

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal, progressive neurodegenerative disease. ALS selectively causes degeneration in upper and lower (spinal) motor neurons, leading to muscle weakness, paralysis and death by ventilatory failure. Although ventilatory failure is generally the cause of death in ALS, little is known concerning the impact of this disorder on respiratory motor neurons, the consequences of respiratory motor neuron cell death, or the ability of the respiratory control system to “fight back” via mechanisms of compensatory respiratory plasticity. Here we review known effects of ALS on breathing, including possible effects on rhythm generation, respiratory motor neurons, and their target organs: the respiratory muscles. We consider evidence for spontaneous compensatory plasticity, preserving breathing well into disease progression despite dramatic loss of spinal respiratory motor neurons. Finally, we review current and potential therapeutic approaches directed toward preserving the capacity to breathe in ALS patients.
Keywords: breathing, neurodegenerative disease, motor neuron disease, compensation, plasticity, ventilatory control
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a degenerative motor neuron disease first characterized in 1869 by Charcot (Charcot and Joffroy, 1869). ALS is characterized by rapid and progressive loss of muscle control, ending in paralysis and death by catastrophic respiratory failure (Cleveland and Rothstein, 2001;Boillee et al., 2006; Krakora et al., 2012). The lifetime risk of developing ALS is 1:400 (Hardiman et al., 2011), with an annual incidence from 0.4 to 8 per 100,000 individuals (Mayeux, 2003; Raibon et al., 2008; Hardiman et al., 2011). Typical ALS onset is between 50–60 years of age. Death typically occurs within 2–5 years of diagnosis in ALS patients, in part because there is often a delay between symptom onset and diagnosis (Ludolph and Knirsch, 1999; Raibon et al., 2008).
Approximately 90% of all ALS cases are of unknown etiology, and are referred to as “sporadic ALS” (Cleveland and Rothstein, 2001; Krakora et al., 2012). Most other ALS cases are familial, resulting from a number of gene mutations. Mutations in the superoxide dismutase 1 (SOD1) gene are associated with some familial ALS cases, and rodent models over-expressing human mutant SOD1 develop symptoms reminiscent of both familial and sporadic ALS (Rosen et al., 1993; Gurney et al., 1994; Wong et al., 1995; Liu et al., 1999; Nagai et al., 2001; Howland et al., 2002; Andersen et al., 2003; Bruijn et al., 2004). Multiple hypotheses concerning the pathogenesis of ALS in transgenic rodents that over-express mutant human SOD1 are still under active debate (Cleveland and Rothstein, 2001; Cluskey and Ramsden, 2001; Bossy-Wetzel et al., 2004; Bruijn et al., 2004; Rothstein, 2009).
Although ventilatory failure is the most frequent cause of death in ALS patients (Lyall et al., 2001), little is known concerning the unique failures and adaptations as respiratory motor neurons die. The major purpose of this brief review is to consider the impact of ALS on overall breathing capacity, followed by discussion of ALS impact on respiratory motor neurons, muscles and rhythm generation (Figures 1 and 2). We review emerging evidence that the respiratory system is able to “fight back,” triggering mechanisms of spontaneous compensatory respiratory plasticity that preserve breathing capacity despite impressive loss of respiratory motor neurons. We also now understand that spontaneous plasticity can be augmented by stimuli known to induce respiratory plasticity (intermittent hypoxia). Understanding the impact of ALS on breathing is crucial to extend the duration and quality of (ventilator independent) life.
Figure 1.
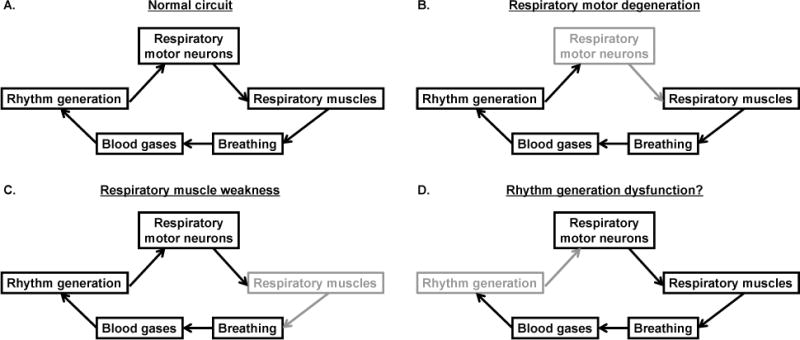
Schematic of the normal respiratory control system, and how it is affected in ALS. A: the normal respiratory system, including brainstem rhythm generating neurons, which activate/modulate respiratory motor neurons. Motor neurons activate respiratory muscles to generate breathing and regulate blood gases. B: respiratory motor neurons (shaded) degenerate in ALS, leading to ventilatory failure. C: respiratory muscles (shaded) become weak and atrophy from decreased activation, thereby contributing to respiratory demise. D: rhythm generating neurons may also become dysfunctional, contributing to central sleep apnea in ALS.
Figure 2.
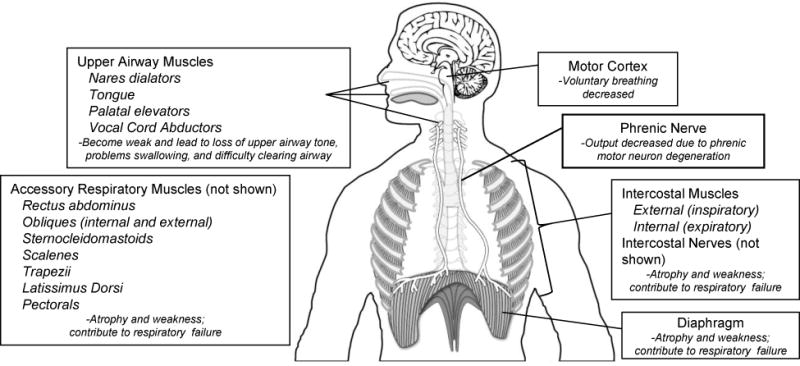
Multiple areas are affected in ALS. Upper motor neurons in the motor cortex have been implicated in affecting voluntary breathing. Upper airway muscles including the nares dilators, tongue, palatal elevators and vocal cord abductors become weak, leading to loss of upper airway tone, problems swallowing and difficulty clearing the airway. As phrenic motor neurons degenerate, diaphragm activity will be compromised, causing the diaphragm to atrophy and weaken. Atrophy and weakness can also occur in accessory respiratory muscles including the intercostal (both inspiratory and expiratory) muscles, rectus abdominus, external obliques, sternocleidomastoids, scalenes, trapezii, latissimus dorsi, and pectorals. Weakness in these muscles contributes to respiratory compromise and, ultimately, failure.
2. Clinical presentation in ALS
Clinical presentation in ALS patients is variable, and can include upper and lower motor neuron signs. Clinical presentation and disease progression depend on the location of initial symptom onset. For example, bulbar onset results from upper motor neuron degeneration, and includes spasticity, hyperreflexia, difficulty swallowing, speaking, or breathing due, at least in part, to degeneration of the corticospinal tract (Cleveland and Rothstein, 2001). Inspiratory (sniffing) related cortical potentials are useful in diagnosing cortical involvement in ALS (Bizovicar et al., 2012). Spinal onset disease results in signs characteristic of alpha motor neuron degeneration, and include generalized muscle weakness, atrophy, slowed motor responses in limbs or appendages and muscle fasciculation (Cleveland and Rothstein, 2001). ALS may first be apparent in: 1) distal extremities or 2) face, tongue or (rarely) respiratory function (Gautier et al., 2010). Interneurons may play a role in ALS, contributing to a loss of inhibitory function in cortical and spinal networks (Turner and Kiernan, 2012).
Patients reporting respiratory problems (e.g. dyspnea or orthopnea) have a poor prognosis versus patients without respiratory symptoms. Patients with bulbar onset ALS typically have earlier respiratory involvement, and these patients experience more rapid disease progression. Motor neuron death is progressive; eventually, paresis develops into overt paralysis. On the other hand, motor neuron death is differential across motor pools. For example, neurons controlling eye movements and the urinary bladder muscle are generally spared at the time of death in ALS (Kanning et al., 2010), even though they exhibit early EMG changes (Palmowski et al., 1995). It is not known why these motor neurons are spared, but it would be interesting to identify cellular/molecular differences between resistant versus susceptible motor pools. Conflicting data have been reported for resistant motor pools; some groups report upregulated calcium-buffering protein expression (possibly conferring resistance to excitotoxic stimuli; Alexianu et al., 1994; Obal et al., 2006), whereas others report that greater calcium-buffering protein expression correlates with increased vulnerability (Sasaki et al., 2006). At this point, the cellular basis for differences in motor neuron resistance during ALS is unknown. Nevertheless, even delayed respiratory muscle weakness greatly affects the quality of life in ALS patients since it may necessitate ventilatory support (Bourke et al., 2001), and often determines a patient’s life span (Lyall et al., 2001).
