GlaxoSmithKline; and has received research support from Bayer, Schering, Merck Serono, Novartis, Department of Health, MS Society, and UK Guthy Jackson Foundation. E. Wassmer has received speaker honoraria and/or travel funding from Biogen, Teva, Genzyme, Shire, UCB, and Merck Serono. M. Tardieu is on the scientific advisory board for Biogen, Genzyme, and Novartis; has received travel funding from Novartis; and is on the editorial board for European Journal of Paediatric Neurology and Multiple Sclerosis Journal. A. Vincent has received travel funding and speaker honoraria from Baxter International Inc and Biogen Inc; is on the editorial board for Neurology; was an associate editor for Brain; holds a patent for LGI1/CASPR2 antibodies and for GABARR antibodies; receives royalties from Athena Diagnostics, Euroimmun, Blackwell Publishing, and Mac Keith Press; has consulted with Athena Diagnostics; and has received research support from NIHR. M. Lim has received travel funding from Merck Serono and has consulted for CSL Behring. P. Waters has received speaker honoraria from Biogen Idec Japan and Euroimmun AG; is a review editor for Frontiers in Molecular Innate Immunity; holds a patent for assays for the detection of antibodies to Igi1, Caspr2, and tag-1; and has submitted a patent on GABAAR. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Hemmer B, Archelos JJ, Hartung HP. New concepts in the immunopathogenesis of multiple sclerosis. Nat Rev Neurosci 2002;3:291–301. [DOI] [PubMed] [Google Scholar]
- 2.Elliott C, Lindner M, Arthur A, et al. Functional identification of pathogenic autoantibody responses in patients with multiple sclerosis. Brain 2012;135:1819–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reindl M, Di Pauli F, Rostasy K, Berger T. The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol 2013;9:455–461. [DOI] [PubMed] [Google Scholar]
- 4.O'Connor KC, McLaughlin KA, De Jager PL, et al. Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat Med 2007;13:211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rostasy K, Reindl M. Role of autoantibodies in acquired inflammatory demyelinating diseases of the central nervous system in children. Neuropediatrics 2013;44:297–301. [DOI] [PubMed] [Google Scholar]
- 6.Kitley J, Woodhall M, Waters P, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 2012;79:1273–1277. [DOI] [PubMed] [Google Scholar]
- 7.Absoud M, Lim MJ, Chong WK, et al. Paediatric acquired demyelinating syndromes: incidence, clinical and magnetic resonance imaging features. Mult Scler 2013;19:76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mikaeloff Y, Suissa S, Vallee L, et al. First episode of acute CNS inflammatory demyelination in childhood: prognostic factors for multiple sclerosis and disability. J Pediatr 2004;144:246–252. [DOI] [PubMed] [Google Scholar]
- 9.Verhey LH, Branson HM, Shroff MM, et al. MRI parameters for prediction of multiple sclerosis diagnosis in children with acute CNS demyelination: a prospective national cohort study. Lancet Neurol 2011;10:1065–1073. [DOI] [PubMed] [Google Scholar]
- 10.Krupp LB, Tardieu M, Amato MP, et al. International Pediatric Multiple Sclerosis Study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Mult Scler 2013;19:1261–1267. [DOI] [PubMed] [Google Scholar]
- 11.Rostasy K, Mader S, Schanda K, et al. Anti-myelin oligodendrocyte glycoprotein antibodies in pediatric patients with optic neuritis. Arch Neurol 2012;69:752–756. [DOI] [PubMed] [Google Scholar]
- 12.Hacohen Y, Absoud M, Woodhall M, et al. Autoantibody biomarkers in childhood-acquired demyelinating syndromes: results from a national surveillance cohort. J Neurol Neurosurg Psychiatry 2014;85:456–461. [DOI] [PubMed] [Google Scholar]
- 13.Banwell B, Bar-Or A, Arnold DL, et al. Clinical, environmental, and genetic determinants of multiple sclerosis in children with acute demyelination: a prospective national cohort study. Lancet Neurol 2011;10:436–445. [DOI] [PubMed] [Google Scholar]
- 14.Baumann M, Sahin K, Lechner C, et al. Clinical and neuroradiological differences of paediatric acute disseminating encephalomyelitis with and without antibodies to the myelin oligodendrocyte glycoprotein. J Neurol Neurosurg Psychiatry Epub 2014 Aug 13; doi: 10.1136/jnnp-2014-308346. [DOI] [PubMed] [Google Scholar]
- 15.Banwell B, Tenembaum S, Lennon VA, et al. Neuromyelitis optica-IgG in childhood inflammatory demyelinating CNS disorders. Neurology 2008;70:344–352. [DOI] [PubMed] [Google Scholar]
- 16.Huppke P, Rostasy K, Karenfort M, et al. Acute disseminated encephalomyelitis followed by recurrent or monophasic optic neuritis in pediatric patients. Mult Scler 2013;19:941–946. [DOI] [PubMed] [Google Scholar]
- 17.Rostasy K, Mader S, Hennes EM, et al. Persisting myelin oligodendrocyte glycoprotein antibodies in aquaporin-4 antibody negative pediatric neuromyelitis optica. Mult Scler 2013;19:1052–1059. [DOI] [PubMed] [Google Scholar]
- 18.Probstel AK, Dornmair K, Bittner R, et al. Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology 2011;77:580–588. [DOI] [PubMed] [Google Scholar]
- 19.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485–1489. [DOI] [PubMed] [Google Scholar]
- 20.Jacob A, McKeon A, Nakashima I, et al. Current concept of neuromyelitis optica (NMO) and NMO spectrum disorders. J Neurol Neurosurg Psychiatry 2013;84:922–930. [DOI] [PubMed] [Google Scholar]
- 21.Palace J, Leite MI, Nairne A, Vincent A. Interferon Beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol 2010;67:1016–1017. [DOI] [PubMed] [Google Scholar]
- 22.Kitley J, Evangelou N, Kuker W, Jacob A, Leite MI, Palace J. Catastrophic brain relapse in seronegative NMO after a single dose of natalizumab. J Neurol Sci 2014;339:223–225. [DOI] [PubMed] [Google Scholar]
- 23.Banwell B, Bar-Or A, Giovannoni G, Dale RC, Tardieu M. Therapies for multiple sclerosis: considerations in the pediatric patient. Nat Rev Neurol 2011;7:109–122. [DOI] [PubMed] [Google Scholar]
- 24.Di Pauli F, Mader S, Rostasy K, et al. Temporal dynamics of anti-MOG antibodies in CNS demyelinating diseases. Clin Immunol 2011;138:247–254. [DOI] [PubMed] [Google Scholar]
- 25.Höftberger R, Sepulveda M, Armangue T, et al. Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler Epub 2014 Oct 24; pii: 1352458514555785. [DOI] [PMC free article] [PubMed]
- 26.Waters P, Woodhall MR, O'Connor KC, et al. MOG cell-based assay detects non-MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm 2015;2:e89 doi:10.1212/NXI.0000000000000089 (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.