

Abstract
Cocaine-mediated neuroadaptations in the prefrontal cortical-nucleus accumbens pathway underlie drug-seeking in animals with a cocaine self-administration (SA) history. Neuroplasticity in the cortico-accumbens pathway is regulated, in part, by the expression and availability of neurotrophic factors, such as BDNF. We have previously demonstrated that infusion of BDNF into the dorsomedial prefrontal cortex (dmPFC) immediately after the last of 10 cocaine SA sessions attenuates contextual, cue- and cocaine prime-induced reinstatement of cocaine-seeking (Berglind et al., 2007) and normalizes cocaine-induced disruption of glutamatergic transmission in the nucleus accumbens (Berglind et al., 2009). In the present study, the suppressive effect of intra-dmPFC BDNF on cocaine-seeking is shown to depend on Trk receptor-mediated activation of extracellular signal-regulated kinase (ERK) signaling in the dmPFC. The tyrosine kinase inhibitor, K252a, and the mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor, U0126 (1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene), prevented BDNF's suppressive effects on cocaine-seeking. Vehicle-infused rats with a cocaine SA history showed significant decreases in ERK and cyclic AMP response element binding protein (CREB), but not Akt, phosphorylation after the final cocaine SA session that were reversed by intra-dmPFC BDNF. Additionally, BDNF's ability to normalize cocaine-mediated decreases in ERK and CREB phosphorylation was blocked by U0126, demonstrating that ERK/MAPK activation mediated the behavioral effects. This study elucidates a mechanism whereby BDNF/TrkB (tropomyosin receptor kinase B) activates ERK-regulated CREB phosphorylation in the dmPFC to counteract the neuroadaptations induced by cocaine SA and subsequent relapse to cocaine-seeking.
Introduction
Cocaine-mediated neuroadaptations in motivational circuits in the CNS underlie cocaine-taking and seeking in animal models of addiction (Koob et al., 1997; White and Kalivas, 1998; Nestler, 2001). Within this circuitry, the cortico-accumbens glutamatergic pathway originating in the dorsomedial prefrontal cortex (dmPFC) and projecting to the nucleus accumbens (NAc) core is critical for the expression and persistence of relapse to cocaine-seeking (McFarland et al., 2003; McLaughlin and See, 2003; Fuchs et al., 2005). Persistent cocaine-mediated neuroplasticity in the cortico-accumbens pathway may be triggered by alterations in BDNF. BDNF mediates plasticity by binding tropomyosin receptor kinase B (TrkB) and activating several intracellular signaling cascades, including mitogen-activated extracellular signal-regulated kinase (ERK), phosphoinositol 3-kinase (PI3K), and phospholipase Cγ (Patapoutian and Reichardt, 2001) that converge on and activate cyclic AMP response element binding protein (CREB) and related transcription factors.
Each of these intracellular signaling cascades has been linked to the effects of cocaine in the CNS. For instance, acute cocaine increased the phosphorylation of ERK (p-ERK), but not Akt (p-Akt), in the mPFC of rats (Fumagalli et al., 2009) and repeated, noncontingent administration of cocaine increased CREB phosphorylation (p-CREB) in the PFC 2 h postinjection (Fumagalli et al., 2007). Extended access to cocaine self-administration (SA) increased p-ERK, but not p-CREB, in the mPFC, NAc (shell and core), immediately after the final SA session (Edwards et al., 2007). Additionally, cocaine sensitization increased PI3K activity in the PFC and NAc shell, but decreased PI3K activity in the NAc core (Zhang et al., 2006). Further, inhibition of PI3K with intraventricular LY294002 decreased cocaine-mediated behavioral sensitization (Izzo et al., 2002). Collectively, these studies implicate ERK, PI3K, and CREB signaling in cocaine-mediated plasticity in the cortico-accumbens pathway.
Exogenous BDNF differentially modifies cocaine-seeking by activating these intracellular pathways in specific brain regions. When infused into the ventral tegmental area(VTA), BDNF enhanced cocaine-seeking in abstinent rats and the ERK kinase inhibitor, U0126, blocked this induction, demonstrating a dependence on ERK signaling (Lu et al., 2004). Similarly, repeated BDNF infusion in the NAc shell increased reinstatement of cocaine-seeking after extinction (Graham et al., 2007). In contrast, intra-dmPFC BDNF suppressed cocaine-seeking following 22 h or 6 d of abstinence, and during tests of cue-induced and cocaine prime-induced reinstatement (Berglind et al., 2007). Further, shRNA-induced BDNF knockdown in the medial PFC increased the cocaine SA breakpoint in a progressive ratio paradigm (Sadri-Vakili et al., 2010). Given that intracellular signaling is necessary for the behavioral effects of BDNF, TrkB-mediated activation of ERK, Akt, and CREB in the dmPFC may provide a means by which BDNF influences cocaine-mediated plasticity and suppresses cocaine-seeking. Therefore, inhibitors of tyrosine kinase and ERK were infused into the dmPFC before BDNF to assess the contribution of each to BDNF's suppression of cocaine-seeking. Then, the interaction between BDNF and cocaine SA on the phosphorylation state of ERK, Akt, and CREB in the dmPFC was evaluated in vivo. The findings support a mechanism by which BDNF/TrkB activates ERK-regulated CREB phosphorylation in the dmPFC to counteract the effects of cocaine SA and subsequent relapse to cocaine-seeking.
Materials and Methods
Animals.
Adult male Sprague Dawley rats (n = 181; Charles River Laboratories) were housed individually on a reverse light/dark cycle. Rats were fed 20–25 g of rat chow daily and water was provided ad libitum. All animal use protocols were approved by the Institutional Animal Care and Use Committee of the Medical University of South Carolina and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23, revised 1996).
Surgery.
Following arrival, rats were given a 3 d acclimation period and food deprived 24 h before surgery. All rats weighed ∼275–325 g at the time of surgery. On the day of surgery, rats were anesthetized with ketamine (66 mg/kg) and xylazine (1.33 mg/kg), followed by equithesin (0.5 ml/kg a solution of 9.72 mg/ml pentobarbital sodium, 42.5 mg/ml chloral hydrate, and 21.3 mg/ml magnesium sulfate heptahydrate dissolved in 44% propylene glycol, 10% ethanol solution, i.p.) and ketorolac (2.0 mg/kg, i.p.), and chronic intravenous catheters were implanted into the right jugular vein. Intravenous catheters were attached to cannulae that were secured to harnesses (Harvard Apparatus). Immediately following catheterization, rats were secured using a stereotaxic device (Stoelting) and bilateral stainless steel guide cannulae precut with 5 mm extending from the threaded plastic end (Plastics One) were aimed 1 mm above the dmPFC infusion target (+3.0 mm anteroposterior relative to bregma, ±0.6 mm mediolateral, −1.6 mm dorsoventral) (Paxinos and Watson, 2005). Guide cannulae were affixed to the skull with cranioplastic cement and anchored with three steel machine screws. Two 10 mm stylets (Plastics One) were placed into the guide cannulae following surgery to prevent blockage. Following surgery, rats were infused intravenously with 0.1 ml each of cefazolin (33.3 mg/kg, 10 mg/0.1 ml/d) and heparinized saline (70 U/ml), as well as twice daily during a 5 d recovery period.
Cocaine SA.