3. Breathing impairment in ALS
Pulmonary function tests (spirometry, blood gas analysis) are of considerable importance in determining the prognosis of ALS patients (Vitacca et al., 1997; Czaplinski et al., 2006; Schmidt et al., 2006; Talakad et al., 2009). Near the end of disease, ALS patients exhibit rapid declines in forced vital capacity, maximum voluntary ventilation and residual volume (Fallat et al., 1979; Stewart et al., 2001; Talakad et al., 2009). Similarly, in the SOD1G93A mouse model, ventilation and the ability to increase ventilation during maximal chemoreceptor stimulation is maintained until late in disease progression, at which time breathing is irreversibly impaired and the mouse dies (Tankersley et al., 2007). SOD1G93A rats similarly maintain their capacity to generate maximal tidal volumes until late in disease progression (20–30% body weight loss; Nichols et al., 2013); it is unknown if SOD1G93A rats would exhibit ventilatory failure if monitored to spontaneous death. Collective evidence suggests that humans with ALS, and rodent models of ALS, preserve inspiratory force generation until end-stage disease. Based on evidence from the SOD1G93A rat model, preservation of breathing capacity occurs in the face of major death in respiratory motor neuron pools (see below). Unfortunately, we do not currently know how many motor neurons are lost in ALS patients at the onset of ventilatory impairment since tissues are available for analysis only on post-mortem analysis. However, eventually in both humans and rodent models of ALS, respiratory motor neuron is too severe for compensation, resulting in inevitable ventilatory failure.
4. Potential sites of breathing impairment in ALS
Breathing is a complex process resulting from the actions of multiple respiratory muscles, each governed by a different motor neuron pool (depicted in Figures 1 & 2). Each motor pool is driven by a complex brainstem network engaged in the processes of rhythm generation, sensory integration and pattern formation (Ramirez et al., 1998; Rekling and Feldman, 1998; McCrimmon et al., 2000; Rekling et al., 2000; Feldman et al., 2013;). Since the primary insult in ALS is motor neuron cell death, the extent of compromise in each respiratory muscle may be heterogeneous; thus, relatively spared respiratory motor neuron pools may be in a position to compensate for failure in others.
In humans, expiration is largely passive during quiet resting conditions, although these muscles are recruited during physical activity, and are essential for expulsive respiratory defense mechanisms such as coughing (Hegland et al., 2012). Thus, even with adequate inspiration, deficits in expiratory muscle activity leave the lung/respiratory system susceptible to infections, similar to spinal cord injury (Corcia et al., 2008; Bolser et al., 2009; Reid et al., 2010; Hardiman, 2011;). Since the primary problem in ALS is motor neuron cell death, the main breathing impairment is expected to be limited capacity to generate inspiratory and expiratory force, thereby limiting the capacity to generate inspiratory volume and expulsive maneuvers needed to defend the respiratory system.
Cortical influences also influence respiratory control (Banzett et al., 1981; Waldrop et al., 1982; Banzett et al., 2000;); since ALS can attack upper respiratory motor neurons, this site may contribute to impairment.
Finally, since central sleep apnea is a prominent feature in ALS, there may be pathology in brainstem regions associated with respiratory rhythm generation. In a sense, brainstem respiratory neurons are “upper motor neurons” for spinal, respiratory motor neurons. Thus, it is important to consider the impact of ALS on these important neuronal groups. Below, we consider what is known concerning impaired respiratory motor neuron function, respiratory muscles and respiratory rhythm generation.
4.1 Respiratory motor neuron degeneration
4.1.1 Upper motor neurons
Motor cortical neuron loss has been implicated in deteriorating voluntary breathing control in ALS patients, particularly motor cortical regions associated with the diaphragm (Shimizu et al., 2010). In ALS patients, short-latency evoked diaphragm responses to transcranial magnetic stimulation of the motor cortex are significantly diminished. Loss of short-latency diaphragm motor evoked potentials may reflect loss of cortico-respiratory pathways, potentially contributing to respiratory compromise during voluntary acts, such as speaking (Shimizu et al., 2010). However, the diaphragm motor cortex (and voluntary influences on breathing) may be affected prior to the onset of symptom onset due to spinal, respiratory motor neurons (Miscio et al., 2006). Indeed, it has been suggested that deterioration of motor cortex pathways may predict imminent respiratory compromise (Miscio et al., 2006).
4.1.2 Spinal respiratory motor neurons
Although respiratory motor neuron degeneration occurs in ALS patients, the onset and progression of motor neuron death is not known (Su et al., 1996; Motoi et al., 1998; Tsuchiya et al., 2002; Hashimoto et al., 2008; Cifra et al., 2011; Kobayashi et al., 2011). In SOD1G93A rats, Llado and colleagues (2006) reported profound degeneration of phrenic (but not hypoglossal) motor neurons, progressive reduction of evoked compound diaphragm action potentials, phrenic nerve fiber loss, and diaphragm atrophy. Thus, there is clear evidence for potential respiratory compromise in a rat model of ALS. On the other hand, these authors did not confirm functional deficits in the ability to generate inspiratory volume. Tankersley and colleagues (2007) assessed the ability to generate tidal volume in SOD1G93A mice; these mice preserved tidal volume generating capacity until late in disease progression, and then exhibited precipitous declines over the final two days of life. Unfortunately, Tankersley and colleagues did not report the extent of respiratory motor neuron death at any point in disease progression. We recently observed major deficits in phrenic and intercostal motor neuron cell counts at disease end-stage in SOD1G93A rats (20–30% decrease in body mass) (Nichols et al., 2013). At this same end-stage, deficits are observed in the capacity to generate phrenic motor output, but not in the capacity to generate maximal tidal volumes, which is remarkably well preserved (Figure 3A; Nichols et al., 2013).
Figure 3.
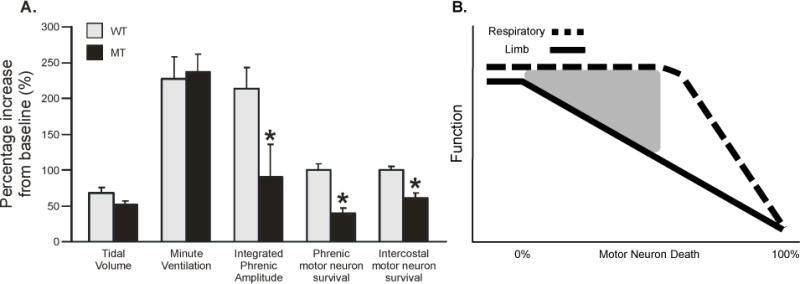
Ventilation is preserved in end-stage SOD1G93A rats. A. Percentage increase from baseline for tidal volume, minute ventilation, integrated phrenic amplitude, and phrenic and intercostal motor neuron survival in wild-type (WT; gray bars) and SOD1G93A (mutant, MT; black bars) rats at disease end-stage (20–30% decrease in body mass). Despite decreased respiratory motor neuron survival and output, ventilation is not affected in MT rats. B. Schematic of postulated respiratory (dashed black line) and limb (solid black line) function as a function of motor neuron death in MT rats. Respiratory function appears to be preserved long after symptoms of somatic motor weakness are observed, despite significant respiratory motor neuron death; the gray triangle represents a zone of protection for respiratory function through as yet unknown mechanisms of compensatory respiratory plasticity (see figure 4). Eventually, respiratory motor neurons death will be too extreme, causing inevitable respiratory failure. Values are means ± SEM and * = different from WT where p < 0.05 (original data described in Nichols et al., 2013 and re-analyzed here for percentage increase from baseline).
Diaphragm activity in response to phrenic nerve stimulation also decreases rapidly in ALS patients, an effect that correlates with pulmonary function tests (Pinto et al., 2009). On the other hand, the extreme inspiratory maneuvers (e.g. maximal force during inspiratory sniffing) necessary to demonstrate functional respiratory deficits prior to late-stage disease suggest that respiratory motor function is somehow protected in humans with ALS long after symptoms of somatic motor weakness begin (see Figure 3B; Lyall et al., 2001).
4.2 Mechanisms of compensatory respiratory plasticity in ALS
Although it is not known how many respiratory motor neurons are necessary to preserve breathing capacity in ALS, there appears to be a mechanism (or suite of mechanisms; Johnson and Mitchell, ibid) preserving breathing capacity despite major respiratory motor neuron cell death (Figure 3B). Possible mechanisms preserving breathing capacity despite progressive respiratory motor neuron death (Figure 4) include: 1) brainstem plasticity, increasing descending central respiratory drive from medullary pre-motor neurons and over-riding the impact of decreased respiratory motor neuron numbers (Figure 4A); 2) plasticity in spared respiratory motor neurons (Figure 4B and D), amplifying an unchanging (or increased) central respiratory drive; 3) plasticity at the neuromuscular junction, enabling spared respiratory motor neurons to activate more muscle fibers (i.e. larger motor units; Figure 4C and E); and 4) greater activation of accessory respiratory muscles, compensating for losses of phrenic/diaphragm function (Figure 4D and E). Changes in descending respiratory drive from medullary pre-motor neurons are not likely to account for full preservation of breathing capacity because 1) the apneic CO2 threshold is unchanged (Nashold et al., 2006; Nichols et al. unpublished) in SOD1G93A rats at end-stage, and 2) chemoreflex responses in the hypoglossal nerve are normal in SOD1G93A rats, even though they receive input from the same brainstem respiratory neurons (Nashold et al., 2006). There is some evidence for spinal motor plasticity since growth/trophic factors associated with respiratory motor neuron plasticity are up-regulated in spared phrenic motor neurons (Satriotomo and Mitchell, unpublished observations). Further, estimates of phrenic motor neuron death exceed deficits in the capacity to increase phrenic motor output, suggesting at least some amplification of bulbospinal respiratory drive at the spinal cord. In rodent ALS models, increased neuromuscular junction numbers per spared motor neuron have been described in other motor systems during ALS disease progression (Gordon et al., 2004); unfortunately, we know virtually nothing concerning this possibility in respiratory motor neurons. By increasing the size of respiratory motor units, respiratory muscle contractions may be preserved despite declining numbers of respiratory motor neurons innervating that muscle. The potential to shift the balance from phrenic/diaphragm to accessory respiratory muscles is another interesting possibility that has not yet been adequately studied. However, this mechanism, if it exists would: 1) require brainstem and/or respiratory motor neuron plasticity, redistributing central respiratory drive among spared respiratory motor pools; and 2) is not likely to be fully effective since intercostal motor neurons degenerate (albeit less than phrenic motor neurons) during disease progression in SOD1G93A rats (Nichols et al., 2013). Further, sternomastoid muscle is unlikely to compensate effectively for loss of diaphragm function since significant denervation and atrophy are observed at disease end-stage (Smittkamp et al., 2010). Studies of mechanisms giving rise to spontaneous compensatory respiratory plasticity during ALS disease progression may yield important insights concerning new strategies to promote neuroprotection and functional recovery in ALS.
