

Abstract
Background
The novel bispecific ligand-directed toxin (BLT) DT2219 consists of a recombinant fusion between the catalytic and translocation enhancing domain of diphtheria toxin (DT) and bispecific single chain variable fragments (scFV) of antibodies targeting human CD19 and CD22. We conducted a phase 1 dose escalation study to assess the safety, maximum tolerated dose (MTD), and preliminary efficacy of DT2219 in patients with relapsed/refractory B cell lymphoma or leukemia.
Methods
DT2219 was administered intravenously over 2 hours every other day for 4 total doses. Dose was escalated from 0.5 μg/kg/day to 80 μg/kg/day in nine dose cohorts until a dose limiting toxicity (DLT) was observed.
Results
Twenty-five patients with mature or precursor B-cell lymphoid malignancies expressing CD19 and/or CD22 enrolled to the study. Patients received median 3 prior lines of chemotherapy and 8 failed hematopoietic transplantation. All patients received a single course of DT2219; one patient was retreated. The most common adverse events (AE) including, weight gain, low albumin, transaminitis and fevers were transient grade 1-2 and occurred in patients in higher dose cohorts (≥40 μg/kg/day). Two subjects experienced DLT at dose levels 40 and 60 μg/kg. Durable objective responses occurred in 2 patients; one was complete remission after 2 cycles. Correlative studies showed a surprisingly low incidence of neutralizating antibody (30%).
Conclusions
We have determined the safety of a novel immunotoxin DT2219 and established it's biologically active dose between 40-80 μg/kg/day ×4. A phase II study exploring repetitive courses of DT2219 is planned.
Keywords: diphtheria toxin, refractory lymphoma, ALL, phase 1 trial, immunotherapy
Introduction
DT2219, a recombinant fusion protein contains the catalytic and translocation enhancing domain of diphtheria toxin (DT390) fused with bispecific single chain variable fragments (scFV) of antibodies targeting human CD19 and CD22 cell surface receptors (1). The protein is engineered so that the native binding region of DT is replaced by the more avidly bound scFV. After binding, CD19 and CD22 readily internalize (2, 3) to promote toxin entry into the cytosol, inhibition of protein synthesis and subsequent apoptotic cell death (4). Notably, previous pre-clinical studies showed that the combination of two different scFVs and a toxin on the same single-chain molecule resulted in greater anti-cancer activity compared to monomeric anti-CD19 or anti-CD22 connected with truncated DT (5). In addition, xenograft studies demonstrated significant inhibition of CD22+CD19+ Daudi tumor growth, and an enhanced therapeutic effect with repetitive dosing in vivo (1).
CD19, a 95kDa membrane glycoprotein, is ubiquitously present on the surface of all stages of B lymphocyte development and is also expressed on most B-cell mature lymphoma cells and leukemia cells (6). CD22 is 135-kDa glycoprotein expressed on B lineage lymphoid precursors, including precursor B acute lymphoblastic leukemia, and often is co-expressed with CD19 on mature B cell malignancies (7). DT mediates potent cell-cycle independent cell death and therefore can be particularly effective as an alternative therapy for chemotherapy refractory malignancies (8). We conducted a phase 1 dose escalation study to assess safety, maximum tolerated dose (MTD), and preliminary efficacy in patients with chemorefractory B cell lymphoma or leukemia expressing CD19 and/or CD22.
Patients and Methods
Patients
All patients gave written informed consent to treatment on the institutional review board (IRB)-approved treatment protocol in accordance with Declaration of Helsinki. This clinical trial was registered at clinicaltrials.gov (NCT 00889408). DT2219 was cGMP manufactured at the University of Minnesota under US Food and Drug Administration (FDA) IND-application (IND number 1000780). Inclusion criteria included: age >12 years, CD19 and/or CD22 expressing B-cell lymphoma or leukemia refractory to conventional therapy, and adequate performance and organ function (creatinine ≤1.5 upper limit of normal (ULN), liver function tests <2.5 × ULN; serum albumin>3g/dL; left ventricular ejection fraction≥40%). We excluded patients with active infections, serious concurrent medical problems, history of penicillin allergy and more recently amended the protocol to also exclude patients with history of central nervous malignancy. Patients were treated at the Baylor Scott & White Medical Center, MD Anderson Cancer Center, and Masonic Cancer Center, University of Minnesota.