Following 5 d of recovery from surgery, rats were placed into standard operant chambers (30 × 24 × 30 cm, Med Associates) for 120 min cocaine SA sessions for 10 consecutive days. Each chamber was equipped with two levers (7 cm above floor), and a circular stimulus light 7 cm above each lever. The infusion lines were attached to a liquid swivel (Instech) and mounted on a suspended counterbalance. Cocaine self-administering rats received response-contingent (active lever presses) infusions of cocaine hydrochloride (0.2 mg/infusion; National Institute on Drug Abuse, Research Triangle Park, NC) delivered by a computer-controlled infusion paired with a compound CS (light plus tone) that consisted of illumination of a white stimulus light directly above the active lever and a tone (2 kHz, 15 dB above ambient noise). Each cocaine infusion was followed by a 20 s timeout period in which active lever responses were recorded, but did not result in infusion of cocaine. Responses on the left (inactive) lever had no programmed consequences, but were recorded. The maintenance criterion that rats had to reach for cocaine SA was at least 10 infusions of cocaine per 2 h session for 10 d. All rats were infused with 0.1 ml each of cefazolin (33.3 mg/kg, i.v.) and heparinized saline (70 U/ml) following each SA session. Catheter patency was tested as needed by infusing 0.1 ml of brevital (sodium methohexital, 10 mg/ml, i.v.). Outlier criteria stipulated that data from any experimental animal that fell at least 2 SDs from the mean number of cocaine infusions or active lever responses were removed from the final dataset.
Intracranial infusions.
Immediately following the 10th and final SA session, bilateral infusion cannulae (28 gauge; Plastics One Inc.) were inserted into the guide cannulae so that 1 mm of the injector length extended beyond the guide cannula. Infusions were administered using drug solutions at pH 7.0–7.5 via preloaded tubing attached to Hamilton gas-tight syringes (10 μl) driven by an infusion pump (Harvard Apparatus). Each infusion volume was 0.5 μl per side infused over 2 min at a rate of 0.25 μl/min. Infusion cannulae remained in the guide cannulae for 1 min before and following the infusion. In experiment 1, rats with a history of cocaine SA were randomly assigned to groups that received the following treatments: K252a (35.7 μm in 50% DMSO; Biomol), U0126 (1 μg/μl in 25% DMSO; Promega), or vehicle (50% DMSO) 20 min before BDNF (0.75 μg/side; recombinant human BDNF, R&D Systems) or 10 mm PBS infusion. These rats were then placed directly into home cage abstinence for 6 d as described below. In experiment 2, drug naive rats with bilateral intra-dmPFC cannulae received intracranial infusion of BDNF or PBS as described above and were killed by rapid decapitation 0.5, 2, or 6 h postinfusion. In experiment 3, rats with a cocaine SA or yoked-saline history received intracranial infusion of BDNF or PBS as described above and were killed by rapid decapitation 2 or 22 h postinfusion. Another group of rats with a similar cocaine SA or yoked-saline history received no infusions and were killed 7 d after the final day of cocaine SA. In experiment 4, rats with a cocaine SA or yoked-saline history received pretreatment with U0126 20 min before intracranial infusion of BDNF or PBS as described above and were killed by rapid decapitation 2 h postinfusion. A schematic flowchart of each experiment is displayed in Figure 1.
Figure 1.

Schematic representation of the experimental paradigms used in experiments 1–4.
Abstinence, extinction, and drug-seeking tests.
The testing design (Fig. 1, experiment 1) was conducted as described previously (Berglind et al., 2007). Rats in experiment 1 underwent 6 d of abstinence directly following intracranial infusions. During abstinence, each rat remained in its home cage. Following 6 d of abstinence from cocaine SA, the rats were placed back into their former operant chamber for 2 h and active (right) and inactive (left) lever presses were recorded but had no programmed consequences. Rats underwent extinction training following the post-abstinence extinction test and after the cue-induced reinstatement test. For 7 d before and after the cue test, responding on the active and inactive levers during 2 h daily extinction sessions was recorded but had no programmed consequences and rats received no infusions of cocaine or saline. Following 7 d of extinction training, rats were reexposed to the operant chamber for a 2 h cue-induced reinstatement test. Rats received response-contingent presentation of the compound CS (light plus tone complex) for the assessment of cue-induced cocaine-seeking that was measured as active (right) lever presses during the reinstatement test. Inactive (left) lever responses were also recorded but had no programmed consequences. Following the cue-induced reinstatement test, rats reentered extinction training for 7 more days. The day after the end of extinction training, rats were administered a 10 mg/kg i.p. injection cocaine or saline and immediately placed back into the operant chamber for a 2 h test of cocaine prime-induced reinstatement. Cocaine-seeking was defined as active (right) lever presses during the reinstatement test but responding on the inactive (left) lever was also recorded. Reponses made on either lever had no programmed consequences. Immediately following this reinstatement test, rats were decapitated and brains were immediately prepared for verification of cannula placement.
Cannula placement.
Upon completion of the behavioral studies in experiment 1, rats were rapidly decapitated and brains were immediately removed and frozen in isopentane for 30 s and stored at −80°C. Brains were sectioned on a cryostat at 20 μm and placed onto slides for cannulae placement verification by Nissl-staining (Fig. 2A). Rats from experiments 2–4 were rapidly decapitated and cannula placements at the site of injection (dmPFC) were immediately verified by observation as the dmPFC was dissected for protein analysis by Western blotting (Fig. 2B).
Figure 2.

Graphical representation of histological placement from experiments 1–4. A, In experiment 1, bilateral cannula placements within the anterior cingulate and prelimbic regions (dmPFC) are represented by black dots. Rats with tract placements that were not within the dmPFC were excluded from the schematic maps and the subjects' data (n = 2) were excluded from the data analysis. B, Rats from experiments 2, 3, and 4 were decapitated and cannula placement within the dmPFC was verified during dissection.
Immunoblotting.
The immunoblotting method described by Shi and McGinty (2007) was used in experiments 2–4. Protein was extracted from dmPFC tissue by sonication in 1% SDS buffer, boiled for 5 min, centrifuged for 20 min at 10,000 × g in an Eppendorf tabletop centrifuge, and the resulting supernatant was isolated as the soluble fraction. Protein concentrations were measured with the Micro-Bicinchoninic Acid assay kit (Pierce). Equal protein concentrations (15 μg) were separated by SDS-PAGE (4–15% polyacrylamide) and transferred onto polyvinylidene difluoride membranes. Membranes were incubated for 1 h in 5% milk/Tris-buffered saline then overnight at 4°C with primary antibody diluted in 5% milk/Tris-buffered saline plus 0.1% Tween 20. The membranes were incubated with rabbit primary antisera against p-ERK1/2 (1:1000), p-Akt-Thr308 (1:2000) p-CREB (1:1000; Cell Signaling Technology). Each membrane was washed three times for 10 min in 5% milk/Tris-buffered saline plus 0.1% Tween 20 and subsequently incubated with goat anti-rabbit horseradish peroxidase-conjugated secondary antiserum (1:1000–1:5000; Millipore). The membranes were again washed three times for 10 min in Tris-buffered saline plus 0.1% Tween 20 and developed for 1–5 min using the enhanced chemiluminescence (ECL Plus) kit from GE Healthcare Bio-Sciences. ECL-treated membranes were exposed on Hyperfilm (GE Healthcare Bio-Sciences) and developed. After a 5 min application of a stripping solution (Re-blot Plus mild stripping solution; Millipore), the membranes were reprobed for total protein with primary antisera (1:1000) against ERK1/2, Akt, and CREB (Cell Signaling Technology) for quantitative assessment. After establishing that total protein levels did not differ between groups, the integrated band density for each phosphoprotein/total protein was measured using ImageJ software (National Institutes of Health, Bethesda, MD) and analyzed.
Data analysis.
