

Abstract
The exact physiological role of NF-κB-inducing kinase (NIK) in the NF-κB activation pathway has not been defined, although it is an upstream kinase of IKKα. Recent studies have indicated that IKKα is a nucleosomal modifier of NF-κB signaling. We hypothesized that NIK generates a proximal signal that contributes to IKKα modification of nucleosomal structure through phosphorylation of histone H3 and enhancement of target gene expression. By using a chromatin immunoprecipitation assay, our data show that endogenous IKKα is recruited to the promoter site of several NF-κB-dependent genes in macrophages. Our data show that immunoreactive NIK is rapidly recruited to nuclear compartment in macrophages in response to treatment with endotoxin where it augments phosphorylation of histone H3 by inducing phosphorylation and kinase activity of IKKα. A small interfering RNA knockdown of NIK markedly reduces phosphorylation of histone H3 in endotoxin treated macrophages. These data, together, demonstrate a novel role for NIK as a histone H3 modifier, through an accessory pathway from NIK to IKKα, that could play an important role in the endotoxin response through modification of nucleosomal structure.
Exposure of macrophages to pro-inflammatory cytokines and bacterial products leads to activation of NF-κB and production inflammatory mediators. In unstimulated cells, NF-κB is sequestrated in the cytoplasm associated with inhibitory κB (IκB).2 Various stimuli induce phosphorylation of IκB followed by ubiquitination and degradation of IκB by the proteasome with liberation of NF-κB to migrate to the nucleus to initiate gene transcription. IκB is phosphorylated by the IκB kinase (IKK) complex or signalsome that contains two functionally distinct kinases, IKKα and IKKβ, and a regulatory subunit IKKγ(NEMO) (1–3). Many studies have showed that the two kinases of the IKK complex have disparate roles in regulation of the NF-κB activation pathway. IKKβ, in conjunction with NEMO/IKK, is directly involved in phosphorylation and degradation of IκB in response to a variety of inflammatory stimuli (4, 5). In contrast, IKKα is not essential for IκBα phosphorylation and degradation and nuclear translocation of RelA (6–8). However, several groups have reported that there is incomplete transcription of NF-κB-dependent gene in IKKα(−/−) cells, which indicates that IKKα has an accessory role in the NF-κB activation pathway (9, 10). In these reports, pro-inflammatory cytokine-induced NF-κB-dependent transcription and promoter activation was markedly decreased in IKKα(−/−) fibroblasts even though IκBα degradation and NF-κB in vitro DNA binding activity was normal in these cells in response to treatment with TNF-α or IL-1 (11). These data suggest that IKKα is not involved in the cytoplasmic phase of the NF-κB activation but is nevertheless required for full activation of NF-κB-dependent gene expression. Recently, using a transient transfection model, two groups have reported that, in response to proinflammatory stimuli, nuclear IKKα is recruited to the promoters of several NF-κB-responsive genes where it phosphorylates histone H3, a component of nucleosome (12, 13).
Nucleosomes are the basic packing unit of chromatin that consist of an octamer of two of each histone (H2A, H2B, H3, and H4) that comprise the histone core. The protruding NH2-terminal tails of nucleosomal histones are covalently modified by a variety of enzymatic activities. A diverse array of modifications of nucleosome regulates genomic function and result in a dynamic regulatory process that is cell and tissue specific (14). For example, serine 10 phosphorylation on histone H3 is linked to transcriptional activation. When mammalian cells are exposed to a mitogen or stress, the time course of phosphorylation of Ser10 histone H3 corresponds to the transient expression of activated immediate-early genes. In many cell types, histone modification is one of many key regulatory steps that are necessary for transcriptional activation (15, 16). Interestingly, only a limited number of kinases are capable of phosphorylating histone H3, such as Rsk-2 and Msk-1 (17, 18). Since IKKα also has histone H3 kinase activity, it is possible that it plays a key role in modulation of transcriptional activity of NF-κB-dependent genes.
NIK is an upstream kinase in the NF-κB activation pathway, preferentially phosphorylates IKKα over IKKβ, leading to the activation of IKKα kinase activity (19, 20). The physiologic function of NIK in the NF-κB signaling is unclear because IKKβ plays a dominant role in the endotoxin induced NF-κB activation. However, gene knock-out studies suggest that NIK regulates the transcriptional activity of NF-κB in a receptor-restricted manner. NIK−/− cells are unable to induce the NF-κB-dependent gene transcription in response to treatment with lymphotoxin-β even though IκBα degradation occurs (21). NIK has both a functional nuclear import and nuclear export signals that result in continuous shuttling between cytoplasm and nucleus, suggesting NIK, like IKKα, has an intranuclear function (22, 23).
Here, we address whether NIK has a role in phosphorylation of histone H3 in macrophages in response to treatment with Escherichia coli endotoxin. Our data show that endogenous IKKα is recruited to the promoter site of several NF-κB-dependent genes in macrophages by treatment with endotoxin. Our experiments indicate that endotoxin induces histone H3 kinase activity of endogenous IKKα in close association with recruitment of immunoreactive NIK into the nucleus. This histone H3 kinase activity is dependent on phosphorylation of IKKα by the upstream kinase NIK, which translocates from the cytoplasmic to the nuclear compartment in response to endotoxin treatment. These data indicate that NIK has an important regulatory role in the endotoxin-induced NK-κB activation pathway through activation of IKKα and phosphorylation of histone H3, which results in modification of nucleosomal structure.