Treatment Plan
In this phase 1 study, patients received DT2219 in a single course at doses ranging from 0.5 μg/kg/day (1/500th of the MTD in rabbits) to 80 μg/kg/day intravenously (IV) over 2 hours (4 hours for 1st dose) every other day for 4 total doses (days 1,3,5 & 8). The dose was escalated in 9 cohorts until a dose limiting toxicity (DLT) was observed (Table 2). The first 15 patients were treated by rapid escalation design (dose cohorts 1-3) or by standard 3+3 dose escalation design (cohorts 4-6). We applied Continual Reassessment Method (9) to the last 10 patients (dose cohorts 8,9) with the goal to identify the dose level which corresponds to a desired toxicity rate of 33% or less using grade 3 or greater DT2219 related toxicity except blood pressure changes and fever as the targeted toxicity (based on NCI Common Terminology Criteria for Adverse Events version (CTCAE) 4). Administration of doses 2-4 was permitted if pre-dose creatinine was <1.5× ULN and absence of DLT. Supportive care included allopurinol (300 mg/day orally); intravenous fluids; and premedication with diphenylhydramine (25 mg IV), acetaminophen (325 mg orally), hydrocortisone (100 mg IV), and ranitidine (50 mg IV) 30 minutes prior to each DT2219 dose.
TABLE 2. Treatment detail and adverse events.
| Cohort | Escalation detail | DT2219 dose μg/kg/day | Doses received | Total dose per cycle in μg | N | Drug related adverse effects (CTCAE v4.03 toxicity grade) | DLT |
| 1 | Rapid escalation | 0.5 | 4 | 2.0 | 1 | None | No |
| 2 | 1.25 | 4 | 5.0 | 1 | None | No | |
| 3 | 2.5 | 4 | 10 | 1 | Gr 1 fever (n=1) | No | |
| 4 | Standard escalation | 5.0 | 4 | 20 | 3 | None | No |
| 5 | 10.0 | 4 | 40 | 4a | None | No | |
| 6 | 20.0 | 4 | 80 | 3 | Gr 1 ALT elevation (n=1) Gr 2 ALT, AST elevation (n=1) |
No | |
| 7 | 40.0 | 4b | 160 | 5 | Gr 1 AST, Gr 2 hypoalbuminemia (n=1) Gr 2 capillary leak syndrome (n=2) Gr 1 fatigue (n=1) Gr 2 hypokalemia (n=1) Gr 3 legs weakness (n=1) |
1 | |
| 8 | Continual Reassessment | 60.0 | 4 c,d | 240 | 5 | Gr 1-2 capillary leak syndrome (n=3) Gr 2 anemia (n=1) Gr 3 thrombocytopenia (n=2) Gr 2 fever (n=2) Gr 4 neutropenia (n=1) Gr 3 capillary leak sy (n=1) Gr 3 neutropenic fever (n=1) Gr 2 hearing loss (n=1) Gr 1 hypocalcemia (n=1) |
1 |
| 9 | 80.0 | 4 | 320 | 3 | Gr 1 hypokalemia (n=1) Gr 1-2 capillary leak syndrome (n=2) Gr 1 vomiting (n=1) Gr 3 hypokalemia (n=1) Gr 1 AST ALT elevation (n=2) Gr 2 fatigue (n=2) |
No |
1 patient at the 10 μg/kg/day was less than 12 years old and enrolled after receiving permission from the local IRB
patient with DLT received 3 doses of DT2219
1 patient at the 60 μg/kg/day was retreated 8 weeks later with 2nd cycle at dose 40 μg/kg/day.
1 patient was dose reduced for 4th injection to 40 μg/kg/day due to capillary leak syndrome
Disease Reassessment and Correlative Studies
Disease assessment included physical examination for lymph node and spleen weekly; blood and marrow evaluation including flow cytometry assessment for CD19 and CD22 expression and assessment for minimal residual disease, and computerized tomography (CT) scan 21-28 days after treatment using Chesson criteria for lymphoma and leukemia staging (10, 11). Adverse event collection focused on targeted and unexpected adverse events before and after each dose at the following time-points: 1-4 hours, 24 hours, and days 9, 15, 22, 29 of the cycle.
