

Abstract
Plasminogen activator inhibitor-1 (PAI-1) promotes or abates fibrotic processes occurring in different organs. Binding of PAI-1 to vitronectin, an extracellular matrix component, may inhibit vitronectin–integrin complex-mediated cellular responses in pathophysiological conditions. To investigate the importance of plasmin suppression vs vitronectin-binding pathways of PAI-1 in cardiac fibrosis, we studied uninephrectomized mice fed a high salt diet and infused with angiotensin II (Ang II) together with different PAI-1 variants, including PAI-1AK (AK) that inhibits plasminogen activators but does not bind vitronectin, PAI-1RR (RR) that binds vitronectin but does not have protease inhibitory effects or control PAI-1 (CPAI), the control mutant that has similar molecular backbone and half-life as AK and RR while retaining all functions of native PAI-1. Compared with RR and CPAI, non-vitronectin-binding AK significantly increased expression of cardiac fibroblast marker, periostin (Ang + AK 8.40± 3.55 vs Ang RR 2.23 ± 0.44 and Ang CPAI 2.33 ± 0.12% positive area, both P <0.05) and cardiac fibrosis (Ang + AK 1.79 ± 0.26% vs Ang + RR 0.91 + 0.18% and Ang + CPAI 0.81 ± 0.12% fibrotic area, both P <0.05), as well as Col1 mRNA (Ang + AK 12.81 ± 1.84 vs Ang + RR 4.04 ± 1.06 and Ang + CPAI 5.23 ± 1.21 fold increase, both P <0.05). To elucidate mechanisms underlying the protective effects of vitronectin-binding PAI-1 against fibrosis, fibroblasts from normal adult human ventricles were stimulated with Ang and different PAI-1 variants. Protease inhibitory AK and CPAI increased supernatant fibronectin, while decreasing plasminogen activator/plasmin activities and matrix metalloproteinase. RR and CPAI variants significantly reduced fibroblast expression of integrin β3, vitronectin level in the supernatant and fibroblast adhesion to vitronectin compared with the non-vitronectin-binding AK. Further, RR and CPAI preserved apoptotic, decreased anti-apoptotic and proliferative activities in fibroblasts. Thus, PAI-1 promotes or protects against development of cardiac fibrosis differentially through the protease inhibitory pathway or through its binding to vitronectin.
Keywords: angiotensin, fibrosis, fibroblast, heart, plasminogen activator inhibitor-1, integrin β3, vitronectin
Elevated levels of plasminogen activator inhibitor 1 (PAI-1) have been associated with increased cardiovascular risk, progression of glomerulosclerosis and fibrosis in the liver and lung.1–4 In cardiac fibrosis, it remains controversial whether PAI-1 is a mediator or inhibitor of cardiac fibrosis.5–7 Our previous studies showed that genetic deficiency of PAI-1 or pharmacological inhibition of PAI-1 potentiated angiotensin II (Ang II) or aldosterone-induced fibrosis in the heart.8,9 Development of Ang II-associated cardiac fibrosis involves proliferation and infiltration of cardiac fibroblasts,10,11 which are the cells primarily responsible for production and remodeling of the extracellular matrix components.12–15
PAI-1 is the primary in vivo inhibitor of plasminogen activators, which convert plasminogen to its active form plasmin. The plasmin/protease system has an important role in matrix degradation, which PAI-1 in turn increases accumulation of extracellular matrix and fibrosis.16 PAI-1 also binds vitronectin, a matrix protein that interacts with cell-surface integrins. The binding of PAI-1 to vitronectin blocks the vitronectin–integrin αVβ3 interaction, and regulates variety of cell activities including adhesion, migration, proliferation and survival.17–19 Ang II has been shown to upregulate PAI-1 and integrin αVβ3 in cardiac fibroblasts.20–22 Interestingly, a recombinant PAI-1 protein variant that only has vitronectin-binding activity but does not inhibit protease attenuates glomerular lesions.23–25 Thus, we hypothesized that PAI-1 differentially impacts Ang II-induced cardiac fibrosis through its vitronectin-binding and protease inhibitory pathways.
To test this hypothesis, three PAI-1 variants were used: PAI-1AK (AK) that retains all protease inhibitory activity but has no effect on binding vitronectin, PAI-1RR (RR) that binds vitronectin normally but does not inhibit protease and control PAI-1 variant (CPAI) that has all native PAI-1 effects. Different effects between the AK and CPAI variants thus are taken to reflect the lack of vitronectin-binding PAI-1, whereas differences between the RR and CPAI reflect the deficiency of protease inhibition.23,26–28 The CPAI, also known as the 14-1B variant, has remarkably increased functional stability compared with the wild-type PAI-1 (wtPAI-1).27 AK, which has further R101A/Q123K mutations on the 14-1B backbone, shows similar functional half-life of ~145 h as the CPAI in vitro. However, owing to the nature of lacking vitronectin-binding capability, AK is eliminated much faster than CPAI in vivo.29 Although RR variant is constructed on the wtPAI-1, its T333R/A335R mutations result in extended half-life both in vitro and in vivo.29,30 Using these PAI-1 protein variants, the present study examined the effects of vitronectin-binding and protease-inhibiting pathways of PAI-1 on cardiac fibrosis in mice with chronic Ang II infusion superimposed on uninephrectomy and high salt intake, a model mimicking human hypertension with reduced nephron number that promotes cardiac fibrosis in both human and rodents. Further studies also explored different effects of PAI-1 variants on cultured cardiac fibroblasts. We revealed that PAI-1 protects against development of cardiac fibrosis through its binding to extracellular vitronectin, which may result in decreased αvβ3 integrin effects.
MATERIALS AND METHODS
Animals
Female C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME, USA) were housed in AAALAC-certified facilities with access to water ad lib. The animal protocols were approved by the Vanderbilt University Institutional Animal Care and Use Committee.
At 12 weeks of age, the mice were uninephrectomized (UNx) under isoflurane anesthesia, and fed a high sodium diet (3.15% Na, Harlan Teklad, Indianapolis, IN, USA). After 2 weeks, the animals were randomized into five experimental groups to receive chronic Ang II infusion and daily intraperitoneal injection of different PAI-1 variants (1 μg/g body weight, Molecular Innovations, Novi, MI, USA) or phosphate-buffered saline (PBS): UNx/salt control + PBS (Cont, n = 4), Ang II/UNx/salt + PBS (Ang + PBS, n = 7), Ang II/UNx/salt + RR (Ang + RR, n = 9), Ang II/UNx/salt + AK (Ang + AK, n = 8) and Ang II/UNx/salt + CPAI (Ang + CPAI, n = 8). The Ang II (1 ng/min/g) was delivered by osmotic mini-pump implanted at week 0 and replaced at 4 weeks (Alzet, Cupertino, CA, USA). Systolic blood pressure (SBP) was measured by tail-cuff plethysmography (BP-2000 Blood Pressure Analysis System, Visitech Systems, Apex, NC, USA) in trained conscious mice. Urine albumin-to-creatinine ratio (UACR) was assessed by ELISA (Exocell, Philadelphia, PA, USA) at weeks 0, 2, 4, 6 and 8. At week 8, plasma and hearts were harvested under isoflurane anesthesia. The ratio of the heart weight-to-body weight was determined as an indicator of cardiac hypertrophy. A transverse section at the base of the left ventricle cut through the cardiac valves was used for histological analyses, and the remaining ventricle tissue was isolated for molecular determinations.