MATERIALS AND METHODS
Cell Culture
A murine macrophage cell line RAW 264.7 and embryonic kidney cell line 293 (ATCC, Manassas, VA) were maintained in Dulbecco’s modified Eagle’s medium (Cellgor) containing 10% fetal bovine serum (Hyclone). The tissue culture medium was supplemented with penicillin (100 units/ml) and streptomycin (100 μg/ml) (Invitrogen).
Reagents
Antibodies for RelA, IKKα, and NIK were from Santa Cruz Biotechnology (Santa Cruz, CA); antibodies for histone and phosphorylated histone H3 (Ser10) were from Upstate Biotechnology (Lake Placid, NY); antibody for FLAG and Myc were from Sigma. Recombinant histone H3 protein was obtained from Upstate Biotechnology. Leptomycin B was a gift from Dr. Yoshida (Department of Biotechnology, University of Tokyo, Tokyo, Japan). Recombinant MSK1, enzymatically active, was purchased from Upstate Biotechnology.
Plasmids and Adenoviral Vectors
Expression vectors for FLAG-tagged wild-type IKKα, kinase-inactive mutant IKKα-KA, Myc-tagged wild-type NIK, and mutant NIK-KKAA were gifts from Tularik (South San Francisco, CA). pGFP-NIK was generated by subcloning NIK cDNA from the expression vector pRK7-S/N into pCDNA3.1-GFP (Invitrogen). IKKα-KA represents lysine to alanine changes at amino acid 44 of IKKα. Adenoviral vectors used in this experiment have been described previously (24). Adenoviral vector encoding constitutively active IKKα (Ad-cIKKα) is in Ser-Glu mutant form in critical serine residues that are phosphorylated in the active kinase (Ser176, Ser180). Adenoviral β-galactosidase was used as a vector control.
Plasmid Transfection
For transient transfection, 293 cells were grown in 60-mm culture dishes to 50–80% confluence and then transfected with expression vectors (total of 2–8 μg) by GenePORTER2 (Gene Therapy Systems, Inc., San Diego, CA), as specified by the manufacturer. Forty-eight hours after transfection, the cells were used for either immunoprecipitation or immunofluorescent imaging.
Immunoprecipitation
Total cell lysate was prepared using radioimmune precipitation assay buffer, and the total amount of proteins was quantified by the Bradford assay. For immunoprecipitation analysis, 1–2 μg of appropriate antibodies was added to precleared cell lysate that had been normalized by protein contents and incubated overnight at 4 °C. Immune complexes were captured with 30 μl of protein A-Sepharose (Zymed Laboratories, Inc.) for 30 min at 4 °C and washed five times with radioimmune precipitation assay buffer. Immunoprecipitates were used for in vitro kinase assay and Western blot.
Kinase Assay
Immunoprecipitates from cell extracts was used for the in vitro kinase reactions in kinase reaction buffer containing 20 mM HEPES, pH 7.6, 1 mM dithiothreitol, 10 mM MgCl2, 20 μM ATP, 10 μCi of [γ-32P]ATP. Recombinant histone H3 (0.5 μg/each) was added as a substrate. Kinase reactions were incubated at 30 °C for 30 min. Reactions were fractionated onto 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes. Phosphorylations of histone H3 and IKKα were analyzed by autoradiogram analysis. Subsequently, the membrane was immunoblotted with specific antibody to determine the protein level. We included the kinase control reactions, which have either no substrate or nonspecific immunoprecipitate.
Immunofluorescence Microscopy
RAW cells were cultured in glass bottom microwell dishes (MatTek) and transfected with pGFP-NIK. The live images of GFP fluorescence were captured using a laser-scanning confocal microscope (Zeiss, Thornwood, NY). During this process, the cells were kept in 37 °C degree and phenol red-free media. The intensity of fluorescence was quantified with Zeiss LSM image viewer (Zeiss).
Chromatin Immunoprecipitation (ChIP) Assay
To detect the in vivo association of NF-κB proteins with the promoters of murine pro-inflammatory genes, ChIP assay was performed by using previously described method (16). The primer sets used in this experiment were described previously, which cover the NF-κB binding site of promoter region of murine pro-inflammatory genes (16, 25). Primer sequence were as follows: cyclooxygenase-2 promoter, 5′-CTAATTCCACCAGTACAGATG-3′ (forward) and 5′-ACTAGGCGAGACTCAGCGAAC-3′ (reverse); IκBα promoter, 5′-CGCTAAGAGGAACAGCCTAG-3′ (forward) and 5′-CAGCTGGCTGAAACATGGC-3′ (reverse); RANTES promoter, 5′-GTGAAGACCAATGGCTTGACC-3′ (forward) and 5′-CATGT-GCTGTCTCAGAGTCCT-3′ (reverse); MnSOD intronic enhancer, 5′-GTGTAAGTGGCCAATCCAAGAG-3′ (forward) and 5′-CCT-GTCCTCAGCCAGGCAAACC-3′ (reverse). The PCR products were analyzed on a 2% agarose gel or 8% polyacrylamide gel.