Figure 4.
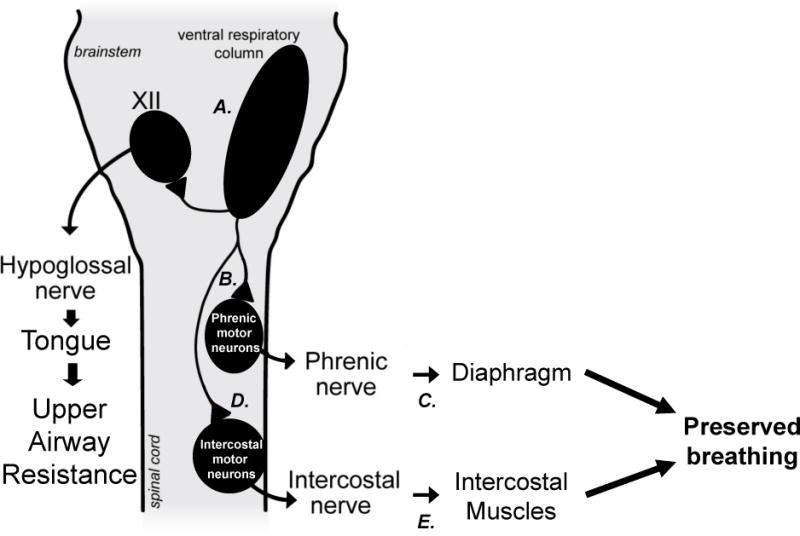
Possible mechanisms of compensatory respiratory plasticity, preserving breathing capacity in ALS. Compensation may arise from greater activation of brainstem respiratory neurons (A.), plasticity in spared phrenic motor neurons (B.), plasticity in the size of motor units (neuromuscular junction plasticity; C.) shifting the balance to motor pools relatively unaffected by disease progression (D. and E.) via motor neuron or NMJ plasticity in (for example) the intercostal muscles.
4.3 Respiratory muscle weakness
Respiratory muscle weakness in ALS (Figures 1 and 2; Bourke et al., 2001; Lyall et al., 2001; Lechtzin et al., 2002) results from loss of diaphragm (de Carvalho et al., 1996; Lechtzin, 2006) and inspiratory intercostal muscle function (Pinto et al., 2007; de Carvalho et al., 2010). Internal intercostal muscle weakness is also linked to weakened expulsive forces during coughing, leaving ALS patients at heightened risk for pneumonia (Corcia et al., 2008; Hardiman, 2011). The external intercostals, and other accessory inspiratory muscles, such as the sternocleidomastoids, scalenes, trapezii, latissimus dorsi, and pectoral muscles, assist in situations of heavy ventilatory demand (e.g. heavy exercise) or in situations leading to impaired diaphragm function (e.g. emphysema, chronic obstructive pulmonary disease, and quadriplegia) (Benditt, 2006; Gregory, 2007).
In ALS patients, inspiratory accessory muscles weaken and fasciculate as the disease progresses. Indeed, increased spontaneous activity (fasciculation) in accessory inspiratory muscle EMGs, including the sternocleidomastoid and trapezius, is thought to be an early indicator of ALS (Li et al., 2002; Xu et al., 2011). Inspiratory muscle weakness exacerbates cough inadequacy by diminishing cough capacity (Smith et al., 1987; Park et al., 2010). Although there is patient variability in spinal-onset ALS patients, muscle weakness in accessory respiratory muscles such as the intercostal and abdominal muscles occur later than muscle weakness in the extremities, but before the muscles of swallowing and speech (Marti-Fabregas et al., 1995; Lechtzin, 2006;). During spinal onset ALS, the muscles maintaining upper airway tone are relatively spared in ALS, but will eventually weaken in late-stage disease (Oliver, 1996). Bulbar onset ALS patients often present with symptoms of dysarthria, dysphagia, and tongue fasciculation, indicating early onset of upper airway muscle weakness (Kuhnlein et al., 2008). Loss of upper airway tone contributes to further complications, such as clearing aspirated particles or secretions (Kuhnlein et al., 2008).
Muscle weakness in ALS is a direct consequence of motor neuron pathology and axonal retraction. The pattern of motor neuron pathology follows the “dying-back” hypothesis, in which motor neuron degeneration progresses from distal to proximal (Krakora et al., 2012). In patients, muscle weakness may not become apparent until a relatively large proportion of motor neurons have retracted from the myofibers (Dadon-Nachum et al., 2011). Axonal retraction of the neuron from the NMJ may occur at different time points in SOD1G93A mouse and rat models (Guillot et al., 2004; Suzuki et al., 2008). Both models eventually lose excitatory input to muscles as the motor axon dies back from the motor endplates of muscle fibers.
In SOD1G93A rats, degeneration initially occurs among fast-twitch fatigable motor units (consisting of fast type IIx and IIb muscle fibers), followed by fast-twitch fatigue resistant (type IIa), and finally a late stage degeneration of the slow twitch fatigue-resistant (type I) motor units (Frey et al., 2000; Hegedus et al., 2007, Hegedus et al., 2008; Hegedus et al., 2009). This fast fatigable to slow fatigue-resistant pattern of motor unit degeneration is the reverse recruitment order of motor units during breathing (Dick et al., 1987; Jodkowski et al., 1987, 1988; Mantilla et al., 2010).
During normal tidal breathing, motor unit recruitment consists mainly of slow fatigue-resistant fibers. During increased ventilatory demand (e.g. hypoxia-hypercapnia), additional fast fatigue-resistant motor units are recruited. Expulsive maneuvers (e.g. coughing or sneezing) require recruitment of fast fatigable motor units. Overall, motor units used least (fast twitch fatigable type IIx/IIb) have the least protection from motor neuron degeneration and axonal retraction. Conversely, motor units used during all respiratory actions (slow twitch fatigue resistant type I) are generally the last to degenerate. Unfortunately, the progression of motor neuron degeneration has not yet been clearly established in human disease.
Motor axon retraction begins with dissolution of NMJs as the axon separates from the muscle fiber motor endplate (Fischer et al., 2004; Dupuis and Loeffler, 2009;). In SOD1 mice, NMJ dissolution is observed prior to any motor axon die back (Frey et al., 2000). Generally, as the motor axon retracts from the muscle fiber, the postsynaptic acetyl choline receptor (AChR) enriched region becomes fragmented and diffuse (Valdez et al., 2012). Without reinnervation, a terminal Schwann cell will cover the entire endplate and likely assists in dissolution of all but the post-synaptic cytoplasmic components of the NMJ on the interior membrane of the muscle fiber (MacIntosh et al., 2006). The mechanism responsible for triggering NMJ dissolution during ALS progression remains unknown. During the last decade, evidence has emerged suggesting that the muscle itself initiates NMJ dissolution prior to motor neuron die back through Nogo-A overexpression in fast-twitch fatigable fibers (Dupuis et al., 2002; Jokic et al., 2005; Jokic et al., 2006; Pradat et al., 2007), hypermetabolism (Dupuis et al., 2004; Mattson et al., 2007), or possibly mSOD1 over expression in muscle, leading to inflammation (Dupuis and Loeffler, 2009).
Whereas breathing capacity is preserved until late stage ALS, studies comparing muscle fibers of diaphragm to limb muscles found diaphragm to be no more or less susceptible to the effects of denervation (Valdez et al., 2012), despite its greater frequency of activation (diaphragm duty cycle ~40% versus 2–15% for other skeletal muscle; Sieck, 1994). The retraction of motor axons and dissolution of the NMJ causes marked changes in myofibers and respiratory muscle wasting. The pattern of muscle wasting once motor axons retract is similar to denervation models, provided that reinnervation does not occur within an as yet undefined period of time (MacIntosh et al., 2006). Generally, in both slow and fast myofibers (including diaphragm and accessory respiratory muscles), the most obvious effects of denervation are a rapid decrease fiber diameter followed by organelle atrophy, mitochondrial shrinkage and/or degeneration, and myonuclear migration to the myofiber center (Pellegrino and Franzini, 1963; Stonnington and Engel, 1973; Schmalbruch and Lewis, 2000; MacIntosh et al., 2006). Approximately 2 months post-denervation, only 20–40% of muscle mass remains (Sunderland and Ray, 1950), largely due to lack of contractile activation(Buffelli et al., 1997). Early changes in gene expression within denervated myofibers leads to AChR expression across the entirety of the membrane (Stya and Axelrod, 1984). Denervation of myofibers upregulates expression of neurotrophic factors, including glial cell-line derived neurotrophic factor (Lie and Weis, 1998), insulin-like growth factors (Czerwinski et al., 1993), brain derived neurotrophic factor (Koliatsos et al., 1993; Sakuma et al., 2001), and nerve growth factor (Amano et al., 1991; Sakuma and Yamaguchi, 2011). Neurotrophic factor expression and diffuse acetylcholine receptor distribution may be important components promoting reinnervation.