Correlative studies included assessment of pharmacokinetics, neutralizing antibody, and immunophenotype of peripheral blood and marrow for CD22, CD19, and CD20. Cell suspensions were stained with the following mAbs: PerCPCy5.5-anti-CD3 (OKT3, Tonbo biosciences65-0037); APC anti-CD45 (HI30. Tonbo biosciences 20-0459),; FITC-anti CD19 (BU-12), FITC-anti CD20 (clone 2H7, eBioscience, 11-0209-42); FITC-anti CD22 (Invitrogen MHCD2201). Phenotypic acquisition of cells was carried out on the BD Accuri C6 and analyzed with BD Accuri C6 software. The presence of DT2219 in serum was measured by the ability of diluted serum to inhibit proliferation of CD22+CD19+ Raji indicator cells and then extrapolating DT2219 concentration using standard curve comparison, as described previously (12). The presence of CD19 and CD22 on lymphoid tumor samples obtained from patients prior to the therapy has been evaluated using standard immunohistochemistry on formalin fixed, paraffin embedded tissues and, where possible, by flow cytometry. We also measured CD19, CD22, CD20 and CD3 expressing peripheral blood cells at weekly intervals. Finally, the presence of neutralizing antibodies was measured with an assay where patient serum was used to block the activity of DT2219 in vitro (5). Peripheral mononuclear cells (MNC) and serum samples were collected pre-treatment and post-treatment at days 1, 8, 15, 21, and 28 and stored at -80C.
Statistical analysis
Patients and disease characteristics were summarized using descriptive statistics. For binary endpoints such as toxicity and clinical response, frequencies and proportions were calculated. For continuous endpoints such as area under the curve (AUC), summary statistic including median and range (minimum and maximum) were used. All statistical analyses were performed with Statistical Analysis System software version 9.3 (SAS Institute, Inc., Cary, NC).
Results
Patients and toxicities
We enrolled 25 patients with a median age of 55 years (range 34-78 years). Patient and disease characteristics are detailed in Table 1. All patients were evaluable for safety and efficacy. Ten patients had pre-B acute lymphoblastic leukemia, 5 had chronic lymphocytic leukemia (CLL), and 10 had non-Hodgkin lymphoma (NHL). All patients were chemo-refractory with a median of 3 (range 2-5) prior therapies. Most patients received prior monoclonal antibody (rituximab, ofatumumab, inotuzumab), none of the patients received blinatumomab and eight failed prior hematopoietic cell transplantation (5 autologous and 3 allogeneic). All tumors were biopsy-confirmed to express CD19 and/or CD22 in at least 20% of malignant cells. Most tumors (89%) had over 60% malignant cells CD19 and/or CD22 positive and 13 expressed both CD19 and CD22 targets.
TABLE 1. Patients and disease characteristics.