In order to test the dosage of the human PAI-1 variants in mice, another 12 normal mice were injected with CPAI, AK or RR (1 μg/g body weight) intraperitoneally every 24 h for 7 days, with 4 mice for each variant. About 100 μl of blood samples were collected through retro-orbital bleeding at 4 to 24 h after the last injection, and plasma isolated for determination of human PAI-1 by ELISA (Molecular Innovations).
Histologic Assessment for Cardiac Fibrosis
Cardiac fibrosis was assessed by Masson trichrome staining on 3-μm paraffin sections and immunohistochemistry for periostin (1:50, Abcam, Cambridge, MA, USA) on 5-μm cryosections.31 Images were acquired by Nikon ECLIPSE E400 system. The percentage of positive area was analyzed by AxioVision software (Carl Zeiss, Gottingen, Germany).
Cardiac Fibroblast Culture
Primary fibroblasts from normal adult human ventricle (NHCF-V, Lonza, Allendale, NJ, USA) were cultured with conditioned fibroblast growth medium (FGM-3, Lonza) containing 10% fetal bovine serum (FBS), hFGF-B, insulin and gentamicin/amphotericin-B, and passaged at 1:3 split. Passage 5–6 cells reached 70% confluence were starved in serum-free medium for 24 h, and then incubated in the experiment medium containing 5% FBS with different PAI-1 variants (1.5 × 10−7 mol/l) with or without Ang II(5 × 10−7 mol/l). The PAI-1 variant was supplemented again at 24 h. Supernatants and cells were collected at 48 h for protein assays by ELISA or western blotting. Each of the eight groups, that is, Cont (without Ang II and any PAI-1 variants), RR, AK, CPAI, Ang + PBS, Ang + RR, Ang + AK and Ang + CPAI, was studied in triplicate.
ELISA
Plasma human PAI-1, active plasmin and total mouse PAI-1 in the heart lysate were determined by ELISA kits (Molecular Innovations). The heart tissue was homogenized in RIPA buffer with EDTA and EGTA (Boston BioProducts, Boston, MA, USA) containing Complete Proteinase Inhibitor Cocktail (Roche Diagnostics, Mannheim, Germany), and 1:5 was diluted before loading onto a microtiter plate. The assay was performed following the manufacturer's instruction. The value was corrected by the heart protein concentration.
Fibronectin in the supernatant of cultured cardiac fibroblasts was measured by QuantiMatrix™ ELISA kit (Millipore, Billerica, MA, USA).
Chromogenic Substrate Assay for Plasmin and Plasminogen Activity
Supernatant plasmin activity was measured using a plasmin-specific chromogenic substrate, Chromozym PL (Roche Molecular Biochemicals).24 Briefly, 100 μl supernatant and 20 μl of 3 mM Chromozym PL were added per well. Absorbance was measured at 405 nm at baseline and 24 h. A standard linear curve was generated with serial dilutions of human plasmin (Sigma-Aldrich, St Louis, MO, USA) ranging 0–100 × 10−4 unit/ml against the increase in the absorbance. Plasmin activity of each sample was calculated and corrected by the supernatant protein concentration. Plasminogen activator activity was measured by a similar method with additional supplement of 4 μM plasminogen (Sigma-Aldrich).
Proliferation and Apoptosis of Fibroblasts
Proliferation of cultured fibroblasts was determined by MTS assay with CellTiter 96® AQueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI, USA). Briefly, 5 × 103 cells in 100 μl conditioned fibroblast medium containing Ang II with or without PAI-1 variants were dispensed into the well and pretreated for 48 h. After adding 20 μl of CellTiter 96 AQueous One Solution Reagent to each well, the plate was incubated at 37 °C for 4 h in a humidified, 5% CO2 atmosphere. Absorbance at 490 nm was recorded.
Apoptosis of fibroblasts was detected by TUNEL method (Millipore). Briefly, 2 × 104 cells were added to each well of four-well culture slides and treated with Ang II and one of the PAI-1 variants as described above. After fixation in 1% paraformaldehyde in PBS, the slides were incubated with working strength TdT enzyme at 37 °C for 1 h, and then stop buffer was added. For each well, 10 images were acquired at x40 magnification, and the fraction of positive cells was evaluated.
Migration Assay
The relative migratory ability of fibroblasts was determined by wound-healing assay. Cells were plated onto wells of six-well plates precoated with collagen type I, and allowed to reach full confluence before a scratch was gently created with a pipet tip. The area devoid of cells was photographed at the time the scratch was created and at 9 h. The relative distance of fibroblasts migrated into the denuded area was measured. Migration experiments were performed in triplicate wells.
Cell Adhesion to Vitronectin
To prepare the culture plates for adhesion assay, 5 μg/ml of human vitronectin (BD Biosciences, Franklin Lakes, NJ, USA) was added to 96-well plate, incubated at 4 °C overnight and the remnant was aspirated. Nonspecific binding to the plate was blocked by incubation with 1% bovine serum albumin at 37 °C for 1 h. After three washes with medium containing 5% FBS, 3 × 104 cells were added to each well in the presence of Ang II and PAI-1 variants. Plates were then incubated at 37 °C for 60 min. Non-adherent cells were washed off with PBS. The attached cells were fixated with 4% paraformaldehyde/PBS for 5 min, stained with 0.5% toluidine blue in 4% paraformaldehyde for 5 min, rinsed with water and solubilized with 100 μl of 1% sodium dodecyl sulfate (SDS). Optical density was read at a wavelength of 595 nm.32
Immunoblot Analysis
Frozen heart tissues and cell samples were homogenized in RIPA buffer with EDTA and EGTA containing proteinase inhibitor. The total protein concentration in each sample was determined by BCA Protein Assay Kit (Thermo Scientific, Pittsburgh, PA, USA). Equal amounts of total protein from each sample (60 μg for heart tissues, 20 μg for cell lysate of cultured fibroblasts and 50 μg for supernatant samples) were separated on 10% SDS-polyacrylamide gel electrophoresis and electrophoretically transferred to nitrocellulose membranes. Membranes were subsequently blocked with 5% powdered non-fat milk in Tris-buffered saline containing 0.1% TWEEN 20 (TBS-T) and incubated with primary antibody at 4 °C overnight. Primary antibodies used in the present studies included anti-vitronectin (1:400, Santa Cruz), anti-Bcl-xL (1:1000, Cell Signaling Technology, Danvers, MA, USA), anti-Bax (1:1000, Cell Signaling Technology), anti-integrin β3 (1:1000, Abcam), anti-integrin αV (1:400, Santa Cruz) and anti-MMP-9 (1:800, Millipore). After washing in TBS-T, horseradish peroxidase-labeled IgG secondary antibodies (1:2500 in 5% non-fat milk/TBS-T) was added and incubated at room temperature for 1 h. Protein bands on western blots were visualized by ECL Plus (Amersham, Arlington Heights, IL, USA) then developed on film. The amount of loading was detected by using anti-tubulin (1:5000, Abcam) for tissue or cell lysate, or Ponceau S stain for culture supernatant samples. The abundance of each protein expression shown as a specific band was analyzed by ImageJ software, normalized by the loading control and expressed by fold changes relative to the Cont group.