4.4 Sleep disordered breathing
ALS patients commonly exhibit sleep disordered breathing, most often due to central versus obstructive apneas (Bourke et al., 2001). Although little is known concerning the mechanistic basis of central sleep apnea in ALS patients, similar disruptions in breathing are observed during sleep in rats with partial lesions of neurons in the pre-Bötzinger complex, a critical site for brainstem respiratory rhythm generation (McKay et al., 2005; Figures 1 and 2). In rhythmogenic brainstem slice preparations from neonatal rodents, selective killing of as few as 18% of pre-Bötzinger neurons stops spontaneous respiratory rhythm, without completely destroying the underlying network (Hayes et al., 2012). Thus, abnormal respiratory behavior can result from even small disruptions in pre-Bötzinger neuron numbers. To date we have no evidence that brainstem interneurons in the region of the pre-Bötzinger complex die during ALS.
Symptoms of fragmented sleep caused in part by sleep disordered breathing carry over into daytime sleepiness, headaches, and short-term memory problems for ALS patients (Bourke et al., 2001). With disease progression, sleep disordered breathing gets worse, and has been attributed by some to periodic disruption of diaphragm function, since the diaphragm is the only active inspiratory muscle during rapid eye movement sleep (Arnulf et al., 2000). Diaphragm dysfunction is a significant predictor of dyspnea (Similowski et al., 2000), but is not always a reliable predictor of sleep disordered breathing since patients with full diaphragm function often experience sleep disordered breathing (Atalaia et al., 2007).
Defects in respiratory chemoreflexes have been suggested to accelerate the progression of ALS, although the abnormal blood gas and pH regulation observed may simply reflect failing respiratory capacity (Morimoto et al., 2011). Hypoglossal motor neurons, which innervate the tongue, are important to preserve upper airway patency and swallowing; swallowing deficits are often seen in ALS patients, suggesting deficits in the hypoglossal motor pool (Cleveland and Rothstein, 2001). However, hypoglossal dysfunction is unlikely to be a major trigger for sleep disordered breathing in ALS, since central are more frequent than upper airway obstruction in ALS (Bourke et al., 2001).
5. Therapeutics
There are no known cures for ALS, and only commonly accepted treatment known to slow disease progression is Riluzole. Unfortunately, Riluzole extends survival only by a matter of months. Riluzole may have a greater survival benefit for patients with bulbar onset ALS, extending the time before patients require ventilatory support. A newer drug, Tirasemtiv (formerly CK-2017357), activates muscle troponin and improves maximum voluntary ventilation and submaximal handgrip endurance in ALS patients (Shefner et al., 2012).
Respiratory muscle rest is frequently used and has shown some promise in ALS patients. For example, bi-level intermittent positive pressure (BIPAP), a form of noninvasive ventilatory support, reduces the work of breathing and improves exercise tolerance and sleep quality (Kleopa et al., 1999; Pinto et al. 1999; de Carvalho et al. 1996). BIPAP slows ventilatory decline and prolongs survival in a subset of ALS patients, namely patients whose FVC has declined below 50% (Kleopa et al. 1999). However, in patients receiving BIPAP support soon after ALS diagnosis, the degradation of respiratory function progresses more rapidly, causing higher mortality versus those not on BIPAP (Cedarbaum and Stambler 2001). The principle behind BIPAP in reducing the work of breathing potentially results in reduced diaphragm loading, reduced motor unit recruitment and less phrenic (and accessory) motor neuron activity. As observed in limb motor neurons/muscles, disuse leaves motor neurons more susceptible to death, and the muscle more susceptible to denervation due to motor axon die-back. BIPAP may be a useful therapeutic tool, but administering BIPAP too early may do more harm than good.
Exercise is effective for delaying respiratory insufficiency in ALS patients (Aboussouan 2009; de Almeida et al. 2012) and deficits in limb function in ALS mice (Carreras et al. 2010). Optimal exercise protocols (aerobic, muscle strength, respiratory muscle training; Aboussouan 2009) are not yet known. Despite potential confounding metabolic, hormonal and oxidative effects, exercise is a useful tool to preserve respiratory and limb function in ALS patients. Increased breathing during exercise may benefit patients due to increased activation of respiratory motor neurons/muscles. Exercise may share some mechanisms with intermittent hypoxia in ALS patients since both therapies increase raphe serotonergic function (Gerin et al., 1995) and spinal BDNF concentrations (Gomez-Pinilla et al., 2001; Gomez-Pinilla et al., 2002), thereby inducing beneficial respiratory motor plasticity. However, as exercise becomes progressively more difficult with disease progression, less stressful stimuli to motor neuron plasticity may be of importance such as intermittent hypoxia (see below).
New therapies that have shown promise in ALS are: 1) intermittent hypoxia, which induces additional respiratory motor plasticity, restoring phrenic motor output to near normal levels in SOD1G93A rats; and 2) implantation of human neural progenitor cells near the phrenic motor nucleus, promoting phrenic motor neuron survival and, thus, the ability to generate phrenic motor output (Nichols et al., 2013; see Figure 5). The combination of therapies may yield further functional improvement. Of interest, intermittent hypoxia may also be a means of enhancing efficacy of stem cell implants since exposure of donor rats to intermittent hypoxia increases the yield of neural progenitor cells (increased cell expansion and differentiation; Ross et al., 2012). Further, intermittent hypoxia after stem cell implantation may enhance their incorporation into spinal tissues since repetitive intermittent hypoxia upregulates growth/trophic factors in respiratory motor nuclei (Satriotomo et al., 2012).
Figure 5.
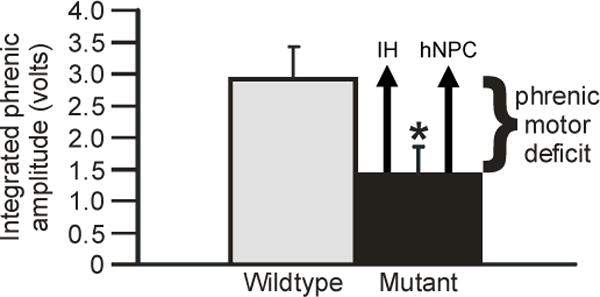
Restoration of phrenic motor output via distinct therapeutic strategies in end-stage SOD1G93A rats. Integrated phrenic burst amplitude during chemoreceptor stimulation was significantly decreased in SOD1G93A (mutant, black bar) versus wild-type rats (gray bar). Phrenic motor output can be restored to near normal levels by exposure to acute intermittent hypoxia (IH) or by human neural progenitor cell transplants (hNPC) near the phrenic motor nucleus (represented by black arrows). Representation adapted from Nichols et al. (2013).
Acute exposure to intermittent hypoxia (AIH) induces spinal respiratory motor plasticity (Feldman et al., 2003; Mahamed and Mitchell, 2008; Mitchell et al., 2001), thereby increasing respiratory motor output by a serotonin and BDNF-dependent mechanism (Baker-Herman et al., 2004). Repetitive AIH restores both respiratory and non-respiratory motor function in rats with cervical spinal injuries (Lovett-Barr et al., 2012), as well as leg strength in humans with chronic, motor incomplete spinal injuries (Trumbower et al., 2012). We investigated the effects of repetitive intermittent hypoxia in ALS rats (exposures began pre-symptomatically, and continued 3x per week until end-stage). Repetitive intermittent hypoxia had not effect on non-respiratory (body mass, limb function) or measured respiratory function (Nashold et al., 2001). However, ventilatory capacity is normally well preserved at the end-stage studied, with no functional deficit for repetitive intermittent hypoxia to “fix” (Nichols et al., 2013). Since we did not record phrenic activity in the former study, it remains possible that intermittent hypoxia preserved or restored lost phrenic motor output, restoring more normal patterns of respiratory muscle recruitment and (possibly) delaying overt respiratory failure.
The therapeutic potential of intermittent hypoxia is demonstrated by observations that: 1) intermittent hypoxia-induced phrenic motor plasticity is enhanced at disease end-stage (20% decrease in body mass) in ALS rats (Nichols et al., 2010; Nichols et al., 2013); and 2) intermittent anoxia elicits long-term facilitation of respiratory burst frequency in vitro (Blitz and Ramirez, 2002). Thus, repetitive AIH may (at least partially) restore respiratory motor function in failing ALS patients. If intermittent hypoxia is implemented before symptoms begin, there do not appear to be detectable detrimental effects. Ultimately, intermittent hypoxia can do little more than prolong the time to respiratory failure since disease progression will eventually overwhelm potential benefits from intermittent hypoxia. Nevertheless, by transiently preserving breathing capacity, intermittent hypoxia may enhance the quality and duration of life at disease end-stage.
6. Conclusions
In ALS, early intervention with non-invasive ventilatory support is beneficial to preserve the quality of life, and prolong survival (Mustfa et al., 2006; Lechtzin et al., 2007). Invasive intervention by tracheostomy and mechanical ventilation can extend survival, but require extensive management of ventilatory support. Further, since ALS does not affect cognitive abilities, mechanical ventilation to prolong life after the loss of voluntary movement presents the very real possibility of entering a “locked-in state.” This occurs when a person is aware of his/her surroundings, but unable to communicate. Thus, the decision regarding whether or not to refuse mechanical ventilation is an important issue.