| Characteristics | Number of subjects N=25 |
|---|---|
| Age median (range) | 55 (34-78) |
| Gender (male/female) | 13/12 |
| Race | |
| Caucasians | 20 |
| Hispanic | 3 |
| Black | 2 |
| Disease | |
| Acute lymphoblastic leukemia | 10 |
| Chronic lymphocytic leukemia | 5 |
| Non-Hodgkin lymphoma | 10 |
| Disease status | |
| Primary refractory | 11 |
| Relapsed refractory | 14 |
| Site of disease | |
| Marrow | 13 |
| Extramedullary ALL | 1 |
| Lymph nodes | 15 |
| Extra lymphatic sites | 3 |
| CD19 and CD22 expression on tumor | |
| CD19 only | 11 |
| CD22 only | 1 |
| CD19 and 22 both | 13 |
| Prior therapy | |
| Lines median (range) | 5 (1-5) |
| Rituximab | 14 |
| Ofatumumab | 1 |
| Inotuzumab | 1 |
| Autologous hematopoietic cell transplantation | 3 |
| Allogeneic hematopoietic cell transplantation | 5 |
All 25 patients received a single course of therapy. One patient attained partial response after the 1st cycle and received an additional 4 dose course after the protocol was amended with FDA and IRB approval. Twelve patients treated at doses ranging from 0.5 ug/kg/day to 20 ug/kg/day exhibited no or minimal adverse reactions (Table 2). All 13 patients treated at dose levels ≥40 ug/kg/day QODx4 experienced adverse events (AE) attributed to drug treatment. No infusion toxicity was observed. The most common transient grade 1-2 AEs included weight gain (range 5-14% of baseline), peripheral edema, and hypoalbuminemia consistent with capillary leak syndrome, grade 1-2 fever and fatigue (Table 2). Seven patients experienced isolated mild elevation of liver function tests (1.1-2.1 × ULN) without hyperbilirubinemia, which resolved within 3-7 days. Thrombocytopenia and anemia occurred in 5 patients; however, marrow involvement by underlying lymphoma or leukemia often contributed to cytopenias. Whereas lactate dehydrogenase (2-2.3-fold) transiently increased in 4 patients after the 1st dose; clinical tumor lysis or acute cytokine release syndrome did not occur. Most AEs were recognized during routine monitoring before the 2nd or 3rd dose of DT2219. All AEs were brief and resolved completely within one week. Two patients experienced DLTs: the first DLT occurred at the 40 μg/kg dose level in a 71-year-old patient with ALL who developed back pain along with acute lower extremity weakness after the 3rd dose of study drug. While the patient had a recent history of CNS leukemia prior to enrollment, brain magnetic resonance imaging and cerebrospinal fluid studies at the time of AE were negative for leukemic CNS involvement. This patient died of rapidly progressive disease. No neurologic adverse effects of any grade occurred in the next 10 patients treated at this or higher doses (40-80 ug/kg). The second DLT event occurred at the 60 μg/kg dose level in a 55-year-old patient who developed grade 3 capillary leak manifested as hypoxemia, hypotension, pulmonary edema, and hypoalbuminemia in combination with febrile neutropenia. The patient was hospitalized and treated with oxygen, IV antibiotics, hydration and diuresis. Her symptoms improved with supportive care to grade 2 after 2-3 days and completely resolved in 10 days.
Pharmacologic and immunologic studies
At the time of enrollment, most patients exhibited low peripheral blood (PB) B-cells counts (median B cell count 3.5% (<0.1 ×106 cells/μL); range 0-52%; n=10) often associated with prior rituximab, corticosteroids and chemotherapy. The effect of DT2219 on B lymphocytes in a patient with an extramedullary ALL relapse shortly after allogeneic HCT was observed with gradual decline in number of PB CD19- and CD22-expressing cells after 4 doses of DT2219 (Figure 1A). The possibility that DT2219 may interfere with fluorochrome-labeled anti-CD19 and anti-CD22 was excluded by examining CD20-positive cells, which also declined over time. The B cell depletion was specific as CD3-positive T cell levels remained constant during the testing interval.
Figure 1. Immunologic and pharmacokinetic studies.