Establishment of NIK Knockdown Cell Line
Short interfering RNA target sequences were designed against the mouse NIK gene using BLOCK-iT RNAi Designer software (Invitrogen). Sequences were determined to be unique to the mouse NIK gene by BLAST searching of GenBank™ data base. Oligonucleotide primers were designed that corresponded to the sense and antisense sequences of the small interfering RNA target sites of interest separated by a hairpin loop sequence. Three test sequences were selected, corresponding to nucleotide positions 1142, 1437, and 2292. The most active sequences of these, Target 2 and 3, are based on the sequence 5′-GCTACAGCAACTGGAGATAGA-3′ and 5′-GGGTCCTGCTTACTGAGAAAC-3′, respectively. We annealed primers and cloned them into pSIREN-RetroQ (Clontech) downstream of the human U6 promoter. RAW cells were transfected with Lipofectamine-2000 (Invitrogen) and selected in the condition of 2.5 μg/ml of puromycin for 1 month. A total of 36 clones were screened for immunoreactive NIK by Western blots of whole cell lysates.
RESULTS
Time Course for the Association of IKKα and RelA to the Promoter Regions of the COX-2 MnSOD, RANTES, and IκBα Genes in Endotoxin-treated Macrophages
We performed a ChIP assays using antibody against IKKα and RelA to identify protein-gene interactions in the promoter regions of specific inflammatory genes that are induced in RAW cells by treatment with endotoxin (Fig. 1, A and B). In this experiment, RAW cells were treated with endotoxin (100 ng/ml) for various time points (0–1 h), and IKKα or RelA was immunoprecipitated. After extensive stringent washing, protein-DNA complexes were extracted and IKKα and RelA binding to the promoter region of various pro-inflammatory cytokine genes were detected by semiquantitative PCR from the immunoprecipitant. In response to endotoxin stimulation, the recruitment of IKKα to the promoter sites of pro-inflammatory genes was detected by 15 min in the IκBα, 30 min at the RANTES gene, and by 60 min at the COX-2 and MnSOD genes (Fig. 1A).
FIGURE 1. ChIP assays with antibody against IKKα (A) or RelA (B) in endotoxin-stimulated RAW cells.

RAW cells were treated with endotoxin (100 ng/ml) for various time points (0–1 h), IKKα and RelA were immunoprecipitated with specific antibody, and binding to the promoter region of various pro-inflammatory cytokine genes was detected by PCR primers for COX-2, MnSOD, RANTES, and IκBα, which flank a κB binding motif. Binding to the various promoters is compared with PCR of the input DNA against using the COX-2 primers. LPS, lipopolysaccharide.
We also performed ChIP assays for RelA to determine the temporal relationship between IKKα and RelA recruitment to the various promoter regions. In response to treatment with endotoxin, RAW cell lysates were immunoprecipitated with specific antibodies against RelA, and RelA recruitment to specific promoters was evaluated by PCR. In these experiment, RelA binding to the MnSOD and IκBα genes occurred by 15 min. Binding to the COX-2 gene occurred by 60 min, and binding to the RANTES gene peaked at 60 min (Fig. 1B).
The relationship between IKKα and RelA recruitment to these inflammatory gene promoters was variable among the selected genes. Recruitment of IκBα and COX-2 closely correlated temporally with the recruitment of IKKα to these promoters. However, recruitment of RelA occurred after recruitment of IKKα to the RANTES genes and preceded recruitment of IKKα to the MnSOD genes.
Identification of the Cellular Location of IKKα by Western Blot in Endotoxin-treated Macrophages
To determine the effect of treatment with endotoxin on the intracellular location of IKKα, we separated the nuclear and cytoplasmic fractions of untreated and endotoxin-treated macrophages and immunoblotted with specific antibodies (Fig. 2). In these experiments, separation of the nuclear and cytoplasmic compartments was identified by showing that α-tubulin was confined to the cytoplasmic compartment and HDAC-1 was only detected in the nuclear compartment (third and fourth row). As expected, we detected an increase in the appearance of RelA in the nuclear compartment (second row) in response to treatment with endotoxin. Using this same membrane, we detected immunoreactive IKKα in the nuclear compartment of untreated cells and this amount of nuclear IKKα was not changed by treatment with endotoxin (first row). We were unable to detect changes in nuclear IKKα immunoreactivity in RAW cells in response to treatment with endotoxin by using immunofluorescent staining and confocal microscopy (data not shown). These data indicate that IKKα is present in the cytoplasmic and nuclear compartments of both untreated and endotoxin-treated cells.
FIGURE 2. The subcellular localization of RelA and IKKα was detected in nuclear and cytoplasmic protein extracts from RAW cells by immunoblot (IB).
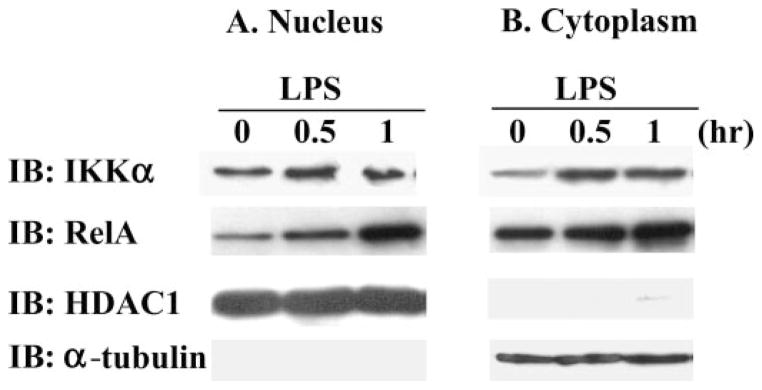
Antibodies specific for cytoplasmic α-tubulin and nuclear HDAC1 demonstrate separation of the nuclear and cytoplasmic protein fractions. RAW cells were treated with 100 ng/ml endotoxin for 0 – 60 min.