A major point of this review is that respiratory weakness and sleep disordered breathing are critical features of ALS, and ultimately determine life span. At some point in disease progression, inspiratory motor neuron death will exceed the capacity for compensation, causing ventilatory failure and death (Figure 3B). However, despite the critical importance of respiratory function in ALS, relatively few studies have addressed how the respiratory system deteriorates, or how it compensates, as it confronts challenges posed by progressive respiratory motor neuron death. Our emerging understanding that there is considerable capacity for spontaneous, compensatory respiratory plasticity, and the ability to induce further plasticity (e.g. AIH) may lead to novel therapies that extend ventilator independence and, thus life-span in ALS.
Highlights.
Ventilatory failure is the main cause of death in ALS.
Few studies have been done to preserve breathing in ALS.
Therapeutic approaches for preservation of breathing in ALS are reviewed.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aboussouan LS. Mechanisms of exercise limitation and pulmonary rehabilitation for patients with neuromuscular disease. Chron Respir Dis. 2009;6(4):231–249. doi: 10.1177/1479972309345927. [DOI] [PubMed] [Google Scholar]
- Alexianu ME, Ho BK, Mohamed AH, La Bella V, Smith RG, Appel SH. The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann Neurol. 1994;36:846–858. doi: 10.1002/ana.410360608. [DOI] [PubMed] [Google Scholar]
- Amano T, Yamakuni T, Okabe N, Sakimura K, Takahashi Y. Production of nerve growth factor in rat skeletal muscle. Neuroscience letters. 1991;132:5–7. doi: 10.1016/0304-3940(91)90418-s. [DOI] [PubMed] [Google Scholar]
- Andersen PM, Sims KB, Xin WW, Kiely R, O’Neill G, Ravits J, Pioro E, Harati Y, Brower RD, Levine JS, Heinicke HU, Seltzer W, Boss M, Brown RH., Jr Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of discoveries, defects and disputes. Amyotrophic lateral sclerosis and other motor neuron disorders : official publication of the World Federation of Neurology, Research Group on Motor Neuron Diseases. 2003;4:62–73. doi: 10.1080/14660820310011700. [DOI] [PubMed] [Google Scholar]
- Arnulf I, Similowski T, Salachas F, Garma L, Mehiri S, Attali V, Behin-Bellhesen V, Meininger V, Derenne JP. Sleep disorders and diaphragmatic function in patients with amyotrophic lateral sclerosis. American journal of respiratory and critical care medicine. 2000;161:849–856. doi: 10.1164/ajrccm.161.3.9805008. [DOI] [PubMed] [Google Scholar]
- Atalaia A, De Carvalho M, Evangelista T, Pinto A. Sleep characteristics of amyotrophic lateral sclerosis in patients with preserved diaphragmatic function. Amyotrophic lateral sclerosis: official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. 2007;8:101–105. doi: 10.1080/17482960601029883. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nature neuroscience. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Banzett R, Strohl K, Geffroy B, Mead J. Effect of transrespiratory pressure on PETCO2-PaCO2 and ventilatory reflexes in humans. J Appl Physiol. 1981;51:660–664. doi: 10.1152/jappl.1981.51.3.660. [DOI] [PubMed] [Google Scholar]
- Banzett RB, Mulnier HE, Murphy K, Rosen SD, Wise RJ, Adams L. Breathlessness in humans activates insular cortex. Neuroreport. 2000;11:2117–2120. doi: 10.1097/00001756-200007140-00012. [DOI] [PubMed] [Google Scholar]
- Benditt JO. The neuromuscular respiratory system: physiology, pathophysiology, and a respiratory care approach to patients. Respiratory care. 2006;51:829–837. discussion 837–829. [PubMed] [Google Scholar]
- Bizovicar N, Zidar I, Koritnik B, Zidar J. Inspiratory- and finger-flexion-related cortical potentials in patients with amyotrophic lateral sclerosis–an exploratory study. Clinical neurology and neurosurgery. 2012;114:455–459. doi: 10.1016/j.clineuro.2012.02.049. [DOI] [PubMed] [Google Scholar]
- Blitz DM, Ramirez JM. Long-term modulation of respiratory network activity following anoxia in vitro. J Neurophysiol. 2002;87(6):2964–2971. doi: 10.1152/jn.2002.87.6.2964. [DOI] [PubMed] [Google Scholar]
- Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Bolser DC, Jefferson SC, Rose MJ, Tester NJ, Reier PJ, Fuller DD, Davenport PW, Howland DR. Recovery of airway protective behaviors after spinal cord injury. Respiratory physiology & neurobiology. 2009;169:150–156. doi: 10.1016/j.resp.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Schwarzenbacher R, Lipton SA. Molecular pathways to neurodegeneration. Nature medicine. 2004;10(Suppl):S2–9. doi: 10.1038/nm1067. [DOI] [PubMed] [Google Scholar]
- Bourke SC, Shaw PJ, Gibson GJ. Respiratory function vs sleep-disordered breathing as predictors of QOL in ALS. Neurology. 2001;57:2040–2044. doi: 10.1212/wnl.57.11.2040. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annual review of neuroscience. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Buffelli M, Pasino E, Cangiano A. Paralysis of rat skeletal muscle equally affects contractile properties as does permanent denervation. Journal of muscle research and cell motility. 1997;18:683–695. doi: 10.1023/a:1018687923929. [DOI] [PubMed] [Google Scholar]
- Carreras I, Yuruker S, Aytan N, Hossain L, Choi JK, Jenkins BG, Kowall NW, Dedeoglu A. Moderate exercise delays the motor performance decline in a transgenic model of ALS. Brain Res. 2010;1313:192–201. doi: 10.1016/j.brainres.2009.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedarbaum JM, Stambler N. Disease status and use of ventilatory support by ALS patients. BDNF Study Group. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2(1):19–22. doi: 10.1080/146608201300079373. [DOI] [PubMed] [Google Scholar]
- Charcot J, Joffroy A. Deux cas d’atrophie musculaire progressive avec lesion de la substance grise et des faisceaux antero-lateraux de la moelle epiniere. Arch Physiol Norm Path. 1869:745–760. [Google Scholar]
- Cifra A, Nani F, Nistri A. Respiratory motoneurons and pathological conditions: lessons from hypoglossal motoneurons challenged by excitotoxic or oxidative stress. Respiratory physiology & neurobiology. 2011;179:89–96. doi: 10.1016/j.resp.2011.03.017. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nature reviews. Neuroscience. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Cluskey S, Ramsden DB. Mechanisms of neurodegeneration in amyotrophic lateral sclerosis. Molecular pathology : MP. 2001;54:386–392. [PMC free article] [PubMed] [Google Scholar]
- Corcia P, Pradat PF, Salachas F, Bruneteau G, Forestier N, Seilhean D, Hauw JJ, Meininger V. Causes of death in a post-mortem series of ALS patients. Amyotrophic lateral sclerosis: official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. 2008;9:59–62. doi: 10.1080/17482960701656940. [DOI] [PubMed] [Google Scholar]
- Czaplinski A, Yen AA, Appel SH. Forced vital capacity (FVC) as an indicator of survival and disease progression in an ALS clinic population. Journal of neurology, neurosurgery, and psychiatry. 2006;77:390–392. doi: 10.1136/jnnp.2005.072660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czerwinski SM, Novakofski J, Bechtel PJ. Modulation of IGF mRNA abundance during muscle denervation atrophy. Med Sci Sports Exerc. 1993;25:1005–1008. [PubMed] [Google Scholar]
- Dadon-Nachum M, Melamed E, Offen D. The “dying-back” phenomenon of motor neurons in ALS. Journal of molecular neuroscience: MN. 2011;43:470–477. doi: 10.1007/s12031-010-9467-1. [DOI] [PubMed] [Google Scholar]
- de Almeida JP, Silvestre R, Pinto AC, deCarvalho M. Exercise and amyotrophic lateral sclerosis. Neurol Sci. 2012;33(1):9–15. doi: 10.1007/s10072-011-0921-9. [DOI] [PubMed] [Google Scholar]
- de Carvalho M, Matias T, Coelho F, Evangelista T, Pinto A, Luis ML. Motor neuron disease presenting with respiratory failure. J Neurol Sci. 1996;139(Suppl):117–122. doi: 10.1016/0022-510x(96)00089-5. [DOI] [PubMed] [Google Scholar]
- de Carvalho M, Pinto S, Swash M. Association of paraspinal and diaphragm denervation in ALS. Amyotrophic lateral sclerosis: official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. 2010;11:63–66. doi: 10.3109/17482960902730080. [DOI] [PubMed] [Google Scholar]
- Dick TE, Kong FJ, Berger AJ. Correlation of recruitment order with axonal conduction velocity for supraspinally driven diaphragmatic motor units. Journal of neurophysiology. 1987;57:245–259. doi: 10.1152/jn.1987.57.1.245. [DOI] [PubMed] [Google Scholar]
- Dupuis L, Gonzalez de Aguilar JL, di Scala F, Rene F, de Tapia M, Pradat PF, Lacomblez L, Seihlan D, Prinjha R, Walsh FS, Meininger V, Loeffler JP. Nogo provides a molecular marker for diagnosis of amyotrophic lateral sclerosis. Neurobiology of disease. 2002;10:358–365. doi: 10.1006/nbdi.2002.0522. [DOI] [PubMed] [Google Scholar]