Reverse Transcription and Quantitative Polymerase Chain Reaction
Snap-frozen heart tissue from the ventricles was homogenized in RLT buffer (Qiagen, Germany) containing 2-mercaptoethanol. The total RNA was extracted by RNeasy Mini Kit (Qiagen), then 2 μg of total RNA with A260/A280 ranging 1.8–2.3 was reverse transcribed by TaqMan Reverse Transcription Kit (Applied Biosystems, Branchburg, NJ, USA) according to the manufacturer's protocol. PCR for Col1 was performed on CFX-96 real-time PCR detection system by using SYBR green master mix (Bio-Rad). The 25 μl PCR reaction mixture contained 0.5 μl of cDNA and 1.25 μmol of each PCR primer (forward: 5′-GGCAGGC GGGAGGTCTT-3′; reverse 5′-ACGGCTGCACGAGTCAC AC-3′).33 The initial denaturation was carried out at 95 °C for 3 min, which was followed by amplification with 40 cycles of 95 °C for 20 s, 60 °C for 20 s and 72 °C for 40 s. Cycle threshold (Ct) values were normalized to 18s rRNA measured on the same plate, and fold differences in gene expression were determined using the 2−ΔΔCt method.34
Statistical Analysis
All results are presented as mean±s.e.m. Repeated-measures ANOVA and post hoc Tukey's test were used to examine the differences among groups and among the different time points. Nonparametric tests were used when data were not normally distributed. A P-value <0.05 for two-tailed tests was considered to be significant.
RESULTS
Plasma Level of Human PAI-1 after Injection with PAI-1 Variant
In mice, normal plasma level of mouse PAI-1 is <25–80 pg/ml,30 and rose to >1 ng/ml in some pathophysiologic conditions.35 In our study, CPAI, AK or RR was given intraperitoneally at a daily dose of 1 μg/g body weight. After 1 week, the mean plasma level of human PAI-1 at 4 h after each injection was 3.2 ng/ml for CPAI, 0.6 ng/ml for AK and 1.2 ng/ml for RR. Those levels were decreased and stabilized after 10 h of injection, ranging 0.15–0.53 ng/ml. There was no significant difference in the plasma human PAI-1 level among CPAI, AK and RR within 10–24 h after injection. The human PAI-1 was not detectable in mice without injection of human PAI-1 variants.
Effects of PAI-1 Variants on SBP, Proteinuria and Cardiac Fibrosis
The body weight of mice did not change significantly throughout the study, and was not significantly different among the groups. Retrospective calculation for the actual amount of Ang II received, based on the body weight and remaining Ang II in the pump, was also similar among the groups, ranging 0.99–1.08 ng/min/g. SBP and heart weight were significantly increased by Ang II infusion in mice with UNx/salt. The various PAI-1 variants did not impact the hypertension or cardiac hypertrophy (Table 1). UACR was significantly increased by Ang II infusion. Compared with the vitronectin-binding PAI-1 variants, RR and CPAI, the non-vitronectin-binding PAI-1 variant, AK, significantly increased UACR (Table 1). Compared with the UNx/salt control mice, Ang II significantly increased cardiac fibrosis, which was further exacerbated by the non-vitronectin-binding PAI-1 variant, AK (Figure 1a). These changes paralleled the results of the periostin staining for fibroblast (Figure 1a) and the mRNA level of collagen type I (Col1) in the ventricles (Figure 1b). Both vitronectin-binding PAI-1 variants, that is, RR without protease inhibition and CPAI with protease inhibition, significantly attenuated the upregulation of periostin and Col1, and reduced cardiac fibrosis compared with the AK.
Table 1.
Effects of different PAI-1 variants on blood pressure, albuminuria and cardiac hypertrophy in mice with Ang II/UNx/salt
| Cont | Ang+PBS | Ang+RR | Ang+AK | Ang II+CPAI | |
|---|---|---|---|---|---|
| N | 4 | 9 | 9 | 10 | 9 |
| Systolic blood pressure (mm Hg) | |||||
| Week 0 | 112±5 | 109±4 | 113±4 | 111±3 | 108±2 |
| Week 2 | 109±5 | 128±6 | 150±5a | 135±7 | 141±5a |
| Week 4 | 102±4 | 157±7a | 149±8a | 166±8a | 162±10a |
| Week 6 | 117±3 | 144±11 | 153±5a | 163±8a | 138±7 |
| Week 8 | 112±6 | 163±12a | 164±6a | 161±7a | 144±4a |
| Urine albumin-to-creatinine ratio (μg/mg) | |||||
| Week 0 | 22.0±7.5 | 14.7±4.2 | 9.2±1.8 | 13.5±4.7 | 38.2±30.5 |
| Week 2 | 6.1±1.2 | 1060.6±5.9a | 522.8±270.4a | 1265.6±358.9a,b,c | 354.3±118.1a |
| Week 4 | 33.9±12.0 | 1117.1±475.9a | 961.0±334.8a | 1618.3±347.0a,b | 496.5±186.4a |
| Week 6 | 28.1±12.9 | 1528.8±1156.4a | 1050.5±390.9a | 2048.0±573.7a | 982.4±305.7a |
| Week 8 | 29.5±22.6 | 1595.3±752.5a | 1165.8±324.1a | 2712.6±573.6a,c | 1365.4±388.9a |
| Heart-to-body weight (mg/g) | |||||
| Week 8 | 5.5±0.2 | 9.8±1.1a | 8.8±0.4a | 8.4±0.5a | 9.2±1.0a |
| Heart rate (beat/min) | |||||
| Week 8 | 685±15 | 637±19 | 656±14 | 639±14 | 671±9 |
Abbreviations: Ang, angiotensin II; AK, PAI-1AK variant; Cont, UNx/salt control; CPAI, control PAI-1 variant; RR, PAI-1RR variant; UNx, uninephrectomy.
Data are shown as mean±s.e.m.
vs Cont.
vs Ang+CPAI.
vs Ang+RR, P<0.05 compared at the same time point.
Figure 1.

Cardiac fibrosis in Ang II/UNx/salt mice treated with PAI-1 variants. (a) Masson trichrome stain (upper panel) showed that, compared with control mice with uninephrectomy and high salt diet (Cont), superimposing chronic infusion of Ang II and PBS (Ang + PBS) increased cardiac fibrosis (shown in blue). The Ang II/UNx/salt mice injected with AK (Ang + AK), the PAI-1 variant without binding capacity to vitronectin, had significantly more cardiac fibrosis than mice injected with RR (Ang + RR), the PAI-1 variant with vitronectin-binding ability but without protease inhibition or CPAI (Ang + CPAI), the control PAI-1 variant with both vitronectin-binding and protease inhibitory capacities. Immunofluorescent staining for periostin, a cardiac fibroblast marker (middle panel, shown in green), showed a significant increase in fibroblasts by Ang II compared with Cont, which was exacerbated by AK and attenuated by RR. (b) The histological changes shown in A were also reflected by the mRNA level of collagen type I (Col1) in the ventricles, which was significantly increased in all Ang II-infused mice and further surged in the Ang + AK group. The RR and CPAI variants attenuated this upregulation. *vs Cont, # vs Ang + PBS, † vs Ang + CPAI, ‡ vs Ang + RR, P <0.05. Scale bar = 50 μm.