Overexpression of IKKα Results in Phosphorylation of Recombinant Histone H3
To test whether overexpressed IKKα could phosphorylate histone H3, we employed an adenoviral vector that expressed a constitutively active form of IKKα (Ad-cIKK1) that was tagged with hemagglutinin. After transfection of Ad-cIKK1 into 293 cells in various amounts, immunoprecipitation was done with anti-hemagglutinin antibodies. An immunoblot of this precipitant showed increasing amounts of both unphosphorylated (Fig. 3A, top row) and phosphorylated IKKα (second row). This precipitant was also used in an in vitro kinase assays to determine whether IKKα can directly phosphorylate recombinant histone H3. Compared with 293 cells that were infected with an adenovirus that over expressed β-galactosidase (100 multiplicity of infectivity) (third row, first lane), there was a dose-dependent increase phosphorylation of histone H3 in the samples transfected with Ad-cIKK1. In these experiments, all lanes contained a similar amount of total histone H3 (fourth row). These data indicate that overexpressed IKKα has kinase activity for recombinant histone H3 under these conditions.
FIGURE 3.

A, we transfected 293 cells with multiple doses of adenoviral vectors that expressed a constitutively active form of hemagglutinin (HA)-tagged IKKα in various amounts (multiplicity of infectivity) and incubated for 24 h. Immunoprecipitation (IP) was done with anti-hemagglutinin antibody, and then kinase reaction was performed using recombinant histone H3 protein as substrate (fourth row). We used adenoviral vector containing the hemagglutinin-tagged constitutive active form of IKKα or β-galactosidase as vector control. Results were detected by immunoblot (IB) using antibody to IKKα (top row), phosphorylated IKKα (second row), and total histone H3 (fourth row). B, we treated RAW cells with endotoxin (100 ng/ml) and harvest cells at different time points (0 – 60 min). Endogenous IKKα was captured by immunoprecipitation with anti-IKKα antibody, and a kinase assay was performed using recombinant histone H3 and [γ-32P]ATP. The results were detected by Western blot with anti-histone H3 (Ser10) antibody and autoradiography.
Endogenous IKKα Kinase Activity for Histone H3 Is Induced in Endotoxin-treated RAW Cells
Next we tested whether there was a sufficient amount of endogenous IKKα in endotoxin-treated macrophages to phosphorylate recombinant histone H3. In this experiment, we treated RAW cells with endotoxin (100 ng/ml) and captured endogenous IKKα by immunoprecipitation with anti-IKKα antibody. The precipitant was incubated with recombinant histone H3 and [γ-32P]ATP, and the results were detected by a Western blot analysis that employed antibody against phosphorylated histone H3 (Ser10). An increase in phosphorylated histone H3 was detected at 30- and 60-min treatment with endotoxin (Fig. 3B, top row), and this was associated with increased kinase activity (second row). In this experiment, an equal amount of immunoreactive recombinant histone H3 substrate was added to each lane (bottom row). These data indicate that treatment of macrophages with endotoxin results in activation of endogenous IKKα kinase activity for histone H3.
Mutation of Kinase Domain of IKKα Has Decreased Capacity to Phosphorylate Histone H3
To investigate whether a kinase-inactive mutant of IKKα (IKKα-KA) can phosphorylates histone H3, we performed an in vitro phosphorylation assay by using an expression vector containing either wild-type IKKα or IKKα-KA, which has an alanine residue at the conserved lysine residue (44 amino acids) in its kinase domain (Fig. 4). FLAG-tagged IKKα was transiently expressed in 293 cells for 24 h and immunoprecipitated with anti-FLAG antibody. The captured IKKα and recombinant histone H3 were incubated with [γ-32P]ATP. In this reaction, we detected both an autophosphorylated form of IKKα (second row, lanes 2, 3, and 6) and the phosphorylated form of histone H3 (third row). The specificity of this reaction was verified by control reactions using no histone H3 (lane 6) and nonspecific immunoprecipitation using control IgG (lane 7). The results of this experiment show that there is a dose-response relationship between IKKα overexpression and accumulation of phosphorylated histone H3 (third row, lanes 2 and 3). In contrast to wild-type IKKα, we were not able detect an autophosphorylated form of the IKKα-KA despite a similar amount of expressed protein and this was associated with minimal phosphorylation of histone H3 (third row, lanes 4 and 5) without a dose response. These data indicate that the conserved lysine residue of kinase domain of IKKα is required for optimal phosphorylation of histone H3.
FIGURE 4. A FLAG-tagged IKKα or a kinase-inactive mutant (IKKα-KA) was transiently expressed in 293 cells for 24 h and immunoprecipitated (IP) with anti-FLAG antibody.

Captured IKKα and IKKα-KA was combined with recombinant histone H3 and [γ-32P]ATP. We used the two types of expression vector, wild-type IKKα and mutant IKKα(K44A), which contained an alanine substitution of a conserved lysine residue in its kinase domain. IB, immunoblot.