- Dupuis L, Loeffler JP. Neuromuscular junction destruction during amyotrophic lateral sclerosis: insights from transgenic models. Current opinion in pharmacology. 2009;9:341–346. doi: 10.1016/j.coph.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Dupuis L, Oudart H, Rene F, Gonzalez de Aguilar JL, Loeffler JP. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11159–11164. doi: 10.1073/pnas.0402026101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallat RJ, Jewitt B, Bass M, Kamm B, Norris FH., Jr Spirometry in amyotrophic lateral sclerosis. Arch Neurol. 1979;36:74–80. doi: 10.1001/archneur.1979.00500380044004. [DOI] [PubMed] [Google Scholar]
- Feldman JL, Del Negro CA, Gray PA. Understanding the rhythm of breathing: so near, yet so far. Annual review of physiology. 2013;75:423–452. doi: 10.1146/annurev-physiol-040510-130049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annual review of neuroscience. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. The Journal of neuroscience. 2000;20:2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier G, Verschueren A, Monnier A, Attarian S, Salort-Campana E, Pouget J. ALS with respiratory onset: clinical features and effects of non-invasive ventilation on the prognosis. Amyotrophic lateral sclerosis: official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. 2010;11:379–382. doi: 10.3109/17482960903426543. [DOI] [PubMed] [Google Scholar]
- Gerin C, Becquet D, Privat A. Direct evidence for the link between monoaminergic descending pathways and motor activity. I. A study with microdialysis probes implanted in the ventral funiculus of the spinal cord. Brain Res. 1995;704(2):191–201. doi: 10.1016/0006-8993(95)01111-0. [DOI] [PubMed] [Google Scholar]
- Gómez-Pinilla F, Ying Z, Opazo P, Roy RR, Edgerton VR. Differential regulation by exercise of BDNF and NT-3 in rat spinal cord and skeletal muscle. Eur J Neurosci. 2004;13(6):1078–1084. doi: 10.1046/j.0953-816x.2001.01484.x. [DOI] [PubMed] [Google Scholar]
- Gómez-Pinilla F, Ying Z, Roy RR, Molteni R, Edgerton VR. Voluntary exercise induces a BDNF-mediated mechanism that promotes neuroplaticity. J Neurophysiol. 2002;88(5):2187–2195. doi: 10.1152/jn.00152.2002. [DOI] [PubMed] [Google Scholar]
- Gordon T, Hegedus J, Tam SL. Adaptive and maladaptive motor axonal sprouting in aging and motoneuron disease. Neurological research. 2004;26:174–185. doi: 10.1179/016164104225013806. [DOI] [PubMed] [Google Scholar]
- Gregory SA. Evaluation and management of respiratory muscle dysfunction in ALS. NeuroRehabilitation. 2007;22:435–443. [PubMed] [Google Scholar]
- Guillot S, Azzouz M, Deglon N, Zurn A, Aebischer P. Local GDNF expression mediated by lentiviral vector protects facial nerve motoneurons but not spinal motoneurons in SOD1(G93A) transgenic mice. Neurobiology of disease. 2004;16:139–149. doi: 10.1016/j.nbd.2004.01.017. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hardiman O. Management of respiratory symptoms in ALS. Journal of neurology. 2011;258:359–365. doi: 10.1007/s00415-010-5830-y. [DOI] [PubMed] [Google Scholar]
- Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nature reviews. Neurology. 2011;7:639–649. doi: 10.1038/nrneurol.2011.153. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Matsubara S, Mochizuki Y, Tsuji S, Mizutani T, Oyanagi K. Forme fruste or incipient form of widespread-type amyotrophic lateral sclerosis, or motor neuron disease with pallido-nigro-luysian atrophy? An autopsy case report. Neuropathology. 2008;28:309–316. doi: 10.1111/j.1440-1789.2007.00838.x. [DOI] [PubMed] [Google Scholar]
- Hayes JA, Wang X, Del Negro CA. Cumulative lesioning of respiratory interneurons disrupts and precludes motor rhythms in vitro. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:8286–8291. doi: 10.1073/pnas.1200912109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Gordon T. Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiology of disease. 2007;28:154–164. doi: 10.1016/j.nbd.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Gordon T. Progressive motor unit loss in the G93A mouse model of amyotrophic lateral sclerosis is unaffected by gender. Muscle Nerve. 2009;39:318–327. doi: 10.1002/mus.21160. [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Tyreman N, Gordon T. Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J Physiol. 2008;586:3337–3351. doi: 10.1113/jphysiol.2007.149286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegland KW, Bolser DC, Davenport PW. Volitional control of reflex cough. J Appl Physiol. 2012;113:39–46. doi: 10.1152/japplphysiol.01299.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proceedings of the National Academy of Sciences of the United States of America. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jodkowski JS, Viana F, Dick TE, Berger AJ. Electrical properties of phrenic motoneurons in the cat: correlation with inspiratory drive. Journal of neurophysiology. 1987;58:105–124. doi: 10.1152/jn.1987.58.1.105. [DOI] [PubMed] [Google Scholar]
- Jodkowski JS, Viana F, Dick TE, Berger AJ. Repetitive firing properties of phrenic motoneurons in the cat. Journal of neurophysiology. 1988;60:687–702. doi: 10.1152/jn.1988.60.2.687. [DOI] [PubMed] [Google Scholar]
- Jokic N, Gonzalez de Aguilar JL, Dimou L, Lin S, Fergani A, Ruegg MA, Schwab ME, Dupuis L, Loeffler JP. The neurite outgrowth inhibitor Nogo-A promotes denervation in an amyotrophic lateral sclerosis model. EMBO reports. 2006;7:1162–1167. doi: 10.1038/sj.embor.7400826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokic N, Gonzalez de Aguilar JL, Pradat PF, Dupuis L, Echaniz-Laguna A, Muller A, Dubourg O, Seilhean D, Hauw JJ, Loeffler JP, Meininger V. Nogo expression in muscle correlates with amyotrophic lateral sclerosis severity. Annals of neurology. 2005;57:553–556. doi: 10.1002/ana.20420. [DOI] [PubMed] [Google Scholar]
- Kanning KC, Kaplan A, Henderson CE. Motor neuron diversity in development and disease. Annu Rev Neurosci. 2010;33:409–440. doi: 10.1146/annurev.neuro.051508.135722. [DOI] [PubMed] [Google Scholar]
- Kleopa KA, Sherman M, Neal B, Romano GJ, Heiman-Patterson T. Bipap improves survival and rate of pulmonary function decline in patients with ALS. J Neurol Sci. 1999;164(1):82–88. doi: 10.1016/s0022-510x(99)00045-3. [DOI] [PubMed] [Google Scholar]
- Kobayashi Z, Tsuchiya K, Kubodera T, Shibata N, Arai T, Miura H, Ishikawa C, Kondo H, Ishizu H, Akiyama H, Mizusawa H. FALS with Gly72Ser mutation in SOD1 gene: report of a family including the first autopsy case. J Neurol Sci. 2011;300:9–13. doi: 10.1016/j.jns.2010.10.030. [DOI] [PubMed] [Google Scholar]
- Koliatsos VE, Clatterbuck RE, Winslow JW, Cayouette MH, Price DL. Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron. 1993;10:359–367. doi: 10.1016/0896-6273(93)90326-m. [DOI] [PubMed] [Google Scholar]
- Krakora D, Macrander C, Suzuki M. Neuromuscular junction protection for the potential treatment of amyotrophic lateral sclerosis. Neurology research international. 2012;379657 doi: 10.1155/2012/379657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnlein P, Gdynia HJ, Sperfeld AD, Lindner-Pfleghar B, Ludolph AC, Prosiegel M, Riecker A. Diagnosis and treatment of bulbar symptoms in amyotrophic lateral sclerosis. Nature clinical practice. Neurology. 2008;4:366–374. doi: 10.1038/ncpneuro0853. [DOI] [PubMed] [Google Scholar]
- Lechtzin N. Respiratory effects of amyotrophic lateral sclerosis: problems and solutions. Respiratory care. 2006;51:871–881. discussion 874–881. [PubMed] [Google Scholar]
- Lechtzin N, Rothstein J, Clawson L, Diette GB, Wiener CM. Amyotrophic lateral sclerosis: evaluation and treatment of respiratory impairment. Amyotrophic lateral sclerosis and other motor neuron disorders: official publication of the World Federation of Neurology, Research Group on Motor Neuron Diseases. 2002;3:5–13. doi: 10.1080/146608202317576480. [DOI] [PubMed] [Google Scholar]
- Lechtzin N, Scott Y, Busse AM, Clawson LL, Kimball R, Wiener CM. Early use of non-invasive ventilation prolongs survival in subjects with ALS. Amyotrophic lateral sclerosis: official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. 2007;8:185–188. doi: 10.1080/17482960701262392. [DOI] [PubMed] [Google Scholar]
- Li J, Petajan J, Smith G, Bromberg M. Electromyography of sternocleidomastoid muscle in ALS: a prospective study. Muscle Nerve. 2002;25:725–728. doi: 10.1002/mus.10115. [DOI] [PubMed] [Google Scholar]
- Lie DC, Weis J. GDNF expression is increased in denervated human skeletal muscle. Neuroscience letters. 1998;250:87–90. doi: 10.1016/s0304-3940(98)00434-0. [DOI] [PubMed] [Google Scholar]