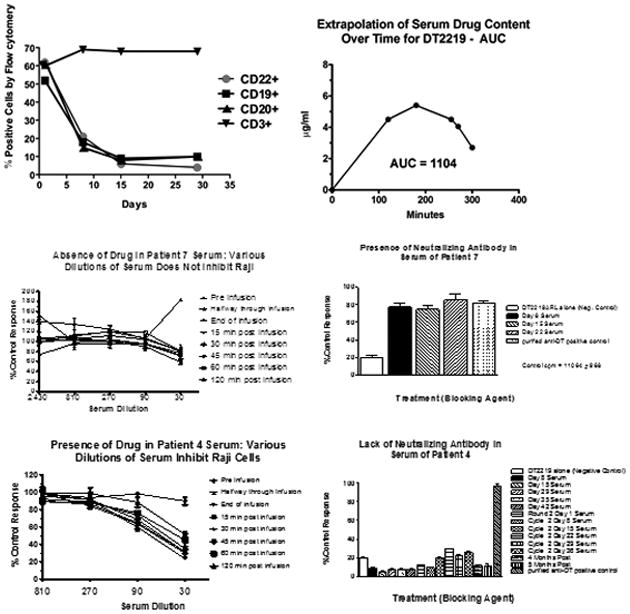
A. Peripheral blood mononuclear cell (PBMC) analysis of a representative patient is shown. PBMCs were enriched from patient blood and collected at various times post-treatment. Flow cytometry was used to count cells expressing CD22, CD19, CD20, or CD3. B. A bioassay was used to determine the area under the curve (AUC) of serum DT2219 levels in serum by measuring the ability of diluted serum to inhibit proliferation of CD22+CD19+ Raji indicator cells. Drug serum levels at various times were analyzed using prism 5.0 software to calculate AUC. A concentration-time curve is shown for our second patient at 60ug/kg. T1/2 was 59 minutes. C. DT2219 serum levels and neutralizing antibodies. Upper panels shows a patient treated at the 80 ug/kg dose level showing no evidence of DT2219 in serum (left) and high levels of neutralizing antibodies at day 8 through 22 (right). In contrast, lower panels shows that a patient treated at 60 ug/kg/day had a serum drug concentration (left) and no detectable neutralizing antibodies (right lower panel). DT2219 serum levels were calculated from assays in which various serum dilutions were tested for their ability to inhibit Raji cell proliferation. Serum collected prior to drug administration served as a negative control. Neutralization assays were performed based on the ability of undiluted patient serum samples to block the killing of a 99% inhibitory dose of DT2219. % neutralization was calculated
We also measured the circulating concentration of DT2219 in a functional pharmacokinetic bioassay. Patients treated at dose levels 0.5-20 μg/kg/day had no detectable drug in serum when sampled on day 1 and 8 at 15, 30, 45, 60, and 120 minutes post-infusion. All evaluable patients at the University of Minnesota treated with ≥40 μg/kg/dose (n=10) demonstrated detectable levels of DT2219 with the exception of one with preexisting antibodies to DT. The median area under the curve (AUC) after the 1st dose (4 hours infusion) was lower at a median of 285 μg/mL × minutes; (range 0-2020; n=8) compared to drug levels after the 4th dose (2 hours infusion; AUC median 1249 μg/ml × minutes; range 0-1692; n=7). A representative AUC is shown in Figure 1B. The drug half-life ranged from 59-110 minutes (n=4).
Because the recombinant immunotoxin contains a bacterial toxin, immunogenicity is expected and can be a major barrier to the potential activity of bacterial toxin-based drugs. We measured serum neutralizing antibodies (NAs) in all patients treated with ≥40 μg/kg/dose at days 1,8,15,29,35 and 42 (n=9). NAs developed in 3 evaluable patients (30%) at dose levels between 40-80 ug/kg at median of one week (range 1-2 weeks) after the 1st dose of DT2219. One patient had pre-formed anti-diphtheria toxin antibody which we detected at screening and attributed to prior DT immunization. In some patients the presence of NA inversely correlated with the serum concentration of DT2219 (Figure 1C), however no consistent pattern was recognized.
Clinical responses
Twenty-five patients were evaluable for response, recognizing that only 9 patients in the highest dose cohorts had measurable drug levels. Three patients had biopsy performed at the time of progression and all 3 demonstrated persistence of one or both CD19/CD22 antigens. Treatment produced an objective tumor response in two of these patients. After a single course of DT2219 at dose level 40 μg/kg/day × 4, a 77-year-old patient with chemotherapy-refractory CD19+ CD22- CLL experienced a 40% reduction in cervical and axillary adenopathy with decrease of an abdominal tumor mass at day 28 after treatment which was sustained for 2 months. (Figure 2A) Patient was in continuous partial remission when she received a salvage ibrutinib therapy. A second response occurred in a 53-year-old patient with relapsed CD19+CD22+ diffuse large B cell lymphoma (dose level 60 μg/kg) who experienced a 75% reduction in size of lymphoma lesion after a single course complicated by a grade 3 capillary leak syndrome. Eight weeks later, after FDA approval, this patient received a second DT2219 course at a reduced dose of 40 μg/kg/dose × 4 which resulted in a complete resolution of a subcutaneous mass and pelvic lymphadenopathy (Figure 2B). Second patient is alive and in complete remission, currently at 8 months after therapy. We observed no correlation between CD19 and CD22 target expression and clinical activity in this small cohort.