NIK Is Translocated to Nucleus in Response to Treatment with Endotoxin
To investigate whether the intracellular location of NIK is affected by the presence of endotoxin, we treated RAW cells with endotoxin for various time points, extracted nuclear protein fraction, and performed Western blots with anti-NIK and RelA antibodies. The equal amount of protein loaded and purity of nuclear fraction were verified by anti-HDAC1 and anti-α-tubulin blots that reside in the nuclear or cytoplasmic compartment, respectively. The total amount of NIK in whole cell lysate was not changed by endotoxin treatment (Fig. 5A, bottom row). In response to endotoxin treatment, NIK was recruited into the nuclear compartment in time dependent manner (first row), which was almost same time with activation of IKKα kinase activity for histone H3 (Fig. 3B). Leptomycin B (10 ng/ml) treatment also induced substantial accumulation of NIK in nuclear compartment (last lane of first row). To investigate a dynamic redistribution of NIK in response to lipopolysaccharide, GFP-NIK was transfected into RAW cells that were treated with endotoxin (100 ng/ml), and a vital image of GFP fluorescence was taken at 5-min intervals under physiologic conditions at 37 °C in phenol red-free media. In these experiments, the intensity of fluorescence in the nuclear compartment was quantified with a Zeiss LSM Image Viewer. As demonstrated in Fig. 5B, the nuclear signal of GFP-NIK greatly increased at 30 min following treatment with endotoxin, which is very similar to the time point for nuclear translocation of immunoreactive NIK that we detected by Western blot.
FIGURE 5.

A, RAW cells were treated with endotoxin for various time points. The nuclear protein was extracted and blotted with anti-NIK, RelA, HDAC1, and α-tubulin antibodies. The whole cell lysate was also blotted with anti-NIK to show the total amount of NIK. B, RAW cells were cultured in glass bottom microwell dishes (MatTek) and transfected with GFP-NIK. After 24 h, the cells were stimulated with lipopolysaccharide (LPS)(100 ng/ml). The live image of GFP fluorescence was taken at 5-min intervals by confocal microscopy. During this process, the cells were kept in 37 °C and phenol red-free media. The intensity of fluorescence in the nucleus was quantified with a Zeiss LSM image viewer (Zeiss). IB, immunoblot; LMB, leptomycin B.
NIK Enhances the Phosphorylation of Histone H3 by IKKα
Since NIK is rapidly recruited to the nucleus by endotoxin treatment and preferentially phosphorylates IKKα, we next investigated the physiologic role of NIK by examining whether NIK potentiates IKKα-mediated phosphorylation of histone H3. This was addressed by using co-expressing FLAG-tagged IKKα and Myc-tagged NIK. In this experiment, we transiently co-transfected 293 cells with Myc-tagged NIK and either FLAG-tagged wild type IKKα or IKKα-KA mutant (Fig. 6A). The FLAG-tagged IKKα and IKKα-KA were immunoprecipitated with an anti-FLAG antibody and incubated with recombinant histone H3 protein and [γ-32P]ATP. Wild-type IKKα was autophosphorylated (row 3, lane 2), and total phosphorylation of IKKα was greatly enhanced by co-transfection with NIK (row 3, lane 3). Overexpression of wild-type IKKα resulted in phosphorylation of histone H3 (row 4, lane 2), and this was greatly enhanced by co-transfection with NIK (row 4, lane 3). In contrast, overexpressed IKKα-KA was not autophosphorylated (row 3, lane 4) and was slightly phosphorylated when overexpressed in the presence of NIK (row 3, lane 5). Despite a low level of phosphorylation of IKKα-KA by NIK, there was no enhancement of histone H3 phosphorylation. Although NIK enhanced phosphorylation of histone H3 by IKKα, it does not have direct kinase activity for histone H3 (Fig. 6B). Compared with MSK1, which is a known kinase for histone H3 (26), NIK alone did not phosphorylate histone H3 (Fig. 6B, row 2, lane 2).
FIGURE 6.

A, 293 cells were transiently transfected with Myc-tagged NIK and either FLAG-tagged wild-type IKKα or IKKα-KA mutant. The FLAG-tagged IKKα and IKKα-KA were immunoprecipitated (IP) with an anti-FLAG antibody and incubated with recombinant histone H3 protein and [γ-32P]ATP. B, NIK does not directly phosphorylate histone H3. 293 cells were transfected with empty vector, Myc-tagged NIK wild-type, or NIK(kkaa) mutant. Myc-tagged NIK and mutant NIK were pulled down with anti-Myc antibody. MSK1 was used as positive control for kinase assay because it was known to directly phosphorylate histone H3. IB, immunoblot.
NIK Knockdown Cells Have Reduced Binding of Phosphorylated Histone H3 to the COX-2 Promoter in Response to Endotoxin Treatment
Since NIK enhances the IKKα-induced histone H3 phosphorylation of the in vitro assay, we assessed whether endogenous NIK activity was related to histone H3 phosphorylation and binding to the promoter region of COX-2 gene. To address this, we raised several stable macrophage cell lines with knockdown NIK using a short interfering RNA sequence targeting against mouse NIK gene (Fig. 7). To examine the physiologic consequence of endotoxin-induced histone H3 phosphorylation in these knockdown cells, we performed a ChIP assay for the relevant portion of the COX-2 promoter using an antibody specific to phosphorylated histone H3 (Ser10) at 30 min after endotoxin stimulation. Our previous data show that phosphorylation of histone H3 is necessary for binding to the COX-2 promoter and enhancement of COX-2 gene expression (18). In this experiment, NIK knockdown cells have much less association of phosphorylaton of histone H3 with the COX-2 promoter in response to endotoxin stimulation. Our ChiP assay detected a dose-dependent effect of NIK on histone H3 phosphorylation, suggesting that NIK is a rate-limiting factor for phosphorylation of histone H3. In combination, these data indicate that treatment with endotoxin is associated with nuclear translocation of NIK, which results in phosphorylation of IKKα and subsequent phosphorylation of histone H3 (Fig. 8).