Changes in Cardiac PAI-1, Plasmin and Vitronectin
ELISA showed that the non-vitronectin-binding PAI-1 variant, AK, significantly increased total mouse PAI-1 protein in the heart compared with its protease inhibitory control variant, CPAI, which also binds vitronectin. As expected, mice injected with protease inhibitory AK and CPAI, numerically lowered active plasmin in the heart compared with the mice treated with non-protease inhibitory RR and the UNx/Salt Cont. Although immunoblotting showed that Ang II significantly increased cardiac vitronectin, this change was numerically alleviated by the vitronectin-binding PAI-1 variants RR and CPAI (Table 2).
Table 2.
Cardiac PAI-1, plasmin and vitronectin in mice with Ang II/UNx/salt and PAI-1 variants
| Cont | Ang+PBS | Ang+RR | Ang+AK | Ang+CPAI | |
|---|---|---|---|---|---|
| N | 4 | 9 | 9 | 10 | 9 |
| PAI-1 (ng/mg) | 0.73±0.06 | 1.10±0.18 | 1.17±0.19a | 1.40±0.23a,b | 0.93±0.09 |
| Active plasmin (ng/mg) | 6.19±3.41 | 3.60±0.88 | 4.84±0.70 | 3.31±0.38 | 3.68±0.83 |
| Vitronectin (a.u.) | 0.21±0.02 | 0.31±0.01a | 0.24±0.03 | 0.27±0.05 | 0.22±0.07 |
Abbreviations: Ang, angiotensin II; AK, PAI-1AK variant; a.u., arbitrary unit by western blotting; Cont, UNx/salt control; CPAI, control PAI-1 variant; RR, PAI-1RR variant; UNx, uninephrectomy.
Data are shown as mean±s.e.m.
vs Cont.
vs Ang+CPAI, P<0.05.
Effects of PAI-1 Variants on Plasminogen Activator Activity, Plasmin Activity and Fibronectin in the Supernatant of Cultured Cardiac Fibroblasts
Without Ang II, the RR variant significantly increased plasminogen activator activity in the supernatant, and RR and CPAI increased plasmin activity compared with the Cont. The plasminogen activator activity was significantly lower in the supernatant of AK- and CPAI-treated fibroblasts than that of RR (Figure 2a). With Ang II, AK and CPAI significantly reduced plasmin generation compared with RR. The supernatant level of MMP-9 was not significantly different in non-Ang II-exposed cells, but significantly increased by vitronectin-binding variants, RR and CPAI in Ang II-exposed fibroblasts compared with those of Cont, Ang + PBS and Ang + AK (Figure 2b).
Figure 2.

Effects of PAI-1 variants on plasmin and fibronectin in the supernatant of primary cardiac fibroblasts. (a) Chromogenic substrate assay: compared with the control fibroblasts (Cont), non-protease inhibitory but vitronectin-binding PAI-1 variant, RR, significantly increased plasminogen activator activity, which was also significantly higher than that of AK or CPAI. RR and CPAI variant also increased plasmin activity in the supernatant. Addition of AK or CPAI to Ang II-exposed cardiac fibroblasts (Ang + AK, or Ang + CPAI) significantly decreased plasminogen activator activity and plasmin activity compared with RR (Ang + RR). (b) Western blot: the supernatant level of MMP-9 was not significantly changed by different PAI-1 variant at baseline, but significantly increased by vitronectin-binding RR and CPAI in Ang II-exposed fibroblasts. (c) ELISA: different PAI-1 variant did not change supernatant fibronectin level at baseline. Ang II numerically increased supernatant fibronectin, which was significantly increased by AK or CPAI in Ang II-exposed fibroblasts. These results suggest that protease inhibition by PAI-1 has a significant role in extracellular matrix accumulation, which may be protected by vitronectin-bound PAI-1. * vs Cont, Δ vs RR, # vs Ang + PBS, ‡ vs Ang + CPAI, ‡ vs Ang + RR, P <0.05. N = 3 for each group.
In non-Ang II-exposed fibroblasts, PAI-1 variants did not significantly change the level of fibronectin in the supernatant. Although Ang II did not significantly increased fibronectin production in the primary cardiac fibroblasts indicating a phenotype of fibroblasts rather than myofibroblasts, addition of AK or CPAI significantly increased supernatant fibronectin (Figure 2c). These results also indicate that the differential effects of PAI-1 variants on fibroblasts are at least partially dependent of Ang II.
Effects of PAI-1 Variants on Vitronectin, αVβ3 Integrin in Ang II-Exposed Cardiac Fibroblasts and Adhesion of Fibroblasts to Vitronectin
Immunoblotting showed that, compared with AK, RR treatment significantly decreased vitronectin level in the supernatant of Ang II-exposed fibroblasts, indicating reduced vitronectin availability for interaction with αVβ3 integrin. Although immunoblotting did not show any difference in protein expression of the αV integrin (Cont 0.36±0.09, Ang + PBS 0.37±0.04, Ang + RR 0.27±0.12, Ang + AK 0.26±0.08 and Ang + CPAI 0.25±0.01), the β3 integrin was significantly higher in the AK-treated than in the RR-treated fibroblasts exposed to Ang II, indicating that binding of PAI-1 to vitronectin downregulates cellular expression of β3 integrin. The adhesion of Ang II-exposed fibroblasts to vitronectin was significantly inhibited by vitronectin-binding RR and CPAI but not by the AK variant, which only has protease inhibitory effects (Figure 3). These results suggested decreased formation of vitronectin–αVβ3 integrin complex by the vitronectin-binding PAI-1.
Figure 3.

Changes in integrin β3 expression, supernatant vitronectin and adhesion of cardiac fibroblasts to vitronectin. In cardiac fibroblasts exposed to Ang II, non-vitronectin-binding AK (Ang + AK) significantly upregulated cellular expression of integrin β3 (western blot, a) compared with Ang II alone (Ang + PBS) and the vitronectin-binding PAI-1 variants, RR (Ang + RR) and CPAI (Ang + CPAI). This AK-caused increase in integrin β3 was accompanied by higher level of supernatant vitronectin than with RR treatment (western blot, b) and greater adhesion to vitronectin than those in response to RR and CPAI (c). These results suggest a decreased vitronectin–αVβ3 interaction by vitronectin-bound PAI-1. * vs Cont, # vs Ang + PBS, † vs Ang + CPAI, † vs Ang + RR, P <0.05. N = 3 for each group.
Effects of PAI-1 Variants on Apoptosis and Proliferation of Ang II-Exposed Cardiac Fibroblasts
The number of TUNEL-positive apoptotic fibroblasts was numerically lower in Ang II-exposed fibroblasts than Cont. Compared with Cont and Ang + PBS, addition of RR and CPAI did not significantly affect the number of apoptotic cells. However, the number of apoptotic fibroblasts was significantly reduced by non-vitronectin-binding AK compared with that of Ang + PBS and Ang + RR. Expression of pro-apoptotic Bax was not significantly different among the Cont and Ang II groups; however, expression of anti-apoptotic Bcl-xL was significantly decreased by the vitronectin-binding RR and CPAI compared with the Ang + PBS, but not by AK (Figure 4). These results indicate that binding of PAI-1 to vitronectin protects apoptotic activity of fibroblasts in response to Ang II.
Figure 4.