The behavioral data were analyzed with a two-way mixed-factor ANOVA followed by Student–Newman–Keuls (SNK) multiple-comparison tests when a significant interaction or main effect was found. Alterations in phosphoprotein expression were analyzed using a mixed factor ANOVA followed by SNK comparisons as above or when only two groups were compared, a Student's t test was performed. Statistically significant differences were determined at a p-value of 0.05.
Results
Histology
Cannula placements from rats in experiment 1 are illustrated in Figure 2. Tract placements that did not fall within the dmPFC were excluded from the schematic maps and the subjects' data (n = 2) were excluded from analysis. After placement verification, the number of rats per experimental group were as follows: experiment 1: vehicle followed by PBS (V-P; n = 16), vehicle followed by BDNF (V-B; n = 15), K252a followed by PBS (K-P; n = 11), K252a followed by BDNF (K-B; n = 10), U0126 followed by PBS (U-P; n = 9), U0126 followed by BDNF (U-B; n = 10); experiment 2: 30 min post-PBS infusion (n = 6), 0.5 h post-BDNF infusion (n = 6), 2 h post-PBS infusion (n = 6), 2 h post-BDNF infusion (n = 6), 6 h post-PBS infusion (n = 5), 6 h post-BDNF infusion (n = 6); experiment 3: yoked-saline 2 h post-PBS infusion (S-P; n = 8), yoked-saline 2 h post-BDNF infusion (S-B; n = 8), cocaine SA 2 h post-PBS infusion (C-P; n = 7), cocaine SA 2 h post-BDNF infusion (S-B; n = 8), yoked-saline 22 h post-PBS infusion (S-P; n = 6), yoked-saline 22 h post-BDNF infusion (S-B; n = 6), cocaine SA 22 h post-PBS infusion (C-P; n = 6), cocaine SA 22 h post-BDNF infusion (C-B; n = 6), yoked-saline with 7 d abstinence (S; n = 8), cocaine SA with 7 d abstinence (C; n = 8); experiment 4: yoked-saline 2 h post-U0126-PBS infusion (S/U-P; n = 8), yoked-saline 2 h post-U0126-BDNF infusion (S/U-B; n = 8), cocaine SA 2 h post-U0126-PBS infusion (C/U-P; n = 8), cocaine SA 2 h post-U0126-BDNF infusion (C/U-B; n = 8).
Experiment 1: BDNF's suppressive effect on cocaine-seeking depends on Trk receptor-mediated ERK/mitogen-activated protein kinase, not PI3K, signaling in the dmPFC
In experiment 1, rats were randomly assigned to groups that received pretreatment with K252a, U0126, or vehicle 20 min before receiving BDNF or PBS infusion directly following the final cocaine SA session to test the roles of tyrosine kinase and ERK signaling in BDNF's ability to suppress cocaine-seeking. All rats reached stable active (right) lever responding during the final 3 sessions of cocaine SA and mean responding for cocaine did not differ between groups before intracranial infusions (F(7,84) = 1.169, p > 0.05). Immediately following intracranial infusions, rats were placed directly into 6 d of home cage abstinence.
Test 1: Post-abstinence relapse test
The suppressive effect of intra-dmPFC BDNF was evaluated during a post-abstinence (PA) relapse test in which active lever responses had no programmed consequences (Fig. 3A). There was a significant treatment by time point interaction (F(1,29) = 7.73, p < 0.01). SNK tests revealed that V-P-treated (p < 0.001), but not V-B-treated (p > 0.05), rats responded significantly more during the PA test than during the final 3 d of cocaine SA. Further testing revealed that V-B rats responded significantly less on the active lever than V-P rats during the PA test (p < 0.05). For K252a, there also was a significant treatment by time point interaction (F(3,48) = 2.887, p < 0.05). SNK tests revealed that K-P- and K-B-treated rats both showed enhanced responding during the PA test compared with the final 3 d of cocaine SA (p < 0.01). Further SNK testing revealed a significant difference between the V-B and K-B groups (p < 0.05) during the PA test, demonstrating a full reversal of BDNF's suppressive effect on post-abstinence relapse by K252a. For U0126, there was a significant treatment by time point interaction (F(3,46) = 2.896, p < 0.05). SNK tests revealed that U-P-treated and U-B-treated rats both showed enhanced responding during the PA test compared with the final 3 d of cocaine SA (p < 0.05). SNK tests revealed a significant difference between the V-B and U-B groups (p < 0.05) during the PA test, demonstrating a reversal of BDNF's suppressive effect on post-abstinence drug-seeking by U0126.
Figure 3.
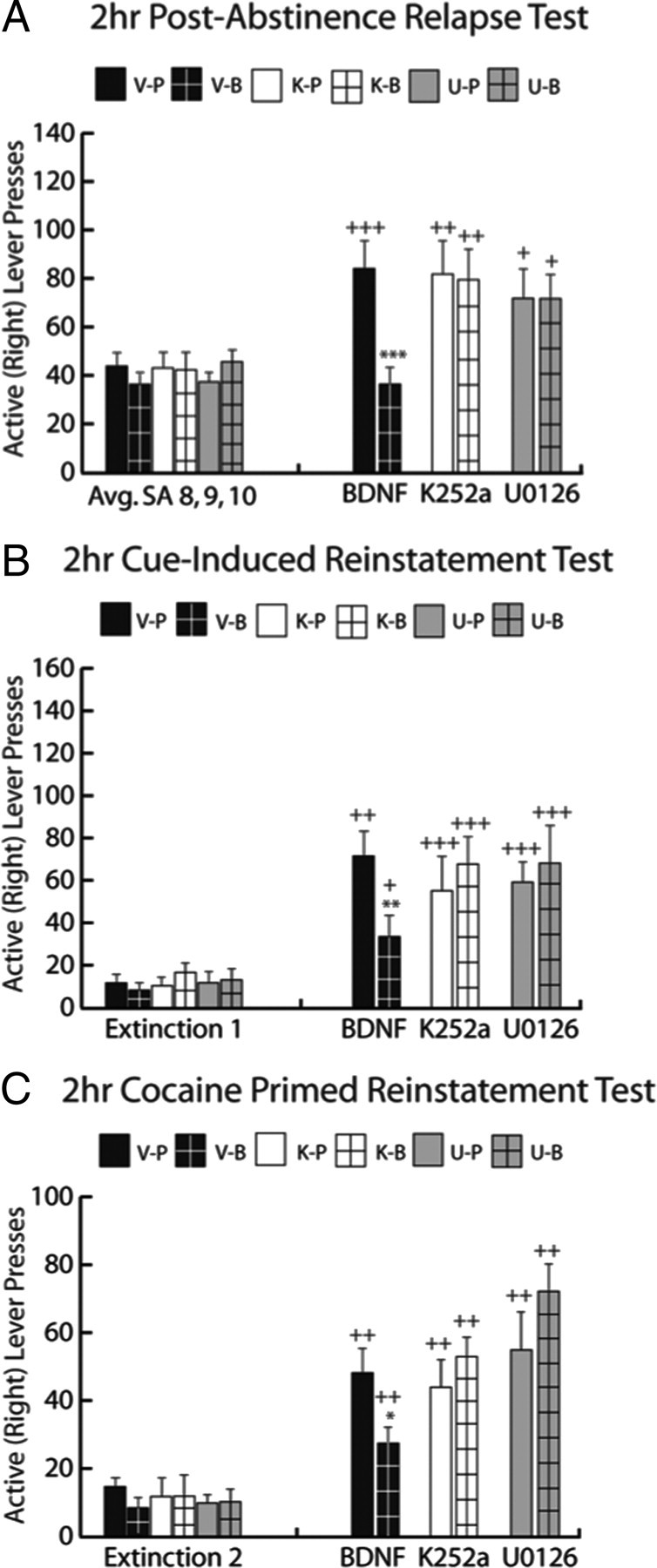
K252a and U0126 blocked BDNF's suppressive effect on cocaine-seeking. A, Post-abstinence test. Left, Before intra-dmPFC infusions, all rats show similar responding during the last 3 d of cocaine SA. On the final day of cocaine SA, rats received one of the following treatments: DMSO vehicle followed by PBS (V-P, black bar), DMSO vehicle followed by BDNF (V-B, black bar with grid pattern), K252a followed by PBS (K-P, white bar), K252a followed by BDNF (K-B, white bar with grid pattern), U0126 followed by PBS (U-P, gray bar), or U0126 followed by BDNF (U-B, gray bar with grid pattern). Right, Rats receiving V-B treatment showed significantly less cocaine seeking during the test compared with V-P controls (***p < 0.001). All other treatment groups demonstrated significantly greater cocaine-seeking in the post-abstinence test compared with cocaine SA responding (+p < 0.05, ++p < 0.01, +++p < 0.001). B, Cue-induced reinstatement test. Left, All rats extinguished lever pressing and reached a common baseline of responding. Right, Rats receiving V-B treatment showed significantly less cocaine seeking during the cue test than V-P controls (**p < 0.01). All treatment groups demonstrated greater cocaine-seeking in the cue test compared with extinction responding (+p < 0.05, ++p < 0.01, +++p < 0.001). C, Cocaine prime-induced reinstatement test. Left, All rats were brought back to a common baseline of responding during extinction. Right, Rats receiving V-B treatment showed significantly less cocaine seeking during the cocaine prime test than V-P controls (*p < 0.05). All treatment groups demonstrated greater cocaine-seeking in the cocaine prime test compared with extinction responding (+p < 0.05, +++p < 0.01). n = 9–16 per group.