- Liu R, Narla RK, Kurinov I, Li B, Uckun FM. Increased hydroxyl radical production and apoptosis in PC12 neuron cells expressing the gain-of-function mutant G93A SOD1 gene. Radiation research. 1999;151:133–141. [PubMed] [Google Scholar]
- Llado J, Haenggeli C, Pardo A, Wong V, Benson L, Coccia C, Rothstein JD, Shefner JM, Maragakis NJ. Degeneration of respiratory motor neurons in the SOD1 G93A transgenic rat model of ALS. Neurobiology of disease. 2006;21:110–118. doi: 10.1016/j.nbd.2005.06.019. [DOI] [PubMed] [Google Scholar]
- Lovett-Barr MR, Satriotomo I, Muir GD, Wilkerson JE, Hoffman MS, Vinit S, Mitchell GS. Repetitive intermittent hypoxia induces respiratory and somatic motor recovery after chronic cervical spinal injury. The Journal of neuroscience. 2012;32:3591–3600. doi: 10.1523/JNEUROSCI.2908-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludolph AC, Knirsch U. Problems and pitfalls in the diagnosis of ALS. J Neurol Sci. 1999;165(Suppl 1):S14–20. doi: 10.1016/s0022-510x(99)00021-0. [DOI] [PubMed] [Google Scholar]
- Lyall RA, Donaldson N, Polkey MI, Leigh PN, Moxham J. Respiratory muscle strength and ventilatory failure in amyotrophic lateral sclerosis. Brain. 2001;124:2000–2013. doi: 10.1093/brain/124.10.2000. [DOI] [PubMed] [Google Scholar]
- MacIntosh BR, Gardiner PF, McComas AJ. Skeletal Muscle: Form and Function. 2. Human Kinetics; 2006. [Google Scholar]
- Mahamed S, Mitchell GS. Respiratory long-term facilitation: too much or too little of a good thing? Advances in experimental medicine and biology. 2008;605:224–227. doi: 10.1007/978-0-387-73693-8_39. [DOI] [PubMed] [Google Scholar]
- Mantilla CB, Seven YB, Zhan WZ, Sieck GC. Diaphragm motor unit recruitment in rats. Respiratory physiology & neurobiology. 2010;173:101–106. doi: 10.1016/j.resp.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti-Fabregas J, Dourado M, Sanchis J, Miralda R, Pradas J, Illa I. Respiratory function deterioration is not time-linked with upper-limb onset in amyotrophic lateral sclerosis. Acta neurologica Scandinavica. 1995;92:261–264. doi: 10.1111/j.1600-0404.1995.tb01699.x. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cutler RG, Camandola S. Energy intake and amyotrophic lateral sclerosis. Neuromolecular medicine. 2007;9:17–20. doi: 10.1385/nmm:9:1:17. [DOI] [PubMed] [Google Scholar]
- Mayeux R. Epidemiology of neurodegeneration. Annual review of neuroscience. 2003;26:81–104. doi: 10.1146/annurev.neuro.26.043002.094919. [DOI] [PubMed] [Google Scholar]
- McCrimmon DR, Ramirez JM, Alford S, Zuperku EJ. Unraveling the mechanism for respiratory rhythm generation. BioEssays: news and reviews in molecular, cellular and developmental biology. 2000;22:6–9. doi: 10.1002/(SICI)1521-1878(200001)22:1<6::AID-BIES3>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- McKay LC, Janczewski WA, Feldman JL. Sleep-disordered breathing after targeted ablation of preBotzinger complex neurons. Nature neuroscience. 2005;8:1142–1144. doi: 10.1038/nn1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miscio G, Gukov B, Pisano F, Mazzini L, Baudo S, Salvadori A, Mauro A. The cortico-diaphragmatic pathway involvement in amyotrophic lateral sclerosis: neurophysiological, respiratory and clinical considerations. J Neurol Sci. 2006;251:10–16. doi: 10.1016/j.jns.2006.05.059. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB., Jr Invited review: Intermittent hypoxia and respiratory plasticity. J Appl Physiol. 2001;90:2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- Morimoto N, Deguchi K, Sato K, Yunoki T, Deguchi S, Ohta Y, Kurata T, Takao Y, Ikeda Y, Matsuura T, Abe K. Correlation of cerebral spinal fluid pH and HCO3- with disease progression in ALS. J Neurol Sci. 2011;307:74–78. doi: 10.1016/j.jns.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Motoi Y, Satoh K, Matsumine H, Wakiya M, Mori H, Shirai T, Kondo T, Mizuno Y. A 49-year-old man with progressive bulbar palsy and respiratory failure. No to shinkei = Brain and nerve. 1998;50:93–100. [PubMed] [Google Scholar]
- Mustfa N, Walsh E, Bryant V, Lyall RA, Addington-Hall J, Goldstein LH, Donaldson N, Polkey MI, Moxham J, Leigh PN. The effect of noninvasive ventilation on ALS patients and their caregivers. Neurology. 2006;66:1211–1217. doi: 10.1212/01.wnl.0000208957.88534.11. [DOI] [PubMed] [Google Scholar]
- Nagai M, Aoki M, Miyoshi I, Kato M, Pasinelli P, Kasai N, Brown RH, Jr, Itoyama Y. Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. The Journal of neuroscience. 2001;21:9246–9254. doi: 10.1523/JNEUROSCI.21-23-09246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashold LJ. PhD dissertation. University of Wisconsin; Madison, WI: 2011. Respiratory motor neuron survival and plasticity in a rodent model of familial amyotrophic lateral sclerosis. [Google Scholar]
- Nashold LJ, Wilkerson JE, Satriotomo I, Dale EA, Svendsen CN, Mitchell GS. Phrenic, but not hypoglossal, motor output is diminished in a rat model of amyotrophic lateral sclerosis (ALS) FASEB J. 2006;20:A1212. [Google Scholar]
- Nichols NL, Gowing G, Satriotomo I, Nashold LJ, Dale EA, Suzuki M, Avalos P, Mulcrone PL, McHugh J, Svendsen CN, Mitchell GS. Intermittent hypoxia and stem cell implants preserve breathing capacity in a rodent model of amyotrophic lateral sclerosis. American journal of respiratory and critical care medicine. 2013;187:535–542. doi: 10.1164/rccm.201206-1072OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols NL, Mitchell GS. Enhanced phrenic but not XII long-term facilitation (LTF) following acute intermittent hypoxia (AIH) in a rat model of amyotrophic lateral sclerosis (ALS) FASEB J. 2010;24:799.9. [Google Scholar]
- Obal I, Engelhardt JI, Siklos L. Axotomy induces contrasting changes in calcium and calcium-binding proteins in oculomotor and hypoglossal nuclei of Balb/c mice. J Comp Neurol. 2006;499:17–32. doi: 10.1002/cne.21041. [DOI] [PubMed] [Google Scholar]
- Oliver D. The quality of care and symptom control–the effects on the terminal phase of ALS/MND. J Neurol Sci. 1996;139(Suppl):134–136. doi: 10.1016/0022-510x(96)00087-1. [DOI] [PubMed] [Google Scholar]
- Palmowski A, Jost WH, Prudlo J, Osterhage J, Kasmann B, Schimrigk K, Ruprecht KW. Eye movement in amyotrophic lateral sclerosis: a longitudinal study. German journal of ophthalmology. 1995;4:355–362. [PubMed] [Google Scholar]
- Park JH, Kang SW, Lee SC, Choi WA, Kim DH. How respiratory muscle strength correlates with cough capacity in patients with respiratory muscle weakness. Yonsei medical journal. 2010;51:392–397. doi: 10.3349/ymj.2010.51.3.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino C, Franzini C. An Electron Microscope Study of Denervation Atrophy in Red and White Skeletal Muscle Fibers. J Cell Biol. 1963;17:327–349. doi: 10.1083/jcb.17.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto S, Geraldes R, Vaz N, Pinto A, de Carvalho M. Changes of the phrenic nerve motor response in amyotrophic lateral sclerosis: longitudinal study. Clinical neurophysiology : official journal of the International Federation of Clinical Neurophysiology. 2009;120:2082–2085. doi: 10.1016/j.clinph.2009.08.025. [DOI] [PubMed] [Google Scholar]
- Pinto S, Pinto A, De Carvalho M. Do bulbar-onset amyotrophic lateral sclerosis patients have an earlier respiratory involvement than spinal-onset amyotrophic lateral sclerosis patients? Europa medicophysica. 2007;43:505–509. [PubMed] [Google Scholar]
- Pinto AC, Alves M, Nogueira A, Evangelista T, Carvalho J, Coelho A, deCarvalho M, Sales-Luís ML. Can amyotrophic lateral sclerosis patients with respiratory insufficiency exercise? J Neurol Sci. 1999;169(1–2):69–75. doi: 10.1016/s0022-510x(99)00218-x. [DOI] [PubMed] [Google Scholar]
- Pradat PF, Bruneteau G, Gonzalez de Aguilar JL, Dupuis L, Jokic N, Salachas F, Le Forestier N, Echaniz-Laguna A, Dubourg O, Hauw JJ, Tranchant C, Loeffler JP, Meininger V. Muscle Nogo-A expression is a prognostic marker in lower motor neuron syndromes. Annals of neurology. 2007;62:15–20. doi: 10.1002/ana.21122. [DOI] [PubMed] [Google Scholar]
- Raibon E, Todd LM, Moller T. Glial cells in ALS: the missing link? Physical medicine and rehabilitation clinics of North America. 2008;19:441–459. vii–viii. doi: 10.1016/j.pmr.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Ramirez JM, Schwarzacher SW, Pierrefiche O, Olivera BM, Richter DW. Selective lesioning of the cat pre-Botzinger complex in vivo eliminates breathing but not gasping. J Physiol. 1998;507(Pt 3):895–907. doi: 10.1111/j.1469-7793.1998.895bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid WD, Brown JA, Konnyu KJ, Rurak JM, Sakakibara BM. Physiotherapy secretion removal techniques in people with spinal cord injury: a systematic review. The journal of spinal cord medicine. 2010;33:353–370. doi: 10.1080/10790268.2010.11689714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekling JC, Feldman JL. PreBotzinger complex and pacemaker neurons: hypothesized site and kernel for respiratory rhythm generation. Annual review of physiology. 1998;60:385–405. doi: 10.1146/annurev.physiol.60.1.385. [DOI] [PubMed] [Google Scholar]