Apoptosis of Ang II-exposed cardiac fibroblast. Treatment with non-vitronectin-binding AK significantly reduced apoptosis, suggesting proapoptotic effects of PAI-1 by binding to vitronectin on Ang II-exposed cardiac fibroblasts (TUNEL, a). This effect was further shown by significantly downregulated Bcl-xL, an anti-apoptotic marker, but not pro-apoptotic Bax, in vitronectin-binding RR and CPAI-treated fibroblasts (western blot, b). These results indicate that the pro-apoptotic effect of vitronectin-binding PAI-1 involves decreased activity of anti-apoptotic factors. * vs Cont, # vs Ang + PBS, ‡ vs Ang + RR, P <0.05. N = 3 for each group.
Although Ang II did not increase proliferation of cultured fibroblasts, vitronectin-binding RR and CPAI significantly decreased the number of viable fibroblasts that were exposed to Ang II (Figure 5).
Figure 5.
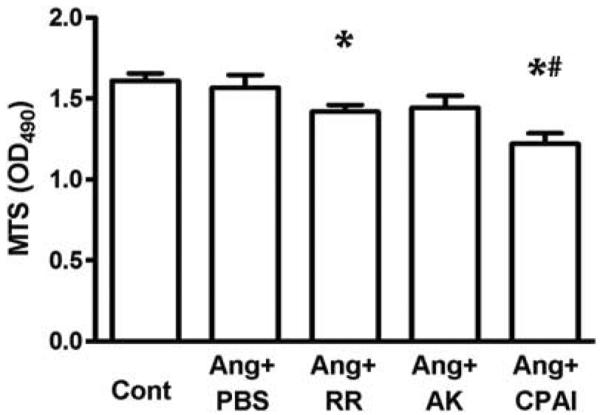
Proliferation of cardiac fibroblasts (MTS assay). Proliferation of Ang II-exposed cardiac fibroblasts was significantly decreased by vitronectin-binding PAI-1 variants, RR and CPAI, but not by AK, which only inhibits protease activity. * vs Cont, # vs Ang + PBS, P <0.05. N = 3 for each group.
Effects of PAI-1 Variants on Migration of Ang II-Exposed Cardiac Fibroblasts
Compared with fibroblasts incubated with Ang II alone, protease inhibitory AK and CPAI significantly increased fibroblast migration determined by wound-healing assay. RR-treated fibroblasts migrated into the scratched region significantly more slowly than fibroblasts incubated with Ang II alone, or treated with Ang II with AK or CPAI (Figure 6).
Figure 6.

Migration of Ang II-treated cardiac fibroblasts (wound-healing assay). Compared with Ang II treatment alone, protease inhibitory AK and CPAI significantly increased cardiac fibroblast migration, whereas vitronectin-binding RR without protease inhibition significantly decreased migration. These results indicate that the migration of cardiac fibroblasts is impacted by both protease inhibition and vitronectin-binding of PAI-1. * vs Cont, # vs Ang + PBS, ‡ vs Ang + RR, P <0.05. N = 3 for each group.
DISCUSSION
Our in vivo results show that cardiac fibrosis caused by chronic infusion of Ang II was exacerbated by a protease inhibitory but non-vitronectin-binding PAI-1 variant, the AK variant. However, development of cardiac fibrosis induced by Ang II was attenuated by vitronectin-binding PAI variants, RR and CPAI, no matter whether it inhibits activation of plasmin (CPAI) or not (RR). In vitro studies on Ang II-exposed cardiac fibroblasts revealed that protease-inhibiting PAI-1 variants, AK and CPAI, increased supernatant fibronectin. On the other hand, fibroblasts treated with vitronectin-binding PAI-1 variants, RR and CPAI, had higher expression level of MMP-9, as well as greater apoptotic and fewer proliferative activities than the cells treated with non-vitronectin-binding AK variant. Further, expression of integrin β3 and cellular adhesion to vitronectin were significantly lower in RR-treated fibroblasts compared with the AK, indicating that the protective effects of vitronectin-binding PAI-1 involves reduced vitronectin–integrin αVβ3 reactions in fibroblasts.
Divergent roles of PAI-1 in development of fibrosis are associated with the complexity of PAI-1 effects on inflammation, cellular adhesion, migration, proliferation and survival, which differ in varying pathological conditions.1,8,9,24,30,36–38 The protective or injurious effects of PAI-1 are mediated not only by its inhibition of plasmin/protease system, but also by PAI-1:vitronectin binding that affects interactions with cell-surface integrins.17–19 Indeed, recent studies suggest that the pro-fibrotic effects of PAI-1 may be independent of plasmin activity.39,40 Vitronectin is an abundant adhesive extracellular glycoprotein that has binding cites for PAI-1, urokinase-type plasminogen activator receptor (uPAR), αVβ3 and αVβ5 integrins. Dynamic binding with these factors regulates many pathophysiological processes, such as hemostasis, wound healing, angiogenesis, tissue remodeling and tumor metastasis.41–46 The vitronectin-bound PAI-1 competes for uPAR and integrins binding to vitronectin, thereby inhibiting uPAR- or integrin-mediated downstream cellular effects such as proliferation, adhesion and migration.46–48 Results of our in vivo studies showed that supplement of vitronectin-binding PAI-1 variants attenuated fibrosis markers, whereas the variant without vitronectin-binding capacity but with protease inhibitory activity significantly exacerbated cardiac fibrosis. These results are the first to differentiate the vitronectin-binding effects of PAI-1 from its protease-inhibitory effects on cardiac fibrosis. Binding to vitronectin also stabilizes PAI-1 from cleavage in locations of injury areas.49,50 In our study, the vitronectin level in the heart was not significantly different among the groups with different PAI-1 variants. However, AK significantly increased endogenous mouse PAI-1 level in the heart of Ang II-infused mice, possibly because of more binding to vitronectin and less cleavage of native mouse PAI-1 than those in RR- or CPAI-treated mice.
Cardiac fibroblasts are the predominant sources of the extracellular matrix protein in the heart.12–15,51 Marked phenotypic alterations in fibroblast adhesion and migration have been correlated with myocardial disease.52–55 Cardiac fibroblasts can also secrete vitronectin and express vitronectin receptor αVβ3 integrin.20,56 Ang II increases adhesion of cardiac fibroblasts to the extracellular matrix, which is associated with upregulated integrins αV and β3.21 Integrin β3−/− cardiac fibroblasts exhibited a significant reduction in cell-matrix adhesion, cell spreading, proliferation and migration.57 In our study with primary cardiac fibroblasts that were exposed to Ang II, the levels of supernatant vitronectin and αVβ3 integrin expression were significantly reduced by the non-protease-inhibiting but vitronectin-binding RR variant, indicating decreased vitronectin–integrin αVβ3 interaction by vitronectin-binding PAI-1.58 We also found that vitronectin-binding PAI-1 variants prevented the adhesion of fibroblasts, whereas the non-vitronectin-binding AK variant did not. This result is consistent with other studies on different cell types.59
Ang II promotes fibroblast proliferation and infiltration, as well as increased PAI-1 level in the heart. Vitronectin is a key determinant of the anti-proliferative effects of PAI-1.60 PAI-1 binding to vitronectin on vascular cells also correlates with apoptosis,61 and vitronectin–integrin complex is linked to prevention of cultured cells from undergoing apoptosis.62,63 Our in vitro study shows that vitronectin-binding PAI-1 variants caused more apoptosis but less proliferation in Ang II-exposed cardiac fibroblasts. These findings complement the in vivo findings of fewer fibroblasts in the heart with the vitronectin-binding variants.