Test 2: Cue-induced reinstatement test
Following extinction training, there were no significant differences in the average amount of lever pressing between groups during the final 2 d of extinction. The suppressive effect of intra-dmPFC BDNF was evaluated during a cue-induced reinstatement test, in which active lever responses resulted in the presentation of the light + tone compound CS (Fig. 3B). For BDNF, there was a significant treatment by time point interaction (F(1,29) = 9.234, p < 0.01). SNK tests revealed that V-P- and V-B-treated rats responded significantly more during the cue-induced reinstatement test than during the final 2 d of extinction training (p < 0.01 and p < 0.05, respectively). Further SNK tests revealed that V-B rats responded significantly less on the active lever than V-P rats during the cue-induced reinstatement test (p < 0.01). For K252a, there was a significant treatment by time point interaction (F(3,48) = 2.867, p < 0.05). SNK tests revealed that K-P- and K-B-treated rats both showed enhanced responding during the cue-induced reinstatement test compared with the final 2 d of extinction training (p < 0.001). Further SNK tests revealed a significant difference between the V-B and K-B groups (p < 0.05) during the cue test, demonstrating a full reversal of BDNF's suppressive effect on cue-induced reinstatement by K252a. For U0126, there was a significant treatment by time point interaction (F(3,46) = 2.949, p < 0.05). SNK tests revealed that U-P- and U-B-treated rats both showed enhanced responding during the cue-induced reinstatement test compared with the final 2 d of extinction training (p < 0.001). Further SNK tests revealed a significant difference between the V-B and U-B groups (p < 0.05) during the cue test, demonstrating a full reversal of BDNF's suppressive effect on cue-induced reinstatement by U0126.
Test 3: Cocaine prime-induced reinstatement test
Following extinction training, again there were no significant differences in the average amount of lever pressing between groups during the final 2 d of extinction. The suppressive effect of intra-dmPFC BDNF was evaluated during a cocaine-prime-induced reinstatement test, in which rats received a cocaine injection directly before being placed back into the operant context where active lever responses had no programmed consequences (Fig. 3C). For BDNF, there was a significant treatment by time point interaction (F(1,29) = 5.01, p < 0.05). SNK tests revealed that V-P- and V-B-treated rats responded significantly more during the cocaine prime-induced reinstatement test than during the final 2 d of extinction training (p < 0.01). SNK tests also revealed that V-B rats responded significantly less on the active lever than V-P controls during the cocaine prime-induced reinstatement test (p < 0.05). For K252a, there was a significant treatment by time point interaction (F(3,48) = 2.96, p < 0.05). SNK tests revealed that K-P- and K-B-treated rats both showed enhanced responding during the cocaine prime-induced reinstatement test compared with the final 2 d of extinction training (p < 0.01). Further SNK tests also revealed a significant difference between the V-B and K-B groups (p < 0.05) during the cocaine prime test, demonstrating a full reversal of BDNF's suppressive effect on cocaine prime-induced reinstatement by K252a. For U0126, there was a significant treatment by time point interaction (F(3,46) = 7.429, p < 0.001). SNK tests revealed that U-P- and U-B-treated rats both showed enhanced responding during the cocaine prime-induced reinstatement compared with the final 2 d of extinction training (p < 0.01). SNK tests also revealed a significant difference between the V-B and U-B groups (p < 0.05) during the cocaine-prime test.
Experiment 2: Intra-PFC BDNF activates phosphoprotein signaling in vivo in the dmPFC of drug naive rats
In experiment 2, drug-naive rats with bilateral intra-dmPFC cannulae received an intracranial infusion of BDNF or PBS and were killed by rapid decapitation 0.5 h, 2 h, or 6 h postinfusion to demonstrate the time course over which intra-dmPFC BDNF would increase p-ERK, p-CREB, and p-Akt in vivo in the dmPFC (Fig. 4). There was a significant main effect of BDNF treatment on p-ERK (ANOVA, F(1,29) = 24.96, p < 0.001), p-Akt (F(1,29) = 33.68, p < 0.001), and p-CREB (F(1,29) = 48.62, p < 0.001) expression in the dmPFC. Pairwise SNK comparisons revealed that BDNF significantly increased p-ERK expression 0.5 h (p < 0.05) and 2 h (p < 0.05), but not 6 h (p > 0.05), postinfusion (Fig. 4A), p-CREB expression 0.5 h (p < 0.05), 2 h (p < 0.05), and 6 h (p < 0.05) postinfusion (Fig. 4B), and p-Akt expression 2 h (p < 0.05), but not 30 min or 6 h (p > 0.05) postinfusion. There were no differences in total protein expression of t-ERK, t-Akt, and t-CREB across all treatment conditions (supplemental Table 1, available at www.jneurosci.org as supplemental material).
Figure 4.