Figure 2. Imaging studies in patients attaining objective response on Phase 1 study.
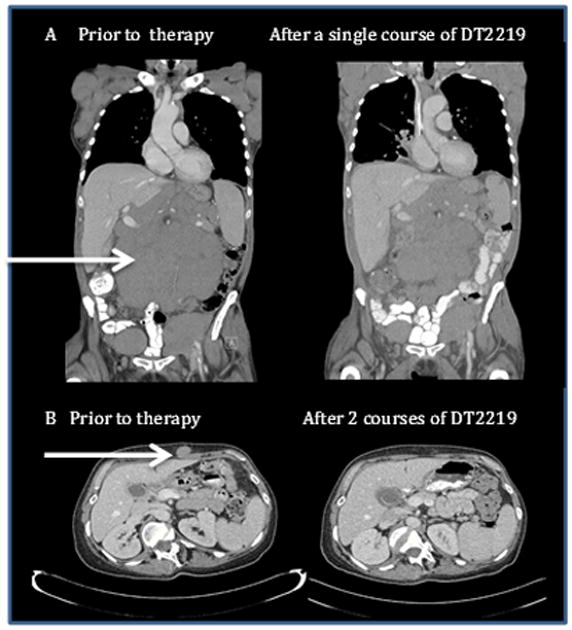
A. Abdominal CT imaging of 77-year-old patient with rituximab and chemotherapy-refractory CLL treated with DT2219 at dose level 40 μg/kg every other day × 4 doses before and at day 28 after therapy is shown. The 40% reduction in the abdominal tumor mass was observed after a single course of therapy. B. CT images of a 53-year-old female with CD22+CD19+ relapsed chemotherapy refractory marginal zone lymphoma. The patient was treated at dose level 60 μg/kg/day QODx4 and experienced a DLT of capillary leak syndrome and neutropenia. After regulatory approval, the patient received a second treatment course 8 weeks later at a reduced dose of 40 μg/kg/day QODx4 which resulted in complete resolution of the tumor mass. CT images were taken prior to therapy and after the second course of DT2219. Arrows indicate a tumor mass.
Discussion
We have established the safety and dosing feasibility of a novel CD19/CD22 bispecific ligand-directed toxin DT2219. We also demonstrated that the current dosing schedule and route of administration achieves drug levels capable of biological and clinical response against CD19/22-expressing lymphoid malignancies refractory to standard therapies with a surprisingly low incidence of neutralizing antibody responses. The present phase I study shows that, although MTD was not reached, the drug can be administered safely up to 80 ug/kg/day at days 1,3,5 & 8 for total of 4 doses. The first dose infused over 4 hours as a safety precaution was always well tolerated. All other doses were administered over 2 hours. Interestingly, the AUC measured for the first dose was almost always lower than the AUC measured for the 4th dose suggesting the importance of shorter infusion time for immunotoxins with brief half-life. Early on-target saturation also may play a role in low AUC at the onset of therapy, yet the DT2219 dosing in 4 infusions 1-2 days apart resulted to adequate drug levels, biological effectiveness, and tolerable toxicity. Although clinical responses to DT2219 were observed at doses 40 and 60 ug/kg/day, the 4 doses as administered in this trial maybe inadequate to induce deeper remissions. In one patient who achieved partial remission after 1 cycle, an additional cycle led to complete tumor elimination. The rationale for improved efficacy with repetitive dosing is supported by others who are developing immunotoxin conjugates using bacterial toxins, such as the anti-CD22 moxerumomab pasudotox for hairy cell leukemia or SL-401, an interleukin 3 receptor-diphteria toxin fusion protein for myeloid malignancies (12-14).
In our experience, increasing the number of consecutive doses per cycle is unlikely to be tolerated, however the treatment schedule with repetitive cycles of four every other day doses at least a week apart should be explored in future studies.
An important observation in this study is the lack of neutralizing antibodies formation in 7 of 10 of the evaluable patients treated at the 3 highest dose cohorts. In other trials involving DT-related immunotoxins in non B-cell malignancies, neutralizing antibody responses have been frequent. One potential explanation is that prior rituximab therapy and B-cell lymphopenia contributed to a blunted humoral response which can last up to 1 year (4).