PAI-1 inhibits cell migration through its vitronectin-binding19,64–66 and plasmin-inhibiting effects.67–69 Our in vitro study supported that the inhibitory effects of PAI-1 on cell migration through the vitronectin-binding mechanisms, as the vitronectin-binding only variant, RR, significantly reduced the migration of cardiac fibroblasts. However, the protease inhibitory PAI-1 variants, AK and CPAI, increased cardiac fibroblast migration. This may also be influenced by the decrease in apoptosis, and/or the increase in proliferation, thus promoting increased cellularity in the cultured fibroblasts.
Although the protective effects of RR may be partially through preservation of plasmin activity that reflects competition with endogenous native PAI-1 to inhibit proteases,25 similar effects observed with the CPAI variant, which has both protease-inhibiting and vitronectin-binding capacities indicate a predominant role of vitronectin binding-mediated protection. In align with this, a most recent study with cultured primary human corneal fibroblasts revealed that vitronectin-bound PAI-1 protected against uPA-induced fibroblast-to-myofibroblast transdifferentiation was independent of plasmin activity.70
Ang II can induce PAI-1 expression in various cell types, including not only fibroblasts, but also endothelial cells, myocytes, vascular smooth muscle cells, etc. Compared with the wild-type, genetic vitronectin-deficient mice have delayed wound healing,71 or similar renal interstitial fibrosis induced by ureteral obstruction.71,72 In addition, cardiac fibroblasts are not unique sources of extracellular matrix and PAI-1 production. These notions prompt future studies using inducible tissue- or cell-specific PAI-1 expression models.
In summary, we used different PAI-1 variants to investigate the relative contributions of protease-inhibiting and vitronectin-binding pathways of PAI-1 in cardiac fibrosis. Our in vivo and in vitro data showed protective effects of vitronectin-binding PAI-1 against development of Ang II-induced cardiac fibrosis. By binding with vitronectin, PAI-1 may block integrin αVβ3 action, induce apoptosis, and suppress proliferation, adhesion and migration of cardiac fibroblasts. Our study results suggest therapeutic implications of vitronectin-binding PAI-1 for cardiac fibrosis.
ACKNOWLEDGMENTS
This study is supported by National Institute of Diabetes and Digestive and Kidney Diseases (DK037868 and DK044757), National Institute of Child Health and Human Development (R21HD069935) and American Heart Association (09BGIA2261364).
Footnotes
DISCLOSURE/CONFLICT OF INTEREST The authors declare no conflict of interest.
References
- 1.Ha H, Oh EY, Lee HB. The role of plasminogen activator inhibitor 1 in renal and cardiovascular diseases. Nat Rev Nephrol. 2009;5:203–211. doi: 10.1038/nrneph.2009.15. [DOI] [PubMed] [Google Scholar]
- 2.Thogersen AM, Jansson JH, Boman K, et al. High plasminogen activator inhibitor and tissue plasminogen activator levels in plasma precede a first acute myocardial infarction in both men and women: evidence for the fibrinolytic system as an independent primary risk factor. Circulation. 1998;98:2241–2247. doi: 10.1161/01.cir.98.21.2241. [DOI] [PubMed] [Google Scholar]
- 3.Zhang LP, Takahara T, Yata Y, et al. Increased expression of plasminogen activator and plasminogen activator inhibitor during liver fibrogenesis of rats: role of stellate cells. J Hepatol. 1999;31:703–711. doi: 10.1016/s0168-8278(99)80351-1. [DOI] [PubMed] [Google Scholar]
- 4.Olman MA, Mackman N, Gladson CL, et al. Changes in procoagulant and fibrinolytic gene expression during bleomycin-induced lung injury in the mouse. J Clin Invest. 1995;96:1621–1630. doi: 10.1172/JCI118201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takeshita K, Hayashi M, Iino S, et al. Increased expression of plasminogen activator inhibitor-1 in cardiomyocytes contributes to cardiac fibrosis after myocardial infarction. Am J Pathol. 2004;164:449–456. doi: 10.1016/S0002-9440(10)63135-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaman AK, Fujii S, Schneider DJ, et al. Deleterious effects of lack of cardiac PAI-1 after coronary occlusion in mice and their pathophysiologic determinants. Histochem Cell Biol. 2007;128:135–145. doi: 10.1007/s00418-007-0300-z. [DOI] [PubMed] [Google Scholar]
- 7.Moriwaki H, Stempien-Otero A, Kremen M, et al. Overexpression of urokinase by macrophages or deficiency of plasminogen activator inhibitor type 1 causes cardiac fibrosis in mice. Circ Res. 2004;95:637–644. doi: 10.1161/01.RES.0000141427.61023.f4. [DOI] [PubMed] [Google Scholar]
- 8.Ma J, Weisberg A, Griffin JP, et al. Plasminogen activator inhibitor-1 deficiency protects against aldosterone-induced glomerular injury. Kidney Int. 2006;69:1064–1072. doi: 10.1038/sj.ki.5000201. [DOI] [PubMed] [Google Scholar]
- 9.Weisberg AD, Albornoz F, Griffin JP, et al. Pharmacological inhibition and genetic deficiency of plasminogen activator inhibitor-1 attenuates angiotensin II/salt-induced aortic remodeling. Arterioscler Thromb Vasc Biol. 2005;25:365–371. doi: 10.1161/01.ATV.0000152356.85791.52. [DOI] [PubMed] [Google Scholar]
- 10.Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010;106:1675–1680. doi: 10.1161/CIRCRESAHA.110.217737. [DOI] [PubMed] [Google Scholar]
- 11.Brown RD, Ambler SK, Mitchell MD, et al. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–687. doi: 10.1146/annurev.pharmtox.45.120403.095802. [DOI] [PubMed] [Google Scholar]
- 12.Baudino TA, Carver W, Giles W, et al. Cardiac fibroblasts: friend or foe? Am J Physiol Heart Circ Physiol. 2006;291:H1015–H1026. doi: 10.1152/ajpheart.00023.2006. [DOI] [PubMed] [Google Scholar]
- 13.Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–278. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 14.Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009;105:1164–1176. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010;225:631–637. doi: 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loskutoff DJ, Sawdey M, Mimuro J. Type 1 plasminogen activator inhibitor. Prog Hemost Thromb. 1989;9:87–115. [PubMed] [Google Scholar]
- 17.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 18.Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- 19.Stefansson S, Lawrence DA. The serpin PAI-1 inhibits cell migration by blocking integrin alpha V beta 3 binding to vitronectin. Nature. 1996;383:441–443. doi: 10.1038/383441a0. [DOI] [PubMed] [Google Scholar]