- Rekling JC, Shao XM, Feldman JL. Electrical coupling and excitatory synaptic transmission between rhythmogenic respiratory neurons in the preBotzinger complex. The Journal of neuroscience. 2000;20:RC113. doi: 10.1523/JNEUROSCI.20-23-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Ross HH, Sandhu MS, Cheung TF, Fitzpatrick GM, Sher WJ, Tiemeier AJ, Laywell ED, Fuller DD. In vivo intermittent hypoxia elicits enhanced expansion and neuronal differentiation in cultured neural progenitors. Exp Neurol. 2012;235:238–245. doi: 10.1016/j.expneurol.2012.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Annals of neurology. 2009;65(Suppl 1):S3–9. doi: 10.1002/ana.21543. [DOI] [PubMed] [Google Scholar]
- Sakuma K, Watanabe K, Sano M, Uramoto I, Nakano H, Li YJ, Kaneda S, Sorimachi Y, Yoshimoto K, Yasuhara M, Totsuka T. A possible role for BDNF, NT-4 and TrkB in the spinal cord and muscle of rat subjected to mechanical overload, bupivacaine injection and axotomy. Brain research. 2001;907:1–19. doi: 10.1016/s0006-8993(01)02288-0. [DOI] [PubMed] [Google Scholar]
- Sakuma K, Yamaguchi A. The recent understanding of the neurotrophin’s role in skeletal muscle adaptation. Journal of biomedicine & biotechnology. 2011;2011:201696. doi: 10.1155/2011/201696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Warita H, Komori T, Murakami T, Abe K, Iwata M. Parvalbumin and calbindin D-28k immunoreactivity in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 2006;1083:196–203. doi: 10.1016/j.brainres.2006.01.129. [DOI] [PubMed] [Google Scholar]
- Satriotomo I, Dale EA, Dahlberg JM, Mitchell GS. Repetitive acute intermittent hypoxia increases expression of proteins associated with plasticity in the phrenic motor nucleus. Exp Neurol. 2012;237:103–115. doi: 10.1016/j.expneurol.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalbruch H, Lewis DM. Dynamics of nuclei of muscle fibers and connective tissue cells in normal and denervated rat muscles. Muscle Nerve. 2000;23:617–626. doi: 10.1002/(sici)1097-4598(200004)23:4<617::aid-mus22>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Schmidt EP, Drachman DB, Wiener CM, Clawson L, Kimball R, Lechtzin N. Pulmonary predictors of survival in amyotrophic lateral sclerosis: use in clinical trial design. Muscle Nerve. 2006;33:127–132. doi: 10.1002/mus.20450. [DOI] [PubMed] [Google Scholar]
- Shefner J, Cedarbaum JM, Cudkowicz ME, Maragakis N, Lee J, Jones D, Watson ML, Mahoney K, Chen M, Saikali K, Mao J, Russell AJ, Hansen RL, Malik F, Wolff AA. Safety, tolerability and pharmacodynamics of a skeletal muscle activator in amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis: official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. 2012;13:430–438. doi: 10.3109/17482968.2012.684214. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Komori T, Kugio Y, Fujimaki Y, Oyanagi K, Hayashi H. Electrophysiological assessment of corticorespiratory pathway function in amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis: official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. 2010;11:57–62. doi: 10.1080/17482960903207385. [DOI] [PubMed] [Google Scholar]
- Sieck GC. Physiological effects of diaphragm muscle denervation and disuse. Clinics in chest medicine. 1994;15:641–659. [PubMed] [Google Scholar]
- Similowski T, Attali V, Bensimon G, Salachas F, Mehiri S, Arnulf I, Lacomblez L, Zelter M, Meininger V, Derenne JP. Diaphragmatic dysfunction and dyspnoea in amyotrophic lateral sclerosis. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology. 2000;15:332–337. doi: 10.1034/j.1399-3003.2000.15b19.x. [DOI] [PubMed] [Google Scholar]
- Smith PE, Calverley PM, Edwards RH, Evans GA, Campbell EJ. Practical problems in the respiratory care of patients with muscular dystrophy. The New England journal of medicine. 1987;316:1197–1205. doi: 10.1056/NEJM198705073161906. [DOI] [PubMed] [Google Scholar]
- Smittkamp SE, Spalding HN, Brown JW, Gupte AA, Chen J, Nishimune H, Geiger PC, Stanford JA. Measures of bulbar and spinal motor function, muscle innervation, and mitochondrial function in ALS rats. Behavioural brain research. 2010;211:48–57. doi: 10.1016/j.bbr.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart H, Eisen A, Road J, Mezei M, Weber M. Electromyography of respiratory muscles in amyotrophic lateral sclerosis. J Neurol Sci. 2001;191:67–73. doi: 10.1016/s0022-510x(01)00621-9. [DOI] [PubMed] [Google Scholar]
- Stonnington HH, Engel AG. Normal and denervated muscle. A morphometric study of fine structure. Neurology. 1973;23:714–724. doi: 10.1212/wnl.23.7.714. [DOI] [PubMed] [Google Scholar]
- Stya M, Axelrod D. Mobility of extrajunctional acetylcholine receptors on denervated adult muscle fibers. The Journal of neuroscience. 1984;4:70–74. doi: 10.1523/JNEUROSCI.04-01-00070.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su M, Wakabayashi K, Tanno Y, Inuzuka T, Takahashi H. An autopsy case of amyotrophic lateral sclerosis with concomitant Alzheimer’s and incidental Lewy body diseases. No to shinkei = Brain and nerve. 1996;48:931–936. [PubMed] [Google Scholar]
- Sunderland S, Ray LJ. Denervation changes in mammalian striated muscle. Journal of neurology, neurosurgery, and psychiatry. 1950;13:159–177. doi: 10.1136/jnnp.13.3.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, McHugh J, Tork C, Shelley B, Hayes A, Bellantuono I, Aebischer P, Svendsen CN. Direct muscle delivery of GDNF with human mesenchymal stem cells improves motor neuron survival and function in a rat model of familial ALS. Molecular therapy: the journal of the American Society of Gene Therapy. 2008;16:2002–2010. doi: 10.1038/mt.2008.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talakad NS, Pradhan C, Nalini A, Thennarasu K, Raju TR. Assessment of pulmonary function in amyotrophic lateral sclerosis. The Indian journal of chest diseases & allied sciences. 2009;51:87–91. [PubMed] [Google Scholar]
- Tankersley CG, Haenggeli C, Rothstein JD. Respiratory impairment in a mouse model of amyotrophic lateral sclerosis. J Appl Physiol. 2007;102:926–932. doi: 10.1152/japplphysiol.00193.2006. [DOI] [PubMed] [Google Scholar]
- Trumbower RD, Jayaraman A, Mitchell GS, Rymer WZ. Exposure to acute intermittent hypoxia augments somatic motor function in humans with incomplete spinal cord injury. Neurorehabilitation and neural repair. 2012;26:163–172. doi: 10.1177/1545968311412055. [DOI] [PubMed] [Google Scholar]
- Tsuchiya K, Takahashi M, Shiotsu H, Akiyama H, Haga C, Watabiki S, Taki K, Nakano I, Ikeda K. Sporadic amyotrophic lateral sclerosis with circumscribed temporal atrophy: a report of an autopsy case without dementia and with ubiquitinated intraneuronal inclusions. Neuropathology: official journal of the Japanese Society of Neuropathology. 2002;22:308–316. doi: 10.1046/j.1440-1789.2002.00451.x. [DOI] [PubMed] [Google Scholar]
- Turner MR, Kiernan MC. Does interneuronal dysfunction contribute to neurodegeneration in amyotrophic lateral sclerosis? Amyotrophic lateral sclerosis: official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. 2012;13:245–250. doi: 10.3109/17482968.2011.636050. [DOI] [PubMed] [Google Scholar]
- Valdez G, Tapia JC, Lichtman JW, Fox MA, Sanes JR. Shared resistance to aging and ALS in neuromuscular junctions of specific muscles. PloS one. 2012;7:e34640. doi: 10.1371/journal.pone.0034640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitacca M, Clini E, Facchetti D, Pagani M, Poloni M, Porta R, Ambrosino N. Breathing pattern and respiratory mechanics in patients with amyotrophic lateral sclerosis. The European respiratory journal: official journal of the European Society for Clinical Respiratory Physiology. 1997;10:1614–1621. doi: 10.1183/09031936.97.10071614. [DOI] [PubMed] [Google Scholar]
- Waldrop TG, Eldridge FL, Millhorn DE. Prolonged post-stimulus inhibition of breathing following stimulation of afferents from muscle. Respiration physiology. 1982;50:239–254. doi: 10.1016/0034-5687(82)90021-4. [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Xu YS, Zheng JY, Zhang S, Fan DS. Upper trapezius electromyography aids in the early diagnosis of bulbar involvement in amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis: official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. 2011;12:345–348. doi: 10.3109/17482968.2011.582647. [DOI] [PubMed] [Google Scholar]