FIGURE 7. NIK knockdown cells have less endotoxin-induced histone H3 phosphorylation.
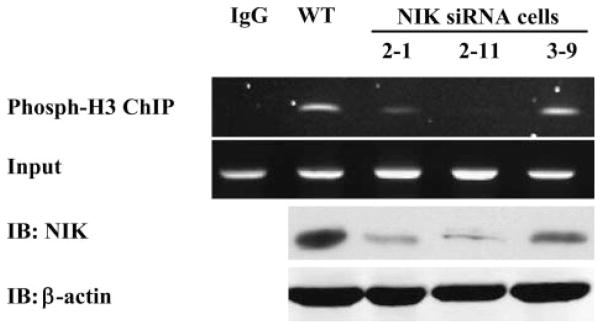
NIK knockdown RAW cell lines were established as described under “Materials and Methods.” NIK content of total cell lysates was measured by Western blots with anti-NIK and β-actin. A ChIP assay was performed using an antibody against phosphorylated histone H3 (Ser10) on RAW cells stimulated with endotoxin for 30 min. PCR was performed on the proximal promoter of the murine COX-2 gene flanking the putative NF-κB binding sites. IB, immunoblot; IP, immunoprecipitate; WT, wild-type.
FIGURE 8. A hypothetical diagram of NIK involvement in nucleosomal regulation of NF-κB pathway.
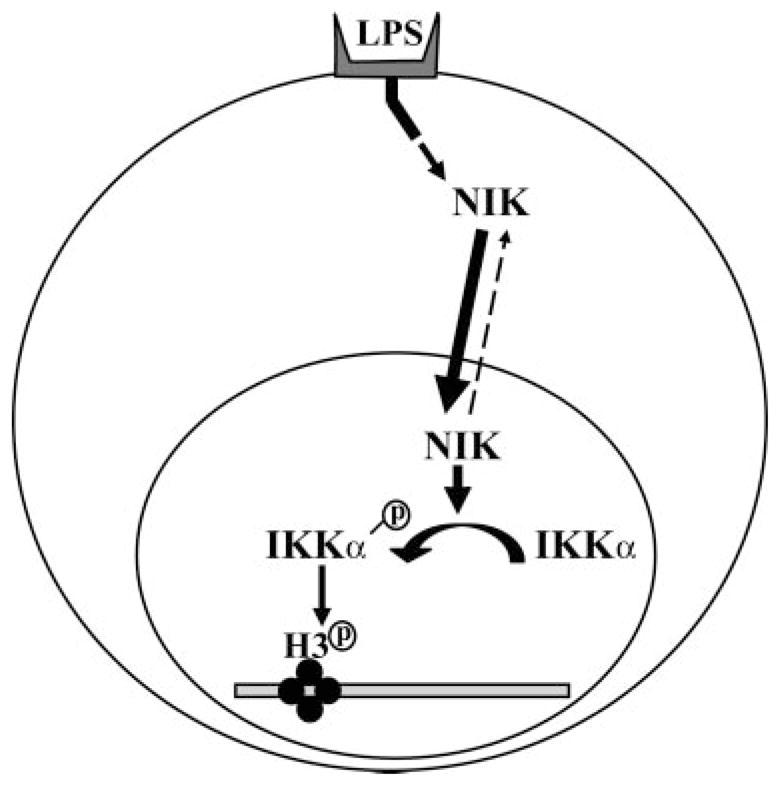
Our results indicate that the treatment of endotoxin is associated with nuclear tranlocation of NIK, which results in phosphorylation of IKKα and subsequent phosphorylation of histone H3.
DISCUSSION
We have reported that histone H3 phosphorylation plays a important role in regulating the endotoxin-induced COX-2 gene expression (16). Here, we examined the molecular pathway that links treatment with endotoxin to histone H3 phosphorylation. Our data identify a novel signaling pathway for nucleosomal modification in macrophages that are treated with endotoxin stimulation that involves induction of nuclear translocation of NIK.
We investigated the role of NIK and IKKα in the phosphorylation of histone H3. Our data show that endogenous IKKα is recruited to the promoter site of multiple pro-inflammatory genes and is activated by treatment with endotoxin to phosphorylate histone H3. Additionally, we have shown that NIK accumulates in the nuclear compartment in response to treatment with endotoxin where it has a role in activation of IKKα, which results in histone H3 phosphorylation. The physiologic consequence of nuclear translocation of NIK is shown by using a small interfering RNA knockdown of NIK that reduces endotoxin-induced association of phosphorylated histone H3 to the COX-2 promoter. Many studies have shown that NIK is not necessary for TNF-α-mediated NF-κB activation even though NIK has some a role in processing of the NF-κB2 subunit p100 (21, 27, 28). Recent papers (22, 23) indicate that NIK shuttles between the cytoplasm and nucleus with relatively rapid kinetics, but the functional significance of this process has not been addressed previously. Our data indicate that NIK has an important role in phosphorylation of IKKα, which enhances the kinase activity of IKKα for histone H3 indicating that NIK indirectly modulates the nucleosomal structure, indirectly, by enhancing histone H3 kinase activity of IKKα.