- 20.Graf K, Neuss M, Stawowy P, et al. Angiotensin II and alpha(v)beta(3) integrin expression in rat neonatal cardiac fibroblasts. Hypertension. 2000;35:978–984. doi: 10.1161/01.hyp.35.4.978. [DOI] [PubMed] [Google Scholar]
- 21.Kawano H, Cody RJ, Graf K, et al. Angiotensin II enhances integrin and alpha-actinin expression in adult rat cardiac fibroblasts. Hypertension. 2000;35(1 Pt 2):273–279. doi: 10.1161/01.hyp.35.1.273. [DOI] [PubMed] [Google Scholar]
- 22.Kawano H, Do YS, Kawano Y, et al. Angiotensin II has multiple profibrotic effects in human cardiac fibroblasts. Circulation. 2000;101:1130–1137. doi: 10.1161/01.cir.101.10.1130. [DOI] [PubMed] [Google Scholar]
- 23.Huang Y, Border WA, Yu L, et al. A PAI-1 mutant, PAI-1R, slows progression of diabetic nephropathy. J Am Soc Nephrol. 2008;19:329–338. doi: 10.1681/ASN.2007040510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Y, Haraguchi M, Lawrence DA, et al. A mutant, noninhibitory plasminogen activator inhibitor type 1 decreases matrix accumulation in experimental glomerulonephritis. J Clin Invest. 2003;112:379–388. doi: 10.1172/JCI18038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Y, Border WA, Lawrence DA, et al. Noninhibitory PAI-1 enhances plasmin-mediated matrix degradation both in vitro and in experimental nephritis. Kidney Int. 2006;70:515–522. doi: 10.1038/sj.ki.5000353. [DOI] [PubMed] [Google Scholar]
- 26.Xu Z, Balsara RD, Gorlatova NV, et al. Conservation of critical functional domains in murine plasminogen activator inhibitor-1. J Biol Chem. 2004;279:17914–17920. doi: 10.1074/jbc.M314197200. [DOI] [PubMed] [Google Scholar]
- 27.Berkenpas MB, Lawrence DA, Ginsburg D. Molecular evolution of plasminogen activator inhibitor-1 functional stability. Embo J. 1995;14:2969–2977. doi: 10.1002/j.1460-2075.1995.tb07299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jensen JK, Gettins PG. High-resolution structure of the stable plasminogen activator inhibitor type-1 variant 14-1B in its proteinase-cleaved form: a new tool for detailed interaction studies and modeling. Protein Sci. 2008;17:1844–1849. doi: 10.1110/ps.036707.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang Y, Border WA, Lawrence DA, et al. Mechanisms underlying the antifibrotic properties of noninhibitory PAI-1 (PAI-1R) in experimental nephritis. Am J Physiol Renal Physiol. 2009;297:F1045–F1054. doi: 10.1152/ajprenal.00024.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Courey AJ, Horowitz JC, Kim KK, et al. The vitronectin-binding function of PAI-1 exacerbates lung fibrosis in mice. Blood. 2011;118:2313–2321. doi: 10.1182/blood-2010-12-324574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stansfield WE, Andersen NM, Tang RH, et al. Periostin is a novel factor in cardiac remodeling after experimental and clinical unloading of the failing heart. Ann Thorac Surg. 2009;88:1916–1921. doi: 10.1016/j.athoracsur.2009.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liaw L, Almeida M, Hart CE, et al. Osteopontin promotes vascular cell adhesion and spreading and is chemotactic for smooth muscle cells in vitro. Circ Res. 1994;74:214–224. doi: 10.1161/01.res.74.2.214. [DOI] [PubMed] [Google Scholar]
- 33.Kenyon NJ, Ward RW, McGrew G, et al. TGF-beta1 causes airway fibrosis and increased collagen I and III mRNA in mice. Thorax. 2003;58:772–777. doi: 10.1136/thorax.58.9.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 35.Schafer K, Muller K, Hecke A, et al. Enhanced thrombosis in atherosclerosis-prone mice is associated with increased arterial expression of plasminogen activator inhibitor-1. Arterioscler Thromb Vasc Biol. 2003;23:2097–2103. doi: 10.1161/01.ATV.0000097766.36623.DF. [DOI] [PubMed] [Google Scholar]
- 36.Hertig A, Berrou J, Allory Y, et al. Type 1 plasminogen activator inhibitor deficiency aggravates the course of experimental glomerulonephritis through overactivation of transforming growth factor beta. Faseb J. 2003;17:1904–1906. doi: 10.1096/fj.03-0084fje. [DOI] [PubMed] [Google Scholar]
- 37.Otsuka G, Agah R, Frutkin AD, et al. Transforming growth factor beta 1 induces neointima formation through plasminogen activator inhibitor-1-dependent pathways. Arterioscler Thromb Vasc Biol. 2006;26:737–743. doi: 10.1161/01.ATV.0000201087.23877.e1. [DOI] [PubMed] [Google Scholar]
- 38.Gharaee-Kermani M, Hu B, Phan SH, et al. The role of urokinase in idiopathic pulmonary fibrosis and implication for therapy. Expert Opin Investig Drugs. 2008;17:905–916. doi: 10.1517/13543784.17.6.905. [DOI] [PubMed] [Google Scholar]
- 39.Oda T, Jung YO, Kim HS, et al. PAI-1 deficiency attenuates the fibrogenic response to ureteral obstruction. Kidney Int. 2001;60:587–596. doi: 10.1046/j.1523-1755.2001.030002587.x. [DOI] [PubMed] [Google Scholar]
- 40.Heymans S, Pauschinger M, De Palma A, et al. Inhibition of urokinase-type plasminogen activator or matrix metalloproteinases prevents cardiac injury and dysfunction during viral myocarditis. Circulation. 2006;114:565–573. doi: 10.1161/CIRCULATIONAHA.105.591032. [DOI] [PubMed] [Google Scholar]
- 41.Schar CR, Blouse GE, Minor KH, et al. A deletion mutant of vitronectin lacking the somatomedin B domain exhibits residual plasminogen activator inhibitor-1-binding activity. J Biol Chem. 2008;283:10297–10309. doi: 10.1074/jbc.M708017200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waltz DA, Natkin LR, Fujita RM, et al. Plasmin and plasminogen activator inhibitor type 1 promote cellular motility by regulating the interaction between the urokinase receptor and vitronectin. J Clin Invest. 1997;100:58–67. doi: 10.1172/JCI119521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Izumi M, Yamada KM, Hayashi M. Vitronectin exists in two structurally and functionally distinct forms in human plasma. Biochim Biophys Acta. 1989;990:101–108. doi: 10.1016/s0304-4165(89)80019-4. [DOI] [PubMed] [Google Scholar]
- 44.Cheresh DA, Spiro RC. Biosynthetic and functional properties of an Arg-Gly-Asp-directed receptor involved in human melanoma cell attachment to vitronectin, fibrinogen, and von Willebrand factor. J Biol Chem. 1987;262:17703–17711. [PubMed] [Google Scholar]
- 45.Smith JW, Vestal DJ, Irwin SV, et al. Purification and functional characterization of integrin alpha v beta 5. An adhesion receptor for vitronectin. J Biol Chem. 1990;265:11008–11013. [PubMed] [Google Scholar]
- 46.Deng G, Curriden SA, Hu G, et al. Plasminogen activator inhibitor-1 regulates cell adhesion by binding to the somatomedin B domain of vitronectin. J Cell Physiol. 2001;189:23–33. doi: 10.1002/jcp.1133. [DOI] [PubMed] [Google Scholar]