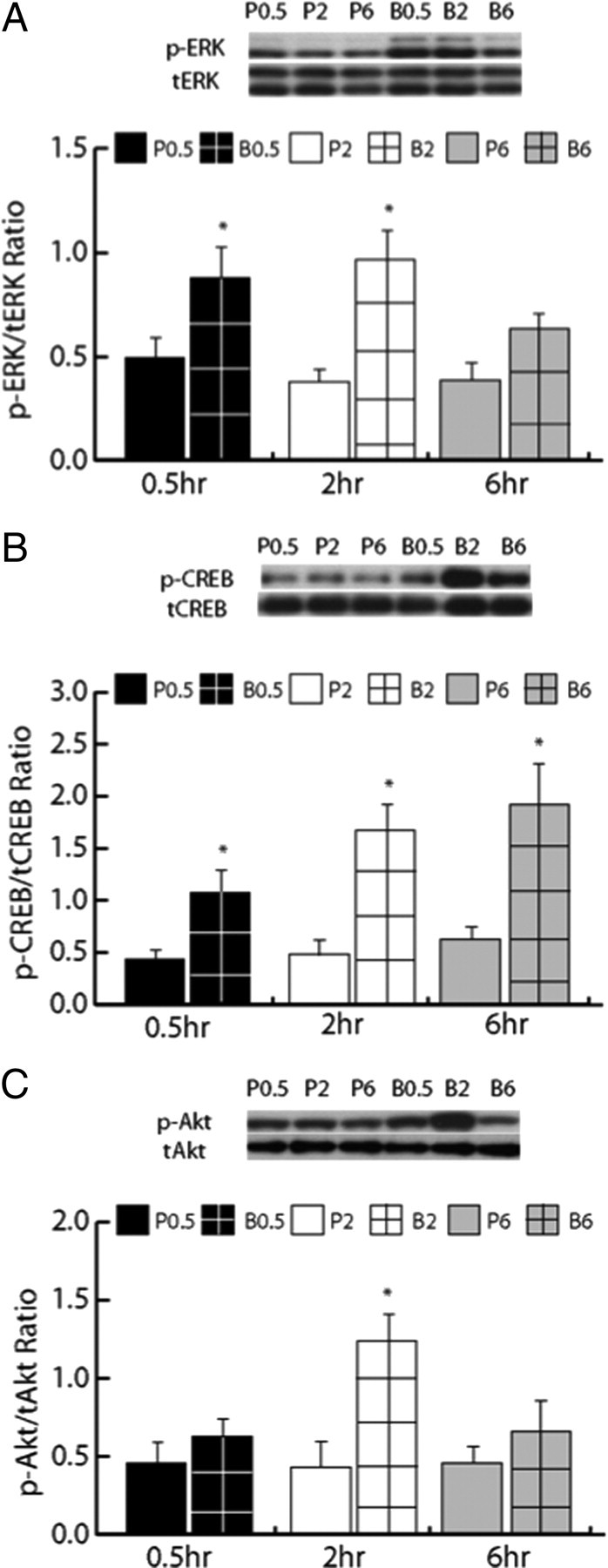
Intra-dmPFC BDNF activates phosphoprotein signaling in the dmPFC of drug-naive rats. A, p-ERK expression was significantly increased in the dmPFC 0.5 h (black bar with grid pattern) and 2 h (white bar with grid pattern), but not 6 h (gray bar with grid pattern), postinfusion in BDNF-infused rats (*p < 0.05) compared with PBS-infused control rats (solid black bar for 0.5 h, white bar for 2 h, and gray bar for 6 h). Above graph, Representative Western blot for p-ERK and total (t)-ERK expression. B, p-CREB expression was significantly increased in the dmPFC 0.5 h (black bar with grid pattern), 2 h (white bar with grid pattern), and 6 h (gray bar with grid pattern) post-BDNF infusion (*p < 0.05) compared with PBS-infused rats (solid black bar for 0.5 h, white bar for 2 h, and gray bar for 6 h). Above graph, Representative Western blot for p-CREB and t-CREB expression. C, p-Akt expression was significantly increased in the dmPFC 2 h (white bar with grid pattern), but not 0.5 h (black bar with grid pattern) or 6 h (gray bar with grid pattern) post-BDNF infusion (*p < 0.05) compared with PBS-infused rats (solid black bar for 0.5 h, white bar for 2 h, and gray bar for 6 h). Above graph, Representative Western blot for p-Akt and t-Akt expression.
Experiment 3: Intra-PFC BDNF counteracts cocaine-mediated abnormalities in phosphoprotein signaling in the dmPFC
Given that BDNF increased the levels of all three phosphoproteins 2 h postinfusion in experiment 2, this time point was used to investigate whether a BDNF infusion would alter cocaine SA-induced changes in these signaling proteins. In experiment 3, rats received intracranial infusion of BDNF or PBS and were killed by rapid decapitation 2 h later to characterize BDNF's effects on phosphoprotein signaling in rats with a cocaine SA or yoked-saline history (Fig. 5A–C). For p-ERK (Fig. 5A), there was a significant main effect of drug treatment (cocaine vs saline; F(1,27) = 6.17, p < 0.05) as well as a significant main effect of infusion (BDNF vs PBS; F(1,27) = 30.12, p < 0.001) but no interaction. SNK tests revealed that PBS-infused rats with a cocaine SA history displayed significantly decreased p-ERK expression in the dmPFC compared with PBS-infused yoked saline rats (p < 0.05). Furthermore, BDNF vs PBS significantly increased p-ERK expression in both yoked-saline (p < 0.001) and cocaine SA rats (p < 0.01). For p-CREB (Fig. 5B), there also was a significant main effect of drug treatment (F(1,27) = 26.15, p < 0.001) and a significant main effect of infusion (F(1,27) = 14.05, p < 0.001). SNK tests revealed that PBS-infused rats with a cocaine SA history displayed significantly decreased p-CREB expression compared with PBS-infused yoked-saline controls (p < 0.001). Additionally, BDNF vs PBS significantly increased p-CREB expression in both yoked saline (p < 0.05) and cocaine SA rats (p < 0.05). For p-Akt (Fig. 5C), there was a significant main effect of infusion only (F(1,27) = 24.19, p < 0.001). There were no differences in total protein expression for t-ERK, t-Akt, and t-CREB (supplemental Table 1, available at www.jneurosci.org as supplemental material) across the treatment conditions.
Figure 5.

Intra-dmPFC BDNF counteracts cocaine-mediated decreases in p-ERK and p-CREB, but not p-Akt, expression 2 h postinfusion, and this normalization of cocaine-mediated decreases in p-ERK and p-CREB expression is blocked by U0126. A, p-ERK expression was suppressed in the cocaine SA group compared with the yoked-saline group (*p < 0.05 C-P vs S-P). Intra-dmPFC BDNF infusion significantly increased p-ERK expression in the yoked-saline group (**p < 0.01 S-B vs S-P) and normalized p-ERK expression in the cocaine SA (+++p < 0.05 C-B vs C-P) group. Above graph, Representative Western blot for p-ERK and t-ERK expression. B, p-CREB expression was suppressed in the cocaine SA group compared with the yoked-saline group (***p < 0.001 C-P vs S-P). Intra-dmPFC BDNF infusion significantly increased p-CREB expression in the yoked-saline group (*p < 0.05 S-B vs S-P) and normalized p-CREB expression in the cocaine SA (+++p < 0.01 C-B vs C-P) group. Above graph, Representative Western blot for p-CREB and t-CREB expression. C, Intra-dmPFC BDNF infusion significantly increased p-Akt expression in the yoked-saline group (***p < 0.001 S-B vs S-P) and in the cocaine SA (***p < 0.001 C-B vs C-P) group. Above graph, Representative Western blot for p-Akt and total Akt expression. D, p-ERK expression was suppressed in both cocaine SA groups compared with the yoked-saline group (***p < 0.001 S/U-P vs C/U-P and C/U-B). Intra-dmPFC BDNF infusion failed to increase p-ERK expression in the presence of U0126 for yoked-saline (S/U-B and C/U-B) or cocaine SA rats. Above graph: Representative Western blot for p-ERK and t-ERK expression. E, p-CREB expression was suppressed in both cocaine SA groups compared with the yoked-saline group (**p < 0.01 S/U-P vs C/U-P and C/U-B). Intra-dmPFC BDNF infusion failed to increase p-CREB expression in the presence of U0126 for yoked-saline or cocaine SA rats. Above graph, Representative Western blot for p-CREB and t-CREB expression. F, Intra-dmPFC BDNF infusion significantly increased p-Akt expression in the yoked-saline group (**p < 0.01 S/U-B vs S/U-P) and in the cocaine SA (**p < 0.01 C/U-B vs C/U-P) groups regardless of U0126 infusion. Above graph, Representative Western blot for p-Akt and total Akt expression.