Like NIK, IKKα is not necessary for TNF-α-induced phosphorylation of IκB, while IKKβ and IKKγ/NEMO are required (29–31). However, despite normal induction of IKK kinase activity for IκBα and IκB degradation, IKKα-null mouse embryo fibroblasts have a diminished array of IL-1- and TNF-induced NF-κB-dependent pro-inflammatory gene transcription (10). This functional activity of IKKα is not dependent on IκBα phosphorylation and generation of in vitro RelA DNA binding activity (9, 11), indicating that more downstream or accessory pathway might be involved that results in transactivation of NF-κB-dependent genes. This effect may depend on a nuclear location of IKKα as suggested by a recent report that showed that nuclear, but not cytoplasm, IKKα is required for terminal differentiation of keratinocyte, and this is dependent on a nuclear localization signal since a nuclear localization signal mutant of IKKα was incapable of inducing terminal differentiation of IKKα−/− keratinocytes (32). Our study shows that immunoreactive IKKα resides in both the nuclear and cytoplasmic compartments in macrophages, and the distribution is not altered by treatment with endotoxin within 1 h. Other reports (23) have shown that the nucleocytoplasmic shuttling of IKKα occurs slowly over a 24-h period in fibroblasts, but this is long after the effects that we have observed in macrophages occur. It is unclear whether translocation of IKKα from the cytoplasm to the nuclear compartment would be an important event at a later time point. By using a ChIP assay, we demonstrate that IKKα is recruited to multiple endogenous pro-inflammatory genes in time-dependent manner by 1 h, and this is correlated with recruitment of RelA, although the temporal relationship is complex.
We demonstrated that endogenous IKKα is capable of phosphorylating histone H3 during in vitro condition. This kinase activity is induced by treatment with endotoxin within 30 min, and this is temporally associated with recruitment of IKKα recruitment to the nucleosome of a panel of pro-inflammatory genes. Because histone phosphorylation is linked to transcriptional activation in many genes (15), it is possible that the endotoxin-inducible histone kinase activity of IKKα is important in regulating expression of NF-κB-dependent gene.
Kinase activity for IκBα is dependent on IKKβ phosphorylation, whereas phosphorylation of IKKα is not essential (6, 8). In this study, we showed the phosphorylated IKKα has greater histone H3 kinase activity compared with that of mutant IKKα that is not phosphorylated. Our data show that nuclear translocation of NIK, in response to treatment with endotoxin, results in phosphorylation of IKKα, which is subsequently involved in phosphorylation of histone H3. To determine the necessity of NIK in enhancing histone kinase activity of IKKα in vivo biologic condition, we developed the NIK knockdown cells by using small interfering RNA strategies and measured binding of phosphorylated histone H3 to the promoter of the COX-2 gene in response to treatment with endotoxin. Even though NIK does not have direct histone H3 kinase activity (data not shown), the NIK knockdown cells had a marked decrease in histone H3 phosphorylation. These data, taken together, indicate that NIK is involved in phosphorylation of histone H3 and distal effects on chromatin structure, through effect on the phosphorylation status of IKKα. Post-translational modification by phosphorylation has important regulatory effects on the NF-κB activation pathway (33–35). In this report, the upstream kinases of NF-κB pathway modulate both the molecules involved in NF-κB signaling pathway and the target binding site of NF-κB-dependent genes through modification of chromatin structure.
In summary, our data suggest that NIK is involved in modification of nucleosomal structure through phosphorylation of endogenous IKKα and subsequent phosphorylation of histone H3 in macrophages. The histone H3 kinase activity of IKKα is strongly induced by endotoxin. Our data suggest that an accessory pathway from NIK to IKKα that results in phosphorylation of histone H3 has a role in the endotoxin induced NF-κB activation pathway through modification of nucleosomal structure.
Footnotes
This work was supported by the Department of Veterans Affairs and National Institutes of Health Grants HL 075557 and HL 66196.
The abbreviations used are: IκB, inhibitory κB; IKK, IκB kinase; TNF, tumor necrosis factor; IL, interleukin; NIK, NF-κB-inducing kinase; GFP, green fluorescent protein; ChIP, chromatin immunoprecipitation; RANTES, regulated on activation normal T cell expressed and secreted; SOD, superoxide dismutase; Ad-cIKKα, adenoviral vector encoding constitutively active IKKα.