As is typical for most immunotoxins, the potential toxicity of greatest concern at higher doses was capillary leak syndrome. The underlying mechanism at least in part involves pinocytosis of the immunotoxin by endothelial cells which is dose-dependent and thus of a particular concern at higher drug concentrations (15). Drug-development strategies to engineer toxins that do not induce capillary leak syndrome are underway (16, 17)). However, despite capillary leak in many patients at the higher dose levels (40-80 ug/kg/day), this side effect was manageable and fully reversible. In contrast to recently approved anti-CD19 targeting bispecific antibody blinatumomab which produce neurotoxicity in 11% of patients, DT2219 therapy caused no grade 1-2 neurotoxicity and only a single grade 3 paraparesis of an uncertain drug-causality.(18) Importantly, other complications inherent in the use of many experimental immunotherapeutic agents such as infusion-related reactions, pyrexia, tumor lysis, or cytokine release syndrome were not observed in this study.(19)
In conclusion, we have demonstrated safety, dosing feasibility and preliminary clinical activity of a bispecific ligand-directed toxin in chemotherapy refractory B cell lymphoid malignancies. In contrast to cytostatic chemotherapy, DT2219-mediated tumor cell killing is cell cycle and p53 independent (8), making it a particularly attractive therapy for overcoming resistance to standard chemotherapeutics in lymphoma.
A phase 1/2 clinical study designed to administer sequential cycles of this unique heterodimeric bispecific antibody toxin conjugate is underway.
Statement of translational relevance.
In a phase 1 clinical trial, we report the safety, dosing feasiblity, biological activity, and clinical efficacy of DT2219, a novel recombinant protein engineered by fusing the truncated diphtheria toxin (DT390) with bispecific single chain variable fragments of antibodies targeting human CD19 and CD22. Immunotoxins represent a novel therapeutic strategy targeting tumor-specific antigens while limiting systemic toxicity. DT2219 will be further developed for therapy of mature or precursor B cell lymphoid malignancies. In the future, DT2219 can be used in combinations with other targeted agents providing a safer and non-genotoxic alternative to chemotherapy.
Acknowledgments
Research reported in this publication was supported US Public Health Service grant R01CA36725 (DV), the Randy Shaver Foundation, the Lion's Children's Cancer Fund, the William Lawrence and Blanche Hughes Fund and by the National Center for Advancing Translational Sciences of the National Institutes of Health Award Number UL1TR000114 (VB). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was also supported in part by NIH P30 CA77598 utilizing the Biostatistics and informatics core, Masonic Cancer Center, University of Minnesota shared resource. We would like to thank Julie Curtsinger from University of Minnesota Cancer Center Translational Therapy Lab for assistance with blood samples retrieval and Michael Franklin for editorial assistance.
Funding: US Public Health Service grant R01CA36725 (DV), the Randy Shaver Foundation, the Lion's Children's Cancer Fund, the William Lawrence and Blanche Hughes Fund and by the National Center for Advancing Translational Sciences of the National Institutes of Health Award Number UL1TR000114 (VB).
Footnotes
Conflict of Interests: Authors have no relevant conflits of interest.
Trial registration is NCT00889408 at clinicaltrials.gov.