- 47.Stefansson S, Lawrence DA. Old dogs and new tricks: proteases, inhibitors, and cell migration. Sci STKE. 2003;2003:pe24. doi: 10.1126/stke.2003.189.pe24. [DOI] [PubMed] [Google Scholar]
- 48.Okumura Y, Kamikubo Y, Curriden SA, et al. Kinetic analysis of the interaction between vitronectin and the urokinase receptor. J Biol Chem. 2002;277:9395–9404. doi: 10.1074/jbc.M111225200. [DOI] [PubMed] [Google Scholar]
- 49.Stoop AA, Lupu F, Pannekoek H. Colocalization of thrombin, PAI-1, and vitronectin in the atherosclerotic vessel wall: a potential regulatory mechanism of thrombin activity by PAI-1/vitronectin complexes. Arterioscler Thromb Vasc Biol. 2000;20:1143–1149. doi: 10.1161/01.atv.20.4.1143. [DOI] [PubMed] [Google Scholar]
- 50.Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. J Cell Physiol. 2012;227:493–507. doi: 10.1002/jcp.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsuruda T, Imamura T, Hatakeyama K, et al. Stromal cell biology—a way to understand the evolution of cardiovascular diseases. Circ J. 2010;74:1042–1050. doi: 10.1253/circj.cj-10-0024. [DOI] [PubMed] [Google Scholar]
- 52.Burgess ML, Terracio L, Hirozane T, et al. Differential integrin expression by cardiac fibroblasts from hypertensive and exercise-trained rat hearts. Cardiovasc Pathol. 2002;11:78–87. doi: 10.1016/s1054-8807(01)00104-1. [DOI] [PubMed] [Google Scholar]
- 53.Marganski WA, De Biase VM, Burgess ML, et al. Demonstration of altered fibroblast contractile activity in hypertensive heart disease. Cardiovasc Res. 2003;60:547–556. doi: 10.1016/j.cardiores.2003.09.021. [DOI] [PubMed] [Google Scholar]
- 54.Squires CE, Escobar GP, Payne JF, et al. Altered fibroblast function following myocardial infarction. J Mol Cell Cardiol. 2005;39:699–707. doi: 10.1016/j.yjmcc.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 55.Flack EC, Lindsey ML, Squires CE, et al. Alterations in cultured myocardial fibroblast function following the development of left ventricular failure. J Mol Cell Cardiol. 2006;40:474–483. doi: 10.1016/j.yjmcc.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 56.Neuss M, Regitz-Zagrosek V, Hildebrandt A, et al. Isolation and characterisation of human cardiac fibroblasts from explanted adult hearts. Cell Tissue Res. 1996;286:145–153. doi: 10.1007/s004410050683. [DOI] [PubMed] [Google Scholar]
- 57.Balasubramanian S, Quinones L, Kasiganesan H, et al. Beta3 integrin in cardiac fibroblast is critical for extracellular matrix accumulation during pressure overload hypertrophy in mouse. PLoS One. 2012;7:e45076. doi: 10.1371/journal.pone.0045076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hertig A, Rondeau E. Plasminogen activator inhibitor type 1: the two faces of the same coin. Curr Opin Nephrol Hypertens. 2004;13:39–44. doi: 10.1097/00041552-200401000-00006. [DOI] [PubMed] [Google Scholar]
- 59.Stefansson S, Su EJ, Ishigami S, et al. The contributions of integrin affinity and integrin-cytoskeletal engagement in endothelial and smooth muscle cell adhesion to vitronectin. J Biol Chem. 2007;282:15679–15689. doi: 10.1074/jbc.M702125200. [DOI] [PubMed] [Google Scholar]
- 60.Wu J, Peng L, McMahon GA, et al. Recombinant plasminogen activator inhibitor-1 inhibits intimal hyperplasia. Arterioscler Thromb Vasc Biol. 2009;29:1565–1570. doi: 10.1161/ATVBAHA.109.189514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Al-Fakhri N, Chavakis T, Schmidt-Woll T, et al. Induction of apoptosis in vascular cells by plasminogen activator inhibitor-1 and high molecular weight kininogen correlates with their anti-adhesive properties. Biol Chem. 2003;384:423–435. doi: 10.1515/BC.2003.048. [DOI] [PubMed] [Google Scholar]
- 62.Meredith JE, Jr, Fazeli B, Schwartz MA. The extracellular matrix as a cell survival factor. Mol Biol Cell. 1993;4:953–961. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Felding-Habermann B, Mueller BM, Romerdahl CA, et al. Involvement of integrin alpha V gene expression in human melanoma tumorigenicity. J Clin Invest. 1992;89:2018–2022. doi: 10.1172/JCI115811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Redmond EM, Cullen JP, Cahill PA, et al. Endothelial cells inhibit flow-induced smooth muscle cell migration: role of plasminogen activator inhibitor-1. Circulation. 2001;103:597–603. doi: 10.1161/01.cir.103.4.597. [DOI] [PubMed] [Google Scholar]
- 65.Proia RR, Nelson PR, Mulligan-Kehoe MJ, et al. The effect of endothelial cell overexpression of plasminogen activator inhibitor-1 on smooth muscle cell migration. J Vasc Surg. 2002;36:164–171. doi: 10.1067/mva.2002.123687. [DOI] [PubMed] [Google Scholar]
- 66.Degryse B, Neels JG, Czekay RP, et al. The low density lipoprotein receptor-related protein is a motogenic receptor for plasminogen activator inhibitor-1. J Biol Chem. 2004;279:22595–22604. doi: 10.1074/jbc.M313004200. [DOI] [PubMed] [Google Scholar]
- 67.Meissauer A, Kramer MD, Schirrmacher V, et al. Generation of cell surface-bound plasmin by cell-associated urokinase-type or secreted tissue-type plasminogen activator: a key event in melanoma cell invasiveness in vitro. Exp Cell Res. 1992;199:179–190. doi: 10.1016/0014-4827(92)90423-6. [DOI] [PubMed] [Google Scholar]
- 68.Liu G, Shuman MA, Cohen RL. Co-expression of urokinase, urokinase receptor and PAI-1 is necessary for optimum invasiveness of cultured lung cancer cells. Int J Cancer. 1995;60:501–506. doi: 10.1002/ijc.2910600413. [DOI] [PubMed] [Google Scholar]
- 69.Carmeliet P, Collen D. Molecular genetics of the fibrinolytic and coagulation systems in haemostasis, thrombogenesis, restenosis and atherosclerosis. Curr Opin Lipidol. 1997;8:118–125. doi: 10.1097/00041433-199704000-00010. [DOI] [PubMed] [Google Scholar]
- 70.Wang L, Ly CM, Ko CY, et al. uPA binding to PAI-1 induces corneal myofibroblast differentiation on vitronectin. Invest Ophthalmol Vis Sci. 2012;53:4765–4775. doi: 10.1167/iovs.12-10042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jang YC, Tsou R, Gibran NS, et al. Vitronectin deficiency is associated with increased wound fibrinolysis and decreased microvascular angiogenesis in mice. Surgery. 2000;127:696–704. doi: 10.1067/msy.2000.105858. [DOI] [PubMed] [Google Scholar]
- 72.Lopez-Guisa JM, Rassa AC, Cai X, et al. Vitronectin accumulates in the interstitium but minimally impacts fibrogenesis in experimental chronic kidney disease. Am J Physiol Renal Physiol. 2011;300:F1244–F1254. doi: 10.1152/ajprenal.00701.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]