References
- 1.Nakano H, Shindo M, Sakon S, Nishinaka S, Mihara M, Yagita H, Okumura K. Proc Natl Acad Sci U S A. 1998;95:3537–3542. doi: 10.1073/pnas.95.7.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pomerantz JL, Baltimore D. Mol Cell. 2002;10:693–695. doi: 10.1016/s1097-2765(02)00697-4. [DOI] [PubMed] [Google Scholar]
- 3.Regnier C, Song H, Gao X, Goeddel D, Cao Z, Rothe M. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 4.Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Science. 1999;284:321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka M, Fuentes ME, Yamaguchi K, Durnin MH, Dalrymple SA, Hardy KL, Goeddel DV. Immunity. 1999;10:421–429. doi: 10.1016/s1074-7613(00)80042-4. [DOI] [PubMed] [Google Scholar]
- 6.Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- 7.Matsushima A, Kaisho T, Rennert PD, Nakano H, Kurosawa K, Uchida D, Takeda K, Akira S, Matsumoto M. J Exp Med. 2001;193:631–636. doi: 10.1084/jem.193.5.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeda K, Takeuchi O, Tsujimura T, Itami S, Adachi O, Kawai T, Sanjo H, Yoshikawa K, Terada N, Akira S. Science. 1999;284:313–316. doi: 10.1126/science.284.5412.313. [DOI] [PubMed] [Google Scholar]
- 9.Li Q, Lu Q, Hwang JY, Buscher D, Lee KF, Izpisua-Belmonte JC, Verma IM. Genes Dev. 1999;13:1322–1328. doi: 10.1101/gad.13.10.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li X, Massa PE, Hanidu A, Peet GW, Aro P, Savitt A, Mische S, Li J, Marcu KB. J Biol Chem. 2002;277:45129–45140. doi: 10.1074/jbc.M205165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sizemore N, Lerner N, Dombrowski N, Sakurai H, Stark GR. J Biol Chem. 2002;277:3863–3869. doi: 10.1074/jbc.M110572200. [DOI] [PubMed] [Google Scholar]
- 12.Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS. Nature. 2003;423:659–663. doi: 10.1038/nature01648. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB. Nature. 2003;423:655–659. doi: 10.1038/nature01576. [DOI] [PubMed] [Google Scholar]
- 14.Khorasanizadeh S. Cell. 2004;116:259–272. doi: 10.1016/s0092-8674(04)00044-3. [DOI] [PubMed] [Google Scholar]
- 15.Berger SL. Curr Opin Genet Dev. 2002;12:142–148. doi: 10.1016/s0959-437x(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 16.Park GY, Joo M, Pedchenko T, Blackwell TS, Christman JW. Am J Physiol. 2004;286:L956–L962. doi: 10.1152/ajplung.00338.2003. [DOI] [PubMed] [Google Scholar]
- 17.Clayton AL, Mahadevan LC. FEBS Lett. 2003;546:51–58. doi: 10.1016/s0014-5793(03)00451-4. [DOI] [PubMed] [Google Scholar]
- 18.Sassone-Corsi P, Mizzen CA, Cheung P, Crosio C, Monaco L, Jacquot S, Hanauer A, Allis CD. Science. 1999;285:886–891. doi: 10.1126/science.285.5429.886. [DOI] [PubMed] [Google Scholar]
- 19.Ling L, Cao Z, Goeddel DV. Proc Natl Acad Sci U S A. 1998;95:3792–3797. doi: 10.1073/pnas.95.7.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woronicz JD, Gao X, Cao Z, Rothe M, Goeddel DV. Science. 1997;278:866–869. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- 21.Yin L, Wu L, Wesche H, Arthur CD, White JM, Goeddel DV, Schreiber RD. Science. 2001;291:2162–2165. doi: 10.1126/science.1058453. [DOI] [PubMed] [Google Scholar]
- 22.Birbach A, Bailey ST, Ghosh S, Schmid JA. J Cell Sci. 2004;117:3615–3624. doi: 10.1242/jcs.01224. [DOI] [PubMed] [Google Scholar]
- 23.Birbach A, Gold P, Binder BR, Hofer E, Martin Rd, Schmid JA. J Biol Chem. 2002;277:10842–10851. doi: 10.1074/jbc.M112475200. [DOI] [PubMed] [Google Scholar]
- 24.Sadikot RT, Han W, Everhart MB, Zoia O, Peebles RS, Jansen ED, Yull FE, Christman JW, Blackwell TS. J Immunol. 2003;170:1091–1098. doi: 10.4049/jimmunol.170.2.1091. [DOI] [PubMed] [Google Scholar]
- 25.Saccani S, Pantano S, Natoli G. J Exp Med. 2001;193:1351–1360. doi: 10.1084/jem.193.12.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomson S, Clayton AL, Hazzalin CA, Rose S, Barratt MJ, Mahadevan LC. EMBO J. 1999;18:4779–4793. doi: 10.1093/emboj/18.17.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saito N, Courtois G, Chiba A, Yamamoto N, Nitta T, Hironaka N, Rowe M, Yamamoto N, Yamaoka S. J Biol Chem. 2003;278:46565–46575. doi: 10.1074/jbc.M302549200. [DOI] [PubMed] [Google Scholar]
- 28.Xiao G, Harhaj EW, Sun SC. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 29.Muller JR, Siebenlist U. J Biol Chem. 2003;278:12006–12012. doi: 10.1074/jbc.M210768200. [DOI] [PubMed] [Google Scholar]
- 30.Nasuhara Y, Adcock IM, Catley M, Barnes PJ, Newton R. J Biol Chem. 1999;274:19965–19972. doi: 10.1074/jbc.274.28.19965. [DOI] [PubMed] [Google Scholar]
- 31.Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 32.Sil AK, Maeda S, Sano Y, Roop DR, Karin M. Nature. 2004;428:660–664. doi: 10.1038/nature02421. [DOI] [PubMed] [Google Scholar]
- 33.Chen L-f, Fischle W, Verdin E, Greene WC. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 34.Kiernan R, Bres V, Ng RWM, Coudart MP, El Messaoudi S, Sardet C, Jin DY, Emiliani S, Benkirane M. J Biol Chem. 2003;278:2758–2766. doi: 10.1074/jbc.M209572200. [DOI] [PubMed] [Google Scholar]
- 35.Zhou H, Wertz I, O’Rourke K, Ultsch M, Seshagiri S, Eby M, Xiao W, Dixit VM. Nature. 2004;427:167–171. doi: 10.1038/nature02273. [DOI] [PubMed] [Google Scholar]