References
- 1.Vallera DA, Chen H, Sicheneder AR, Panoskaltsis-Mortari A, Taras EP. Genetic alteration of a bispecific ligand-directed toxin targeting human CD19 and CD22 receptors resulting in improved efficacy against systemic B cell malignancy. Leuk Res. 2009;33(9):1233–42. doi: 10.1016/j.leukres.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pulczynski S. Antibody-induced modulation and intracellular transport of CD10 and CD19 antigens in human malignant B cells. Leuk Lymphoma. 1994;15(3-4):243–52. doi: 10.3109/10428199409049720. [DOI] [PubMed] [Google Scholar]
- 3.Chan CH, Wang J, French RR, Glennie MJ. Internalization of the lymphocytic surface protein CD22 is controlled by a novel membrane proximal cytoplasmic motif. J Biol Chem. 1998;273(43):27809–15. doi: 10.1074/jbc.273.43.27809. [DOI] [PubMed] [Google Scholar]
- 4.Keppler-Hafkemeyer A, Brinkmann U, Pastan I. Role of caspases in immunotoxin-induced apoptosis of cancer cells. Biochemistry. 1998;37(48):16934–42. doi: 10.1021/bi980995m. [DOI] [PubMed] [Google Scholar]
- 5.Vallera DA, Todhunter DA, Kuroki DW, Shu Y, Sicheneder A, Chen H. A bispecific recombinant immunotoxin, DT2219, targeting human CD19 and CD22 receptors in a mouse xenograft model of B-cell leukemia/lymphoma. Clin Cancer Res. 2005;11(10):3879–88. doi: 10.1158/1078-0432.CCR-04-2290. [DOI] [PubMed] [Google Scholar]
- 6.Scheuermann RH, Racila E. CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk Lymphoma. 1995;18(5-6):385–97. doi: 10.3109/10428199509059636. [DOI] [PubMed] [Google Scholar]
- 7.Cesano A, Gayko U. CD22 as a target of passive immunotherapy. Semin Oncol. 2003;30(2):253–7. doi: 10.1053/sonc.2003.50057. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez R, Lim HY, Bartkowski LM, Simons JW. Identification of diphtheria toxin via screening as a potent cell cycle and p53-independent cytotoxin for human prostate cancer therapeutics. Prostate. 1998;34(4):259–69. doi: 10.1002/(sici)1097-0045(19980301)34:4<259::aid-pros3>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 9.Yuan Y, Yin G. Bayesian hybrid dose-finding design in phase I oncology clinical trials. Stat Med. 2011;30(17):2098–108. doi: 10.1002/sim.4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheson BD. New staging and response criteria for non-hodgkin lymphoma and hodgkin lymphoma. Radiol Clin North Am. 2008;46(2):213–23. vii. doi: 10.1016/j.rcl.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 11.Cheson BD, Bennett JM, Grever M, Kay N, Keating MJ, O'Brien S, et al. National cancer institute-sponsored working group guidelines for chronic lymphocytic leukemia: Revised guidelines for diagnosis and treatment. Blood. 1996;87(12):4990–7. [PubMed] [Google Scholar]
- 12.Kreitman RJ, Tallman MS, Robak T, Coutre S, Wilson WH, Stetler-Stevenson M, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol. 2012;30(15):1822–8. doi: 10.1200/JCO.2011.38.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frankel AE, Woo JH, Ahn C, Pemmaraju N, Medeiros BC, Carraway HE, et al. Activity of SL-401, a targeted therapy directed to interleukin-3 receptor, in blastic plasmacytoid dendritic cell neoplasm patients. Blood. 2014;124(3):385–92. doi: 10.1182/blood-2014-04-566737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kreitman RJ, Pastan I. Antibody fusion proteins: Anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin Cancer Res. 2011;17(20):6398–405. doi: 10.1158/1078-0432.CCR-11-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buser A, Stern M, Arber C, Medinger M, Halter J, Rovo A, et al. Impaired B-cell reconstitution in lymphoma patients undergoing allogeneic HSCT: An effect of pretreatment with rituximab? Bone Marrow Transplant. 2008;42(7):483–7. doi: 10.1038/bmt.2008.229. [DOI] [PubMed] [Google Scholar]
- 16.Smallshaw JE, Ghetie V, Rizo J, Fulmer JR, Trahan LL, Ghetie MA, et al. Genetic engineering of an immunotoxin to eliminate pulmonary vascular leak in mice. Nat Biotechnol. 2003;21(4):387–91. doi: 10.1038/nbt800. [DOI] [PubMed] [Google Scholar]
- 17.Wayne AS, Fitzgerald DJ, Kreitman RJ, Pastan I. Immunotoxins for leukemia. Blood. 2014;123(16):2470–7. doi: 10.1182/blood-2014-01-492256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Topp MS, Gokbuget N, Stein AS, Zugmaier G, O'Brien S, Bargou RC, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: A multicentre, single-arm, phase 2 study. Lancet Oncol. 2014 doi: 10.1016/S1470-2045(14)71170-2. [DOI] [PubMed] [Google Scholar]
- 19.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]