A separate group of rats received intra-dmPFC infusion of BDNF or PBS and was killed by rapid decapitation 22 h later to characterize BDNF's prolonged effects on phosphoprotein signaling in rats with a cocaine SA or yoked-saline history (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). For p-ERK (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material), there were no significant main effects of drug treatment (cocaine vs saline; F(1,20) = 0.96, p > 0.05) or infusion (BDNF vs PBS; F(1,20) = 2.73, p > 0.05). For p-CREB (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material), there were no significant main effects of drug treatment (cocaine vs saline; F(1,20) = 0.16, p > 0.05) or infusion (BDNF vs PBS; F(1,20) = 1.7, p > 0.05). For p-Akt (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material), there were no significant main effects of drug treatment (cocaine vs saline; F(1,20) = 0.84, p > 0.05) or infusion (BDNF vs PBS; F(1,20) = 0.65, p > 0.05). There were no differences in total protein expression for t-ERK, t-Akt, and t-CREB (supplemental Table 1, available at www.jneurosci.org as supplemental material) across all treatment conditions.
Another cohort of rats with a cocaine SA or yoked-saline history were killed by rapid decapitation after 7 d of forced abstinence to characterize cocaine SA's prolonged effects on phosphoprotein signaling in dmPFC (supplemental Fig. S1D–F, available at www.jneurosci.org as supplemental material). There was no significant difference in p-ERK (t14 = −0.66, p > 0.05) or p-Akt (t14 = −0.99, p > 0.05) levels between the cocaine and saline groups (supplemental Fig. S1D,F, available at www.jneurosci.org as supplemental material). However, there was significantly more p-CREB expression in the dmPFC of the cocaine vs saline group (supplemental Fig. S1E, available at www.jneurosci.org as supplemental material; t14 = 3.99, p = 0.05). There were also no differences in total protein expression for t-ERK, t-Akt, and t-CREB in cocaine vs saline groups (supplemental Table 1, available at www.jneurosci.org as supplemental material).
Experiment 4: The normalizing effects of intra-dmPFC BDNF on p-CREB depend on ERK mitogen-activated protein kinase signaling
In experiment 4, rats with a cocaine SA or yoked-saline history received pretreatment with U0126 followed by infusion of BDNF or PBS and were killed 2 h later to determine whether BDNF's ability to rescue p-CREB levels in the dmPFC depends on ERK mitogen-activated protein kinase (MAPK) signaling (Fig. 5). For p-ERK (Fig. 5D) and p-CREB (Fig. 5E), there was a significant main effect of drug treatment (cocaine vs saline; p-ERK: F(1,26) = 58.28, p < 0.001; p-CREB: (F(1,26) = 24.837, p < 0.05) but no main effect of infusion. Thus, BDNF treatment failed to significantly increase p-ERK and p-CREB expression in yoked-saline controls or cocaine SA history rats that received BDNF in the presence of U0126. For p-Akt (Fig. 5F), there was a significant main effect of infusion only (F(1,26) = 10.63, p < 0.01). This result demonstrates that U0126 did not affect BDNF's ability to activate p-Akt in saline or cocaine SA rats. To ensure that U0126 did not influence basal expression of p-ERK, p-Akt, and p-CREB, we compared the expression of each phosphoprotein between yoked-saline rats from experiment 3 that received PBS infusion and yoked-saline rats from experiment 4 that received U0126 followed by PBS infusion. There were no significant main effects of U0126 for p-ERK (t14 = −1.27, p > 0.05), p-Akt (t14 = −0.14, p > 0.05), or for p-CREB (t14 = −0.79, p > 0.05, p > 0.05), demonstrating that there was no effect of U0126 treatment on basal phosphoprotein expression. Furthermore, there were no differences in total protein expression for t-ERK, t-Akt, and t-CREB across all treatment conditions (supplemental Table 1, available at www.jneurosci.org as supplemental material).
Discussion
The current study demonstrated that an intra-dmPFC infusion of the tyrosine kinase inhibitor, K252a, and the MEK inhibitor, U0126, blocked the ability of intra-dmPFC BDNF, when infused immediately after the last cocaine SA session, to suppress enduring cocaine-seeking after abstinence and in subsequent reinstatement testing after extinction. Moreover, BDNF reversed a cocaine-mediated dephosphorylation of ERK and CREB in the dmPFC 2 h after intra-dmPFC infusion. Consistent with its ability to block BDNF's enduring suppressive effects on cocaine-seeking, U0126 blocked BDNF's ability to restore the cocaine-induced decrease in p-ERK and p-CREB in the dmPFC. These data suggest that there is a critical, time-restricted window immediately after the end of chronic cocaine exposure in which intra-PFC BDNF is able to suppress cocaine-induced neuroadaptations that lead to drug-seeking.
The suppressive effects of BDNF on cocaine-seeking were blocked by K252a
Intra-dmPFC BDNF's ability to persistently suppress cocaine-seeking after forced abstinence, as well as during cue- and cocaine prime-induced reinstatement, was blocked by K252a, suggesting that BDNF binds endogenous TrkB receptors locally in the dmPFC to exert its behavioral effects. Although K252a is not selective for Trk receptors, it is a high-affinity tyrosine kinase inhibitor and it did not alter cocaine-seeking in the absence of BDNF. This finding indicates that the concentration of K252a used did not alter basal, but only BDNF-stimulated, signaling in the dmPFC. Since BDNF does not directly stimulate other Trk receptor populations (TrkA, TrkC) or ser-thr kinase cascades (PKA, PKC, CAMK), it is unlikely that K252a's blockade of BDNF's effects was mediated by these non-TrkB signaling molecules. This interpretation is supported by the fact that BDNF's ability to suppress cocaine-seeking 22 h after the last SA session was previously demonstrated to be prevented by the BDNF scavenging compound, TrkB-Fc (McGinty et al., 2010).
The suppressive effects of BDNF on cocaine-seeking were blocked by U0126
Intra-dmPFC BDNF's ability to persistently suppress cocaine-seeking after forced abstinence, as well as during cue- and cocaine prime-induced reinstatement, was also blocked by the selective MEK inhibitor, U0126, suggesting that after BDNF binds to TrkB receptors, it activates ERK MAPK signaling locally in the dmPFC. This result is consistent with evidence that implicates ERK signaling in the behavioral effects of BDNF on cocaine-seeking when it is infused into the VTA (Lu et al., 2004). In addition, the ability of BDNF to modify amygdala-dependent fear conditioning (Ou and Gean, 2006), hippocampal-dependent memory (Igaz et al., 2006), and mood stability (Einat et al., 2003) also depends on ERK MAPK signaling. The ability of K252a, TrkB-Fc (McGinty et al., 2010), and the selective MEK inhibitor, U0126, to block BDNF's behavioral effects reinforces the conclusion that the TrkB-ERK cascade mediates the suppressive effects of BDNF on cocaine-seeking. Indeed, PI3K inhibition with intra-PFC wortmannin failed to significantly reverse BDNF's suppressive effect on cocaine-seeking in each test (data not shown), indicating that the behavioral effects of BDNF occur via a PI3K-independent mechanism. Although wortmannin has been previously shown to facilitate acute D1 agonist-induced stimulation of ERK phosphorylation in vitro (Brami-Cherrier et al., 2002), there was no greater suppression of cocaine-seeking by BDNF in animals that received wortmannin and BDNF. These data, along with a lack of effect of cocaine on p-Akt in this study, demonstrate that BDNF's behavioral effects on cocaine-seeking selectively require ERK/MAPK, not PI3K, signaling in the dmPFC.
Cocaine SA caused a significant dephosphorylation of p-ERK and p-CREB, but not p-Akt, in the dmPFC
The significant decrease in p-ERK and p-CREB, but not p-Akt, levels in the dmPFC during early withdrawal was remarkable given that cocaine is generally considered to increase ERK, CREB, and Akt phosphorylation in the CNS (Berhow et al., 1996; Mattson et al., 2005; Valjent et al., 2004). However, the latter conclusion has been drawn mostly from the effects of acute and repeated noncontingent administration of cocaine. Despite few studies that have investigated the response of phosphoproteins in the mPFC to chronic cocaine SA, a recent study by Edwards et al. (2007) showed that p-ERK, but not p-CREB, was significantly elevated in the mPFC immediately following the last of 18 daily 4 h cocaine SA sessions. However, our data indicate that p-ERK and p-CREB are rapidly dephosphorylated, with their levels plummeting below control value during the first hours of withdrawal from cocaine SA. We hypothesize that this “crash” early in withdrawal triggers a cascade of deleterious events, possibly affecting CREB targets in the prefrontal-accumbens pathway, that promote relapse to cocaine-seeking if not immediately reversed. Given that p-ERK and p-CREB levels in the PFC had normalized 22 h after cocaine SA, it appears that critical events in the dmPFC underlying persistent drug-seeking occur quickly and transiently after repeated cocaine exposure. If they are not reversed, then other long-term neuroadaptations are allowed to occur if abstinence continues. Under the conditions of this experiment, 7 d after the end of cocaine exposure, p-ERK levels in the PFC remained normal but p-CREB levels were elevated, suggesting that CREB phosphorylation was no longer ERK-dependent. This interpretation is consistent with BDNF-independence of cocaine-seeking after 7 d of abstinence because when infusion is delayed for 6 d, BDNF does not alter cocaine-seeking on day 7 (Berglind et al., 2007).
Although it is unknown how cocaine withdrawal suppresses ERK and CREB phosphorylation, chronic exposure to psychostimulants upregulates the expression of dual specificity MAP-kinase phosphatases (MKPs), essential regulators of p-ERK, in the PFC and striatum (Takaki et al., 2001). Given that upregulation of MKPs corresponds with decreased p-ERK in the hippocampus (Dwivedi et al., 2001), increased MKP activity may underlie suppressed p-ERK levels during early withdrawal from cocaine SA. However, future studies will be needed to explore this question because there are several MKPs that dephosphorylate ERK and they are not the only phosphatases to do so (Farooq and Zhou, 2004).
U0126 prevented BDNF's ability to activate p-ERK and p-CREB, whereas U0126 had no effect on BDNF's ability to phosphorylate Akt, nor did this dose of U0126 affect basal phosphoprotein expression as previously described (Shi and McGinty, 2006). This result is consistent with other reports indicating that BDNF's ability to activate p-CREB-dependent structural and synaptic plasticity is reversed by U0126 (Ying et al., 2002; Alonso et al., 2004). These studies also reinforce the finding that BDNF counteracts cocaine-mediated neuroadaptations by activating CREB in an ERK MAPK-dependent, not PI3K-dependent, manner in the dmPFC.
BDNF is critically involved in the neuroplasticity of the dmPFC, as well as the propensity to engage in drug-seeking behavior in rats with a cocaine SA history
The finding that the expression of p-ERK and p-CREB in the dmPFC were suppressed during early abstinence from cocaine SA positively correlates with reports that chronic cocaine SA alters prefrontal cortical excitability and neuroplasticity. For instance, acute cocaine induces BDNF mRNA and protein in the mPFC 24 h postinjection; however after 2 or 72 h of withdrawal from repeated cocaine injections, BDNF protein levels in the PFC were significantly decreased (Fumagalli et al., 2007). Similarly, the expression of BDNF mRNA, along with other immediate early genes in the dmPFC, is decreased 22 h after 10 d of cocaine SA (McGinty et al., 2010), signifying a decline in cortical activity during early abstinence. When rats are reexposed to the cocaine-associated operant context after 22 h or 15 d of abstinence and allowed to press levers that previously resulted in cocaine delivery, BDNF and other immediate early gene mRNA was induced in the mPFC, signifying an increase in cortical activation (Hearing et al., 2008). Prefrontal cortical excitability follows a similar pattern, in which basal excitability of the PFC decreases over the course of cocaine SA, while burst firing during exposure to cocaine and cocaine-related stimuli is enhanced (Sun and Rebec, 2006). Together, these data indicate that acute cocaine activates PFC BDNF transcription and translation in a manner consistent with the sensitizing effects of cocaine, but that repeated cocaine administration and cocaine SA suppresses prefrontal cortical plasticity and excitability, in part due to suppression of endogenous BDNF expression. Reversal of this cocaine-mediated suppression in the dmPFC with BDNF infusion restores balance to plasticity-associated signaling locally in the PFC. As a result, cocaine-induced abnormalities in glutamate transmission in the NAc, which have been shown to regulate cocaine-seeking during relapse (McFarland et al., 2003), are reversed (Berglind et al., 2009).
Together, these studies have begun to characterize the mechanism of BDNF's suppressive effect on cocaine relapse. Exogenous BDNF's ability to recover suppressed p-ERK and p-CREB signaling in the dmPFC during early withdrawal leads to a persistent attenuation of cocaine-seeking. The implication from this finding is that early intervention during withdrawal from chronic cocaine administration may be a critical way to facilitate long-term therapeutic outcomes. Further, the current work solidifies the site-specific role of BDNF in cocaine-mediated plasticity in the CNS. Future studies will evaluate the ability of novel therapeutic interventions to restore normal BDNF expression and activity in the dmPFC to counteract cocaine's deleterious effects and suppress addictive cocaine-seeking.
Footnotes
This work was supported by National Institutes of Health Grants P50 DA015369, T32 DA07288, F31 DA023743, and C06 RR015455. We thank Adrian Gomez, Phong Do, and Shannon Ghee for technical assistance.
References
- Alonso M, Medina JH, Pozzo-Miller L. ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn Mem. 2004;11:172–178. doi: 10.1101/lm.67804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglind WJ, See RE, Fuchs RA, Ghee SM, Whitfield TW, Jr, Miller SW, McGinty JF. A BDNF infusion into the medial prefrontal cortex suppresses cocaine seeking in rats. Eur J Neurosci. 2007;26:757–766. doi: 10.1111/j.1460-9568.2007.05692.x. [DOI] [PubMed] [Google Scholar]
- Berglind WJ, Whitfield TW, Jr, LaLumiere RT, Kalivas PW, McGinty JF. A single intra-PFC infusion of BDNF prevents cocaine-induced alterations in extracellular glutamate within the nucleus accumbens. J Neurosci. 2009;29:3715–3719. doi: 10.1523/JNEUROSCI.5457-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berhow MT, Hiroi N, Nestler EJ. Regulation of ERK (extracellular signal regulated kinase), part of the neurotrophin signal transduction cascade, in the rat mesolimbic dopamine system by chronic exposure to morphine or cocaine. J Neurosci. 1996;16:4707–4715. doi: 10.1523/JNEUROSCI.16-15-04707.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brami-Cherrier K, Valjent E, Garcia M, Pagès C, Hipskind RA, Caboche J. Dopamine induces a PI3-kinase-independent activation of Akt in striatal neurons: a new route to cAMP response element-binding protein phosphorylation. J Neurosci. 2002;22:8911–8921. doi: 10.1523/JNEUROSCI.22-20-08911.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi Y, Rizavi HS, Roberts RC, Conley RC, Tamminga CA, Pandey GN. Reduced activation and expression of ERK1/2 MAP kinase in the post-mortem brain of depressed suicide subjects. J Neurochem. 2001;77:916–928. doi: 10.1046/j.1471-4159.2001.00300.x. [DOI] [PubMed] [Google Scholar]
- Edwards S, Graham DL, Bachtell RK, Self DW. Region-specific tolerance to cocaine-regulated cAMP-dependent protein phosphorylation following chronic self-administration. Eur J Neurosci. 2007;25:2201–2213. doi: 10.1111/j.1460-9568.2007.05473.x. [DOI] [PubMed] [Google Scholar]
- Einat H, Yuan P, Gould TD, Li J, Du J, Zhang L, Manji HK, Chen G. The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J Neurosci. 2003;23:7311–7316. doi: 10.1523/JNEUROSCI.23-19-07311.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooq A, Zhou MM. Structure and regulation of MAPK phosphatases. Cell Signal. 2004;16:769–779. doi: 10.1016/j.cellsig.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Fuchs RA, Evans KA, Ledford CC, Parker MP, Case JM, Mehta RH, See RE. The role of the dorsomedial prefrontal cortex, basolateral amygdala, and dorsal hippocampus in contextual reinstatement of cocaine seeking in rats. Neuropsychopharmacology. 2005;30:296–309. doi: 10.1038/sj.npp.1300579. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Di Pasquale L, Caffino L, Racagni G, Riva MA. Repeated exposure to cocaine differently modulates BDNF mRNA and protein levels in rat striatum and prefrontal cortex. Eur J Neurosci. 2007;26:2756–2763. doi: 10.1111/j.1460-9568.2007.05918.x. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Caffino L, Racagni G, Riva MA. Repeated stress prevents cocaine-induced activation of BDNF signaling in rat prefrontal cortex. Eur Neuropsychopharmacol. 2009;19:402–408. doi: 10.1016/j.euroneuro.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Graham DL, Edwards S, Bachtell RK, DiLeone RJ, Rios M, Self DW. Dynamic BDNF activity in nucleus accumbens with cocaine use increases self-administration and relapse. Nat Neurosci. 2007;10:1029–1037. doi: 10.1038/nn1929. [DOI] [PubMed] [Google Scholar]
- Hearing MC, Miller SW, See RE, McGinty JF. Relapse to cocaine seeking increases activity-regulated gene expression differentially in the prefrontal cortex of abstinent rats. Psychopharmacology (Berl) 2008;198:77–91. doi: 10.1007/s00213-008-1090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaz LM, Winograd M, Cammarota M, Izquierdo LA, Alonso M, Izquierdo I, Medina JH. Early activation of extracellular signal-regulated kinase signaling pathway in the hippocampus is required for short-term memory formation of a fear-motivated learning. Cell Mol Neurobiol. 2006;26:989–1002. doi: 10.1007/s10571-006-9116-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo E, Martin-Fardon R, Koob GF, Weiss F, Sanna PP. Neural plasticity and addiction: PI3-kinase and cocaine behavioral sensitization. Nat Neurosci. 2002;5:1263–1264. doi: 10.1038/nn977. [DOI] [PubMed] [Google Scholar]
- Koob GF, Caine SB, Parsons L, Markou A, Weiss F. Opponent process model and psychostimulant addiction. Pharmacol Biochem Behav. 1997;57:513–521. doi: 10.1016/s0091-3057(96)00438-8. [DOI] [PubMed] [Google Scholar]
- Lu L, Dempsey J, Liu SY, Bossert JM, Shaham Y. A single infusion of brain-derived neurotrophic factor into the ventral tegmental area induces long-lasting potentiation of cocaine seeking after withdrawal. J Neurosci. 2004;24:1604–1611. doi: 10.1523/JNEUROSCI.5124-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson BJ, Bossert JM, Simmons DE, Nozaki N, Nagarkar D, Kreuter JD, Hope BT. Cocaine-induced CREB phosphorylation in nucleus accumbens of cocaine-sensitized rats is enabled by enhanced activation of extracellular signal-related kinase, but not protein kinase A. J Neurochem. 2005;95:1481–1494. doi: 10.1111/j.1471-4159.2005.03500.x. [DOI] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23:3531–3537. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty JF, Whitfield TW, Jr., Berglind WJ. Brain-derived neurotrophic factor and cocaine addiction. Brain Res. 2010;1314C:183–193. doi: 10.1016/j.brainres.2009.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin J, See RE. Selective inactivation of the dorsomedial prefrontal cortex and the basolateral amygdala attenuates conditioned-cued reinstatement of extinguished cocaine-seeking behavior in rats. Psychopharmacology (Berl) 2003;168:57–65. doi: 10.1007/s00213-002-1196-x. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–128. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- Ou LC, Gean PW. Regulation of amygdala-dependent learning by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol-3-kinase. Neuropsychopharmacology. 2006;31:287–296. doi: 10.1038/sj.npp.1300830. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol. 2001;11:272–280. doi: 10.1016/s0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Burlington, MA: Elsevier Academic; 2005. [Google Scholar]
- Sadri-Vakili G, Kumaresan V, Schmidt HD, Famous KR, Chawla P, Vassoler FM, Overland RP, Xia E, Bass CE, Terwilliger EF, Pierce RC, Cha JHC. Cocaine-induced chromatin remodeling increases brain-derived neurotrophic factor transcription in the rat medial prefrontal cortex, which alters the reinforcing efficacy of cocaine. J Neurosci. 2010;30:11735–11744. doi: 10.1523/JNEUROSCI.2328-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, McGinty JF. Extracellular signal-regulated mitogen-activated protein kinase inhibitors decrease amphetamine-induced behavior and neuropeptide gene expression in the striatum. Neuroscience. 2006;138:1289–1298. doi: 10.1016/j.neuroscience.2005.12.024. [DOI] [PubMed] [Google Scholar]
- Shi X, McGinty JF. Repeated amphetamine treatment increases phosphorylation of extracellular signal-regulated kinase, protein kinase B, and cyclase response element-binding protein in the rat striatum. J Neurochem. 2007;103:706–713. doi: 10.1111/j.1471-4159.2007.04760.x. [DOI] [PubMed] [Google Scholar]
- Sun W, Rebec GV. Repeated cocaine self-administration alters processing of cocaine-related information in rat prefrontal cortex. J Neurosci. 2006;26:8004–8008. doi: 10.1523/JNEUROSCI.1413-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaki M, Ujike H, Kodama M, Takehisa Y, Nakata K, Kuroda S. Two kinds of mitogen-activated protein kinase phosphatases, MKP-1 and MKP-3, are differentially activated by acute and chronic methamphetamine treatment in the rat brain. J Neurochem. 2001;79:679–688. doi: 10.1046/j.1471-4159.2001.00615.x. [DOI] [PubMed] [Google Scholar]
- Valjent E, Pagès C, Hervé D, Girault JA, Caboche J. Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur J Neurosci. 2004;19:1826–1836. doi: 10.1111/j.1460-9568.2004.03278.x. [DOI] [PubMed] [Google Scholar]
- White FJ, Kalivas PW. Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alcohol Depend. 1998;51:141–153. doi: 10.1016/s0376-8716(98)00072-6. [DOI] [PubMed] [Google Scholar]
- Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV, Bramham CR. Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci. 2002;22:1532–1540. doi: 10.1523/JNEUROSCI.22-05-01532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Mi J, Wetsel WC, Davidson C, Xiong X, Chen Q, Ellinwood EH, Lee TH. PI3 kinase is involved in cocaine behavioral sensitization and its reversal with brain area specificity. Biochem Biophys Res Commun. 2006;340:1144–1150. doi: 10.1016/j.bbrc.2005.12.114. [DOI] [PubMed] [Google Scholar]